Back to Journals » Blood and Lymphatic Cancer: Targets and Therapy » Volume 6
Waldenström macroglobulinemia: biology, genetics, and therapy
Received 3 May 2016
Accepted for publication 21 June 2016
Published 26 July 2016 Volume 2016:6 Pages 49—58
DOI https://doi.org/10.2147/BLCTT.S84157
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor David Dingli
Jonas Paludo,1,2 Stephen M Ansell,1
1Division of Hematology, 2Division of Medical Oncology, Mayo Clinic, Rochester, MN, USA
Abstract: Waldenström macroglobulinemia (WM) is a distinct clinicopathologic entity characterized by the presence of a lymphoplasmacytic lymphoma, a non-Hodgkin lymphoma, and IgM monoclonal gammopathy. WM is an indolent, uncommon malignancy mostly affecting the elderly. Patient outcomes have modestly improved since the introduction of rituximab to conventional cytotoxic chemotherapy more than 20 years ago. However, the pivotal discovery of the somatic MYD88 L265P mutation, harbored by most patients with WM, and the somatic CXCR4 WHIM mutations, similar to germline CXCR4 mutations seen in the warts, hypogammaglobulinemia, immunodeficiency, and myelokathexis (WHIM) syndrome, present in approximately one-third of patients with WM, has fundamentally changed our understanding of this disease and expanded the potential therapeutic targets. Within this new paradigm, ibrutinib emerged as a promising new drug. Ibrutinib targets Bruton’s tyrosine kinase, a downstream protein in the B-cell receptor pathway that is overactivated by the MYD88 L265P mutation. A seminal Phase II trial of ibrutinib in previously treated WM patients showed impressive response rates and confirmed the effects of MYD88 L265P and CXCR4 WHIM mutations in response to therapy. Ibrutinib is the first and only US Food and Drug Administration–approved drug specifically for the treatment of WM. However, before ibrutinib can be established as the standard of care for WM, long-term data regarding efficacy and safety are required. Further research to address ibrutinib resistance and cost-effectiveness is also imperative before ibrutinib can gain widespread acceptance. This review will cover the present pathophysiologic understanding of WM in light of the recent MYD88 and CXCR4 discovery, as well as current and emergent treatment regimens with focus on ibrutinib.
Keywords: Bruton’s tyrosine kinase, ibrutinib, lymphoplasmacytic lymphoma, Waldenström macroglobulinemia
Introduction
Waldenström macroglobulinemia (WM) was described by Jan Waldenström, a Swedish physician, in 1944.1 The World Health Organization categorizes WM as a subset of low-grade non-Hodgkin lymphoma, characterized by the presence of IgM monoclonal gammopathy and infiltration of the bone marrow by lymphoplasmacytic lymphoma (LPL).2
The clonal lymphoplasmacytic cells in WM usually express global B-cell markers, such as CD19, CD20, CD22, and CD79a, but do not express CD5 (as in mantle cell lymphoma [MCL] and chronic lymphocytic leukemia [CLL]), CD10 (as in follicular lymphoma), or CD56 (as in the majority of multiple myeloma [MM] cases).3–5 In rare instances, WM lymphocytes may express CD5, but not as much as in MCL or CLL.6 The phenotype of LPL cells suggests that they are in a late stage of B-cell differentiation, which probably resembles an IgM+ B-cell that has undergone somatic hypermutation but not isotype class switching.7 Table 1 compares the features of various IgM monoclonal gammopathies.
 | Table 1 Comparison of IgM monoclonal gammopathiesa Notes: aThe table lists a few important differential characteristics of IgM monoclonal gammopathies. IgM paraprotein can be present in virtually all B-cell lymphoproliferative disorders; bMayo Clinic criteria requires at least 10% of bone marrow involvement by LPL, whereas the Second International Workshop on WM (IWWM-2) eliminated the requirement for a minimum amount of marrow involvement. cConstitutional symptoms: hepatosplenomegaly, lymphadenopathy, anemia, hyperviscosity, solid organ involvement, rarely lytic lesions. dCRAB features (hyperCalcemia, Renal failure, Anemia, and Bone lesions); eOrgans typically involved are kidneys, heart, nerves, tongue, gastrointestinal tract, and liver. Patients with IgM amyloid light-chain (AL) amyloidosis have higher frequency of pulmonary, lymph node, peripheral nerve involvement, and lower cardiac involvement. Concentration of free light chain tends to be lower than in non-IgM AL amyloidosis; fPrimarily involves spleen; lymphadenopathy is rare. Reprinted from Blood Rev, 29(5), Kapoor P, Paludo J, Vallumsetla N, Greipp PR, Waldenstrom macroglobulinemia: what a hematologist needs to know, 301–319,15 copyright 2015, with permission from Elsevier. Abbreviations: DLBCL, diffuse large B-cell lymphoma; IgM, immunoglobulin M; LPL, lymphoplasmacytic lymphoma; MGUS, monoclonal gammopathy of undetermined significance; N/A, not applicable; PC, plasma cell; WM, Waldenström macroglobulinemia; IgH, immunoglobulin heavy locus. |
Features required for the diagnosis of WM include 1) presence of an IgM monoclonal gammopathy of any size in serum, 2) bone marrow biopsy findings showing LPL infiltration, and 3) exclusion of other lymphoproliferative disorders, including CLL and MCL. The Mayo Clinic criteria for WM diagnosis require at least 10% marrow involvement by clonal lymphoplasmacytic cells in asymptomatic patients,8 whereas the Second International Workshop on WM eliminated the requirement for a minimum amount of marrow involvement.6
Clinical features
WM represents only 2% of all hematologic malignancies, with an age-adjusted incidence of 3.8 cases per million persons per year.9 WM is predominantly a disease of elderly persons, with a median age at diagnosis ranging from 63 to 73 years in various studies, and a 95-fold higher incidence among octogenarians than among those <50 years.10 Although the estimated median disease-specific survival is 11 years,11 the median overall survival (OS) of Waldenström’s patients is approximately 7 years and reflects associated comorbidities seen in these elderly population.12 Young patients with WM (<50 years) have a longer median OS (13–14.8 years).12,13
Approximately 25% of patients with WM are asymptomatic at the time of diagnosis, but symptoms will develop in up to 70% of them within 10 years of diagnosis.14 The initial presentation of WM is usually nonspecific constitutional symptoms such as fatigue, weight loss, fever, and night sweats. Ultimately, symptoms develop secondary to hematopoietic tissue infiltration by LPL, IgM paraprotein deposition, and/or IgM autoimmune activity.15 The most common clinical features are lymphadenopathy, thrombocytopenia, splenomegaly, anemia, bleeding due to hyperviscosity, and peripheral neuropathy. Table 2 shows the clinical features of WM.
