Back to Journals » OncoTargets and Therapy » Volume 13
Von Hippel-Lindau Disease: Current Challenges and Future Prospects
Authors Gläsker S, Vergauwen E, Koch CA, Kutikov A, Vortmeyer AO
Received 14 February 2020
Accepted for publication 20 May 2020
Published 16 June 2020 Volume 2020:13 Pages 5669—5690
DOI https://doi.org/10.2147/OTT.S190753
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Leo Jen-Liang Su
Sven Gläsker,1,2 Evelynn Vergauwen,2,3 Christian A Koch,4 Alexander Kutikov,4 Alexander O Vortmeyer5
1Neurosurgical Practise Lake Constance, Singen (Hohentwiel), Germany; 2Department of Neurosurgery, VUB University Medical Center Brussels, Brussels, Belgium; 3Department of Neurology, University Hospital Antwerp, Antwerp, Belgium; 4Fox Chase Cancer Center, Philadelphia, PA, USA; 5Department of Pathology, Indiana University-Purdue University, Indianapolis, IN, USA
Correspondence: Sven Gläsker
Department of Neurosurgery, VUB University Medical Center Brussels, Laarbeeklaan 101, Brussels 1090, Belgium
Email [email protected]
Abstract: Understanding of molecular mechanisms of tumor growth has an increasing impact on the development of diagnostics and targeted therapy of human neoplasia. In this review, we summarize the current knowledge on molecular mechanisms and their clinical implications in von Hippel-Lindau (VHL) disease. This autosomal dominant tumor syndrome usually manifests in young adulthood and predisposes affected patients to the development of benign and malignant tumors of different organ systems mainly including the nervous system and internal organs. A consequent screening and timely preventive treatment of lesions are crucial for patients affected by VHL disease. Surgical indications and treatment have been evaluated and optimized over many years. In the last decade, pharmacological therapies have been evolving, but are largely still at an experimental stage. Effective pharmacological therapy as well as detection of biomarkers is based on the understanding of the molecular basis of disease. The molecular basis of von Hippel-Lindau disease is the loss of function of the VHL protein and subsequent accumulation of hypoxia-inducible factor with downstream effects on cellular metabolism and differentiation. Organs affected by VHL disease may develop frank tumors. More characteristically, however, they reveal multiple separate microscopic foci of neoplastic cell proliferation. The exact mechanisms of tumorigenesis in VHL disease are, however, still not entirely understood and knowledge on biomarkers and targeted therapy is scarce.
