")
Back to Journals » The Application of Clinical Genetics » Volume 13
Voretigene Neparvovec and Gene Therapy for Leber’s Congenital Amaurosis: Review of Evidence to Date
Authors Padhy SK, Takkar B, Narayanan R , Venkatesh P, Jalali S
Received 6 August 2020
Accepted for publication 6 November 2020
Published 25 November 2020 Volume 2020:13 Pages 179—208
DOI https://doi.org/10.2147/TACG.S230720
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Srikanta Kumar Padhy,1 Brijesh Takkar,2,3 Raja Narayanan,2 Pradeep Venkatesh,4 Subhadra Jalali2,5
1Vitreoretina and Uveitis Services, L V Prasad Eye Institute, Mithu Tulsi Chanrai Campus, Bhubaneswar, India; 2Srimati Kanuri Santhamma Center for Vitreoretinal Diseases, Kallam Anji Reddy Campus, L.V. Prasad Eye Institute, Hyderabad, India; 3Center of Excellence for Rare Eye Diseases, Kallam Anji Reddy Campus, L V Prasad Eye Institute, Hyderabad, India; 4Dr RP Centre for Ophthalmic Sciences, All India Institute of Medical Sciences, New Delhi, India; 5Jasti V. Ramanamma Childrens’ Eye Care Centre, Kallam Anji Reddy Campus, L V Prasad Eye Institute, Hyderabad, India
Correspondence: Brijesh Takkar
Srimati Kanuri Santhamma Center for Vitreoretinal Diseases, Kallam Anji Reddy Campus, L.V. Prasad Eye Institute, L V Prasad Marg, Banjara Hills, Hyderabad 500034, Telangana, India
Tel +91-9868092215
Fax +91-40-23548271
Email [email protected]
Abstract: Gene therapy has now evolved as the upcoming modality for management of many disorders, both inheritable and non-inheritable. Knowledge of genetics pertaining to a disease has therefore become paramount for physicians across most specialities. Inheritable retinal dystrophies (IRDs) are notorious for progressive and relentless vision loss, frequently culminating in complete blindness in both eyes. Leber’s congenital amaurosis (LCA) is a typical example of an IRD that manifests very early in childhood. Research in gene therapy has led to the development and approval of voretigene neparvovec (VN) for use in patients of LCA with a deficient biallelic RPE65 gene. The procedure involves delivery of a recombinant virus vector that carries the RPE65 gene in the subretinal space. This comprehensive review reports the evidence thus far in support of gene therapy for LCA. We explore and compare the various gene targets including but not limited to RPE65, and discuss the choice of vector and method for ocular delivery. The review details the evolution of gene therapy with VN in a phased manner, concluding with the challenges that lie ahead for its translation for use in communities that differ much both genetically and economically.
Keywords: gene therapy, voretigene neparvovec, Luxturna, Leber’s congenital amaurosis, retinal dystrophy
Introduction
Inherited retinal dystrophies (IRD) include a diverse group of bilateral and often progressive retinal diseases which cause functional loss of vision and may subsequently progress to legal blindness. IRDs encompass both non-syndromic conditions like Leber’s congenital amaurosis (LCA), retinitis pigmentosa (RP), etc., as well as syndromic conditions involving multiple organs in addition to the eye such as Usher syndrome, Bardet-Biedel syndrome, etc.1 Before the detection of gene defects, IRDs were classified according to clinical features, age of onset, and Mendelian inheritance pattern. With the leap forward in the field of genetics and subsequent identification of causative genes and mutations over the past three decades, a much better understanding of disease pathologies in IRD is now present. The current approach is now towards classifying IRDs on the basis of genotype and not phenotype.2
In 1869, German ophthalmologist Dr Theodor Leber first described LCA.3,4 He mentioned the progressive course of retinal degeneration in his initial description of young children with LCA, but later noted non-progressive variants too.5,6 Treatment of both RP and LCA has long been controversial, with poor outcomes marked by relentless disease progression and not uncommonly, though not universally, an eventual complete blindness. Varied therapies had been attempted, including pharmacological, surgical, electrical, ozone therapy, and stem cells, before LCA became the first eye disorder successfully treated with gene therapy.