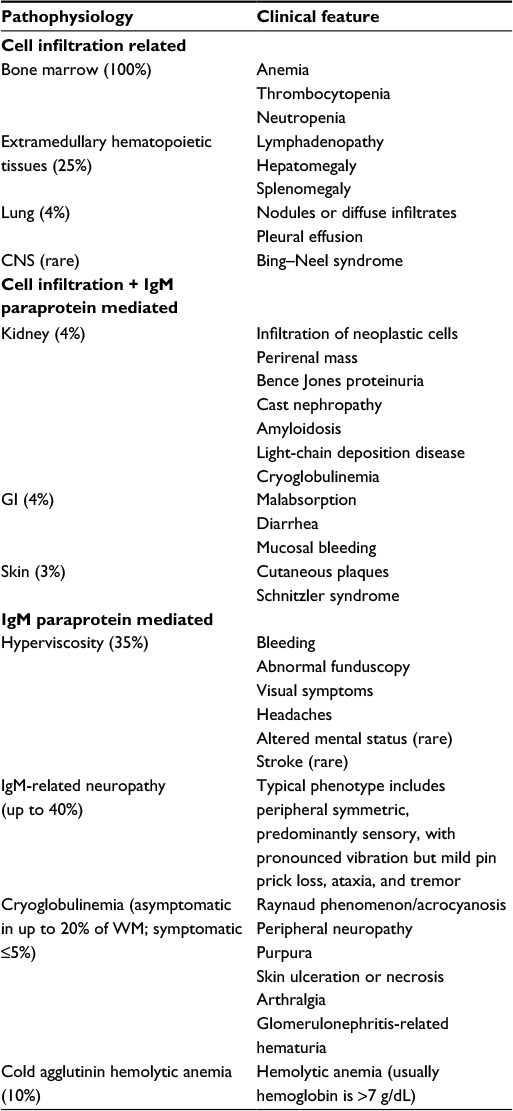 | Table 2 Characteristics of IgM or direct Waldenström macroglobulinemia cell infiltration–related morbidities Notes: Reprinted from Blood Rev, 29(5), Kapoor P, Paludo J, Vallumsetla N, Greipp PR, Waldenstrom macroglobulinemia: what a hematologist needs to know, 301–319,15 copyright 2015, with permission from Elsevier. Abbreviations: CNS, central nervous system; GI, gastrointestinal; WM, Waldenström macroglobulinemia; IgM, immunoglobulin M. |
In patients with a history of IgM monoclonal gammopathy of undetermined significance (MGUS), a known precursor of WM, the risk of WM is increased by 46-fold.16 IgM MGUS patients carrying the MYD88 L265P mutation have higher risk of progressing to WM than those with wild-type MYD88 (odds ratio 4.7, 95% confidence interval [CI] 0.8–48.7).17 WM has been known to show familial clustering; about 20% of patients with WM have at least one first degree relative with WM or other B-cell disorder.18 There is abundant evidence to support a genetic predisposition in familial cases of WM.19 In a genome-wide analysis of families at high risk for WM, the authors found strong evidence for involved genes on 1q and 4q, and evidence suggestive of gene linkage on 3q and 6q.20 The pattern of affected patients in familial WM, which is usually seen in multiple generations and equally distributed between men and women, favors autosomal dominant or codominant inheritance.
Patients with WM are at increased risk for secondary malignancies. The most common sex-adjusted malignancies are prostate (2%–9.4%), breast (0.9%–8%), nonmelanoma skin cancer (7.1%), melanoma (2.2%), and lung (1.4%–2.1%).19,21 The cumulative incidence of solid cancer was shown to be 6%–9.5% at 5 years, 11%–16.1% at 10 years, and 17% at 15 years in one study.21,22 Hematologic malignancies, mainly diffuse large B-cell lymphoma and myelodysplastic syndrome/acute myeloid leukemia, were seen in 3.7%–5% of patients with WM, but these were mostly in patients previously treated with alkylating agents and nucleoside analogs.21,22 Second malignancies were seen more frequently in sporadic WM than in familial WM. Interestingly, five times more cases of lung cancer were seen in familial WM, and two times more cases of prostate cancer were seen in sporadic WM.19 First-degree relatives of patients with WM are at significantly increased risk for MGUS, WM, or other non-Hodgkin lymphoma.23
Genetic mutations associated with WM
WM has been associated with numerous chromosomal abnormalities and somatic mutations, but no causal relationship has been shown. An abnormality on cytogenetic analysis or fluorescence in situ hybridization is found in about half of patients with WM; the most common are 6q deletion (22%–46%), 13q14 deletion (13%–15%), trisomy 18 (11%–23%), trisomy 4 (4%–12%), and p53 deletion (4%–23%). A rearrangement of the IgH genes as part of a translocation is uncommon in WM, unlike in MM.24,25 Deletion of 6q is common but not specific to WM (it is found in other B-cell disorders), but trisomy 4 is unique to WM and occasionally is the only abnormality seen in these patients.26 No cytogenetic abnormality has been shown to be diagnostic, predictive, or prognostic in WM patients.