Keywords: Von Hippel-Lindau, VHL, tumor suppressor gene, neuroendocrine tumor, pancreatic tumor, pheochromocytoma, tumor formation, second hit, hemangioblastoma, renal cancer
Overview of VHL Disease
Von Hippel-Lindau (VHL) disease is an autosomal dominantly inherited tumor syndrome. The disease usually manifests in young adulthood and predisposes affected patients to the development of benign and malignant tumors of different organ systems, mainly including nervous system and internal organs. The incidence of VHL disease has been assessed as one in 36,0001,2 and the penetrance is higher than 90%.3
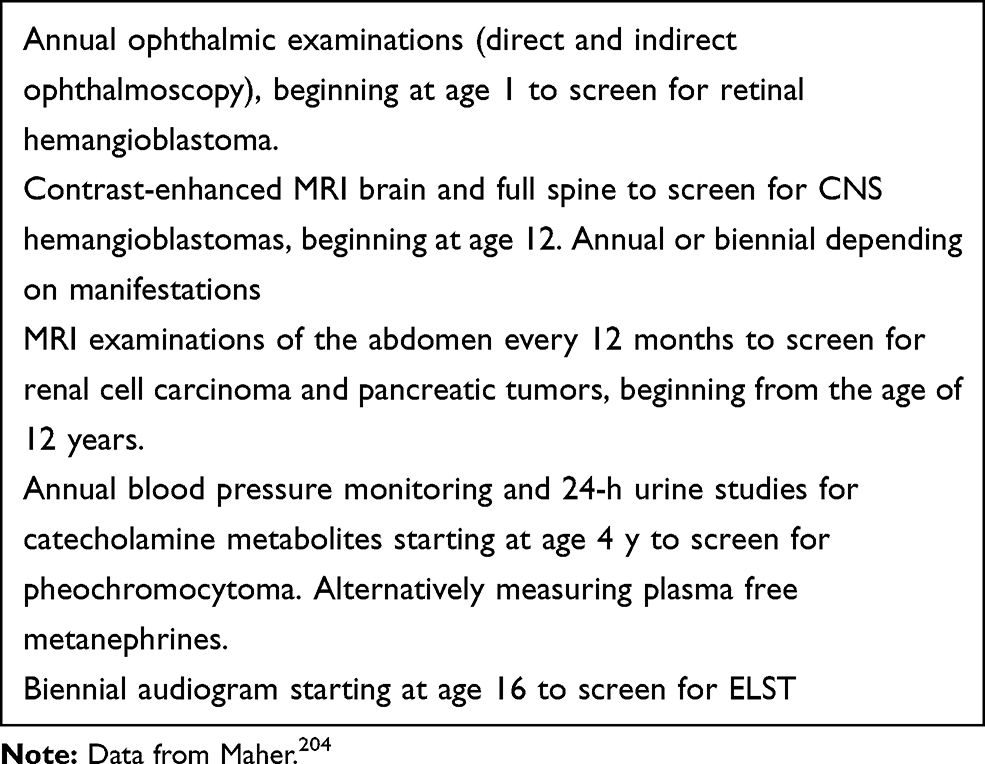 |
Box 1 Example of a Routine Surveillance Protocol for Von Hippel–Lindau Disease |
VHL disease is named after German ophthalmologist Eugen von Hippel, who identified and described retinal hemangioblastomas,4 and Arvid Lindau, a Swedish Pathologist, who discovered the coincidental occurrence of retinal and cerebellar hemangioblastoma with tumors and cysts in internal organs. He published the clinical spectrum of VHL disease.5,6
Clinically, the patients are divided into different groups: Patients with VHL type 1 predominantly without pheochromocytoma, and VHL type 2 predominantly with pheochromocytoma.7 VHL type 2 is further subdivided into type 2A (with renal cancer) and type 2B (without renal cancer). In type 2C, affected patients develop solely pheochromocytomas.8
Molecular and Histomorphological Basis of VHL Disease
VHL inactivation has a variety of different effects on human tissue on molecular as well as on histomorphological levels. Since there is no animal model available to date, which has the full VHL phenotype, most knowledge is based on restricted knockout models or on observations in human tissues.
Molecular Basis of VHL Disease
Patients affected by VHL disease carry a germline mutation of the VHL tumor suppressor gene.9 Five-hundred different pathogenic germline mutations have been identified in families with VHL disease.10 The VHL protein (pVHL) interacts with elongins B, C and Cullin-2 to form the VBC complex, an E3 ubiquitin ligase.11 This complex mediates ubiquitin-mediated degradation.12–14 Biallelic inactivation of VHL is thought to be the basis of tumorigenesis in VHL disease. Reintroduction of the VHL function can reverse some effects of inactivation.15 The consequences of VHL inactivation can be divided into HIF-dependent and HIF-independent effects (Figure 1).
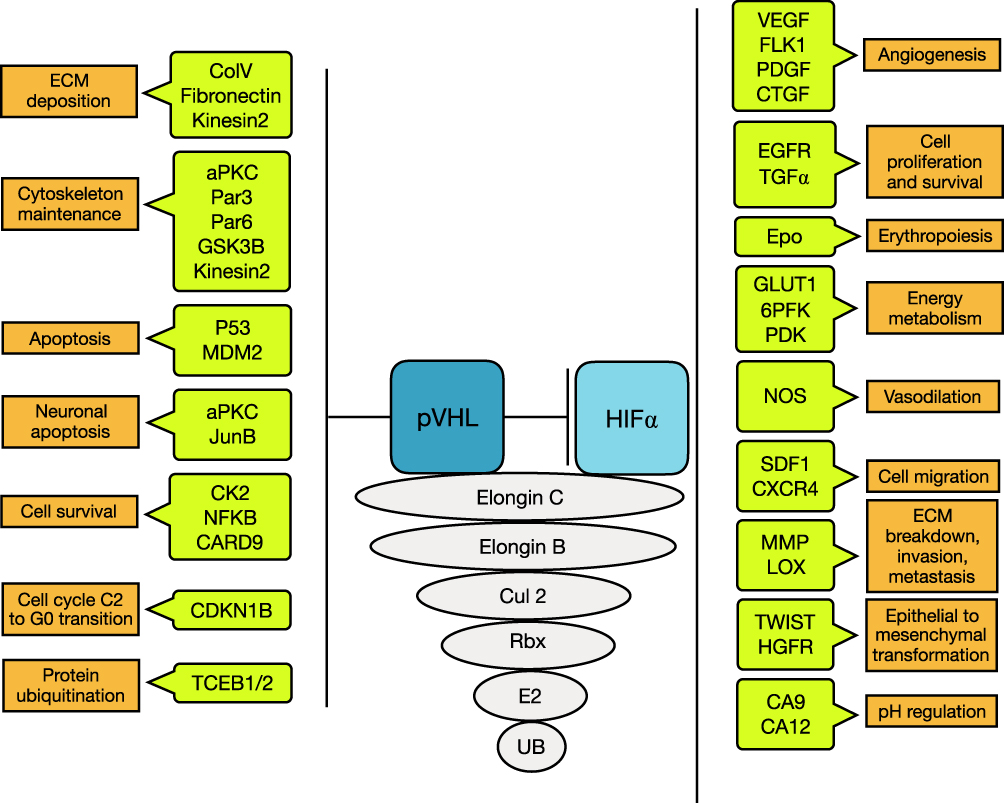 |