Anderson et al7 launched human gene therapy in 1990 by treating a child suffering from severe combined immunodeficiency. The triumph of that gene therapy was short-lived, but acknowledged by significant media coverage. The gene therapy did not completely cure the patient’s condition and she had to continue previous forms of therapy. As further clinical trials of gene therapy were launched, the potential risks involved with such were revealed, including immune mechanisms driven mortality and neoplasia,8 necessitating more research and refinement. Based on the cell targeted, gene therapy can be classified into
Somatic cell gene therapy (SCGT); in which therapeutic genes are transferred to somatic cells which are not inherited to future generations and germline gene therapy (GGT); here therapeutic genes are introduced into germ cells, therefore the changes are heritable and passes to later generations.9–11
The vehicles used for transfer of desired genes to target cells are called vectors, broadly classified into viral and non-viral types. The essential features of an ideal vector comprise cellular specificity (induction of only targeted cells), large cloning specificity, low immunogenicity, and the feasibility of being produced at a high titre.12 Different viruses have been used as vectors in humans, and adeno-associated viruses (AAV) are the most common and most widely used.12–15 Gene therapy for LCA with voretigene neparvovec (VN) (LUXTURNA, Novartis AG, Basel, Switzerland) is SCGT based and dependent on an AAV vector, and is the first such therapy approved for IRD by the US-FDA in 2017 following publications from Phase III trials (NCT00999609). It was developed by Spark Therapeutics as AAV2-hRPE65v2.16 Other similar developments in earlier phases of research include rAAV2-CB-hRPE65 by Applied Genetic Technologies Corporation (5 year results of Phase I/II trial published, NCT00749957), and AAV2/5-OPTIRPE65 by MeiraGTx (Phase I/II study completed in December 2018, NCT02781480). The latter two are not yet approved for human use.
With approval of VN, the targets are now shifting to other human diseases – from rare ones with no available therapy to common disorders with many other therapeutic options. Gene therapies not only signify a successful alternative in potentially curative medicine, but they pose the next affordability dilemma too. For example, gene therapy with VN currently costs around US$ 850,000. However, in some situations a one-time gene therapy may be useful in the longer run.17 This review focuses on the evolution and evidence of gene therapy for LCA especially with VN, bringing out the challenges physicians might face towards its application to population at large.
Exploring Gene Targets in LCA
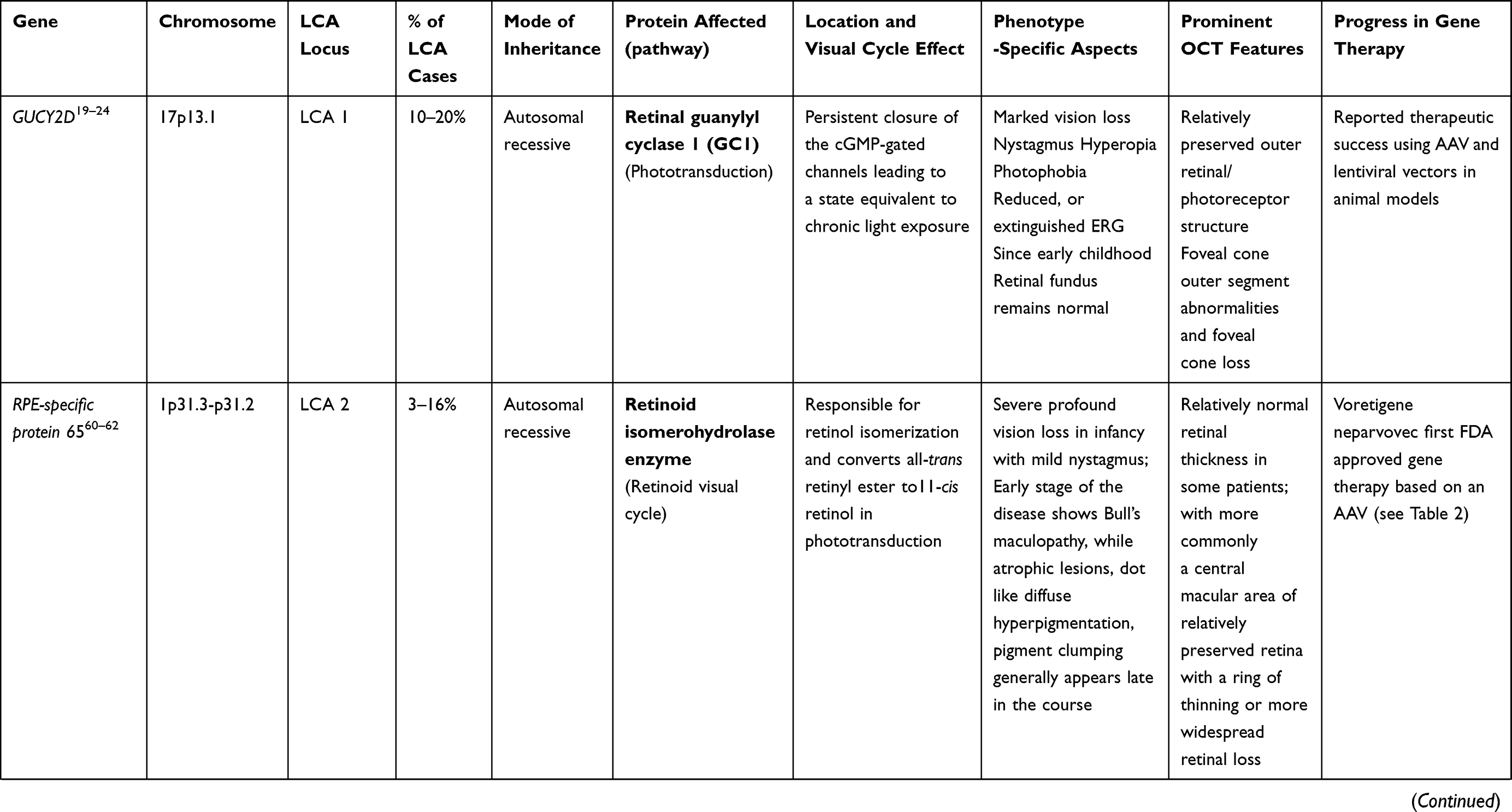
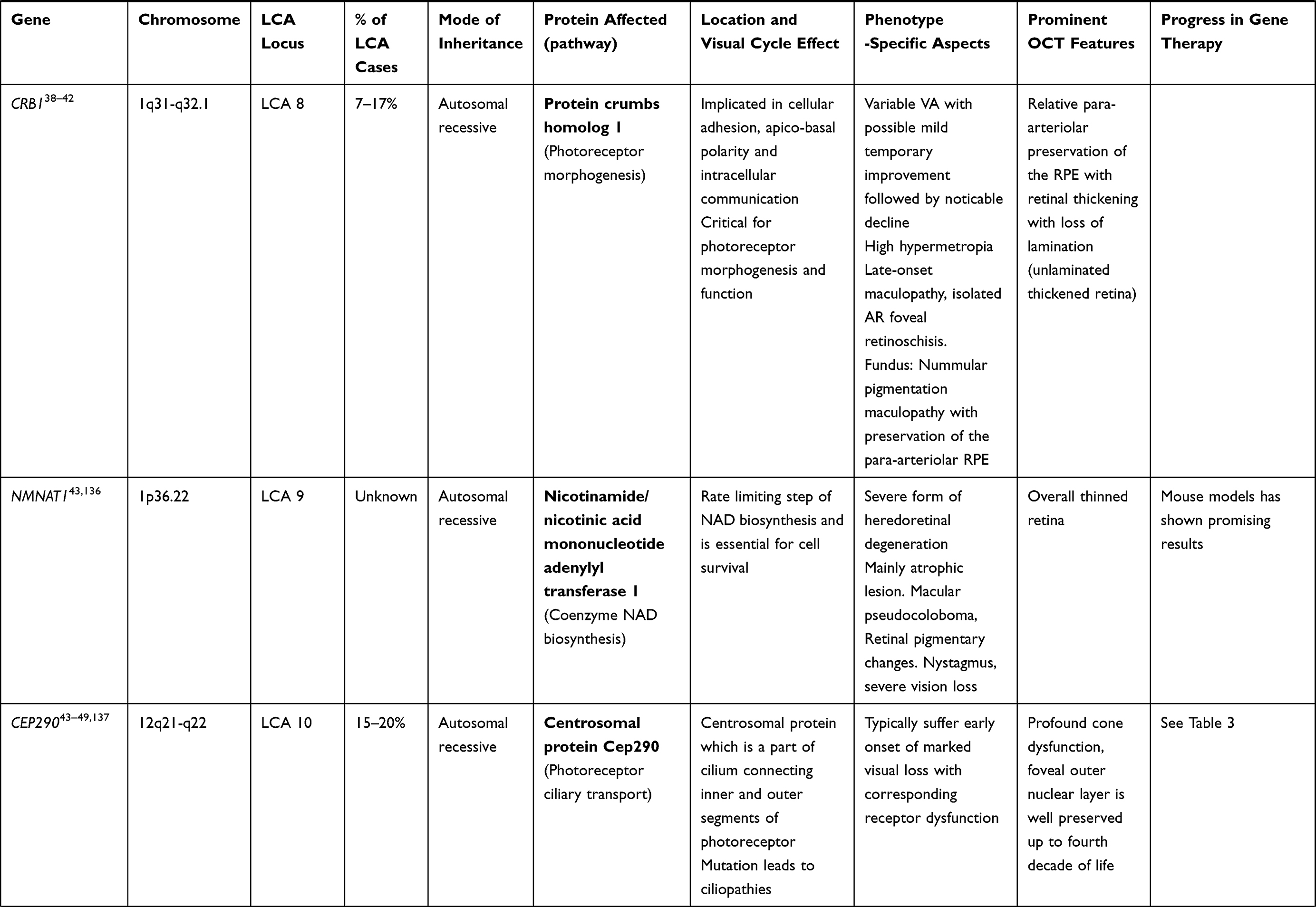
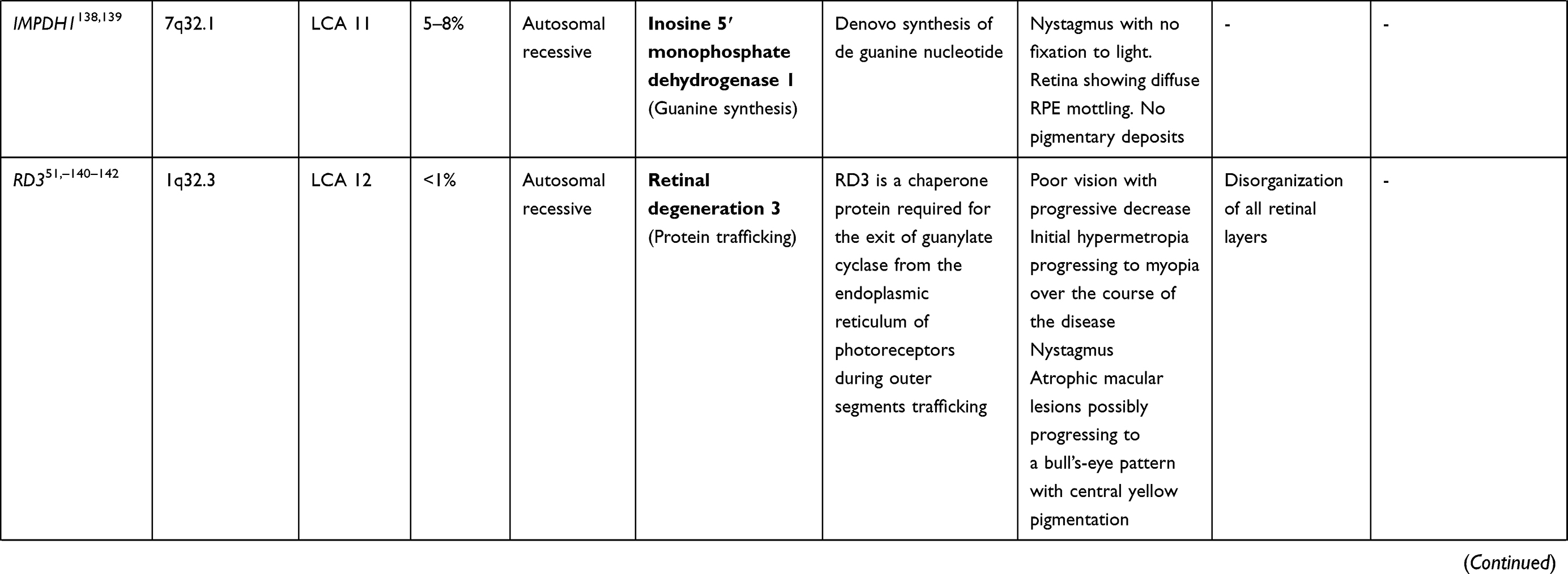
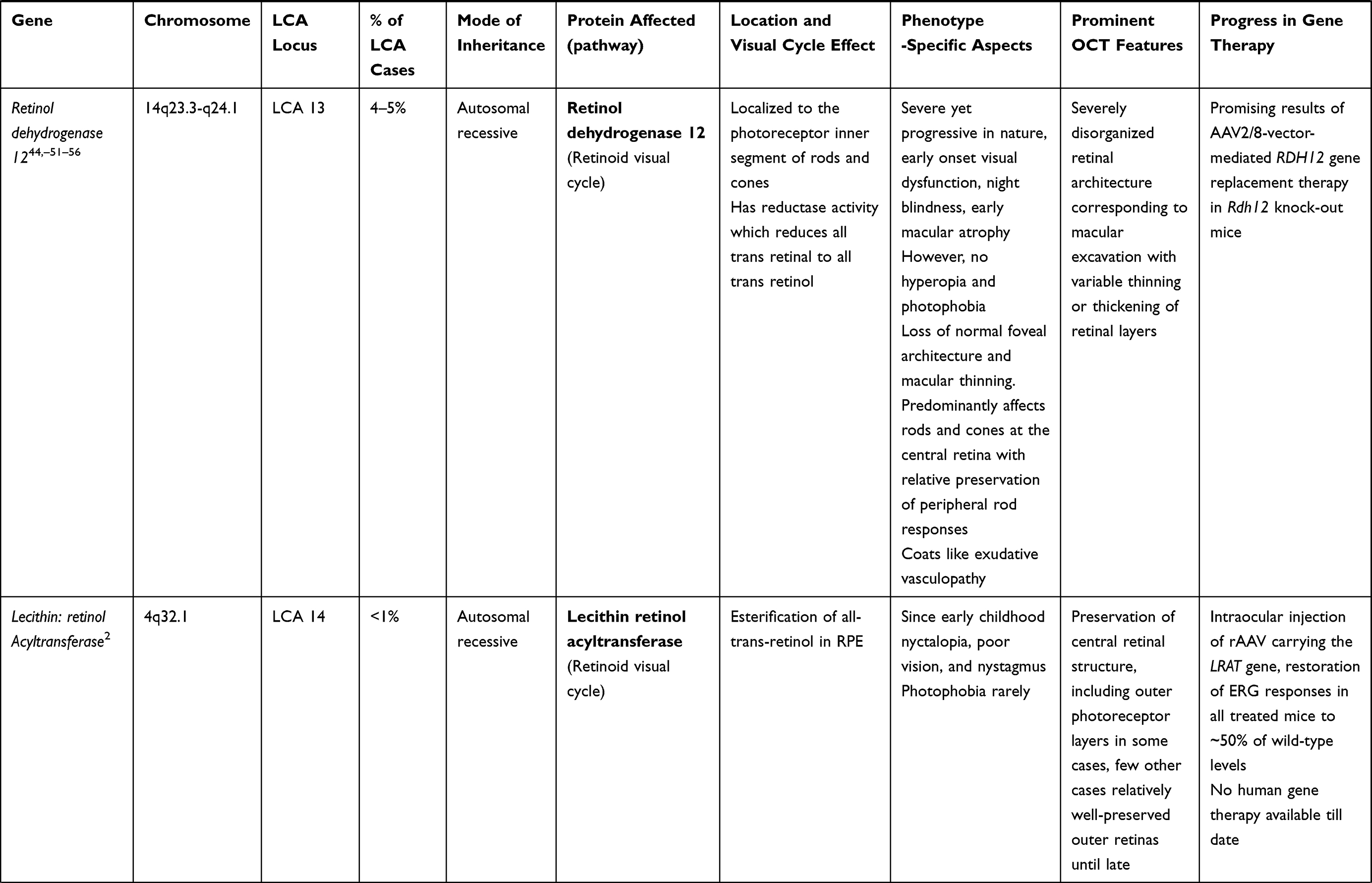
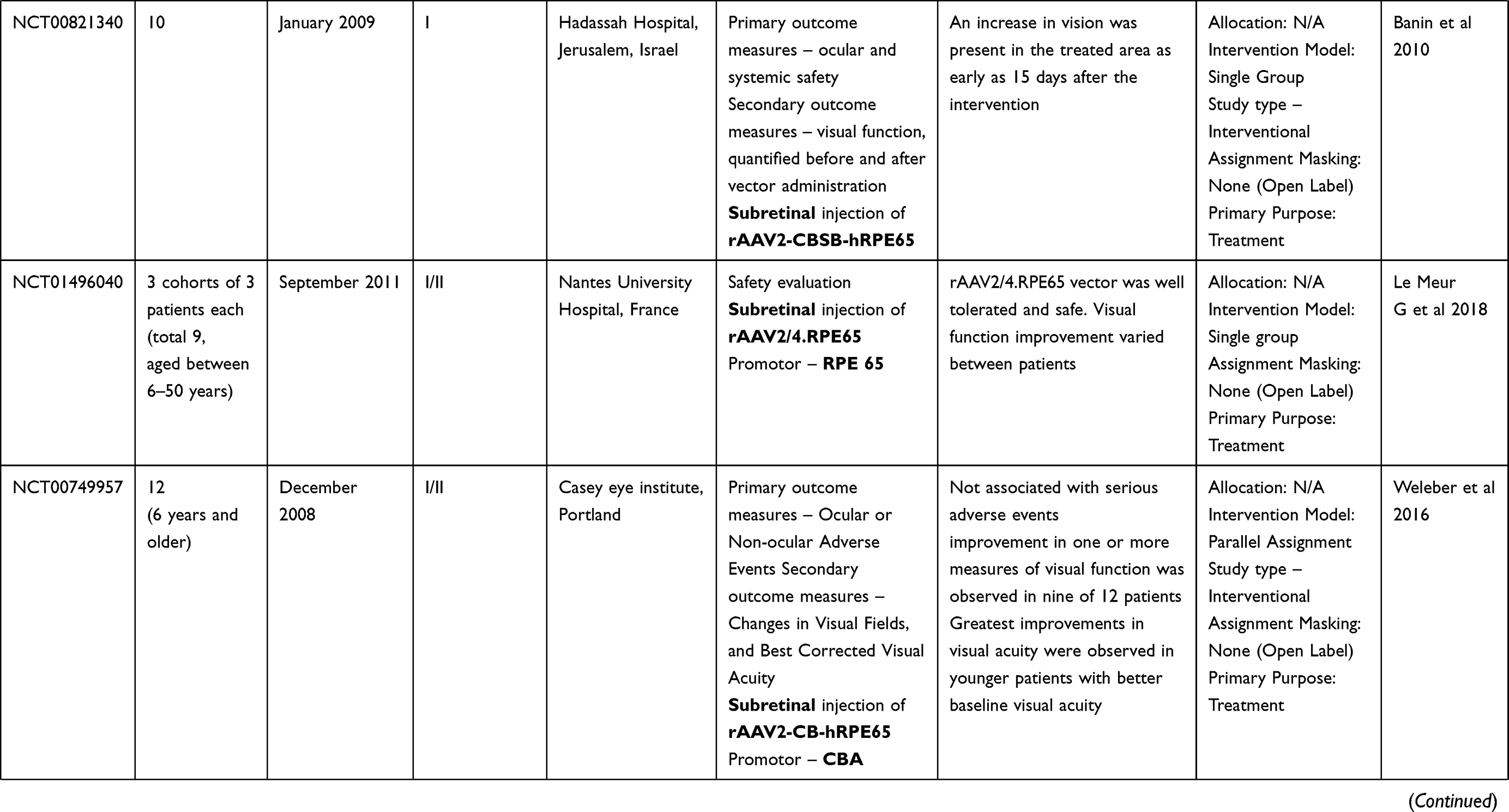
LCA is a genetically heterogenous ocular condition where the primary mode of inheritance is autosomal recessive, though a few dominant traits are also reported. There are as many as 28 genes implicated in the pathogenesis of the disease. Mutation in these genes account for almost 75% of the cases.18 The more common and well characterized ones, where attempts at gene therapy have been made, are discussed below (Table 1).
 |
 |
 |
 |
 |
 |
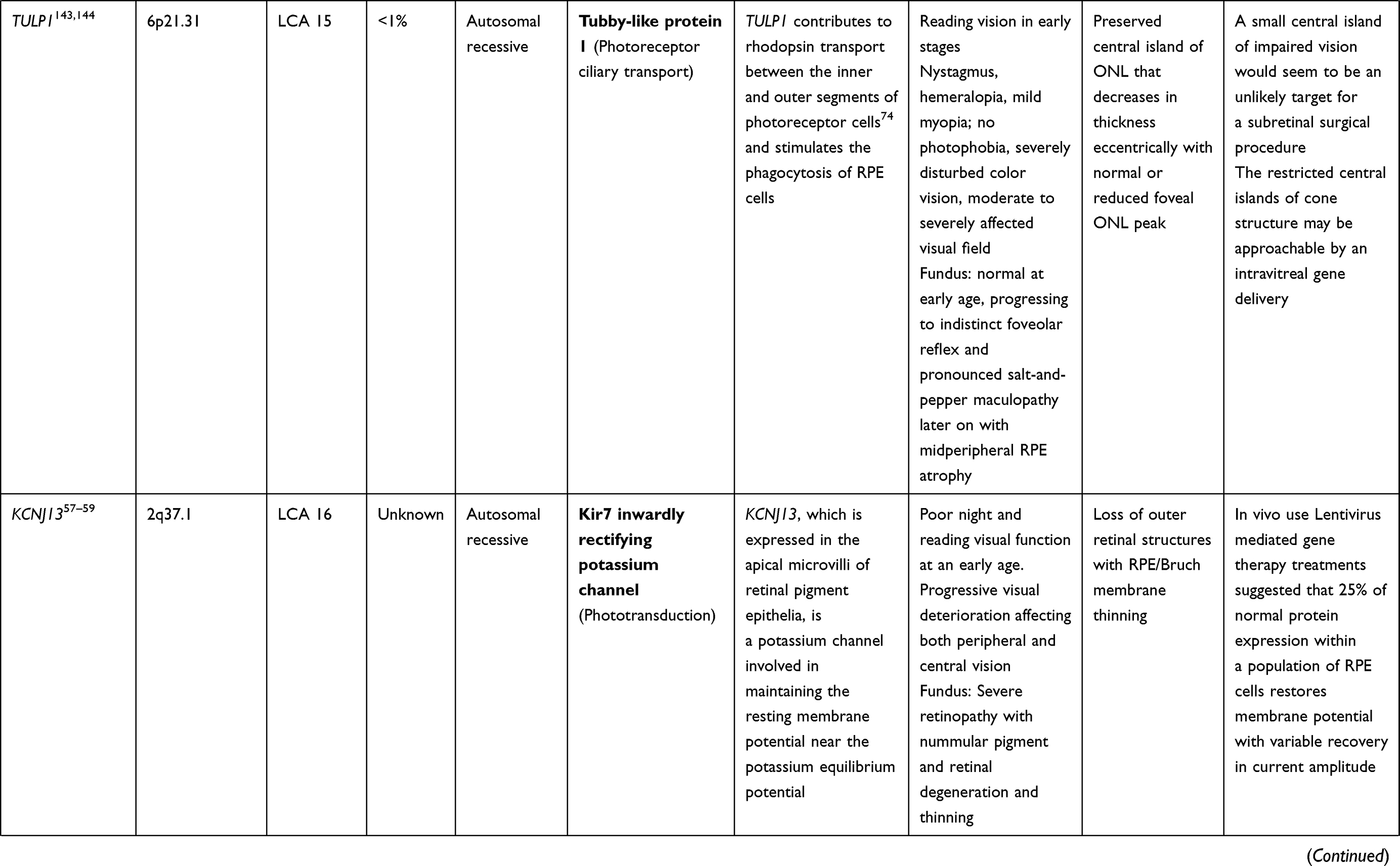 |
Table 1 Targets of Gene Therapy for Leber’s Congenital Amaurosis |
 |
 |
 |
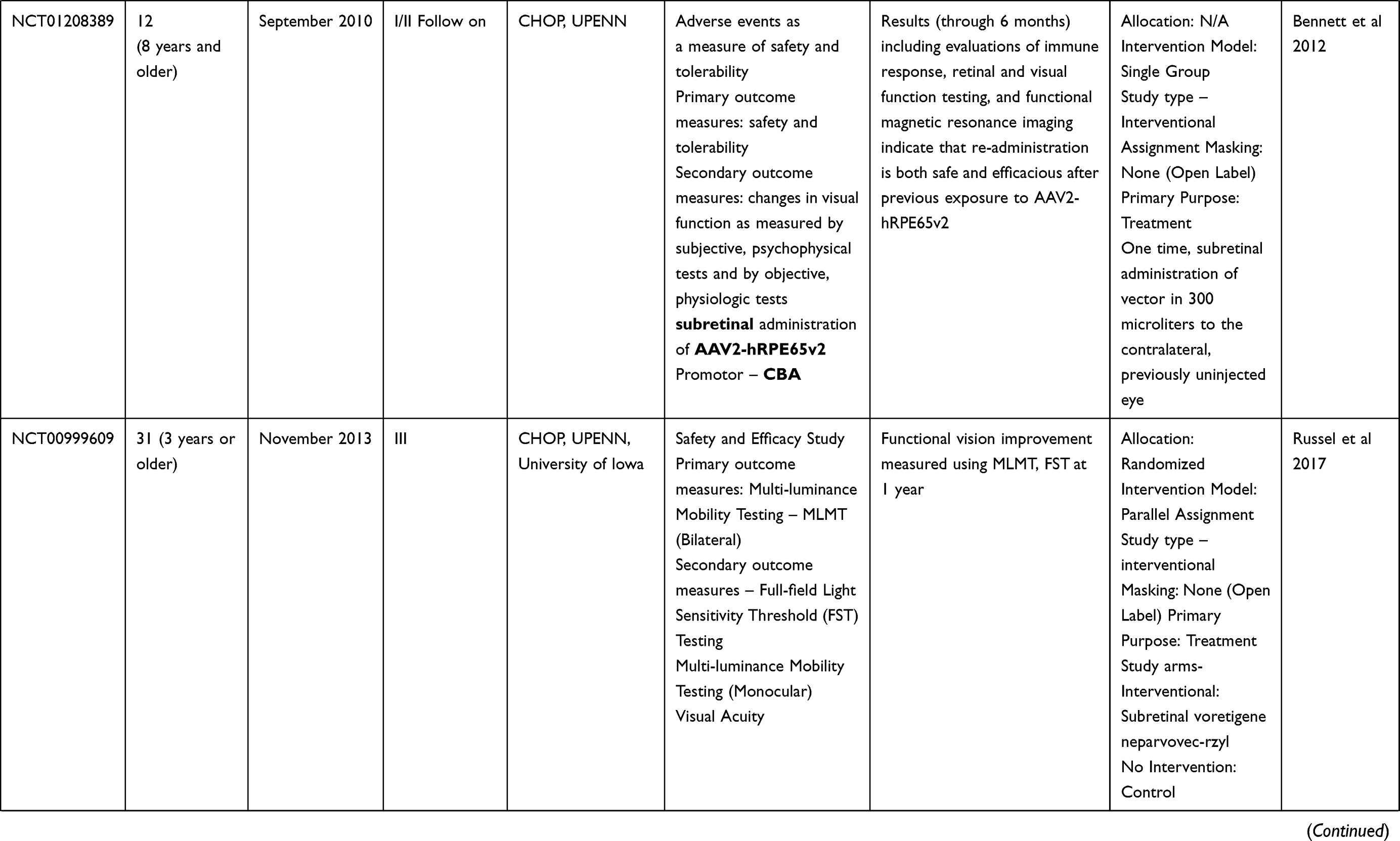 |
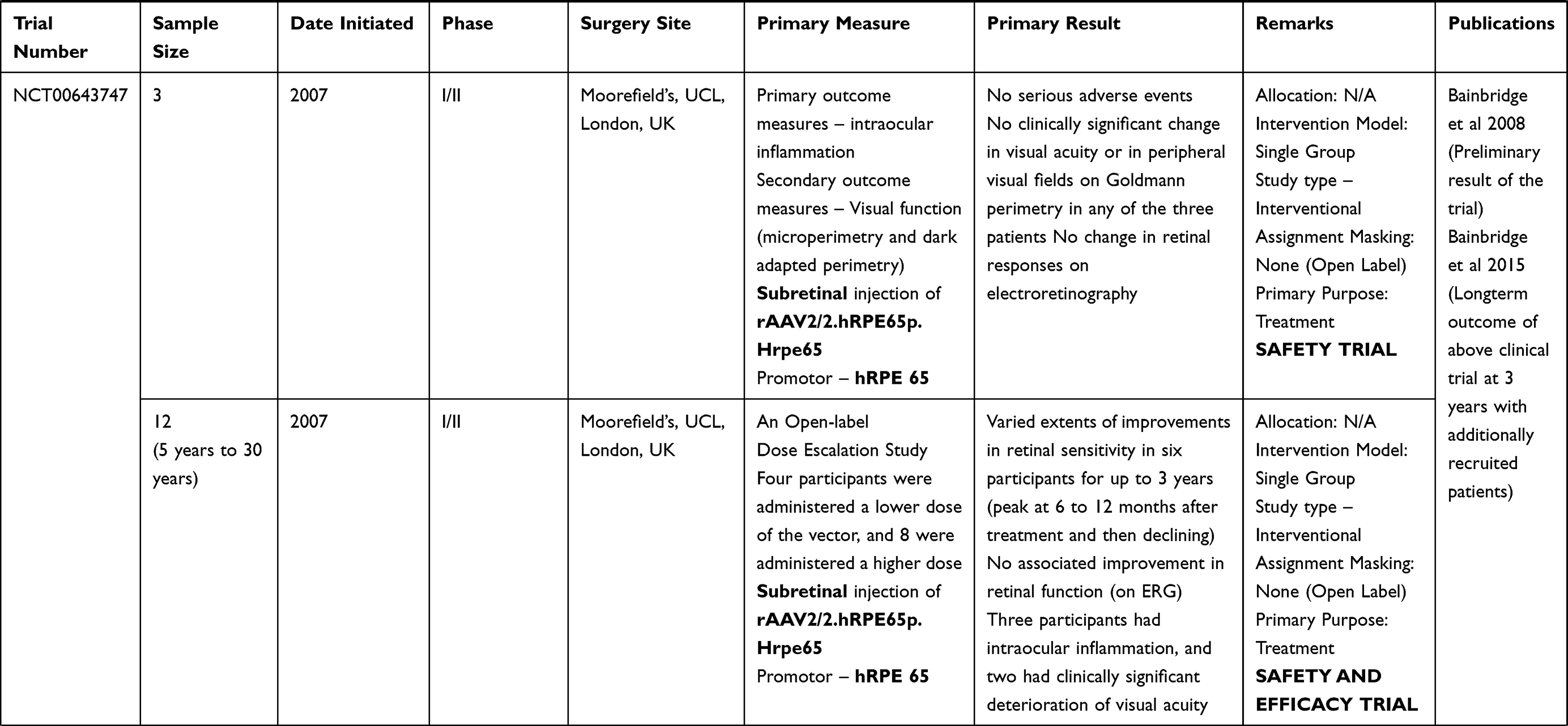
Table 2 Outcome Measures and Results of Human Trials on Gene Therapy for LCA 2 (RPE65) |
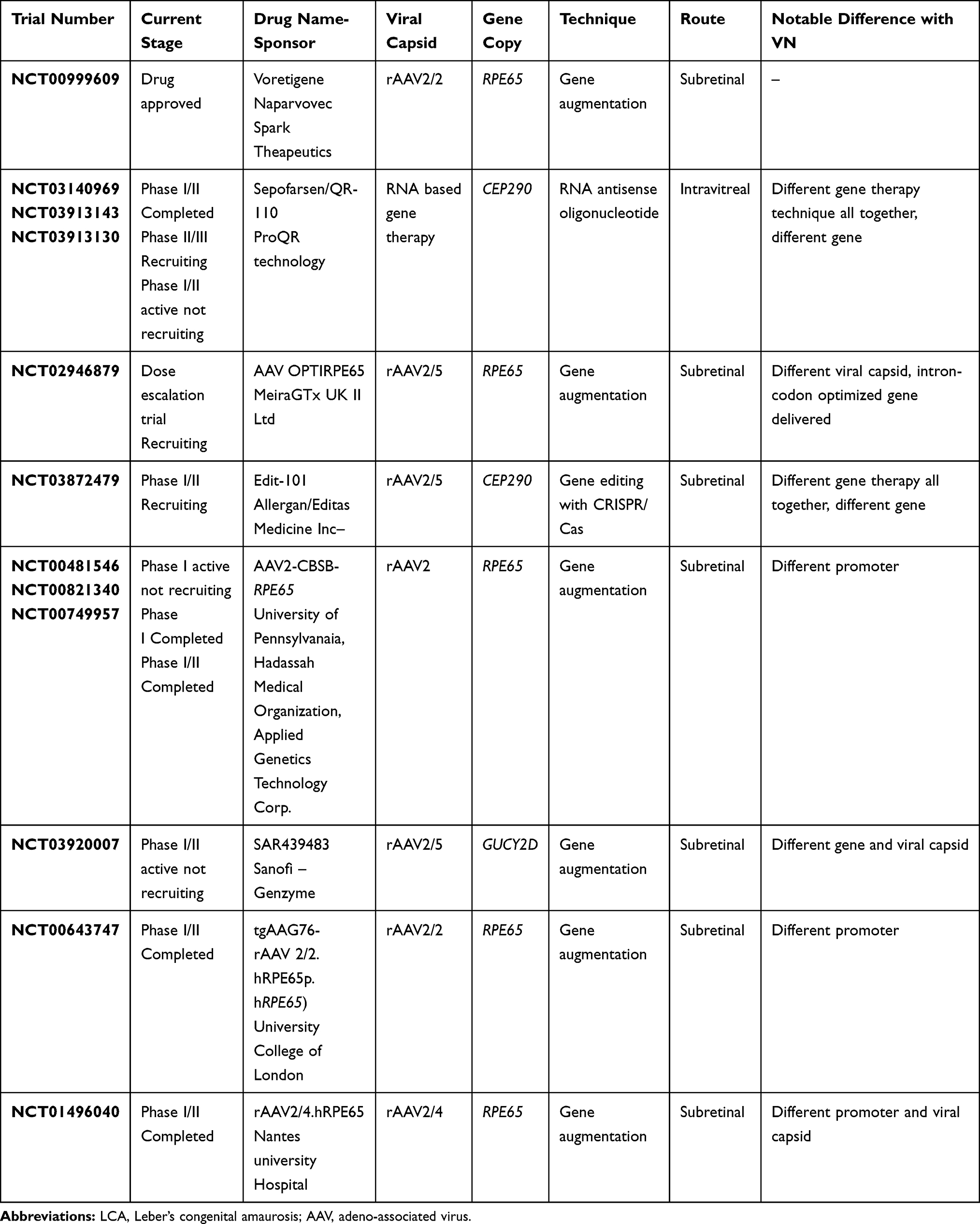 |
Table 3 Gene Cassettes Under Trials for LCA in Comparison to Voretigene Naparvovec |
GUCY2D Gene (LCA1 Locus)