The WM tumoral clone shows wide morphologic heterogeneity, from clonal B lymphocytes (BLs) to clonal plasma cells (PCs), and includes a lymphoplasmacytic population. WM-PC shows a pattern of cytogenetic abnormalities and gene rearrangement similar to that in WM-BL, which supports the idea that the WM clonal PC results from an incomplete maturation process of a WM clonal BL. Furthermore, the gene expression profile of PCs and BLs in WM shows complex clones that are similar to their counterparts in CLL and MM, but each still represents a singular entity. Interestingly, the PAX5 gene is significantly upregulated in WM-PC compared with MM-PC; PAX5 expression must be repressed to allow final PC differentiation. The BLIMP1 gene, which is also needed for differentiation from BLs to PCs, is downregulated in WM-PC.27 This evidence indicates that WM clonal cells are in a later stage of B-cell differentiation into PCs that express IgM, but have not undergone final isotype class switching. In contrast to MM-PC, WM clonal PCs do not express DKK1 and FRZB, which cause bone disease in MM, and may explain the lack of bone lesions in WM.28
The protein MYD88 is an adaptor in the Toll-like receptor and interleukin-1 receptor signaling pathway. The pivotal discovery of the L265P mutation in MYD88 considerably improved our understanding of WM pathogenesis.29 The gain-of-function MYD88 L265P mutation strongly promotes WM cell growth and survival through downstream activation of the above-mentioned pathways, with the transcription of NF-κB.15 The MYD88 L265P mutation has been detected in 86% of sporadic WM cases and in all familial WM cases.29 Even though the MYD88 L265P mutation was seen uniformly in familial WM, a recent study did not support the presence of germline MYD88 mutations in familial WM or IgM MGUS.30
The MYD88 L265P mutation is not exclusive to WM. It can be identified in up to 87% of patients with IgM MGUS and in smaller proportions of patients with activated B-cell type diffuse large B-cell lymphomas (29%), mucosa-associated lymphoid tissue lymphoma (9%), splenic marginal zone lymphoma (10%–21%), and CLL (4%–10%).31–33 The role of MYD88 L265P mutation in the development of WM is not clear; it may be a driver mutation in IgM MGUS transformation to WM or may only indicate a subset of patients with IgM MGUS who are predisposed to WM.4,29
In addition to the L265P mutation of MYD88, mutation of the C-X-C chemokine receptor type 4 (CXCR4) was found in 29% of patients with WM.34 The somatic CXCR4 mutations found in WM are similar to germline mutations in CXCR4 described in the rare warts, hypogammaglobulinemia, immunodeficiency, and myelokathexis (WHIM) syndrome. CXCR4 WHIM mutations lead to permanent activation of CXCR4 by its ligand stromal cell-derived factor 1/C-X-C motif chemokine ligand 12 (CXCL12), stimulate the proliferation, migration, and homing of WM cells to bone marrow niches, and therefore promote cell survival.35 Even though CXCR4 WHIM mutations have been associated with more aggressive disease features, such as higher IgM levels and bone marrow involvement, their presence has not affected the OS.36 The CXCR4 WHIM mutations seem to mediate drug resistance and have been shown to affect response to novel treatments, such as Bruton’s tyrosine kinase (BTK), mammalian target of rapamycin, and phosphoinositide 3-kinase (PI3K) inhibitors, as described below.37,38
Risk stratification and prognosis of WM
In patients with IgM MGUS or smoldering WM, which by definition have no disease-related symptoms, the OS is similar to that in the general population of comparable age and sex,39 but these patients carry an increased risk of disease progression to WM. The overall average risk of progression in patients with IgM MGUS is approximately 1.5% per year,40 whereas patients with smoldering WM have an average risk of progression to symptomatic WM of 12% per year for the first 5 years, after which the risk decreases to rates similar to those for IgM MGUS.14 The rate of progression is affected by IgM levels, hemoglobin levels, and degree of bone marrow involvement by LPL. Most patients with smoldering WM with more than 50% marrow involvement will progress to WM within 5 years.14
The International Prognostic Scoring System for WM (IPSSWM) is widely used for stratification of patients with WM. Five adverse parameters are included in the IPSSWM score (age >65 years, hemoglobin ≤11.5 g/dL, platelets ≤100×109/L, β2-microglobulin >3 mg/L, and IgM level >7 g/dL); age >65 years is assigned two points because of its higher prognostic value, and the others are assigned one point. The total score at initiation of therapy stratifies patients into low- (score ≤1), intermediate- (score 2), or high-risk (score ≥3) categories. Five-year survival rates are 87%, 68%, and 36% for low, intermediate, and high risk, respectively.41 The IPSSWM score was independently validated by a large Greek study.42 Increased lactate dehydrogenase level (>250 IU/L) has been shown to further stratify patients with a high-risk IPSSWM score – an increased value was associated with a median OS of 37 months, compared with 104 months for a lower value.42 The presence of MYD88 wild-type was also associated with an increased risk of death in WM patients (hazard ratio 10.6, 95% CI 2.4–46.2).43 In the era of BTK inhibitors, further prognostication can be derived from MYD88 and CXCR4 mutation status as predictors of response to therapy, as discussed below.
Current and emerging treatments
Since WM remains an incurable disease, the aims of treatment are to relieve symptoms and decrease the risk of end-organ damage. Treatment is usually reserved for symptomatic patients and those with severe cytopenias (hemoglobin <10 -g/dL or platelet count <100×109/L). When possible, clinical trials should be considered for patients with newly diagnosed WM or in the relapsed/refractory setting. Table 3 shows a summary of selected clinical trials in WM.
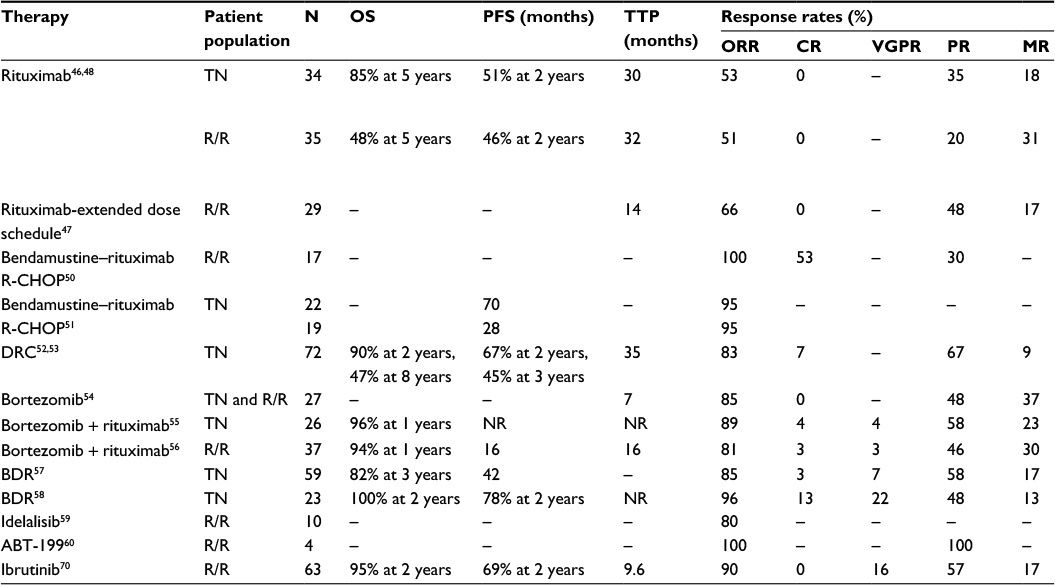 | Table 3 Selected trials in Waldenström macroglobulinemia Abbreviations: BDR, bortezomib, dexamethasone, rituximab; CR, complete response; DRC, dexamethasone, rituximab, cyclophosphamide; m, months; MR, minor response; ORR, overall response rate; OS, overall survival; PFS, progression free survival; PR, partial response; R-CHOP, rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; R/R, relapsed or refractory; TN, treatment naïve; TTP, time to progression; VGPR, very good partial response; NR, not reached. |
Rituximab, a monoclonal anti-CD20 antibody, is widely used in the treatment of WM, either in combination with other agents or as monotherapy. A recent population-based study showed single-agent rituximab as the frontline choice in approximately 55% of WM patients.44 The low toxicity profile and substantial response rates as a single agent make rituximab monotherapy suitable for minimally symptomatic WM patients or those with IgM-related neuropathy, hemolytic anemia, or mild cytopenias. An IgM “flare” is seen in approximately half of patients after starting rituximab.45 Physicians should be aware of this phenomenon as it does not indicate treatment failure, but could aggravate IgM-mediated symptoms/hyperviscosity. Plasma exchange should be considered in patients with symptomatic hyperviscosity before starting rituximab. Single-agent rituximab has consistently shown overall response rates (ORRs) from 50% to 60% in treatment-naïve and relapsed/refractory patients, with a median progression-free survival (PFS) approaching 2 years.46–48 The role of rituximab maintenance therapy was assessed in a retrospective study of 238 rituximab-naïve WM patients of whom 68 patients received maintenance therapy. Rituximab maintenance therapy was associated not only with improved median PFS (56 months vs 29 months, P=0.0001) and OS (not reached vs 116 months, P=0.0095) but also with increased infectious complications.49 The superiority of rituximab maintenance therapy has not been evaluated in randomized, prospective studies, therefore not recommended outside clinical trials.