Figure 1 VHL protein functions: HIF independent and HIF dependent. Abbreviations: aPKC, atypical protein kinase C; CA9/12, carbonic anhydrase 9/12; CARD9, caspase recruitment domain-containing protein 9; CDKN1B, cyclin-dependent kinase inhibitor 1B; CK2, protein kinase CK2; CoV, type V collagen; CTGF, connective tissue growth factor; Cul2, Cullin 2; CXCR4, CX chemokine receptor type 4; ECM, extracellular matrix; EGFR, epidermal growth factor; FLK1, fetal liver kinase 1; GLUT1, glucose transporter 1; GSK3B, glycogen synthase kinase 3 beta; HIF, hypoxia-inducible factor; HGFR, hepatocyte growth factor; LOX, lysol oxidase; MDM2, mouse double minute 2 homolog; MMP, matrix metalloproteinases; NFKB, nuclear factor kappa-light-chain-enhancer of activated B cells; NOS, nitric oxide synthase; PDGF, platelet-derived growth factor; RBX1, ring box protein 1; SDF1, stromal cell-derived factor 1; TCEB1/2, transcription elongation factor B1/2; TGF, transforming growth factor; TWIST, twist related protein; VEGF, vascular endothelial growth factor; 6PFK, 6 phosphofructokinase. |
HIF-Dependent Effects
HIF is a heterodimeric transcription factor consisting of an unstable α and a stable β subunit. Different HIFα genes have been identified in the human genome.16 HIF1α and HIF2α do not appear to be fully redundant in function. Although germline knock-out of HIF1α and HIF2α results in embryonic lethality the timing and cause of death appear to differ.17 Moreover, post-natal inactivation of HIF1α and HIF2α leads to differing phenotypes as well. The two proteins are also differentially expressed in VHL lesions, where immature cells show exclusive activation of HIF2α in contrast to frank tumors that show activation of both HIF2α and HIF1α.18,19
Many of the HIF-dependent proteins are involved in oxygen sensation and regulation and include genes involved in the uptake and metabolism of glucose (GLUT-1, 6-PFK, PDK), angiogenesis (VEGF, PDGF, CTGF), control of extracellular pH (CA9), mitogenesis (TGFα), and erythropoiesis (erythropoietin).20–23 EGFR and TGFα promote cell proliferation and survival. CXCR4 and its ligand SDF1 stimulate chemotaxis and may also contribute to tumor cell invasion and metastases. MMP1 and lysyl oxidase (encoded by LOX) are implicated in ECM breakdown and tumor cell invasion/migration. Finally, dysregulation of TWIST and activation of HGFR (encoded by c-MET) are involved in epithelial‐to‐mesenchymal transition (EMT).
Deciphering of the molecular mechanisms of VHL functioning has had a large impact on the understanding of oxygen sensation in mammalian cells and the development of neoplasia in general.24 The Nobel Prize in Physiology or Medicine 2019 was awarded jointly to William G. Kaelin, Peter J. Ratcliffe and Gregg L. Semenza “for their discoveries of how cells sense and adapt to oxygen availability.”