GUCY2D mutation (located on chromosome 17) accounts for 10–20% of cases of autosomal recessive LCA and 40% cases of autosomal dominant cone rod dystrophy.19 The GUCY2Dgene is expressed in both types of photoreceptors (rods and cones), predominant in disc membranes of cone receptor outer segments. The gene codes a protein, retinyl guanylate cyclase 1 (GC-1) that is important for the synthesis of c-GMP. This c-GMP, the intracellular messenger of photoreceptor excitation, in turn regulates intracellular calcium levels, a vital step in phototransduction (conversion of light to neuronal signals) in the recovery phase. It allows the resurrection of cGMP-gated channels by replenishing cGMP stores within the cell, thus regaining the resting depolarized state after the activation stimulus.20 Mutation of GUCY2D leads to a state which is comparable to chronic light exposure. GUCY2D-associated LCA patients usually show marked vision loss, photophobia, hyperopia, nystagmus, and defective ERG since early childhood.21,22 Regardless of the severe vision loss, clinical retinal changes are minimal.
Animal model gene therapy for GUCY2D mutants has used both AAV and lentiviral vectors. Preserved ellipsoid zone integrity on OCT, despite damaged receptor functions, opens the door for gene therapy trials.23–25
AIPL1 Gene (LCA 4 Locus)
Mutation of AIPL1 gene (Aryl-hydrocarbon-interacting-protein-like 1, chromosome 17) accounts for 4–8% cases of recessive LCA.26,27 It exclusively expresses in rod, cone photoreceptors, and the pineal gland. AIPL1 acts as a photoreceptor-specific co-chaperone of phosphodiesterase 6, a crucial enzyme effector in the phototransduction pathway.28 So, a mutation in AIPL1 gene prevents assembly of the PDE6 holoenzyme. This results in an upsurge in intracellular cGMP leading to a prolonged opening of the cyclic nucleotide-gated channels. These patients develop pigmentary changes and maculopathy at an early age.29–31 In addition to photophobia, light gazing, and night blindness, they are also commonly affected with nystagmus, keratoconus, hyperopia, extinguished ERG, and cataract.32 Fascinatingly, hand-held OCT imaging of four patients (all of them of age less than 4 years) showed a relative preservation of central outer retinal structure, making them candidates for gene therapy.21 Replacement of the AIPL1 gene with an AAV vector in a mouse model has revealed promising results in terms of restoring cellular function. The AIPL1 sequence is small in size (~1.2 kb) so it can be proficiently packaged. Due to the rapid progression and early onset, there is only a narrow window period to intervene in order to rescue this form of retinopathy.31
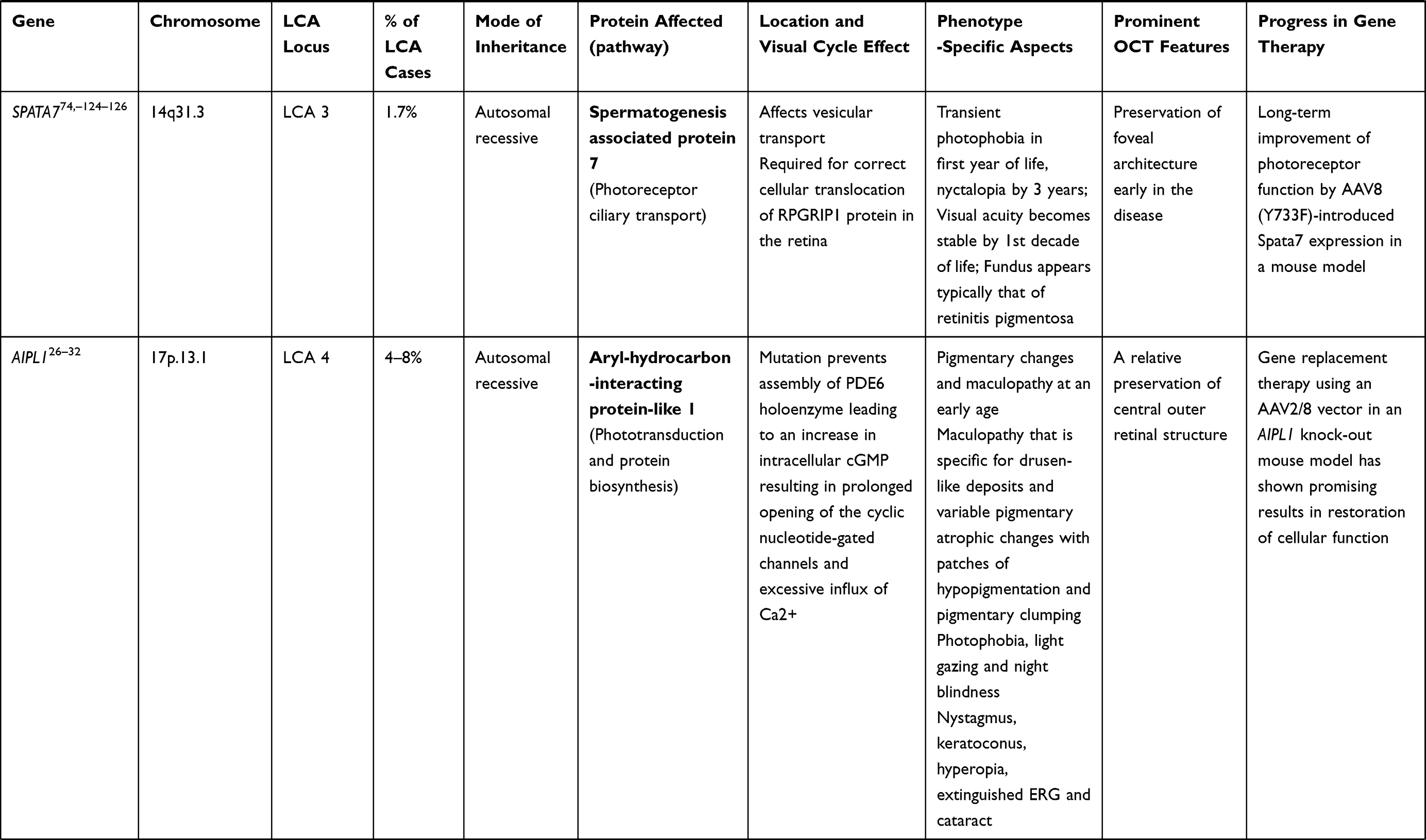
RPGRIP1 (LCA 6 Locus)
Mutation of RPGRIP1 gene (chromosome 14q11) accounts for 5% of cases of LCA.33 RPGRIP1 encodes a 1287 amino acid protein that binds with retinitis pigmentosa GTPase regulator (RPGR) and anchors it to connecting cilia present between the two segments of photoreceptors.34,35 In contrast to other variants, RPGRIP1-LCA trails a relatively non-progressive course after an initial rapid deterioration of visual acuity. In addition, the photoreceptors in the central retina remain preserved for a longer period of time following visual deterioration (which later progress to pigmentary retinopathy), providing a wide window of opportunity for intervention.21 Studies have been done in RPGRIP1 knock-out mice and canine models, showing improvement in both photoreceptor function and structure.36,37
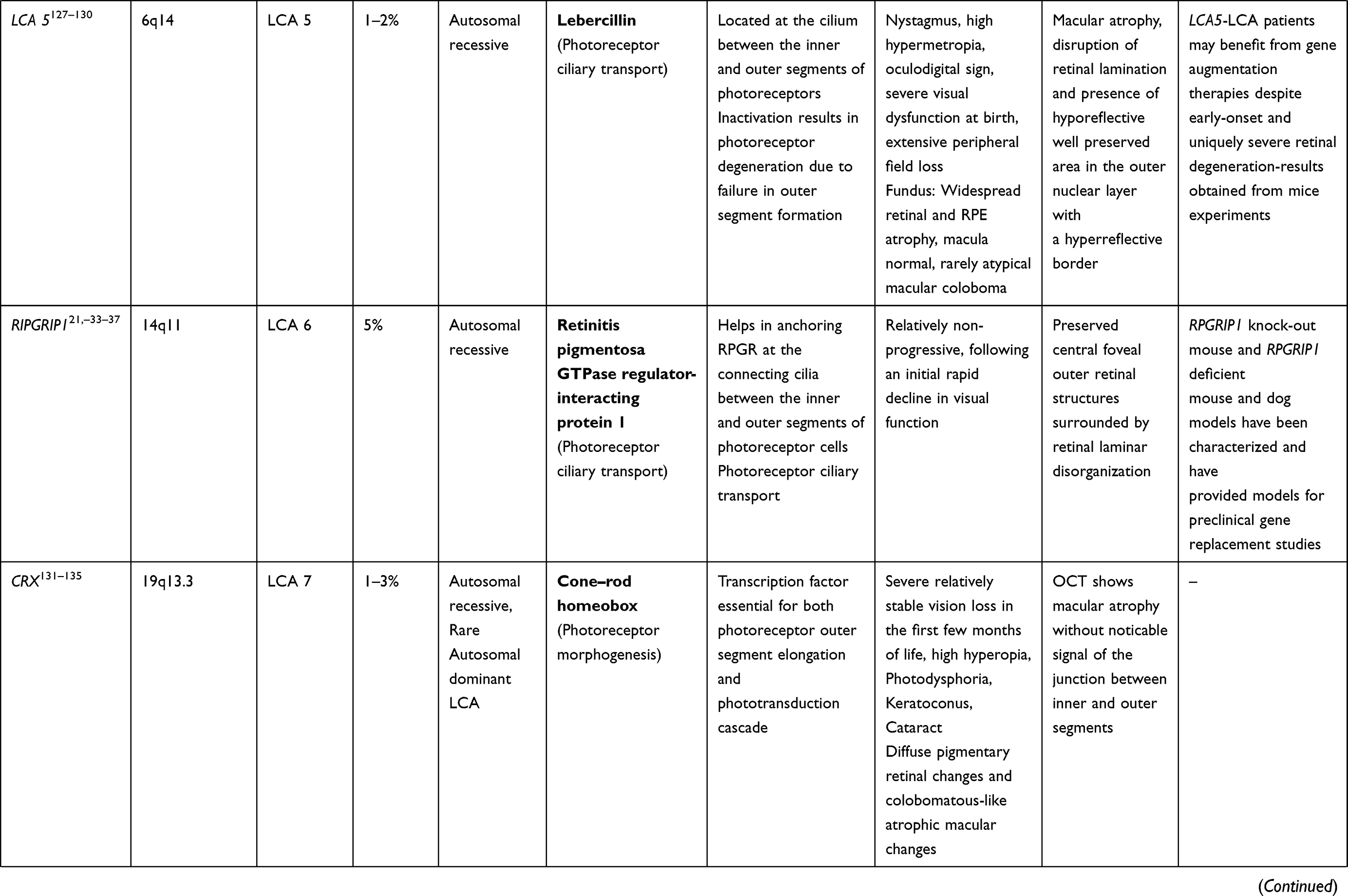
CRB1 Gene (LCA 8 Locus)
CRB1 is a “human homologue of the Drosophila melanogaster gene” responsible for the protein Crumbs. Mutations in it are associated with RP, LCA (7–17% of all cases), and some other IRDs.38–40 CRB1 is implicated in cellular adhesion, maintenance of apico-basal polarization, and cellular communication. It is considered critical for the structure and function of photoreceptors.41 In the developing retina, these proteins are present at the apex of the RPE, Muller cell, and photoreceptor inner segments which will eventually result in the junction between receptor cells and glial cells, creating the external limiting membrane. Characteristic ocular findings comprise macular thinning, pigmentation, relative preservation of para-vascular retinal pigment epithelium with increased retinal thickness along with altered laminar organization and loss of external limiting membrane.38,39 The occurrence of CRB1 mutation diverges considerably in different geographic regions (between specific populations) because of the founder effect of genetic variation or consanguinity, and ranges as high as 17% in Spain to possibly 0% (absent) in India.