The combination of bendamustine, an alkylating agent, with rituximab in treatment-naïve patients with WM demonstrated an ORR of 100% and complete response (CR) of 53% in a Phase II trial.50 The primary adverse effect was myelosuppression (leukopenia Grade ≥3: 16%; thrombocytopenia Grade ≥3: 3%). A Phase III trial comparing bendamustine plus rituximab (BR) with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) in patients with indolent lymphomas, including 41 with WM, showed a similar ORR (95%) in both treatment arms. BR, however, had a significantly longer median PFS (70 months vs 28 months for R-CHOP; hazard ratio 0.33; P=0.003), fewer relapses and adverse events.51 Because of these results, BR became a suitable first-line regimen for WM, particularly for patients with bulky disease in need of rapid disease control.
A Phase II trial of dexamethasone, rituximab, and cyclophosphamide (DRC) in treatment-naïve patients with WM demonstrated an ORR of 83%, including a CR rate of 7%. DRC was well tolerated, with a less than 9% rate of Grade 3 or greater adverse reactions.52 The recently reported long-term outcomes of this Phase II trial showed a median PFS of approximately 3 years after DRC and an estimated 10-year OS of 53%.53 DRC remains an active, well-tolerated, and non-stem-cell toxic treatment option in WM.
Proteasome inhibitors have also been shown to be effective against WM. Bortezomib has been studied as monotherapy, in combination with rituximab, and as a triplet regimen with rituximab and dexamethasone. Single-agent bortezomib in treatment of relapsed/refractory WM achieved an ORR of 85%. Responses occurred at a median of 1.4 months, but no CRs were seen.54 The addition of rituximab to weekly bortezomib has been investigated in the treatment-naïve55 and relapsed/refractory56 settings, showing ORRs of 89% and 81%, as well as CR rates of 4% and 3%, respectively. Median PFS was 16 months in the relapsed/refractory setting. Neuropathy Grade 3 or higher was seen in 5% of patients with this weekly regimen of bortezomib and rituximab. The combination of bortezomib/rituximab plus dexamethasone in treatment-naïve WM patients has yielded ORRs ranging from 85% to 96%, with CR rates as high as 13%. This triplet combination showed a median PFS of 42 months and 7%–30% incidence of Grade 3 or higher neuropathy.57,58
A recent Phase II trial of idelalisib, a selective oral inhibitor of PI3Kδ, in patients with indolent lymphomas (including ten with WM) refractory to alkylating agents and rituximab, showed promising results. The subset of WM patients achieved an ORR of 80% with this oral agent. The most common Grade 3 or higher adverse events were neutropenia (27%), increased liver enzyme values (21%), and diarrhea (13%).59
A Phase I study showed activity of ABT-199, a small-molecule BCL-2 inhibitor recently named venetoclax, in patients with relapsed/refractory non-Hodgkin lymphoma, including four patients with WM who achieved a partial response.60 Preclinical data showed a synergistic effect of ABT-199 with ibrutinib or idelalisib in WM cell models, independent of CXCR4 mutation status61 and may support its further investigation in combination strategies.
The role of autologous stem cell transplant (ASCT) in WM is not well established, but it was deemed safe and effective in multiple retrospective studies.62–65 The largest cohort of patients with WM who underwent ASCT included 158 patients and showed a nonrelapse mortality of only 3.8% at 1 year.64 ASCT achieved an ORR of approximately 95% and a major response rate (MRR) of 78%–80%, with an estimated 5-year OS of approximately 69% and 5-year time-to-next-therapy of 48%.64,65 Survival was affected by number of prior treatment regimens and disease chemosensitivity at the time of transplant.64 Potential ASCT-eligible patients with otherwise long life expectancy could have stem cells harvested and stored early in the disease course before they are exposed to multiple stem-cell toxic treatments.13,64 In contrast to ASCT, allogeneic stem cell transplant is not recommended in patients with WM outside clinical trials due to the prohibitively high transplant-related mortality (23%–44% at 1 year) in this indolent disease.63,66
Comparative safety, efficacy, and tolerability of ibrutinib
BTK, a downstream cytoplasmic enzyme in the MYD88 signaling pathway, has been targeted by several drugs, most notably ibrutinib. The gain-of-function MYD88 L265P mutation, almost ubiquitous in WM patients, is thought to be the main driver of BTK activation in WM, which leads to increased cell survival and proliferation through NF-κB pathways.67
Ibrutinib is an oral, small-molecule, selective, irreversible inhibitor of BTK that triggers apoptosis in WM cells with the MYD88 L265P mutation.68 A Phase I trial of ibrutinib in 56 patients with relapsed/refractory B-cell lymphomas, including four patients with WM, showed an objective response in 60% of the patients and a median PFS of 16 months.69 The encouraging results and acceptable tolerability led to a pivotal Phase II trial of ibrutinib in previously treated WM patients.70 A total of 63 consecutive patients received daily ibrutinib, 420 mg, until disease progression or unacceptable adverse effects. The ORR was 90%, with a median treatment duration of 19 months.