HIF-Independent Effects
In addition to HIF degradation, the VHL protein is involved in HIF-independent cellular processes, which may be connected with tumorigenesis. It regulates the proper deposition of fibronectin and collagen IV within the extracellular matrix.25 It furthermore stabilizes microtubules and maintains the primary cilium. The VHL protein also activates and stabilizes p53 and, in neuronal cells, induces apoptosis by downregulation of Jun-B.25 Acute loss of VHL protein causes a senescent-like phenotype. It appears that it increases p400 activity, which results in inactivation (hypophosphorylation) of the retinoblastoma protein (pRb) and prevents senescence.26 The VHL protein can also act as an adaptor to bind CK2, which inactivates the NF-kB agonist CARD9, leads to inhibition of NF-kB signaling and overall inhibits cell survival. It is furthermore needed for primary cilium function by microtubule stabilization and binding with aPKC and the polarity proteins Par3 and Par6.
The different HIF dependent and HIF-independent molecular functions imply several possible ways of how VHL related tumorigenesis may occur and which molecular targets could possibly be used for therapy. Despite the broad understanding of VHL functioning, it remains largely unclear, how a “normal” cell of a VHL patient which carries one mutated VHL allele is transformed into a tumor cell. Simple inactivation of the second VHL allele seems insufficient for tumor growth27–29 and does also not explain the peculiar organ distribution of the disease with mainly CNS and internal organs involved. Some keys to these unsolved questions are discussed in the next section on histomorphological tumorigenesis.
Histomorphological Basis of VHL Disease
Organs affected by VHL disease may develop frank tumors. More characteristically, however, they reveal multiple separate microscopic foci of neoplastic cell proliferation. Detailed histologic studies – combined with molecular analysis – were performed on autopsy material after processing the entire anatomic structure (eg studies on entire spinal cord (Figures 2 and 3), cerebellum, epididymis (Figures 4 and 5), or vestibular aqueduct) allowing for variably precise quantitation of these neoplastic foci. Studies on both the entire VHL spinal cord and surgically resected spinal cord tumors revealed these microscopic neoplastic foci to represent early, VHL-inactivated precursor lesions. This chapter describes histologic changes occurring organs affected by VHL disease with special consideration of early tumorigenesis.