Gene replacement therapy for CRB1 related retinopathies is challenging because of its large size of cDNA which approaches the limits of packaging capacity of AAV. However, techniques like vector and codon optimizations have facilitated the packaging of CRB1 cDNA in adeno viral vectors with in vivo expression. The other challenge in CRB1 gene therapy is simultaneous expression of the therapeutic vector in dissimilar cell types that demand CRB function.42
CEP290 Gene (LCA 10 Locus)
Of the LCA cases, 15–20% harbor CEP290 gene mutation, making it the most common gene involved.43 CEP290 is a centrosomal protein present at the connecting cilium of photoreceptors, connecting its outer and inner segments. Mutation of genes encoding ciliary protein leads to a spectrum disorder called ciliopathies.44,45 In addition to other features, CEP290-associated LCA patients typically suffer early onset of marked visual loss with corresponding receptor dysfunction.44 Studies using optical coherence tomography have revealed that, even with profound dysfunction of cones, the foveal outer nuclear layer remains preserved up to the fourth decade of life, though thinner as compared to the perifoveal region.46,47
After the advent of RPE65 based gene therapy for LCA, there has been a substantial curiosity and encouragement to devise effective gene transfer methods for the treatment of CEP290-related retinal dystrophy. However, the barrier to its AAV mediated delivery of CEP290 is the size of its cDNA (~8 kb), which surpasses the maximum capacity of traditional vectors (~4.7–4.9 kb). Hence, use of the lentivirus (having larger packaging limit of 8–10 kb) vector may be advantageous.23 Other viable interventions which are currently under investigation are antisense oligonucleotide, minigene transfer (truncated CEP290 domain – miniCEP290580–1180) and CRISPR/Cas9-based techniques.48,49
RDH 12 Gene (LCA 13 Locus)
RDH 12 gene mutation accounts for about 4–5% of recessive LCA. RDH12 gene (approximately 12kb length) is localised to chromosome 14q and consists of seven exons which encode enzyme retinol dehydrogenase-12. The enzyme is present in the inner segment of photoreceptors and has reductase activity which reduces the all-trans retinal to all-trans retinol.50,51 These mutations lead to reduced expression and activity of the enzyme, in turn affecting photopigment regeneration (Figures 1 and 2). The enzyme naturally protects against excess build-up of retinaldehyde and subsequent cytotoxicity. Loss of function of the gene is highly detrimental early in life, particularly for the macula.52,53 Fundus shows extensive atrophy of the RPE and retina. The unique fundus picture in RDH12 mutation is it's watercolour like appearance, where there is a clear demarcation pattern at the boundary between the preserved and affected retina, which enlarges with the advancement of disease and becomes less apparent at the end stage of the dystrophy.40 There is loss of macular autofluorescence and typical peripapillary sparing is clearly evident on autofluorescence. Spectral domain OCT shows loss of normal foveal architecture and macular thinning.44,54,55
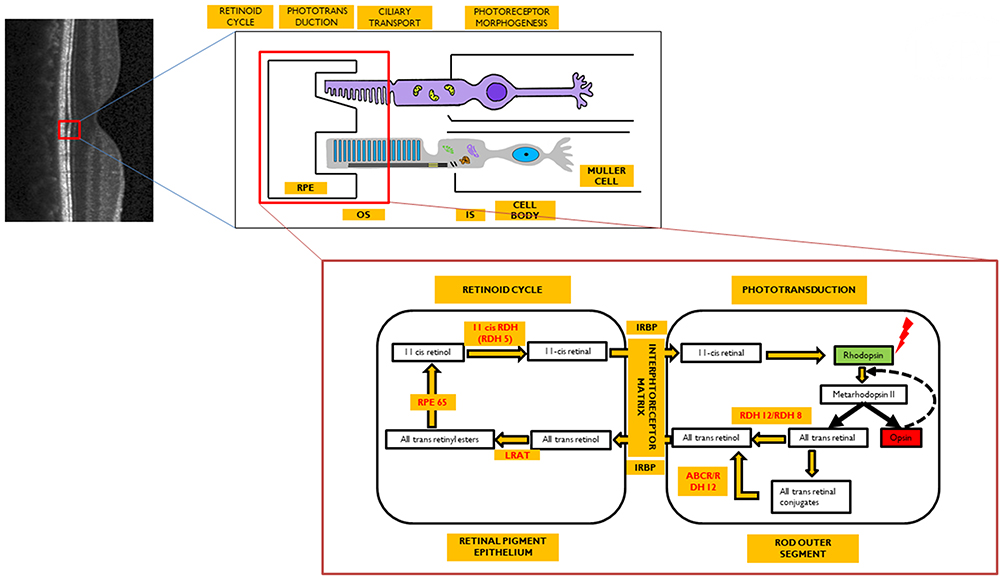 |
Figure 1 Image representing the visual cycle. Enzymes (in red) are localized to their site of action. Optical coherence tomography image (grey scale) represents normal structure of retina with its neural layers. Red colored square bracket is the localization of the retinal pigment epithelium (RPE)-photoreceptor complex within the retina which is further expanded to show that the photoreceptors (rods/cones) have two segments: outer segment (OS) and inner segment (IS) connected by a cilium. The OS interdigitates with the RPE, while the IS is connected to the cell body that relays further to the neural retina. The retinoid cycle occurs within the RPE cell, whereas the phototransduction occurs within the OS of the photoreceptor. Both of these are further expanded to reflect the sequential changes in the visual pigment after exposure to light. |
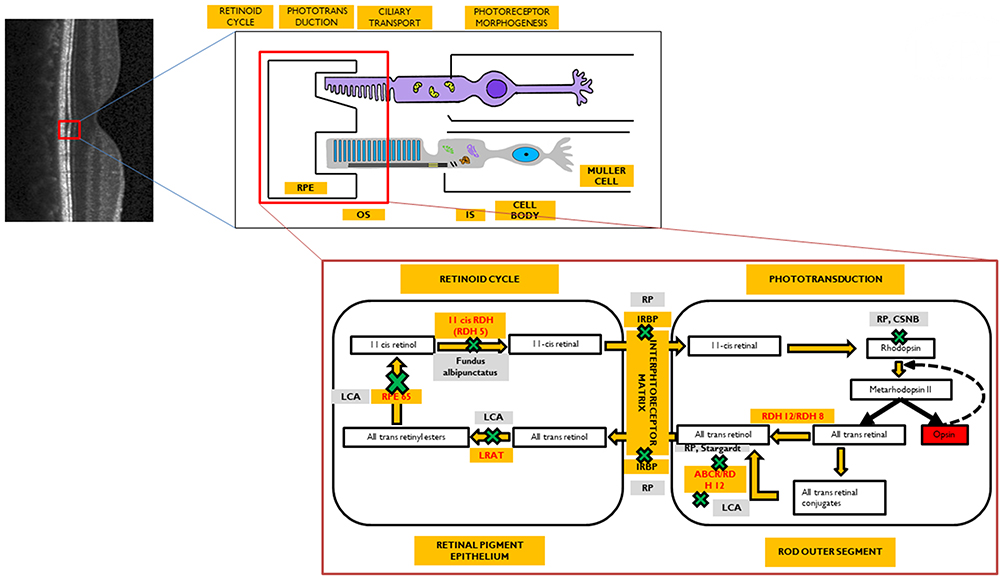 |
Figure 2 Phenotypic disorders corresponding to the enzyme defects shown in Figure 1. The various enzyme deficiencies can lead on to disorders like Leber’s congenital amaurosis (LCA), Fundus Albipunctus, Retinitis pigmentosa (RP), Congenital stationary night blindness (CSNB), and Stargardt's disease, as indicated. |
Thompson et al56 reported AAV-vector mediated RDH 12 gene therapy by evaluating outcomes in wild-type and RDH12-knockout mice. Subretinal injection of an AAV2/8 vector produced recombinant human RDH12 at the desired location without disrupting the structural integrity.
KCNJ13 (LCA 16 Locus)
Mutation of the KCNJ13 gene (chromosome 2q37) also accounts for cases of autosomal recessive LCA 16. Mutations in the KCNJ13gene which encode the inwardly rectifying potassium channel Kir7.1, causing snowflake vitreoretinal degeneration and LCA. In the retina, Kir7.1 is localized exclusively in the RPE apical processes, where it controls retinal function and health. Light activation of photoreceptors reduces the RPE extracellular K+ concentration. The conductance of the Kir7.1 channel increases when extracellular K+ decreases and vice versa. Thus, KCNJ13 loss-of-function directly impacts K+ buffering in the tight subretinal space and thereby alters photoreceptor function.57 Retinal examination in these cases revealed arteriolar abnormalities, pigmentation of the retina in the macular region, and RPE abnormalities in one case,58 along with visually significant cataract in another report.59 In vivo studies for use of Lentivirus mediated gene therapy with this gene has suggested that membrane potential of RPE cells can be restored with 25% of normal protein expression. Accomplishing functional rescue in vivo could be less challenging, and residual mutant protein product is not expected to negatively influence functional outcomes.57
RPE65 (LCA 2 Locus)
RPE65 mutation accounts for both LCA (3–16%) and recessive RP (~2%). The highest prevalence of RPE65 variants is seen in Caucasian (16.5%) and Asian Indian populations (16%). These mutations are rarer in other populations. The gene is localized to chromosome 1p31 and consists of 31 exons. RPE 65 codes for retinoid isomerohydrolase (65KD) expressed in RPE.60 It is important for isomerization of photopigment and converts all-trans retinyl ester to 11-cis retinol in phototransduction (Figures 1 and 2). This isomerohydrolase along with LRAT (Lecithin: retinol acyl transferase) works continuously to regenerate visual pigments.61 Hence a mutation (RPE65deficiency) causes deficiency of 11 cis-retinal required for the beginning of a new visual cycle. Subsequently there is accumulation of pigment granules in RPE (Figures 1 and 2). The consequence is progressive retinal degeneration. RPE65 mutation affects rod and cone photoreceptors differently. 11 cis-retinal deficiency in rod photoreceptors causes an early and profound nyctalopia. However, cone photoreceptors do have an alternate retinoid cycle pathway for generation of 11 cis-retinal which do not depend on RPE65, so cone mediated vision persists in younger patients.62 Generally, this mutation culminates in severe profound vision loss in infancy with mild, if any, nystagmus. Early stage of the disease shows bull’s eye maculopathy, while atrophy, diffuse hyperpigmentation, and clumping of pigments generally appears very late in the course. Other associations include myopia and cataract. LRAT (a key enzyme in visual cycle, locus 14 LCA) deficiency also presents with similar phenotypes, though far less common compared to RPE65.2
Early Challenges for Gene Therapy with RPE 65 Gene
Seeing Light – From Mouse, Dog and Monkey to Man
Acland et al63 reported success in gene replacement therapy in Briard dogs with retinopathy caused by a homozygous 4 base pairs deletion in RPE65. The authors used subretinal delivery of recombinant AAV2 vector that expressed the wild type canine RPE65cDNA regulated by the ubiquitous cytomegalovirus chicken beta actin promoter. Subretinal injection dose ranged from 1.5x108 to 4.5x1012 vector genomes (100–150 µL). Injected eyes displayed a dramatically improved ERG response, pupillometry, and dark-adapted flash evoked cortical potentials. These results persisted up to 10 years following a single procedure.64,65 The results attained in the Rpe65−/− Briard dogs produced deep exhilaration in the field of gene therapy owing to more human-like ocular anatomy and immune system of the animal subject. However, when examined serially over years, the outer photoreceptor nuclear layer showed progressive thinning in spite of gene therapy, indicating that the effects may be temporary. Further studies on dogs extended the usage of other serotypes of AAV like 1, 4, and 5.66–70 Afterward, both natural and knockout murine models were researched for gene replacement therapy with RPE65 gene. The dose–response relationship was also demonstrated.71,72 Further, 17 normal cynomolgus monkeys, with human-like retina, were treated with single subretinal injection of rAAV2/2-CBSB-RPE65 vector. No systemic toxicity or grave ocular adverse effects on the retinal structure or function were noted, as confirmed at 3 weeks and 3 months post-treatment.