Patients with WM who had the MYD88 L265P mutation and wild-type CXCR4 had the most benefit from ibrutinib, with an ORR of 100% and MRR of 91%; patients with MYD88 L265P and CXCR4 WHIM had less benefit (ORR of 86% and MRR of 62%), and even less benefit was seen for those with wild-type MYD88 and CXCR4 (ORR of 71% and MRR of 28%). These findings highlight BTK’s activity in proliferation of WM cells and survival in the mutated MYD88 L265P signaling pathway and show the drug resistance, possibly the result of bone marrow homing of WM cells, increased by CXCR4 WHIM mutation.70 A later review by the same authors showed that all patients with wild-type MYD88 who had achieved a major response actually harbored a mutation in the MYD88 gene that was not captured by allele-specific polymerase chain reaction analysis for MYD88 L265P. The updated ORR and MRR for WM patients with wild-type MYD88 and CXCR4 were 60% and 0%, respectively.71
The initial rapid and profound decrease in IgM levels (median time to response, 4 weeks) was not matched by a decrease in bone marrow involvement, which suggests that ibrutinib affects IgM secretion rather than cell killing. Improvement in lymphadenopathy (68%), splenomegaly (57%), and peripheral neuropathy (55%) was seen in most patients after initiation of ibrutinib. All patients in whom treatment was initiated for hyperviscosity symptoms required no additional plasma exchange after two cycles of therapy.70
Overall, ibrutinib was well tolerated. Grade 3 or higher neutropenia and thrombocytopenia were seen in 14% and 13% of the patients, respectively. Most of these patients received three or more prior types of therapy. Grade 2 or higher bleeding complications were seen in 6% of the patients, all of which were associated with concomitant use of fish oil supplements. Atrial fibrillation was reported in 5% of patients, all of whom had a history of paroxysmal atrial fibrillation, which resolved after ibrutinib was withdrawn.70 The effect of BTK on the platelet glycoprotein IV/collagen interaction, a critical component of platelet aggregation, is well known and was first identified in patients with X-linked agammaglobulinemia, which is caused by BTK deficiency.72 The bleeding associated with ibrutinib is therefore thought to be secondary to its inhibitory effect on BTK and collagen-mediated platelet aggregation.73 Platelet aggregation normalizes after 2–3 days of ibrutinib cessation.74
Preliminary results of a multicenter Phase III trial demonstrated activity of ibrutinib in rituximab-refractory WM patients. With an ORR rate of 84%, single-agent ibrutinib shows activity in heavily pretreated rituximab-refractory WM patients. Grade 3 or higher adverse events rate was similar to prior studies of single-agent ibrutinib.75
Despite the promising results, CR was not achieved in any patients with WM with the use of ibrutinib.70 Primary and secondary cases of resistance to ibrutinib have also been described. Although ibrutinib resistance is uncommon, its true incidence is unknown given the relatively short follow-up of available clinical trials. Multiple mechanisms have been proposed to explain acquired resistance to BTK inhibitors, including mutations in BTK that interfere with drug binding and alternative pathways that bypass BTK entirely.76 The BTK C481S mutation has been associated with secondary ibrutinib resistance in patients with CLL and MCL. At the structural level, the BTK C481S mutation disrupts covalent binding and allows for reversible, instead of irreversible, binding of ibrutinib by BTK.77,78 Primary resistance to ibrutinib has also been associated with sustained PI3K–Akt activity, rather than BTK activity, in some patients with MCL.78
In WM, primary ibrutinib resistance is seen in patients harboring CXCR4 WHIM mutations. WHIM-like mutations reduce CXCR4 internalization and allow for sustained enzymatic activity of Akt (protein kinase B) and extracellular signal-regulated kinase, with subsequent increased cell survival, migration, and bone marrow homing.79,34 Plerixafor, an anti-CXCR4 agent used for many years as a stem-cell mobilizing agent, has been shown to effectively inhibit the CXCR4–CXCL12 pathway. Its use in patients with WM carrying a CXCR4 WHIM mutation has been proposed as a way to mobilize WM cells from the prosurvival stem cell niche in bone marrow, thus making them more amenable to cytotoxic chemotherapy.80 Several other CXCR4 inhibitors are currently under investigation.81
The use of BTK inhibitors together with CXCR4 antagonists as a way to decrease intrinsic drug resistance in patients with a CXCR4 WHIM mutation is yet to be studied. This combination has the theoretical potential to show even better response rates in WM patients. Furthermore, the patient’s MYD88 and CXCR4 mutation status could be used for a more individualized treatment selection in patients with WM.
Conclusion
Despite expansion of the therapeutic options in recent years, WM remains an incurable disease. Ibrutinib has become the first drug approved by the US Food and Drug Administration specifically for WM, owing to its impressive clinical efficacy, easy route of administration, and favorable toxicity profile reported in a recent Phase II trial.70 Ibrutinib has also been approved in Europe, Canada, Japan, and is under review in Mexico. Ibrutinib as single agent has activity in WM comparable to that with combination therapies; however, no head-to-head trials have been conducted. Furthermore, the data that led to ibrutinib approval by the US Food and Drug Administration are based only on response rates. Long-term data are required to ascertain end points such as PFS, OS, resistance rates, long-term complications, and quality of life before a definitive conclusion on the role of ibrutinib in WM treatment can be made.
Nonetheless, ibrutinib has inaugurated a new era in the treatment of WM. Clinical trials of ibrutinib in combination with other established or novel agents are under way, and results could further solidify the role of ibrutinib in WM. Additionally, a myriad of strategies has been proposed to overcome ibrutinib resistance, such as ABT-199 (BLC2-ihibitor),82 RP6530 (dual PI3Kδ/γ inhibitor),83 and plerixafor (CXCR4 inhibitor).84 The treatment of WM continues to evolve and expand rapidly, with the potential to affect the course of this disease.
Disclosure
The authors report no conflicts of interest in this work.