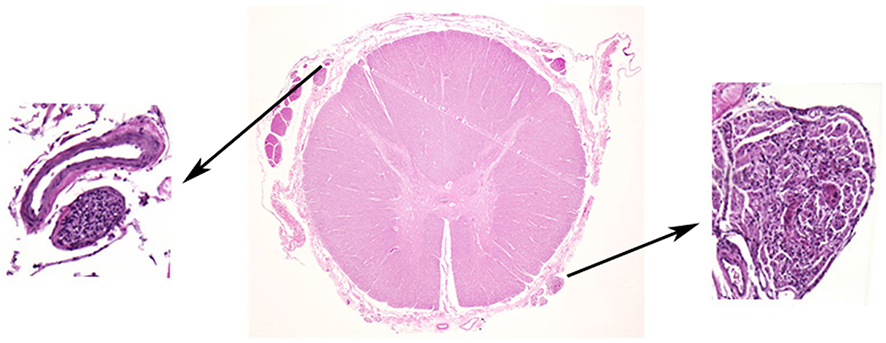 |
Figure 2 Early VHL pathogenesis in the nervous system: Hemangioblastoma precursors. Histologic examination of grossly normal-appearing nerve root tissue of VHL patients reveals numerous microscopic hemangioblastoma precursor structures within nerve roots. Reprinted with permission from Vortmeyer AO, Tran MG, Zeng W, et al Evolution of VHL tumourigenesis in nerve root tissue. J Pathol. 2006;210(3):374–382.18 |
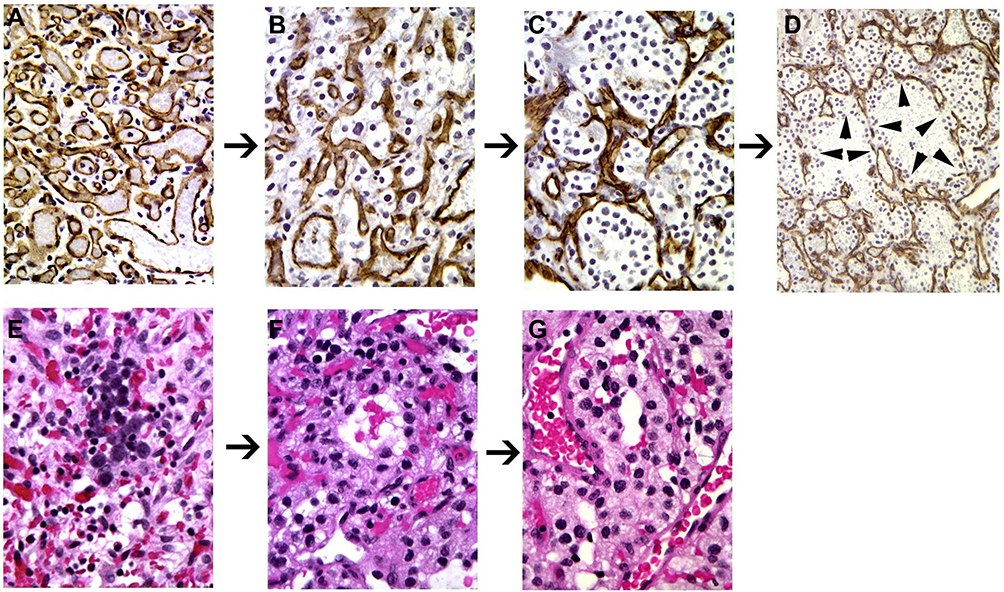 |
Figure 3 Hemangioblastoma progression. Proposed structural progression of hemangioblastoma from mesenchymal (A) to epitheloid (B and C) and vasculogenetic architecture/extramedullary erythropoiesis (D–G); (A–D), immunohistochemical stain for CD31; (E–G), (H and E) stain. Immunohistochemistry for CD31 was performed to better differentiate reactive vascular cells (positively staining cells) from neoplastic cells (negatively staining cells). (H and E) stain was performed to demonstrate erythropoiesis within epitheloid and vasculogenetic structures. Reprinted from Exp Mol Pathol, 96(2), Glasker S, Smith J, Raffeld M, Li J, Oldfield EH, Vortmeyer AO, VHL-deficient vasculogenesis in hemangioblastoma 162-167, Copyright (2014), with permission from Elsevier.205 |
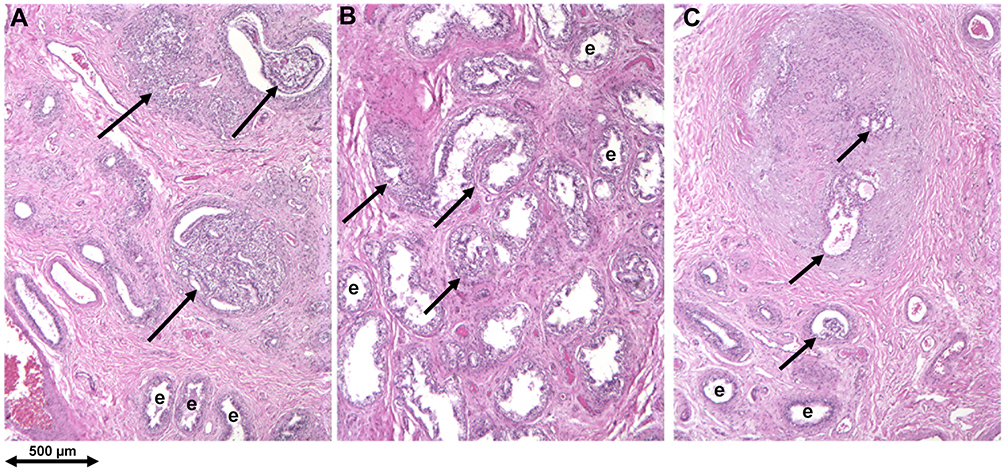 |
Figure 4 Early VHL pathogenesis in the epididymis: cystadenoma precursors. (A–C) Multifocality of cystadenoma precursor structures: “Tumor-free” VHL epididymis contains multifocal microscopic precursors in the efferent ductule compartment of the caput (marked by arrows); e= normal efferent ductules. Republished with permission of John Wiley & Sons-Books, from Epididymal cystadenomas and epithelial tumorlets: Effects of VHL deficiency on human epididymis, Glasker S, Tran MG, Shively SB, et al. J Pathol. 210(1):32–41. permission conveyed through Copyright Clearance Center, Inc. Copyright © 2006 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.48 |
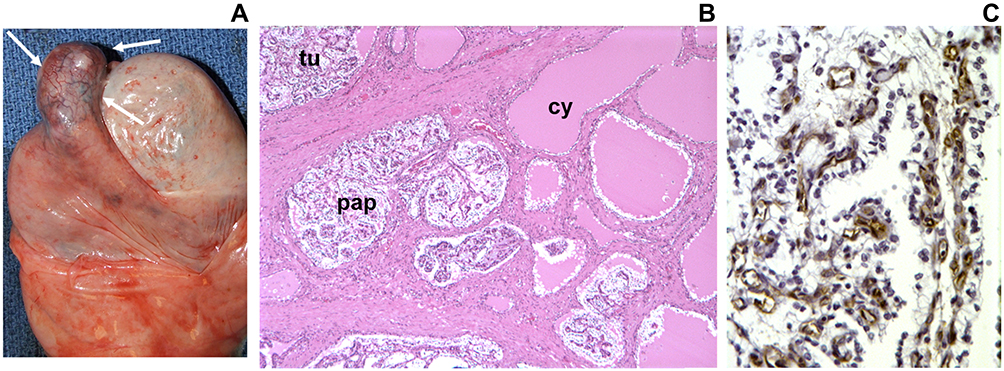 |
Figure 5 Papillary cystadenoma of the epididymis. (A), Gross examination of VHL epididymis shows enlargement of the caput epididymis (white arrows). (B), Cystadenomas reveal papillary (pap), tubular (tu) and cystic (cy) architecture. (C), Immunohistochemical staining with anti-CD31 reveals extensive vascularization of the tumor stroma. Numerous reactive vascular cells are in direct contact with overlying neoplastic epithelial cells. Republished with permission of John Wiley & Sons-Books, from Epididymal cystadenomas and epithelial tumorlets: Effects of VHL deficiency on human epididymis, Glasker S, Tran MG, Shively SB, et al J Pathol. 210(1):32–41. permission conveyed through Copyright Clearance Center, Inc. Copyright © 2006 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.48 |