AAV2/2 mediated subretinal gene transfer in Briard dogs showed substantial morphological and functional salvage of photoreceptors, translating to improvement of functional ERG (around 20–30% of wildtype levels) and behavioral-based vision tests, particularly under photopic conditions.63,66,69 The stable and long-standing visual restoration was noted to be maintained at 4 years and even at 10 years of follow-up post-treatment in different studies.65,68,73,74
Early Challenges for Humanizing the Progress in Gene Therapy
These early experimental trials were reassuring in terms of safety and efficacy, paving the way for human intervention. Apart from dosage-related issues, a major challenge in humans was the heterogeneity of mutations; which obfuscated the genetic and visual correlation. Another noteworthy difference that later arose between the different human trials involved the promoter driving the expression of RPE65; a human RPE65 promoter was used by one group,75 whereas others used a CAG promoter (changed form of ubiquitous chicken β actin promoter).76–78 Though an ubiquitous promoter like CAG offers a more robust and stronger expression pattern, it shows a non-specific cell expression profile other than RPE cells. In contrast, the human RPE65 promoter drives RPE-specific expressions of the transgene shown in preclinical studies. Even though the CAG promoter expresses genes stronger than the human RPE65 promoter, enough transgene expression was driven by the latter to salvage the treated canine phenotypes.69,79 Robust outcomes were also reported after use of the CAG promoter in multiple parameters (discussed later).76,77,80,81 Although these trials (Table 2) established the stability, safety and efficacy persisting up to no less than 3 years following treatment, an age-dependent effect of the treatment remained contentious with contradictory conclusions.80–82 Cideciyan et al,83 in their human study, concluded that, even with therapy, the outcomes followed the anticipated natural history. Surprisingly, they also showed dissimilarities between canine and human models, used for preclinical experiments. Cideciyan et al also reported that rod recovery in humans remained grossly impaired following RPE65 gene therapy, despite improved visual sensitivity. Thus it is obvious that there were several variables which the early human trials needed to account for.84
The Choice of Subretinal Delivery
Gene therapy can be employed for ocular usage for the following reasons: 1) The eye is an immune privileged site, 2) Miniscule quantities of vectors are required to achieve therapeutic targets, 3) It permits local treatment without the need of intravenous dosing, and 4) The safety and effect can be supervised by non-invasive tests. However, compared to other parts of the body, several barriers that shield the eye from toxicants (anatomically and physiologically) also make delivery of drugs difficult.85,86 For targeted retinal gene therapy, the possible routes could be intravitreal, suprachoroidal, or subretinal drug delivery (Figure 3). Subretinal drugs or genes have direct access to cell membranes of photoreceptors and RPE cells, making it a good location for drug delivery, exclusively in patients with diseases that mainly affect RPE cells and photoreceptors.87 Intravitreal injections on the other hand face the whole neural retina as a potential barrier and are useful when the target for therapy lies in the inner retina or the ganglion cell layer. A higher dosage of drug will be needed while treating with intravitreal drug injection. However, in comparison to subretinal delivery, intravitreal injection is an office-based procedure with minimal requirement of training.88 The subretinal route offers a direct route with more specific localization and a lower dose is needed. The subretinal space itself may be approached in multiple ways.88–91 AAV mediated subretinal delivery of genes has been found to be safe by a large number of studies,75,–76,–79,–80,–92–94 although procedure-related potential side-effects can occur. The complications reported with subretinal surgery include macular hole, retinal tears, retinal detachment, endophthalmitis, glaucoma, cataract, etc. There are also concerns regarding the limited locale of retina benefitting from gene therapy, with the improved area corresponding only to the area of the subretinal bleb formed during subretinal delivery. This has been confirmed using dark adapted perimetry. Another concern is photoreceptor degeneration due to a sub-foveal bleb.95
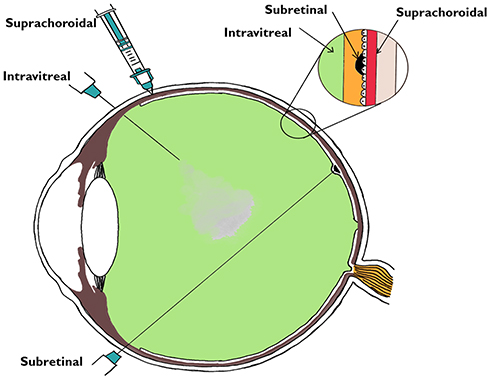 |
Figure 3 Various routes of vector delivery to the eye. The intravitreal, subretinal, and suprachoroidal spaces are shown in the magnified inset corresponding to the sites of the delivery of the drug. |
The other novel mode of drug delivery is the suprachoroidal approach. The suprachoroidal space (nominal thickness of 35 μm) is a potential space found between the sclera and choroid, and, under physiologic conditions (Figure 3), it is mostly collapsed due to the intraocular pressure and fibers that attach the sclera to the choroid. Unlike the subretinal route, the suprachoroidal approach does not require retrobulbar anesthesia or pars plana vitrectomy, and avoids its complications while offering a greater surface area coverage of the posterior segment of the eye. However, the suprachoroidal space is not anatomically immune privileged and the presence of choriocapillaris causes rapid clearance of the administered drug hindering effective transduction of the retina.96
The Choice of AAV Vector
AAV, a parvovirus, has a protein capsid surrounding a DNA genome (single stranded) approximately 4.8 kb in size. The genome contains genes responsible for replication, capsid, and assembly. Through usage of three promoters, alternative translation starts sites, and differential splicing, at least nine gene products are constructed by the virus. The coding sequences are bordered by inverted terminal repeats (ITRs) that are essential for genome replication and packaging. On the other hand, recombinant AAV, a protein-based nanoparticle, lacks viral DNA, can pass through the cell membrane, and eventually deliver genes into the nucleus. In the absence of replication proteins, rAAV forms circular concatemers which accumulate in the nucleus of transduced cells as episomes.97 The peak gene expression is usually reached by around 4–6 weeks in AAV mediated gene therapy. There is no integration between host and episomal DNA, therefore the cargo gene gets diluted over time following mitoses. This ultimately results in diminished gene expression over time, the rate depending on cellular replication. In the perspective of the RPE cells which have very low turnover rates, rAAV is therefore an ideal vector for certain genes.
However, an essential contemplation for using the rAAV vector is the maximum packaging limit, which is less than other vectors like Lentivirus. In the beginning, packaging size under 5 kb was considered adequate.98 As discussed before, Lentivirus has been used for gene therapy for LCA loci other than LCA 2 (RPE65), like loci 1, 10, and 16. Other possible non-viral approaches for gene delivery include bacteria or plasmid based, or others like nanoparticles, electroporation of nucleic acids, antisense oligonucleotides (LCA 10), and liposomes, etc. Efforts in producing rAAV vectors with higher packaging limits resulted in lower viral production yields.99 After delivery of single-stranded AAV-delivered transgenes to the nucleus, it needs to be transformed into a double-stranded form, a rate limiting step at the commencement of transgene expression.100 A substitute is the use of self-complementary AAV (scAAV). Here the single-stranded genome can complement itself to produce its double-stranded form.100,101 Though gene expression becomes faster this way, the packaging capacity can drop significantly to around 3 kb in this strategy. scAAV2,102 scAAV5,103 and scAAV8104 have the advantage of faster onset of expression in retinal cells, though the pattern of expression is identical to that of single-stranded vectors.
An additional constraint to the onset and extent of gene expression is the degradation of AAV vector occurring through phosphorylation of surface-exposed tyrosine residues that enhance proteosome-mediated degradation. A mutation of these tyrosine residues to phenylalanine enables vectors to partially circumvent this pathway leading to increased transduction after subretinal, intravitreal, and intravenous administration compared to their naturally occurring counterparts. Rational design and library selection strategies are considered to be quite useful for achieving improved function of AAV in the retina.105
The factors that decide the choice of AAV factor for gene transfer therefore are: 1) targeted cell/tissue types; 2) the target gene and its safety profile; 3) the selection of route of delivery; and 4) the promoter sequences utilized. AAV matches these for subretinal delivery being very stable to wide pH and temperature changes.106 However, the concentration at which it can be formulated presently is limited to approximately 5×1013 particles per mL, and higher doses pose the risk of inducing inflammatory reactions.107
Promoters and Enhancers for Viral Vectors
Gene therapy requires use of strong ubiquitous promoters such as the cytomegalovirus (CMV) or the chimeric chicken β-actin CMV enhancer and promoter. Other promoters like the human RPE65promoter and other forms of the chicken β-actin (CB) promoter (discussed later) have also been utilized. The choice of promoter is also dependent on the gene and the human cell intended to be transduced. Following viral vector transduction and insertion of genetic material into the target cell, specific promotors permit for the transcription and construction of the anticipated transgene only in designated target cells. By restricting the expression of the gene to a specific target cell type (eg, RPE, rods, or cones), they avert undesirable side-effects from the transgene production. Addition of promoters and enhancers in AAV mediated gene therapy is necessitated because of their absence in the recombinant serotypes. Robust promoters also diminish the minimum dose of viral genomes (and related complications) by augmenting the gene expression. However, the size of the promoter can be a concern as it limits the gene base pairs that can be carried by the drug (discussed earlier).
RPE-specific expression can be driven by the use of RPE65 promoters. RPE specific promoters, the target of LCA gene therapy, include the human RPE65 promoter, NA65 promoter (optimized promoter), VMD2, and Synpiii promoters.108 With these cell-specific promotors, only the cells that typically recognize those particular promotors will produce the transgene product. Although other cell types may be transduced by the viral vector, without recognition of the promotor sequence, the genetic material is not transcribed and the protein is not produced in those cells. As mentioned before, human RPE65 and CAG promoters have been extensively employed for LCA. Theoretically the latter has aore robust gene expression profile, but is not specific for the human RPE cells. In clinical trials, the use of human RPE6 promoter has also resulted in satisfactory gene expression.
Phase I/II Clinical Trials
Early Results
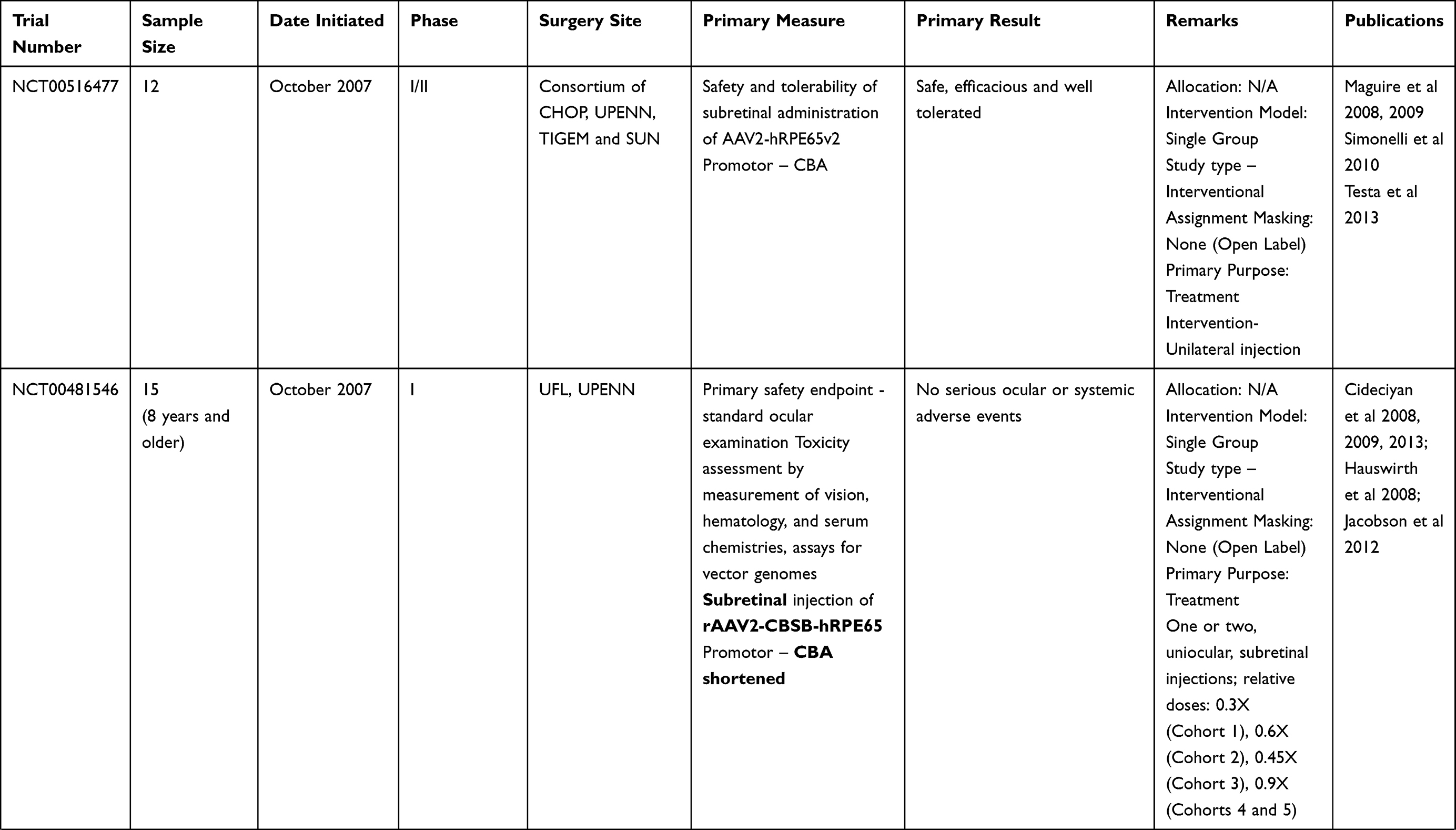
Clinical trials for LCA2 began in 2007 following success in preclinical studies (Table 2). AAV-RPE65 therapy had already been shown to be safe in canine and primate models, as discussed before.109,110 The initial results were stated in 2008 by two independent groups. The group from University College London and Moorfield’s eye hospital (MEH) reported results of treatment of three human subjects with an AAV2.RPE65 construct whose expression was controlled by the human RPE65 promoter. The drug was filled in a buffered saline solution at a titer of 1×1011 vector particles per milliliter and frozen in 1-mL aliquots at −70°C. Up to 1 mL of the vector was introduced, after detachment of approximately a third of the retina, into the subretinal space using a subretinal canula without any serious adverse events. Though statistically significant improvement in vision or visual fields was not seen in any patient, one subject showed significant functional improvement in dark-adapted perimetry and microperimetry. Subjective improvement in visual mobility was also noted in that patient.75
The consortium, led by the Children’s Hospital of Philadelphia (CHOP), including the Telethon Institute of Genetics and Medicine (TIGEM), University of Pennsylvania (UPENN), and the Second University of Naples (SUN), also described the outcomes from treatment of three subjects aged between 19–26 years with AAV2.hRPE65v2 vector.76 A surfactant was used here to avert the loss of the vector in containments. A subretinal injection of 1.5×1010 vector genome of AAV2.hRPE65v2 in 150 μL volume of phosphate-buffered saline was performed, thereby creating a confined dome shaped retinal detachment. The subretinal bleb was seen to resolve after 14 hours of surgery. No adverse event related to viral dissemination was recorded. An asymptomatic macular hole was noted in one patient on postoperative day 5, possibly related to a pre-existing epiretinal membrane. Remarkable improvements were seen in pupillary light responses in all cases. Three times increase in light sensitivity was noted in each treated eye, and even surpassing that of the fellow – previously better – eye. All three subjects revealed an improvement in dim light vision as early as 2 weeks after surgery. A drift toward enlarged visual field areas in each of the three patients was observed.
Several months later, a group from UPENN and the University of Florida (UFL) also stated comparable results.77,111 In that study, three subjects (aged between 21–24 years) were injected with subretinal AAV2-CBSB-hRPE65 containing 5.96x1010 viral genome in 150 μL, in the areas outside the macula in two subjects and within the macular area in the third.109 No local or systemic adverse events were documented. Dark adapted perimetry sensitivities improved in all three eyes. A significant increase in cone and rod sensitivities were demonstrated. However, there was no improvement in visual acuity compared to the baseline. The authors appraised that nearly normal sensitivity to light was reinstated in specific retinal areas, even though the rod recovery time was still higher than normal (8 vs 1 hour).