References
Waldenstrom J. Incipient myelomatosis or essential hyperglobulinemia with fibrinogenopenia – a new syndrome? Acta Med Scand. 1944;117(3–4):216–247. | ||
Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood. 2011;117(19):5019–5032. | ||
Lin P, Medeiros LJ. Lymphoplasmacytic lymphoma/Waldenstrom macroglobulinemia: an evolving concept. Adv Anat Pathol. 2005;12(5):246–255. | ||
Kapoor P, Paludo J, Ansell SM. Waldenstrom macroglobulinemia: familial predisposition and the role of genomics in prognosis and treatment selection. Curr Treat Options Oncol. 2016;17(3):16. | ||
San Miguel JF, Vidriales MB, Ocio E, et al. Immunophenotypic analysis of Waldenstrom’s macroglobulinemia. Semin Oncol. 2003;30(2):187–195. | ||
Owen RG, Treon SP, Al-Katib A, et al. Clinicopathological definition of Waldenstrom’s macroglobulinemia: consensus panel recommendations from the Second International Workshop on Waldenstrom’s Macroglobulinemia. Semin Oncol. 2003;30(2):110–115. | ||
Kriangkum J, Taylor BJ, Treon SP, Mant MJ, Belch AR, Pilarski LM. Clonotypic IgM V/D/J sequence analysis in Waldenstrom macroglobulinemia suggests an unusual B-cell origin and an expansion of polyclonal B cells in peripheral blood. Blood. 2004;104(7):2134–2142. | ||
Ansell SM, Kyle RA, Reeder CB, et al. Diagnosis and management of Waldenstrom macroglobulinemia: Mayo stratification of macroglobulinemia and risk-adapted therapy (mSMART) guidelines. Mayo Clin Proc. 2010;85(9):824–833. | ||
Wang H, Chen Y, Li F, et al. Temporal and geographic variations of Waldenstrom macroglobulinemia incidence. Cancer. 2012;118(15):3793–3800. | ||
Wang H, Chen Y, Li F, et al. Temporal and geographic variations of Waldenstrom macroglobulinemia incidence: a large population-based study. Cancer. 2012;118(15):3793–3800. | ||
Ghobrial IM, Fonseca R, Gertz MA, et al. Prognostic model for disease-specific and overall mortality in newly diagnosed symptomatic patients with Waldenstrom macroglobulinaemia. Br J Haematol. 2006;133(2):158–164. | ||
Castillo JJ, Olszewski AJ, Kanan S, Meid K, Hunter ZR, Treon SP. Overall survival and competing risks of death in patients with Waldenstrom macroglobulinaemia: an analysis of the Surveillance, Epidemiology and End Results database. Br J Haematol. 2015;169(1):81–89. | ||
Vallumsetla N, Paludo J, Ansell SM, et al. Outcomes of young patients with Waldenstrom macroglobulinemia (WM). J Clin Oncol (Meeting Abstracts). 2014;32(15_suppl):Abstract number 8609. | ||
Kyle RA, Benson JT, Larson DR, et al. Progression in smoldering Waldenström macroglobulinemia: long-term results. Blood. 2012;119(19):4462–4466. | ||
Kapoor P, Paludo J, Vallumsetla N, Greipp PR. Waldenstrom macroglobulinemia: what a hematologist needs to know. Blood Rev. 2015;29(5):301–319. | ||
Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med. 2002;346(8):564–569. | ||
Varettoni M, Arcaini L, Zibellini S, et al. Prevalence and clinical significance of the MYD88 (L265P) somatic mutation in Waldenstrom’s macroglobulinemia and related lymphoid neoplasms. Blood. 2013;121(13):2522–2528. | ||
Treon SP, Hunter ZR, Aggarwal A, et al. Characterization of familial Waldenstrom’s macroglobulinemia. Ann Oncol. 2006;17(3):488–494. | ||
Hanzis C, Ojha RP, Hunter Z, et al. Associated malignancies in patients with Waldenstrom’s macroglobulinemia and their kin. Clin Lymphoma Myeloma Leuk. 2011;11(1):88–92. | ||
McMaster ML, Goldin LR, Bai Y, et al. Genomewide linkage screen for Waldenström macroglobulinemia susceptibility loci in high-risk families. Am J Hum Genet. 2006;79(4):695–701. | ||
Castillo JJ, Olszewski AJ, Hunter ZR, Kanan S, Meid K, Treon SP. Incidence of secondary malignancies among patients with Waldenstrom macroglobulinemia: an analysis of the SEER database. Cancer. 2015;121(13):2230–2236. | ||
Morra E, Varettoni M, Tedeschi A, et al. Associated cancers in Waldenstrom macroglobulinemia: clues for common genetic predisposition. Clin Lymphoma Myeloma Leuk. 2013;13(6):700–703. | ||
Kristinsson SY, Landgren O. What causes Waldenstrom’s macroglobulinemia: genetic or immune-related factors, or a combination? Clin Lymphoma Myeloma Leuk. 2011;11(1):85–87. | ||
Nguyen-Khac F, Lambert J, Chapiro E, et al. Chromosomal aberrations and their prognostic value in a series of 174 untreated patients with Waldenstrom’s macroglobulinemia. Haematologica. 2013;98(4):649–654. | ||
Braggio E, Philipsborn C, Novak A, Hodge L, Ansell S, Fonseca R. Molecular pathogenesis of Waldenstrom’s macroglobulinemia. Haematologica. 2012;97(9):1281–1290. | ||
Braggio E, Fonseca R. Genomic abnormalities of Waldenstrom macroglobulinemia and related low-grade B-cell lymphomas. Clin Lymphoma Myeloma Leuk. 2013;13(2):198–201. | ||
Shapiro-Shelef M, Calame K. Regulation of plasma-cell development. Nat Rev Immunol. 2005;5(3):230–242. | ||
Gutierrez NC, Ocio EM, de las Rivas J, et al. Gene expression profiling of B lymphocytes and plasma cells from Waldenstrom’s macroglobulinemia: comparison with expression patterns of the same cell counterparts from chronic lymphocytic leukemia, multiple myeloma and normal individuals. Leukemia. 2007;21(3):541–549. | ||
Treon SP, Xu L, Yang G, et al. MYD88 L265P somatic mutation in Waldenstrom’s macroglobulinemia. N Engl J Med. 2012;367(9):826–833. | ||
Pertesi M, Galia P, Nazaret N, et al. Rare circulating cells in familial Waldenstrom macroglobulinemia displaying the MYD88 L265P mutation are enriched by Epstein-Barr virus immortalization. PLoS ONE. 2015;10(9):e0136505. | ||