Tumors Involving CNS
The histology of neurosurgically resected hemangioblastomas shows a high degree of variability. Cytologically, hemangioblastomas are predominantly composed of two types of cells. The first cytologic component is characterized by conspicuous neoplastic and VHL-inactivated clear cells that are conventionally called “stromal” cells. “Stromal” cells do not exist in normal nervous system tissue, and their origin has been controversial. While “stromal” cells are lipid- and glycogen-rich cells with abundant clear or bubbly cytoplasm in larger tumors, they are smaller and less conspicuous in small tumors or precursor structures. The other cytologic component of hemangioblastomas is represented by abundant blood vessels. The blood vessels do not show VHL inactivation30,31 and therefore represent reactive angiogenesis. Intense reactive angiogenesis is a characteristic feature of hemangioblastoma and explained by the expression of HIF, VEGF and other angiogenic factors in stromal cells.32,33
Hemangioblastomas show not only cytological but also marked architectural variation. Tumor cells may be scattered within a matrix of abundant capillaries, referred to as “reticular”34 or “mesenchymal”35 architecture. Tumors may also reveal epitheloid tumor cell clustering, referred to as “cellular”34 or “epitheloid”35 architecture. A transition from mesenchymal to epitheloid architecture occurs gradually with tumor growth. Hemangioblastomas smaller than 8 mm3 in size show exclusively mesenchymal architecture, while epitheloid features may be developed in tumors larger than 6 mm3; epitheloid features have been consistently observed in tumors larger than 737 mm3.35 A markedly elevated Ki67 proliferation index in epitheloid areas suggests that epitheloid features are associated with accelerated tumor growth.35
Biologically, the profound cytologic and architectural transformation from mesenchymal to epitheloid tumor closely resembles stages of embryonic hemangioblastic transformation.36 The capacity of “stromal” cells to differentiate into red blood cells has been subsequently demonstrated37,38 and a shared protein expression profile with hemangioblast precursor cells has been identified.28 Therefore, multiple lines of evidence have confirmed the “stromal” cells to represent hemangioblastic precursor cells.