Long-Term Results
At the beginning of 2009, extended results from these early phase studies were issued. Collectively the reports suggested an age dependent response to gene therapy in LCA with functional improvement in treated eyes.
Dose Escalation Trials: The CHOP-TIGEM-UPENN-SUN group assessed the retinal and visual function in 12 patients (aged 8–44 years) with RPE65-mutation related LCA up to 2 years.80 The subjects were given either of the three doses, ie, 1.5×1010 (low), 4.8×1010 (medium), or 1.5×1011 (high) vector genomes delivered in 150 or 300 µL solution. The previous surgical procedure was slightly modified by including surgical peeling of epiretinal membranes if present. Injections below the macula were avoided in three patients with macular atrophy. High dose injections were given after buttressing fovea with perfluoro-octane liquid (aspirated subsequently).80 The treatment was well tolerated in all patients showing a continuous enhancement in vision both subjectively and objectively. Pupillary light responses improved by at least 2 log units in all patients when measured using pupillometry (amplitude and velocity of constriction) as early as 7 days following injection. There was also a corresponding increase in full-field threshold sensitivities. Ambulatory vision was attained by children. An 8-year old patient attained near normal levels of light sensitivity.
Additional reports with an up to 3-year follow-up period revealed maximal improvement in vision within the initial 6 months of therapy and later stabilized.82,112 Functional MRI measurements revealed functioning in the visual pathway in previously long-standing blind eyes. This indicated a greater elasticity in the visual pathway contrary to previous notions.113
The UPENN/UFL group in their longer-term results described outcomes in 15 subjects, all less than 30 years of age, with the AAV2 vector. The investigators evaluated different doses and injection strategies in five cohorts (Table 2). The surgical adverse events included retinal detachment in one subject requiring additional intervention and non-resolving effusion of the choroid in a second patient. Systemic toxicity was not noted. An improvement of visual function, though to variable degree, was noted in all subjects. Photoreceptors became significantly more sensitive in the treated regions of the retina as early as 3 months post-injection and were sustained through 3 years. Eyes with the lowest visual acuity at baseline displayed the largest improvement in mean visual acuity.
There have been other such trials in the last decade. Two additional publications, one each from Israel and France, describe the outcomes in peer reviewed journals. The immediate results from the Israel study group stated improvement in function as early as 2 weeks in the treated retinal area (Table 2).114 In their report of a phase I/II study performed at Nantes University Hospital, the French study group described the safety of AAV mediated RPE 65 treatment for LCA. However, the improvements in visual function were variable (Table 2).115
Phase II Clinical Re-Administration Trials – A Step Forward
Phase I/II studies reported the success of unilateral injections. It was unclear whether treatment of the fellow eye secondarily would yield any benefit. The apprehension was that the first injection of virus might serve a role similar to vaccination. Hence, an immunogenic response might also cause injury to the primarily injected eye. Therefore, re-administration studies were first performed in large animal models before testing in the human clinical trial subjects. Sequential subretinal delivery of a high dose (1.5x1011 vector genome) of AAV2-hRPE65v2 was tested in both canine models, ie, six Briard (affected) dogs, besides four unaffected non-human primates that had been formerly exposed systemically to research grade AAV. It was found to be a safe procedure with a lack of systemic and ocular toxicity for both eyes. This was followed by initiation of human re-administration studies.116
In human trials, re-administration of vector was done in the fellow eye of three adults with LCA due to RPE65 gene mutation 1.7–3.3 years after their initial therapy. The area targeted in the re-administration was the region which had sufficient viable retinal cells. All subjects received 1.5×1011 µg in 300 µL for the re-administration study in their previously un-injected eye. The oldest participants were registered foremost. Subjects were assessed every week and a 3-months stagger was maintained between enrolments of each of these patients. There were no toxic immune responses in any of the subjects. After injection, the “contralateral” eyes showed functional improvement in multiple aspects. The results reflected an age effect whereby the younger individuals exhibited larger gains than the older individual.117
In continuation of a previous early phase clinical study, a dose of AAV2-hRPE65v2 (1.5×1011 µg in 300 µl) was administered to formerly un-injected eyes of 11 subjects.118 There were no AAV related adverse events. However, dellen formation occurred in three patients, cataract in two cases, while one case developed endophthalmitis. Compared with baseline, results of 10 subjects displayed developments in sensitivity of light and mobility as early as 1 month, that sustained till 3 years.118
Orphan Drug Status
VN received Orphan Drug designation by US FDA on June 24, 2008, which provided incentives to assist and encourage its development. On April 2, 2012, orphan designation (EU/3/12/981) was granted by the European Commission to Alan Boyd Consultants Ltd, UK, for AAV2.RPE65 gene therapy for treating LCA. Spark Therapeutics Ireland Ltd later acquired the sponsorship in 2017.
Phase III Clinical Trial
Based on the results of Phase I and II trials described above, a Phase III (“pivotal”) trial for RPE65 gene augmentation therapy was started at CHOP and the University of Iowa. Thirty-one patients were registered and randomly allocated to intervention (n=21) or control (n=10) groups between November 15, 2012, and November 21, 2013. Individuals with age ≥3 years with an established diagnosis of biallelic RPE65 mutations were enrolled if a) bilateral eyes had visual acuity of ≤20/60 or visual field <20 degrees in any meridian, or both, b) they had enough viable retinal cells as evidenced by retinal thickness on SD-OCT (>100 microns within the posterior pole), fundus photography, and clinical evaluation; and c) they were able to accomplish a standardized multi-luminance mobility test (MLMT) within the luminance range assessed, but incapable to pass the lowest luminance level tested of 1 lux. Primary intervention was bilateral subretinal injection of AAV2.hRPE65v2 at 1.5x1011 µg in eligible individuals. Randomization of subjects was done to intervention or control group (2:1, respectively). Subjects in both the arms were assessed at the same time intervals for 1 year, and then the control group was crossed to the intervention group. Subjects in the control group, meeting all the inclusion criteria, received VN (bilateral) 1 year after their baseline evaluations.119
To measure results, subjects were requested to navigate a standardized course (discussed later) under seven different progressing illuminations commonly met during the course of a day, “starting from 1 lux (like that of a moonless summer night) to 400 lux (equivalent to a brightly lit office environment)”. The course involved navigation of a path defined by large black arrows on the floor avoiding placed obstacles. A change in multi-luminance mobility test score of ≥2, from baseline to year 1, was considered a meaningful benefit in terms of functional vision.120
The primary measure was defined as bilateral MLMT performance gain at year 1. Secondary measures were white light Full field light sensitivity threshold (FST) testing at 1 year, taking averaged value of two eyes, and averaged change of best-corrected visual acuity over both eyes. FST testing assesses night blindness (rod photoreceptors function) predominantly affected by RPE65mutations. Kinetic and static perimetry, visual function questionnaire, contrast sensitivity, pupil light sensitivity, and domestic mobility assessments were also performed. Safety assessments included immunology testing apart from other standard tests. At 1 year, the mean change in bilateral MLMT score was 1.8 (SD=1.1) light levels in the intervention group, as opposed to 0.2 (1.0) in the control group. No control participant could clear MLMT at 1 lux (lowest level), however 13/20 treated subjects did. This was a very strong indicator of efficacy. Minimal inflammation, temporarily increased intraocular pressure, and retinal tears were the most commonly documented adverse effects.119
Mobility Tests as an Indicator of Efficiency
With time, subjects with untreated RPE65 related LCA lose perception of light at any illumination, restricting independent navigation in early life. Since the traditional metrics of mobility do not account for ambient illumination while judging accuracy and speed of navigation, the MLMT was developed. This test is specifically designed to quantify and ascertain outcomes focused on various facets of vision, like field, acuity, and light sensitivity.121 For visually impaired individuals, the test also divides between higher and lower performers. Even young children (4 years) navigated the standardized course precisely and at a reasonable pace. Most important, the MLMT is a validated test found to be both reliable and reproducible.
Safety Concerns
VN, unlike most FDA-approved injectable treatments, necessitates a surgical procedure for administration. The FDA reviewers analyzed 41 subjects with VN intervention taking into account safety results of both Phase I and Phase III studies. The various treatment-related ocular adverse events, during the follow-up period spanning between 1– 6 years, included maculopathy (5%), macular holes (7%), retinal tears (10%), raised intraocular pressure (IOP) (15%), and cataract (20%). In general, 66% (21) of treated subjects experienced one or more ocular adverse events, mostly mild or moderate, and resolved, primarily ascribed to the surgical procedure.119 However, two (5%) study participants developed irreversible loss of vision, one due to raised IOP related optic atrophy, and the other because of macular atrophy secondary to the procedure. A 15-year follow-up study to evaluate long-term safety and efficacy is ongoing.
Gene Cassette of Voretigene Neparvovec
VN, as developed by Spark therapeutics, uses the rAAV2 capsid. The structure of rAAV gene cassettes is bounded at its ends by two inverted terminal repeats (ITRs). The ITRs of rAAV are the only palindromic repeats carried over from the wild type AAV. These ITRs are required as they allow for episomal concatemerization, and thus action of the drug. However, the ITRs of the AAV serotype lack the promoter and the termination function, which makes the presence of promoters and enhancers necessary. Therefore, following the 5ʹ ITR, a promoter sequence is incorporated in the drug. In the case of VN, it is the ubiquitous CAG promoter (consisting of a CMV enhancer and a chicken beta actin promoter). The job of the promoter and enhancer sequence here is to stabilize and increase the gene expression and transcription. This is followed by the chicken beta actin exon and intron which are necessary for gene regulation. Following this the next portion of the gene cassette consists of the RPE65 DNA which is the dysfunctional target in the case of LCA. In VN, this DNA is a complimentary DNA representing the human RPE65 gene, thus referred to as hRPE65v2. Following the DNA target, the rAAV gene cassette also requires a polyadenylation sequence. The polyadenylation sequence is necessary for the stability and expression of the gene product. In the case of VN, this sequence is derived from the bovine growth hormone. Lastly, the gene rAAV gene cassette contains the 3ʹITR. VN, therefore, pharmacologically reads as ITR.CAG.RPE65v2.bGHpolA.ITR gene sequence harbored in a rAAV2 capsid. Other similar constructs being evaluated for gene therapy for LCA are listed in Table 3.122
FDA Approval
On October 12, 2017, a committee dedicated to cell-tissue-gene therapies discussed the efficacy and safety of VN. Approval was given in December 2017 after all 16 members voted in favor of the drug. Two-year outcomes of MLMT were taken into account to judge the efficacy of VN (Luxturna). The approval was given for patients with retinal dystrophy due to RPE65 mutations.16 It is the first FDA approved gene replacement therapy that uses AAV vectors. The drug was priced at US$425,000 for each eye.
EU Approval
The European Commission (EC) approved VN on November 23, 2018 as a one-time gene replacement therapy for RPE65 mutation related retinal dystrophy, provided some retinal function was present. The EC decision was based on judgment of the committee that evaluates medical products intended for humans. In early 2018, Spark Therapeutics and Novartis entered into an agreement for marketing VN in the rest of the world.123
Conclusion
Gene therapy for LCA with VN has matured to a level of evidence-based success that inspires hopes for other IRDs and non-ocular disorders. However, it also brings with itself new challenges that need immediate addressal. At the level of regional groups, these include individualizing patterns of genetic defects in different ethnicities, training retinal surgeons and ocular geneticists, provision of appropriate visual tests for monitoring, and economical aspects of gene therapy. For researchers, the options of routes of drug delivery is still open ended, especially with the advent of the suprachoroidal approach. Subretinal delivery is sophisticated and not an office-based procedure. Further, due to the involvement of multiple genes, the approach to each genotype needs to be refined, and gene augmentation may not be the only solution. Specifically, in vivo gene editing is likely to find a role in specific genotypes of LCA. While AAV has currently become the approved vector for subretinal gene delivery, its small capacity is a hindrance and the capacity for transduction can be improved further. The perfect promoter-gene combination is yet elusive, and the size of promoter can be decreased to allow more space for the gene cDNA. Some long-term studies have shown visual and structural decline after early improvement, which makes concerns over appropriately timed re-administration in a previously treated eye a reality. As retinal imaging continues to evolve with advances like adaptive optics, outcome measures that are simpler or universally available as against the MLMT may be more pertinent. At the level of the patient, identification of ideal candidates for gene therapy needs to be done very cautiously, with emphasis on the stage of LCA, availability of functioning photoreceptors, and choice of retinal area for sub-retinal delivery. Judicious use of other rehabilitative therapies simultaneously with gene therapy is important, especially in the perspective of patient expectations of outcomes.
Acknowledgments
Hyderabad Eye Research Foundation, India, provides research support to SP, BT, RN and SJ. The authors thank Dr Sohini Mandal, MD for help with figures.
Disclosure
No author has any conflicts of interest to disclose.
References
1. Sahel JA, Marazova K, Audo I. Clinical characteristics and current therapies for inherited retinal degenerations. Cold Spring Harb Perspect Med. 2014;5(2):a017111. doi:10.1101/cshperspect.a017111
2. Cremers FPM, Boon CJF, Bujakowska K, et al. Special issue introduction: inherited retinal disease: novel candidate genes, genotype-phenotype correlations, and inheritance models. Genes (Basel). 2018;9(4):215. doi:10.3390/genes9040215
3. Gregory-Evans K, Pennesi ME, Weleber RG. Retinitis pigmentosa and allied disorders. In: Ryan SJ, editor. Retina. Elsevier; 2013:761–835.
4. Leber T. Ueber retinitis pigmentosa und angeborene amaurose. Graefes Arch Clin Exp Ophthalmol. 1869;15(3):1–25. doi:10.1007/BF02721213
5. Perrault I, Rozet JM, Gerber S, et al. Leber congenital amaurosis. Mol Genet Metab. 1999;68(2):200–208. doi:10.1006/mgme.1999.2906
6. Weleber RG, Michaelides M, Trzupek KM, et al. The phenotype of severe early childhood onset retinal dystrophy (SECORD) from mutation of RPE65 and differentiation from leber congenital amaurosis. Invest Ophthalmol Vis Sci. 2011;52(1):292–302. doi:10.1167/iovs.10-6106
7. Anderson WF, Blaese RM, Culver K. The ADA human gene therapy clinical protocol: points to consider response with clinical protocol. Hum Gene Ther. 1990;1(3):331–362. doi:10.1089/hum.1990.1.3-331
8. Deakin CT, Alexander IE, Kerridge I. Accepting risk in clinical research: is the gene therapy field becoming too risk-averse? Mol Ther. 2009;17(11):1842–1848. doi:10.1038/mt.2009.223
9. Bank A. Human somatic cell gene therapy. Bioessays. 1996;18(12):999–1007. doi:10.1002/bies.950181210
10. Mathews QL, Curiel DT. Gene therapy: human germline genetics modifications-assessing the scientific, socioethical, and religious issues. South Med J. 2007;100(1):98–100. doi:10.1097/SMJ.0b013e31802e645f
11. Gonçalves GAR, Paiva RMA. Gene therapy: advances, challenges and perspectives. Einstein (Sao Paulo). 2017;15(3):369–375. doi:10.1590/S1679-45082017RB4024
12. Zeng Y, Takada Y, Kjellstrom S, et al. RS-1 gene delivery to an adult Rs1h knockout mouse model restores ERG b-wave with reversal of the electronegative waveform of X-linked retinoschisis. Investig Ophthalmol Vis Sci. 2004;45(9):3279–3285. doi:10.1167/iovs.04-0576
13. Han Z, Conley SM, Naash MI. Gene therapy for stargardt disease associated with ABCA4 gene. Adv Exp Med Biol. 2014;801:719–724.