Xu L, Hunter ZR, Yang G, et al. MYD88 L265P in Waldenstrom macroglobulinemia, immunoglobulin M monoclonal gammopathy, and other B-cell lymphoproliferative disorders using conventional and quantitative allele-specific polymerase chain reaction. Blood. 2013;121(11):2051–2058. | ||
Ngo VN, Young RM, Schmitz R, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470(7332):115–119. | ||
Jimenez C, Sebastian E, Chillon MC, et al. MYD88 L265P is a marker highly characteristic of, but not restricted to, Waldenstrom’s macroglobulinemia. Leukemia. 2013;27(8):1722–1728. | ||
Hunter ZR, Xu L, Yang G, et al. The genomic landscape of Waldenstrom macroglobulinemia is characterized by highly recurring MYD88 and WHIM-like CXCR4 mutations, and small somatic deletions associated with B-cell lymphomagenesis. Blood. 2014;123(11):1637–1646. | ||
Ngo HT, Leleu X, Lee J, et al. SDF-1/CXCR4 and VLA-4 interaction regulates homing in Waldenstrom macroglobulinemia. Blood. 2008;112(1):150–158. | ||
Cao Y, Hunter ZR, Liu X, et al. CXCR4 WHIM-like frameshift and nonsense mutations promote ibrutinib resistance but do not supplant MYD88(L265P)-directed survival signalling in Waldenstrom macroglobulinaemia cells. Br J Haematol. 2015;168(5):701–707. | ||
Roccaro AM, Sacco A, Jimenez C, et al. C1013G/CXCR4 acts as a driver mutation of tumor progression and modulator of drug resistance in lymphoplasmacytic lymphoma. Blood. 2014;123(26):4120–4131. | ||
Treon S, Tripsas C, Meid K, et al. Ibrutinib in previously treated patients with Waldenstrom’s macroglobulinemia is highly active, produces durable responses, and is impacted by MYD88 and CXCR4 mutation status. Haematologica. 2015;100:311. | ||
Vajdic CM, Landgren O, McMaster ML, et al. Medical history, lifestyle, family history, and occupational risk factors for lymphoplasmacytic lymphoma/Waldenstrom’s macroglobulinemia: the InterLymph Non-Hodgkin Lymphoma Subtypes Project. J Natl Cancer Inst Monogr. 2014;2014(48):87–97. | ||
Kyle RA, Therneau TM, Rajkumar SV, et al. Long-term follow-up of IgM monoclonal gammopathy of undetermined significance. Blood. 2003;102(10):3759–3764. | ||
Morel P, Duhamel A, Gobbi P, et al. International prognostic scoring system for Waldenstrom macroglobulinemia. Blood. 2009;113(18):4163–4170. | ||
Kastritis E, Kyrtsonis MC, Hadjiharissi E, et al. Validation of the International Prognostic Scoring System (IPSS) for Waldenstrom’s macroglobulinemia (WM) and the importance of serum lactate dehydrogenase (LDH). Leuk Res. 2010;34(10):1340–1343. | ||
Treon SP, Cao Y, Xu L, Yang G, Liu X, Hunter ZR. Somatic mutations in MYD88 and CXCR4 are determinants of clinical presentation and overall survival in Waldenstrom macroglobulinemia. Blood. 2014;123(18):2791–2796. | ||
Olszewski AJ, Fallah J, Eaton CB, Treon SP, Castillo JJ. The evolution of management and survival outcomes of Waldenström macroglobulinemia (WM) in the United States (US). Blood. 2015;126(23):882–882. | ||
Ghobrial IM, Fonseca R, Greipp PR, et al. Initial immunoglobulin M ‘flare’ after rituximab therapy in patients diagnosed with Waldenstrom macroglobulinemia. Cancer. 2004;101(11):2593–2598. | ||
Gertz MA, Abonour R, Heffner LT, Greipp PR, Uno H, Rajkumar SV. Clinical value of minor responses after 4 doses of rituximab in Waldenstrom macroglobulinaemia: a follow-up of the Eastern Cooperative Oncology Group E3A98 trial. Br J Haematol. 2009;147(5):677–680. | ||
Treon SP, Emmanouilides C, Kimby E, et al. Extended rituximab therapy in Waldenstrom’s macroglobulinemia. Ann Oncol. 2005;16(1):132–138. | ||
Gertz MA, Rue M, Blood E, Kaminer LS, Vesole DH, Greipp PR. Multicenter phase 2 trial of rituximab for Waldenstrom macroglobulinemia (WM): an Eastern Cooperative Oncology Group Study (E3A98). Leuk Lymphoma. 2004;45(10):2047–2055. | ||
Treon SP, Hanzis C, Manning RJ, et al. Maintenance rituximab is associated with improved clinical outcome in rituximab naïve patients with Waldenstrom macroglobulinaemia who respond to a rituximab-containing regimen. Br J Haematol. 2011;154(3):357–362. | ||
Rummel MJ, Al-Batran SE, Kim SZ, et al. Bendamustine plus rituximab is effective and has a favorable toxicity profile in the treatment of mantle cell and low-grade non-Hodgkin’s lymphoma. J Clin Oncol. 2005;23(15):3383–3389. | ||
Rummel MJ, Niederle N, Maschmeyer G, et al. Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet. 2013;381(9873):1203–1210. | ||
Dimopoulos MA, Anagnostopoulos A, Kyrtsonis MC, et al. Primary treatment of Waldenstrom macroglobulinemia with dexamethasone, rituximab, and cyclophosphamide. J Clin Oncol. 2007;25(22):3344–3349. | ||
Kastritis E, Gavriatopoulou M, Kyrtsonis MC, et al. Dexamethasone, rituximab, and cyclophosphamide as primary treatment of Waldenstrom macroglobulinemia: final analysis of a phase 2 study. Blood. 2015;126(11):1392–1394. | ||
Treon SP, Hunter ZR, Matous J, et al. Multicenter clinical trial of bortezomib in relapsed/refractory Waldenstrom’s macroglobulinemia: results of WMCTG Trial 03-248. Clin Cancer Res. 2007;13(11):3320–3325. | ||
Ghobrial IM, Xie W, Padmanabhan S, et al. Phase II trial of weekly bortezomib in combination with rituximab in untreated patients with Waldenstrom macroglobulinemia. Am J Hematol. 2010;85(9):670–674. | ||
Ghobrial IM, Hong F, Padmanabhan S, et al. Phase II trial of weekly bortezomib in combination with rituximab in relapsed or relapsed and refractory Waldenstrom macroglobulinemia. J Clin Oncol. 2010;28(8):1422–1428. | ||