The histologic evolution of hemangioblastomas is not completely understood. The earliest stages of VHL tumorigenesis are represented by microscopic foci, undetectable radiologically or by the naked eye. Because of their minute size, these foci can only be detected by detailed histological analysis of normal-appearing tissues of VHL patients. Moreover, to obtain information on the number and distribution of microscopic lesions, tissue blocks need to be step-sectioned at 50-micron intervals. Such studies have been performed on tissues of VHL patients that had been obtained at autopsy. Analysis of a series of cases revealed that precursor lesions of spinal cord hemangioblastomas were exclusively located in nerve root tissue, dorsal roots being far more frequently involved than anterior roots. Tumors preferentially developed from dorsal root precursors in close proximity to the spinal cord.29,39 In general, precursor structures are detected in far greater abundance than frank tumors;29,39 therefore, precursor structures have extremely low proliferative potential, and only a small subset of precursor structures will develop into tumors. In the cerebellum, precursors originate in the molecular layer.19 Numerically, precursor structures are far more frequent in dorsal root tissue compared to cerebellar tissue. It, therefore, appears that precursors in the cerebellum have a higher probability to develop into clinically relevant tumor than nerve root precursors.19
Histologically, precursor CNS lesions reveal a sharply demarcated zone containing abundant capillaries and scattered small VHL-deficient cells;29 by immunohistochemistry, VHL-deficient cells have revealed HIF2 activation.39 Significant changes occur during the progression of a precursor structure to tumor.39 Cytologically, the VHL-deficient cells markedly increase in size and develop abundant clear, vacuolated or eosinophilic cytoplasm. While in the earliest stages of tumorigenesis VHL-deficient tumor cells show – similar to the VHL-deficient cells in the precursor lesions – a diffuse distribution pattern (“mesenchymal architecture”), further tumor growth consistently reveals tumor cells to form small clusters (“epitheloid architecture”) in parts of the tumor.35
The retina develops from extensions of the neural tube, providing justification to include retinal hemangioblastomas among the craniospinal hemangioblastomas. Furthermore, histologic features of small and larger retinal tumors are identical to those of nerve root and cerebellar hemangioblastomas. Analogous to CNS tumors, retinal hemangioblastomas are composed of VHL-inactivated tumor cells with intense reactive vascularization40,41 and unrelated to capillary hemangiomas occurring elsewhere in the body.
Endolymphatic sac tumors were originally described by Heffner as adenocarcinomas of probable endolymphatic sac origin.42 Endolymphatic sac tumors in VHL disease are extensively vascularized tumors forming prominent papillary structures, the papillae being lined by a single row of cuboidal epithelial cells. Cysts lined by single rows of cuboidal cells are frequently present, and epitheloid clear cell proliferation is occasionally detected.27 Tumors larger than 10 mm in size usually present with bony erosion, while the exact border between endolymphatic duct and sac is not clearly defined, it is of clinical relevance that tumors appear to originate most frequently from intraosseous portions of endolymphatic sac/duct epithelium.27
Studies on clinically uninvolved endolymphatic sac and duct of VHL patients showed evidence for VHL-inactivated cells lining cysts or papillary projections; in analogy to studies in other tissues these microscopically small cystic or papillary structures may represent early stages of endolymphatic sac tumors.27
Visceral Tumors
The diseased VHL kidney is characterized by renal cysts and clear cell carcinoma. These pathologic changes show wide differences in growth.43 Histologically, solid tumors and cysts are composed of clear cells and associated with intense vascularization.44 There is a histopathologic continuum ranging from benign cysts and atypical cysts to carcinoma.44 Numerically, by far the most pathogenic events in the kidney are represented by microscopic cystic and solid neoplasms.45 VHL gene function is inactivated in the smallest foci of microscopic neoplasia.46,47 All clear cell tumors in the kidney are designated renal cell carcinoma, although they need to reach at least 3 cm size to acquire the capacity to metastasize. Histologically, larger renal clear cell tumors may develop frankly malignant features including tumor necrosis and cytologic anaplasia with mitotic activity.