14. Cashman SM, Gracias J, Adhi M, Kumar-Singh R. Adenovirus-mediated delivery of factor H attenuates complement C3 induced pathology in the murine retina: a potential gene therapy for age-related macular degeneration. J Gene Med. 2015;17(10–12):229–243. doi:10.1002/jgm.2865
15. Matet A, Kostic C, Bemelmans AP, et al. Evaluation of tolerance to lentiviral LV-RPE65 gene therapy vector after subretinal delivery in non-human primates. Transl Res. 2017;188:40–57. doi:10.1016/j.trsl.2017.06.012
16. FDA. FDA Briefing Document: Advisory Committee Meeting. October 12, 2017. BLA 125610 (Voretigene Neparvovec). FDA; 2017.
17. Hampson G, Towse A, Pearson SD, Dreitlein WB, Henshall C. Gene therapy: evidence, value and affordability in the US health care system. J Comp Eff Res. 2017;7(1):15–28. doi:10.2217/cer-2017-0068
18. Takkar B, Bansal P, Venkatesh P. Leber’s congenital amaurosis and gene therapy. Indian J Pediatr. 2018;85(3):237–242. doi:10.1007/s12098-017-2394-1
19. Perrault I, Rozet JM, Gerber S, et al. Spectrum of retGC1 mutations in Leber’s congenital amaurosis. Eur J Hum Genet. 2000;8(8):578–582. doi:10.1038/sj.ejhg.5200503
20. Wimberg H, Lev D, Yosovich K, et al. Photoreceptor guanylate cyclase (GUCY2D) mutations cause retinal dystrophies by severe malfunction of Ca2+-dependent cyclic GMP synthesis. Front Mol Neurosci. 2018;11:348. doi:10.3389/fnmol.2018.00348
21. Jacobson SG, Cideciyan AV, Peshenko IV, et al. Determining consequences of retinal membrane guanylyl cyclase (RetGC1) deficiency in human Leber congenital amaurosis en route to therapy: residual cone-photoreceptor vision correlates with biochemical properties of the mutants. Hum Mol Genet. 2013;22(1):168–183. doi:10.1093/hmg/dds421
22. Boye SE. Leber congenital amaurosis caused by mutations in GUCY2D. Cold Spring Harb Perspect Med. 2014;5(1):a017350. doi:10.1101/cshperspect.a017350
23. Haire SE, Pang J, Boye SL, et al. Light-driven cone arrestin translocation in cones of postnatal guanylate cyclase-1 knockout mouse retina treated with AAV-GC1. Invest Ophthalmol Vis Sci. 2006;47(9):3745–3753. doi:10.1167/iovs.06-0086
24. Mihelec M, Pearson RA, Robbie SJ, et al. Long-term preservation of cones and improvement in visual function following gene therapy in a mouse model of leber congenital amaurosis caused by guanylate cyclase-1 deficiency. Hum Gene Ther. 2011;22(10):1179–1190. doi:10.1089/hum.2011.069
25. Boye SE, Boye SL, Pang J, et al. Functional and behavioral restoration of vision by gene therapy in the guanylate cyclase-1 (GC1) knockout mouse. PLoS One. 2010;5(6):e11306. doi:10.1371/journal.pone.0011306
26. Hidalgo-de-Quintana J, Evans RJ, Cheetham ME, et al. The Leber congenital amaurosis protein AIPL1 functions as part of a chaperone heterocomplex. Invest Ophthalmol Vis Sci. 2008;49(7):2878–2887. doi:10.1167/iovs.07-1576
27. Sohocki MM, Bowne SJ, Sullivan LS, et al. Mutations in a new photoreceptor-pineal gene on 17p cause Leber congenital amaurosis. Nat Genet. 2000;24(1):
28. Liu X, Bulgakov OV, Wen XH, et al. AIPL1, the protein that is defective in Leber congenital amaurosis, is essential for the biosynthesis of retinal rod cGMP phosphodiesterase. Proc Natl Acad Sci U S A. 2004;101(38):
29. Tan MH, Mackay DS, Cowing J, et al. Leber congenital amaurosis associated with AIPL1: challenges in ascribing disease causation, clinical findings, and implications for gene therapy. PLoS One. 2012;7(3):e32330. doi:10.1371/journal.pone.0032330
30. Dharmaraj S, Leroy BP, Sohocki MM, et al. The phenotype of leber congenital amaurosis in patients with AIPL1 mutations. Arch Ophthalmol. 2004;122(7):1029–1037. doi:10.1001/archopht.122.7.1029
31. Aboshiha J, Dubis AM, Spuy J, et al. Preserved outer retina in AIPL1 Leber’s congenital amaurosis: implications for gene therapy. Ophthalmology. 2015;122(4):862–864. doi:10.1016/j.ophtha.2014.11.019
32. Testa F, Surace EM, Rossi S, et al. Evaluation of Italian patients with leber congenital amaurosis due to AIPL1 mutations highlights the potential applicability of gene therapy. Invest Ophthalmol Vis Sci. 2011;52(8):5618–5624. doi:10.1167/iovs.10-6543
33. Dryja TP, Adams SM, Grimsby JL, et al. Null RPGRIP1 alleles in patients with Leber congenital amaurosis. Am J Hum Genet. 2001;68(5):
34. Gerber S, Perrault I, Hanein S, et al. Complete exon-intron structure of the RPGR-interacting protein (RPGRIP1) gene allows the identification of mutations underlying Leber congenital amaurosis. Eur J Hum Genet. 2001;9(8):561–571. doi:10.1038/sj.ejhg.5200689
35. Li T. Leber congenital amaurosis caused by mutations in RPGRIP1. Cold Spring Harb Perspect Med. 2014;5(4):a017384. doi:10.1101/cshperspect.a017384
36. Pawlyk BS, Smith AJ, Buch PK, et al. Gene replacement therapy rescues photoreceptor degeneration in a murine model of Leber congenital amaurosis lacking RPGRIP. Invest Ophthalmol Vis Sci. 2005;46(9):3039–3045. doi:10.1167/iovs.05-0371
37. Pawlyk BS, Bulgakov OV, Liu X, et al. Replacement gene therapy with a human RPGRIP1 sequence slows photoreceptor degeneration in a murine model of Leber congenital amaurosis. Hum Gene Ther. 2010;21:993–1004.
38. Henderson RH, Mackay DS, Li Z, et al. Phenotypic variability in patients with retinal dystrophies due to mutations in CRB1. Br J Ophthalmol. 2011;95:811–817. doi:10.1136/bjo.2010.186882
39. Simonelli F, Ziviello C, Testa F, et al. Clinical and molecular genetics of Leber’s congenital amaurosis: a multicenter study of Italian patients. Invest Ophthalmol Vis Sci. 2007;48(9):4284–4290. doi:10.1167/iovs.07-0068
40. Ahmed Khan S, Richard Nestel A. CRB1 gene mutation causing different phenotypes of leber congenital amaurosis in siblings. J Ophthalmic Vis Res. 2019;14(4):518–524. doi:10.18502/jovr.v14i4.5467
41. van de Pavert SA, Kantardzhieva A, Malysheva A, et al. Crumbs homologue 1 is required for maintenance of photoreceptor cell polarization and adhesion during light exposure. J Cell Sci. 2004;117(18):4169. doi:10.1242/jcs.01301
42. Boon N, Wijnholds J, Pellissier LP. Research models and gene augmentation therapy for CRB1 retinal dystrophies. Front Neurosci. 2020;14:860. doi:10.3389/fnins.2020.00860
43. den Hollander AI, Koenekoop RK, Yzer S, et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am J Hum Genet. 2006;79(3):556–561. doi:10.1086/507318
44. Perrault I, Delphin N, Hanein S, et al. Spectrum of NPHP6/CEP290 mutations in Leber congenital amaurosis and delineation of the associated phenotype. Hum Mutat. 2007;28(4):416. doi:10.1002/humu.9485
45. McAnany JJ, Genead MA, Walia S, et al. Visual acuity changes in patients with leber congenital amaurosis and mutations in CEP290. JAMA Ophthalmol. 2013;131(2):178–182. doi:10.1001/2013.jamaophthalmol.354
46. Yzer S, Hollander AI, Lopez I, et al. Ocular and extra-ocular features of patients with Leber congenital amaurosis and mutations in CEP290. Mol Vis. 2012;18:
47. Cideciyan AV, Rachel RA, Aleman TS, et al. Cone photoreceptors are the main targets for gene therapy of NPHP5 (IQCB1) or NPHP6 (CEP290) blindness: generation of an all-cone Nphp6 hypomorph mouse that mimics the human retinal ciliopathy. Hum Mol Genet. 2011;20(7):1411–1423. doi:10.1093/hmg/ddr022
48. Kong J, Kim S-R, Binley K, et al. Correction of the disease phenotype in the mouse model of stargardt disease by lentiviral gene therapy. Gene Ther. 2008;15(19):1311–1320. doi:10.1038/gt.2008.78
49. Verrier JD, Madorsky I, Coggin WE, et al. Bicistronic lentiviruses containing a viral 2A cleavage sequence reliably co-express two proteins and restore vision to an animal model of LCA1. PLoS One. 2011;6(5):e20553. doi:10.1371/journal.pone.0020553
50. den Hollander AI, Roepman R, Koenekoop RK, et al. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res. 2008;27(4):391–419. doi:10.1016/j.preteyeres.2008.05.003
51. Mackay DS, Dev Borman A, Moradi P, et al. RDH12 retinopathy: novel mutations and phenotypic description. Mol Vis. 2011;17:2706–2716.
52. Parker RO, Crouch RK. Dehydrogenases RDHs in the visual cycle. Exp Eye Res. 2010;91(6):788–792. doi:10.1016/j.exer.2010.08.013
53. Haeseleer F, Jang GF, Imanishi Y, et al. Dual-substrate specificity short chain retinol dehydrogenases from the vertebrate retina. J Biol Chem. 2002;277(47):
54. Schuster A, Janecke AR, Wilke R, et al. The phenotype of early-onset retinal degeneration in persons with RDH12 mutations. Invest Ophthalmol Vis Sci. 2007;48(4):1824–1831. doi:10.1167/iovs.06-0628
55. Valverde D, Pereiro I, Vallespín E, et al. Complexity of phenotype–genotype correlations in Spanish patients with RDH12 mutations. Invest Ophthalmol Vis Sci. 2009;50(3):1065–1068. doi:10.1167/iovs.08-2083
56. Thompson DA, Jia L, Yao J, et al. AAV-mediated expression of human Rdh12 in mouse retina. Invest Ophthalmol Vis Sci. 2012;53:1916.
57. Shahi PK, Hermans D, Sinha D, et al. Gene augmentation and readthrough rescue channelopathy in an iPSC-RPE model of congenital blindness. Am J Hum Genet. 2019;104(2):310–318. doi:10.1016/j.ajhg.2018.12.019
58. Pattnaik BR, Shahi PK, Marino MJ, et al. A novel KCNJ13 nonsense mutation and loss of Kir7.1 channel function causes leber congenital amaurosis (LCA16). Hum Mutat. 2015;36(7):720–727. doi:10.1002/humu.22807
59. Khan AO, Bergmann C, Neuhaus C, Bolz HJ. A distinct vitreo-retinal dystrophy with early-onset cataract from recessive KCNJ13 mutations. Ophthalmic Genet. 2015;36(1):79–84. doi:10.3109/13816810.2014.985846
60. Redmond TM, Poliakov E, Yu S, et al. Mutation of key residues of RPE65 abolishes its enzymatic role as isomerohydrolase in the visual cycle. Proc Natl Acad Sci U S A. 2005;102(38):
61. Hanein S, Perrault I, Gerber S, et al. Leber congenital amaurosis: comprehensive survey of the genetic heterogeneity, refinement of the clinical definition, and genotype-phenotype correlations as a strategy for molecular diagnosis. Hum Mutat. 2004;23(4):306–317. doi:10.1002/humu.20010
62. Dev Borman A, Ocaka LA, Mackay DS, et al. Early onset retinal dystrophy due to mutations in LRAT: molecular analysis and detailed phenotypic study. Invest Ophthalmol Vis Sci. 2012;53(7):3927–3938. doi:10.1167/iovs.12-9548
63. Acland GM, Aguirre GD, Ray J, et al. Gene therapy restores vision in a canine model of childhood blindness. Nat Genet. 2001;28(1):92–95. doi:10.1038/ng0501-92
64. Cideciyan AV, Jacobson SG, Beltran WA, et al. Human retinal gene therapy for Leber congenital amaurosis shows advancing retinal degeneration despite enduring visual improvement. Proc Natl Acad Sci U S A. 2013;110(6):E517–E525. doi:10.1073/pnas.1218933110
65. Swain G, Prociuk M, Bagel J, et al. Adeno-associated virus serotypes 9 and rh10 mediate strong neuronal transduction of the dog brain. Gene Ther. 2014;21(1):28–36. doi:10.1038/gt.2013.54
66. Narfstrom K, Katz ML, Bragadottir R, et al. Functional and structural recovery of the retina after gene therapy in the RPE65 null mutation dog. Invest Ophthalmol Vis Sci. 2003;44(4):1663–1672. doi:10.1167/iovs.02-0595
67. Narfstrom K, Katz ML, Ford M, et al. In vivo gene therapy in young and adult RPE65-/- dogs produces long-term visual improvement. J Hered. 2003;94(1):31–37. doi:10.1093/jhered/esg015
68. Acland GM, Aguirre GD, Bennett J, et al. Long-term restoration of rod and cone vision by single dose rAAV-mediated gene transfer to the retina in a canine model of childhood blindness. Mol Ther. 2005;12(6):1072–1082. doi:10.1016/j.ymthe.2005.08.008
69. Le Meur G, Stieger K, Smith AJ, et al. Restoration of vision in RPE65-deficient briard dogs using an AAV serotype 4 vector that specifically targets the retinal pigmented epithelium. Gene Ther. 2007;14(4):292–303. doi:10.1038/sj.gt.3302861
70. Annear MJ, Bartoe JT, Barker SE, et al. Gene therapy in the second eye of RPE65-deficient dogs improves retinal function. Gene Ther. 2011;18(1):53–61. doi:10.1038/gt.2010.111
71. Wright JF. Manufacturing and characterizing AAV-based vectors for use in clinical studies. Gene Ther. 2008;15(11):840–848. doi:10.1038/gt.2008.65
72. Jacobson SG, Aleman TS, Cideciyan AV, et al. Identifying photoreceptors in blind eyes caused by RPE65 mutations: prerequisite for human gene therapy success. Proc Natl Acad Sci U S A. 2005;102(17):6177–6182. doi:10.1073/pnas.0500646102
73. Narfström K, Seeliger M, Lai CM, et al. Morphological aspects related to long-term functional improvement of the retina in the 4 years following rAAV-mediated gene transfer in the RPE65 null mutation dog. Adv Exp Med Biol. 2008;613:139–146. doi:10.1007/978-0-387-74904-4_15
74. Kumaran N, Moore AT, Weleber RG, et al. Leber congenital amaurosis/early-onset severe retinal dystrophy: clinical features, molecular genetics and therapeutic interventions. Br J Ophthalmol. 2017;101(9):1147–1154. doi:10.1136/bjophthalmol-2016-309975
75. Bainbridge JW, Smith AJ, Barker SS, et al. Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med. 2008;358(21):2231–2239. doi:10.1056/NEJMoa0802268
76. Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med. 2008;358(21):2240–2248. doi:10.1056/NEJMoa0802315
77. Hauswirth WW, Aleman TS, Kaushal S, et al. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther. 2008;19(10):979–990. doi:10.1089/hum.2008.107
78. Miyazaki J, Takaki S, Araki K, et al. Expression vector system based on the chicken beta-actin promoter directs efficient production of interleukin-5. Gene. 1989;79(2):269–277. doi:10.1016/0378-1119(89)90209-6
79. Annear MJ, Mowat FM, Bartoe JT, et al. Successful gene therapy in older Rpe65-deficient dogs following subretinal injection of an adeno-associated vector expressing RPE65. Hum Gene Ther. 2013;24(10):883–893. doi:10.1089/hum.2013.146
80. Maguire AM, High KA, Auricchio A, et al. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: a Phase 1 dose-escalation trial. Lancet. 2009;374(9701):1597–1605. doi:10.1016/S0140-6736(09)61836-5
81. Jacobson SG, Cideciyan AV, Ratnakaram R, et al. Gene therapy for leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol. 2012;130(1):9–24. doi:10.1001/archophthalmol.2011.298
82. Testa F, Maguire AM, Rossi S, et al. Three-year follow-up after unilateral subretinal delivery of adeno-associated virus in patients with Leber congenital amaurosis type 2. Ophthalmology. 2013;120(6):1283–1291. doi:10.1016/j.ophtha.2012.11.048
83. Cideciyan AV, Hauswirth WW, Aleman TS, et al. Human RPE65 gene therapy for Leber congenital amaurosis: persistence of early visual improvements and safety at 1 year. Hum Gene Ther. 2009;20(9):999–1004. doi:10.1089/hum.2009.086
84. Wojno AP, Pierce EA, Bennett J. Seeing the light. Sci Transl Med. 2013;5(175):175–178. doi:10.1126/scitranslmed.3005798