Dimopoulos MA, Garcia-Sanz R, Gavriatopoulou M, et al. Primary therapy of Waldenstrom macroglobulinemia (WM) with weekly bortezomib, low-dose dexamethasone, and rituximab (BDR): long-term results of a phase 2 study of the European Myeloma Network (EMN). Blood. 2013;122(19):3276–3282. | ||
Treon SP, Ioakimidis L, Soumerai JD, et al. Primary therapy of Waldenstrom macroglobulinemia with bortezomib, dexamethasone, and rituximab: WMCTG clinical trial 05-180. J Clin Oncol. 2009;27(23):3830–3835. | ||
Gopal AK, Kahl BS, de Vos S, et al. PI3Kdelta inhibition by idelalisib in patients with relapsed indolent lymphoma. N Engl J Med. 2014;370(11):1008–1018. | ||
Gerecitano JF, Roberts AW, Seymour JF, et al. A Phase 1 study of venetoclax (ABT-199 / GDC-0199) monotherapy in patients with relapsed/refractory non-Hodgkin lymphoma. Blood. 2015;126(23):254. | ||
Cao Y, Yang G, Hunter ZR, et al. The BCL2 antagonist ABT-199 triggers apoptosis, and augments ibrutinib and idelalisib mediated cytotoxicity in CXCR4 wild-type and CXCR4 WHIM mutated Waldenstrom macroglobulinaemia cells. Br J Haematol. 2015;170(1):134–138. | ||
Anagnostopoulos A, Hari PN, Perez WS, et al. Autologous or allogeneic stem cell transplantation in patients with Waldenstrom’s macroglobulinemia. Biol Blood Marrow Transplant. 2006;12(8):845–854. | ||
Gilleece MH, Pearce R, Linch DC, et al. The outcome of haemopoietic stem cell transplantation in the treatment of lymphoplasmacytic lymphoma in the UK: a British Society Bone Marrow Transplantation study. Hematology. 2008;13(2):119–127. | ||
Kyriakou C, Canals C, Sibon D, et al. High-dose therapy and autologous stem-cell transplantation in Waldenstrom macroglobulinemia: the Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. J Clin Oncol. 2010;28(13):2227–2232. | ||
Paludo J, Gertz M, Ansell S, et al. Impact of day-100 response post autologous stem cell transplantation (ASCT) in Waldenstrom macroglobulinemia (WM). Biol Blood Marrow Transplant. 2016;22(3):S130. | ||
Kyriakou C, Canals C, Cornelissen JJ, et al. Allogeneic stem-cell transplantation in patients with Waldenstrom macroglobulinemia: report from the Lymphoma Working Party of the European Group for Blood and Marrow Transplantation. J Clin Oncol. 2010;28(33):4926–4934. | ||
Buggy JJ, Elias L. Bruton tyrosine kinase (BTK) and its role in B-cell malignancy. Int Rev Immunol. 2012;31(2):119–132. | ||
Cameron F, Sanford M. Ibrutinib: first global approval. Drugs. 2014;74(2):263–271. | ||
Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol. 2013;31(1):88–94. | ||
Treon SP, Tripsas CK, Meid K, et al. Ibrutinib in previously treated Waldenstrom’s macroglobulinemia. N Engl J Med. 2015;372(15):1430–1440. | ||
Treon SP, Xu L, Hunter Z. MYD88 mutations and response to ibrutinib in Waldenstrom’s macroglobulinemia. N Engl J Med. 2015;373(6):584–586. | ||
Quek LS, Bolen J, Watson SP. A role for Bruton’s tyrosine kinase (Btk) in platelet activation by collagen. Curr Biol. 1998;8(20):1137–1140. | ||
Kamel S, Horton L, Ysebaert L, et al. Ibrutinib inhibits collagen-mediated but not ADP-mediated platelet aggregation. Leukemia. 2015;29(4):783–787. | ||
Ysebaert L, Levade M, Cedric G, et al. Elucidation of mild bleeding disorders reported under ibrutinib (Imbruvica(R)) therapy: implications for optimal clinical management. Blood. 2014;124(21):3296–3296. | ||
Dimopoulos MA, Trotman J, Tedeschi A, et al. Ibrutinib therapy in rituximab-refractory patients with Waldenström’s macroglobulinemia: initial results from an international, multicenter, open-label phase 3 substudy (iNNOVATETM). Blood. 2015;126(23):2745–2745. | ||
Zhang SQ, Smith SM, Zhang SY, Lynn Wang Y. Mechanisms of ibrutinib resistance in chronic lymphocytic leukaemia and non-Hodgkin lymphoma. Br J Haematol. 2015;170(4):445–456. | ||
Furman RR, Cheng S, Lu P, et al. Ibrutinib resistance in chronic lymphocytic leukemia. N Engl J Med. 2014;370(24):2352–2354. | ||
Chiron D, Di Liberto M, Martin P, et al. Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed by longitudinal functional genomics in mantle cell lymphoma. Cancer Discov. 2014;4(9):1022–1035. | ||
Cao Y, Hunter ZR, Liu X, et al. The WHIM-like CXCR4(S338X) somatic mutation activates AKT and ERK, and promotes resistance to ibrutinib and other agents used in the treatment of Waldenstrom’s macroglobulinemia. Leukemia. 2015;29(1):169–176. | ||
Beider K, Darash-Yahana M, Blaier O, et al. Combination of imatinib with CXCR4 antagonist BKT140 overcomes the protective effect of stroma and targets CML in vitro and in vivo. Mol Cancer Ther. 2014;13(5):1155–1169. | ||
Scala S. Molecular pathways: targeting the CXCR4-CXCL12 axis-untapped potential in the tumor microenvironment. Clin Cancer Res. 2015;21(19):4278–4285. | ||
Cao Y, Yang G, Hunter ZR, et al. The BCL2 antagonist ABT-199 triggers apoptosis, and augments ibrutinib and idelalisib mediated cytotoxicity in CXCR4 wild-type and CXCR4 WHIM mutated Waldenstrom macroglobulinaemia cells. Br J Haematol. 2015;170(1):134–138. | ||
Vakkalanka S, Nyayapathy S, Viswanadha S. Addition of RP6530, a dual PI3Kδ/γ inhibitor, overcomes ibrutinib resistance in DLBCL cells in vitro. Blood. 2014;124(21):4497–4497. | ||
McDermott DH, Liu Q, Velez D, et al. A phase 1 clinical trial of long-term, low-dose treatment of WHIM syndrome with the CXCR4 antagonist plerixafor. Blood. 2014;123(15):2308–2316. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.