85. Yavuz B, Kompella UB. Ocular drug delivery. Handb Exp Pharmacol. 2017;242:57–93.
86. Patel A, Cholkar K, Agrahari V, et al. Ocular drug delivery systems: an overview. World J Pharmacol. 2013;2(2):47–64. doi:10.5497/wjp.v2.i2.47
87. Maia M, Kellner L, de Juan E
88. Stout JT, Francis PJ. Surgical approaches to gene and stem cell therapy for retinal disease. Hum Gene Ther. 2011;22(5):531–535. doi:10.1089/hum.2011.060
89. Qi Y, Dai X, Zhang H, et al. Trans-corneal subretinal injection in mice and its effect on the function and morphology of the retina. PLoS One. 2015;10(8):e0136523. doi:10.1371/journal.pone.0136523
90. Ehlers JP, Petkovsek DS, Yuan A, et al. Intrasurgical assessment of subretinal tPA injection for submacular hemorrhage in the PIONEER study utilizing intraoperative OCT. Ophthalmic Surg Lasers Imaging Retina. 2015;46(3):327–332. doi:10.3928/23258160-20150323-05
91. Parikh S, Le A, Davenport J, et al. An alternative and validated injection method for accessing the subretinal space via a transcleral posterior approach. J Vis Exp. 2016;7(118):e54808. doi:10.3791/54808
92. Georgiadis A, Duran Y, Ribeiro J, et al. Development of an optimized AAV2/5 gene therapy vector for Leber congenital amaurosis owing to defects in RPE65. Gene Ther. 2016;23(12):857–862. doi:10.1038/gt.2016.66
93. Weber M, Rabinowitz J, Provost N, et al. Recombinant adeno-associated virus serotype 4 mediates unique and exclusive long-term transduction of retinal pigmented epithelium in rat, dog, and nonhuman primate after subretinal delivery. Mol Ther. 2003;7(6):774–781. doi:10.1016/S1525-0016(03)00098-4
94. Jacobson SG, Acland GM, Aguirre GD, et al. Safety of recombinant adeno-associated virus type 2- RPE65 vector delivered by ocular subretinal injection. Mol Ther. 2006;13(6):1074–1084. doi:10.1016/j.ymthe.2006.03.005
95. Boye SE, Boye SL, Lewin AS, Hauswirth WW. A comprehensive review of retinal gene therapy. Mol Ther. 2013;21(3):509–519. doi:10.1038/mt.2012.280
96. Kansara V, Muya L, Wan CR, Ciulla TA. Suprachoroidal delivery of viral and nonviral gene therapy for retinal diseases. J Ocul Pharmacol Ther. 2020;36(6):384–392. doi:10.1089/jop.2019.0126
97. Choi VW, McCarty DM, Samulski RJ. Host cell DNA repair pathways in adeno-associated viral genome processing. J Virol. 2006;80(21):10346–10356. doi:10.1128/JVI.00841-06
98. Dong B, Nakai H, Xiao W. Characterization of genome integrity for oversized recombinant AAV vector. Mol Ther. 2010;18(1):87–92. doi:10.1038/mt.2009.258
99. Wu Z, Yang H, Colosi P. Effect of genome size on AAV vector packaging. Mol Ther. 2010;18(1):80–86. doi:10.1038/mt.2009.255
100. McCarty DM, Monahan PE, Samulski RJ. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001;8(16):1248–1254. doi:10.1038/sj.gt.3301514
101. McCarty DM. Self-complementary AAV vectors; advances and applications. Mol Ther. 2008;16(10):1648–1656. doi:10.1038/mt.2008.171
102. Koilkonda RD, Chou T-H, Porciatti V, Hauswirth WW, Guy J. Induction of rapid and highly efficient expression of the human ND4 complex I subunit in the mouse visual system by self-complementary adeno-associated virus. Arch Ophthalmol. 2010;128(7):876–883. doi:10.1001/archophthalmol.2010.135
103. Petersen-Jones SM, Bartoe JT, Fischer AJ, et al. AAV retinal transduction in a large animal model species: comparison of a self-complementary AAV2/5 with a single-stranded AAV2/5 vector. Mol Vision. 2009;15:1835–1842.
104. Natkunarajah M, Trittibach P, McIntosh J, et al. Assessment of ocular transduction using single-stranded and self-complementary recombinant adeno-associated virus serotype 2/8. Gene Ther. 2007;15(6):463–467. doi:10.1038/sj.gt.3303074
105. Day TP, Byrne LC, Schaffer DV, Flannery JG. Advances in AAV vector development for gene therapy in the retina. Adv Exp Med Biol. 2014;801:687–693. doi:10.1007/978-1-4614-3209-8_86
106. Rayaprolu V, Kruse S, Kant R, et al. Comparative analysis of adeno-associated virus capsid stability and dynamics. J Virol. 2013;87(24):13150–13160. doi:10.1128/JVI.0141513
107. Pang JJ, Chang B, Kumar A, et al. Gene therapy restores vision-dependent behavior as well as retinal structure and function in a mouse model of RPE65 Leber congenital amaurosis. Mol Ther. 2006;13(3):565–572. doi:10.1016/j.ymthe.2005.09.001
108. Buck TM, Wijnholds J. Recombinant adeno-associated viral vectors (rAAV)-vector elements in ocular gene therapy clinical trials and transgene expression and bioactivity assays. Int J Mol Sci. 2020;21(12):4197. doi:10.3390/ijms21124197
109. Jacobson SG, Boye SL, Aleman TS, et al. Safety in nonhuman primates of ocular AAV2-RPE65, a candidate treatment for blindness in leber congenital amaurosis. Hum Gene Ther. 2006b;17(8):845–858. doi:10.1089/hum.2006.17.845
110. Pierce EA, Bennett J. The status of RPE65 gene therapy trials: safety and efficacy. Cold Spring Harb Perspect Med. 2015;5(9):a017285. doi:10.1101/cshperspect.a017285
111. Cideciyan AV, Aleman TS, Boye SL, et al. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc Natl Acad Sci. 2008;105(39):15112–15117. doi:10.1073/pnas.0807027105
112. Simonelli F, Maguire AM, Testa F, et al. Gene therapy for Leber’s congenital amaurosis is safe and effective through 1.5 years after vector administration. Mol Ther. 2010;18(3):643–650. doi:10.1038/mt.2009.277
113. Ashtari M, Cyckowski LL, Monroe JF, et al. The human visual cortex responds to gene therapy-mediated recovery of retinal function. J Clin Invest. 2011;121(6):2160–2168. doi:10.1172/JCI57377
114. Banin E, Bandah-Rozenfeld D, Obolensky A, et al. Molecular anthropology meets genetic medicine to treat blindness in the North African Jewish population: human gene therapy initiated in Israel. Hum Gene Ther. 2010;21(12):1749–1757. doi:10.1089/hum.2010.047
115. Le Meur G, Lebranchu P, Billaud F, et al. Safety and long-term efficacy of AAV4 gene therapy in patients with RPE65 leber congenital amaurosis. Mol Ther. 2018;26(1):256–268. doi:10.1016/j.ymthe.2017.09.014
116. Amado D, Mingozzi F, Hui D, et al. Safety and efficacy of subretinal readministration of an AAV2 vector in large animal models: implications for studies in humans. Sci Transl Med. 2010;2(21):21ra16. doi:10.1126/scitranslmed.3000659
117. Bennett J, Ashtari M, Wellman J, et al. AAV2 gene therapy readministration in three adults with congenital blindness. Sci Transl Med. 2012;4(120):120ra115. doi:10.1126/scitranslmed.3002865
118. Bennett J, Wellman J, Marshall KA, et al. Safety and durability of effect of contralateral-eye administration of AAV2 gene therapy in patients with childhood-onset blindness caused by RPE65 mutations: a follow-on phase 1 trial. Lancet. 2016;388(10045):661–672. doi:10.1016/S0140-6736(16)30371-3
119. Russell S, Bennett J, Wellman JA, et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: a randomised, controlled, open-label, Phase 3 trial. Lancet. 2017;390(10097):849–860. doi:10.1016/S0140-6736(17)31868-8
120. Darrow JJ. Luxturna: FDA documents reveal the value of a costly gene therapy. Drug Discov Today. 2019;24(4):949–954. doi:10.1016/j.drudis.2019.01.019
121. Chung DC, McCague S, Yu ZF, et al. Novel mobility test to assess functional vision in patients with inherited retinal dystrophies. Clin Exp Ophthalmol. 2018;46(3):
122. Rodrigues GA, Shalaev E, Karami TK, Cunningham J, Slater NKH, Rivers HM. Pharmaceutical development of AAV-based gene therapy products for the eye. Pharm Res. 2018;36(2):29. doi:10.1007/s11095-018-2554-7
123. Novartis announces landmark EU approval for one-time gene therapy Luxturna® to restore vision in people with rare inherited retinal disease [Internet]. Novartis; [
124. Moiseyev G, Chen Y, Takahashi Y, Wu BX, Ma JX. RPE65 is the isomerohydrolase in the retinoid visual cycle. Proc Nat Acad Sci. 2005;102(35):12413–12418. doi:10.1073/pnas.0503460102
125. Mackay DS, Ocaka LA, Borman AD, et al. Screening of SPATA7 in patients with leber congenital amaurosis and severe childhood-onset retinal dystrophy reveals disease-causing mutations. Invest Ophthal Vis Sci. 2011;52(6):3032–3038. doi:10.1167/iovs.10-7025
126. Li Y, Wang H, Peng J, et al. Mutation survey of known LCA gene and loci in the Saudi Arabian population. Invest Ophthalmol Vis Sci. 2009;50(3):1336–1343. doi:10.1167/iovs.08-2589
127. Boldt K, Mans DA, Won J, et al. Disruption of intraflagellar protein transport in photoreceptor cilia causes Leber congenital amaurosis in humans and mice. J Clin Invest. 2011;121(6):2169–2180. doi:10.1172/JCI45627
128. Dharmaraj S, Li Y, Robitaille JM, et al. A novel locus for Leber congenital amaurosis maps to chromosome 6q. (letter). Am J Hum Genet. 2000;66(1):319–326. doi:10.1086/302719
129. Mohamed MD, Topping NC, Jafri H, Raashed Y, McKibbin MA, Inglehearn CF. Progression of phenotype in Leber’s congenital amaurosis with a mutation at the LCA5 locus. Br J Ophthalmol. 2003;87(4):473–475. doi:10.1136/bjo.87.4.473
130. Jacobson SG, Cideciyan AV, Huang WC, et al. Leber congenital amaurosis: genotypes and retinal structure phenotypes. Adv Exp Med Biol. 2016;854:169–175.
131. Nichols LL
132. Kimura A, Singh D, Wawrousek EF, Kikuchi M, Nakamura M, Both ST. PCE-1/RX and OTX/CRX interactions are necessary for photoreceptor-specific gene expression. J Biol Chem. 2000;14(275):1152–1160. doi:10.1074/jbc.275.2.1152
133. Freund CL, Wang QL, Chen S, et al. De novo mutations in the CRX homeobox gene associated with Leber congenital amaurosis. Nat Genet. 1998;18(4):311–312. doi:10.1038/ng0498-311
134. Swaroop A, Wang QL, Wu W, et al. Leber congenital amaurosis caused by a homozygous mutation (R90W) in the homeodomain of the retinal transcription factor CRX: direct evidence for the involvement of CRX in the development of photoreceptor function. Hum Mol Genet. 1999;8(2):299–305. doi:10.1093/hmg/8.2.299
135. Akagi T, Mandai M, Ooto S, et al. Otx2 homeobox gene induces photoreceptor-specific phenotypes in cells derived from adult iris and ciliary tissue. Invest Ophthal Vis Sci. 2004;45(12):4570–4575. doi:10.1167/iovs.04-0697
136. Koenekoop RK, Wang H, Majewski J, et al. Mutations in NMNAT1 cause Leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat Genet. 2012;44(9):1035–1039. doi:10.1038/ng.2356
137. Burnight ER, Wiley LA, Drack AV, et al. CEP290 gene transfer rescues Leber congenital amaurosis cellular phenotype. Gene Ther. 2014;21(7):
138. Collart FR, Huberman E. Cloning and sequence analysis of the human and Chinese hamster inosine-5-prime-monophosphate dehydrogenase cDNAs. J Biol Chem. 1988;263:15769–15772.
139. Bowne SJ, Sullivan LS, Mortimer SE, et al. Spectrum and frequency of mutations in IMPDH1 associated with autosomal dominant retinitis pigmentosa and Leber congenital amaurosis. Invest Ophthal Vis Sci. 2006;47(1):34–42. doi:10.1167/iovs.05-0868
140. Molday LL, Djajadi H, Yan P, et al. RD3 gene delivery restores guanylate cyclase localization and rescues photoreceptors in the Rd3 mouse model of Leber congenital amaurosis 12. Hum Mol Genet. 2013;22(19):3894–3905. doi:10.1093/hmg/ddt244
141. Preising MN, Hausotter-Will N, Solbach MC, Friedburg C, Rüschendorf F, Lorenz B. Mutations in RD3 are associated with an extremely rare and severe form of early onset retinal dystrophy. Invest Ophthal Vis Sci. 2012;53(7):3463–3472. doi:10.1167/iovs.12-9519
142. Friedman JS, Chang B, Kannabiran C, et al. Premature truncation of a novel protein, RD3, exhibiting subnuclear localization is associated with retinal degeneration. Am J Hum Genet. 2006;79(6):1059–1070. doi:10.1086/510021
143. Xi Q, Pauer GJ, Marmorstein AD, Crabb JW, Hagstrom SA. Tubby-like protein 1 (TULP1) interacts with F-actin in photoreceptor cells. Invest Ophthalmol Vis Sci. 2005;46(12):4754–4761. doi:10.1167/iovs.05-0693
144. Mataftsi A, Schorderet DF, Chachoua L, et al. Novel TULP1 mutation causing leber congenital amaurosis or early onset retinal degeneration. Invest Ophthalmol Vis Sci. 2007;48(11):5160–5167. doi:10.1167/iovs.06-1013
145. Zhang L, Lim SL, Du H, et al. High temperature requirement factor A1 (HTRA1) gene regulates angiogenesis through transforming growth factor-β family member growth differentiation factor 6. J Biol Chem. 2012;287(2):1520–1526. doi:10.1074/jbc.M111.275990
146. Asai-Coakwell M, French CR, Berry KM. GDF6, a novel locus for a spectrum of ocular developmental anomalies. Am J Hum Genet. 2007;80(2):306–315. doi:10.1086/511280
147. The online mendelian inheritance in man website. Available from: www.omim.org.
148. Bascom RA, Connell G, Garcia-Heras J, et al. Molecular and ultrastructural characterization of the products of the human retinopathy candidate genes ROM1 and RDS. (Abstract). Am J Hum Genet. 1990;47(suppl):A101.
149. Wang X, Wang H, Sun V, et al. Comprehensive molecular diagnosis of 179 Leber congenital amaurosis and juvenile retinitis pigmentosa patients by targeted next generation sequencing. J Med Genet. 2013;50(10):674–688. doi:10.1136/jmedgenet-2013-101558
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.