Epididymal cystadenomas are component tumors of VHL disease composed of cuboidal epithelial cells, with papillary, tubular and cystic architecture, and prominent angiogenicity. Numerous tumor precursor elements were detected by a thorough analysis of nontumorous epididymides obtained at autopsy (Figure 4). Topographically, precursor structures were confined to the efferent ductules of the caput epididymis. Precursor structures were characterized by the formation of epithelial cysts and prominent epithelial proliferation into the lumina of cysts and efferent ductules.48 A 3dimensional reconstruction of intraductular papillary neoplastic growth revealed the complex intermingling of pre-existent anatomic structure and slow neoplastic growth creating a hamartomatous appearance.49 Since epididymal cystadenomas are rare neoplasms, the occurrence of bilateral tumors is highly associated with VHL disease.50
Papillary cystadenoma of the broad ligament occurs less frequently than epididymal cystadenoma. Morphological and immunohistochemical features strongly overlap. Morphologic and genetic studies have established this tumor as a VHL component tumor, and it is considered as female counterpart tumor of epididymal cystadenoma.51
Microcystic adenomas of the pancreas (serous cystadenomas) are tumors composed of small cysts lined by flattened or cuboidal glycogen-rich cells.52 After the initial demonstration of VHL gene deletion and mutation in microcystic adenomas,53 a more detailed analysis of the VHL pancreas revealed the presence of numerous small microcystic adenomas as well as VHL gene-inactivated pancreatic cysts; most cysts were of microscopic size suggesting a continuum of development from the pancreatic cyst(s) to microcystic adenoma.54
Pancreatic neuroendocrine tumors were established as VHL component tumors in 1998.55 Compared to sporadic neuroendocrine tumors, VHL-associated neuroendocrine tumors more frequently exhibit clear cell morphology which may be challenging to differentiate from metastatic renal cell carcinoma or pancreatic microcystic adenoma which may occur in the same pancreas.55
Pheochromocytomas and paragangliomas are associated with multiple hereditary disorders. The histologic features are distinguished from other VHL disease-associated clear cell neoplasms by more variable, frequently polyhedral cytology, basophilic and finely granulated cytoplasm and cytoplasmic bodies in both familial and nonfamilial cases.56 In contrast, analysis of VHL disease-associated pheochromocytomas revealed tumor cells with clear or amphophilic, inclusion-free cytoplasm that were more intensely intermixed with vascular cells.57
In conclusion, the characterization of precursor structures has provided more precise insight on the anatomic and cytologic origin of neoplastic processes associated with VHL disease. In all anatomic structures investigated so far, precursor structures were far more numerous than frank tumors. Therefore, VHL inactivation (“second hit”) is necessary, but insufficient for the development of a frank tumor. In the future, comparative molecular analysis of frank tumor and precursor structures may be helpful to identify a “third hit” promoting tumorigenesis from precursor material.
VHL Disease Manifestations and Treatment
Patients affected by VHL disease develop specific types of heavily vascularized tumors in a highly selective subset of organs. Multiple and bilateral tumors occur frequently. Affected organs and lesions include retinal hemangioblastomas, cerebellar, brainstem and spinal cord hemangioblastomas, endolymphatic sac tumors, renal cell carcinomas, pheochromocytomas, pancreatic cysts, microcystic serous adenomas, neuroendocrine tumors, as well as epididymal cystadenomas.58–61
Retinal Hemangioblastomas
Retinal hemangioblastomas are benign tumors that frequently occur in multiplicity and bilaterally in VHL patients. Expression of high levels of VEGF in these tumors causes hypervascularization, vascular leakage and eventually retinal detachment.
Annual screening by fundoscopy and if necessary fluorescence angiography is recommended for VHL patients starting at 1 year of age. Early treatment of asymptomatic lesions is recommended. Laser coagulation or cryotherapy can control the majority of peripheral retinal hemangioblastomas.62,63 Larger tumors can be treated by vitrectomy.64 Optic disc hemangioblastomas should be monitored without treatment. With regular screening and early treatment of detected lesions, visual prognosis for VHL patients is in general good: In one major study approximately 8% of the eyes of VHL patients had poor visual acuity of 20/200 or worse with approx. 8% of these eyes requiring enucleation.65
Pharmacological treatments are in experimental stages and are discussed in the section on targeted therapies.
Craniospinal Hemangioblastomas
Hemangioblastomas are benign slow-growing highly vascularized tumors. They are frequently the first manifestation of VHL disease. These patients frequently develop multiple hemangioblastomas.66 The hemangioblastoma tumor burden in VHL disease is associated with germline deletions and male sex.67 Depending on their size and location hemangioblastomas cause different neurologic symptoms.66,67 It is usually an associated pseudocyst or syrinx and not the tumor itself which causes the symptoms.68 The syrinx is caused by vascular leakage and is frequently larger than the tumor itself.66 Polyglobulia occurs in 10% of patients69 and may cause thrombosis. Removal of the largest hemangioblastoma usually resolves polyglobulia.69
Treatment is primarily surgical and is recommended for all symptomatic tumors and for all tumors causing CSF obstruction.66,70,71 If surgery is not possible, radiation therapy may be an alternative, however, its effectiveness remains controversial.72–74 Pharmacotherapy of CNS hemangioblastoma is at an experimental stage and is discussed in the section on targeted therapy.
Endolymphatic Sac Tumors
These histologically benign tumors originate in the vestibular aqueduct and grow locally invasive into the petrous bone. Patients will typically present with hypoakusis or hearing loss (100%), tinnitus (77%), dysequilibrium (62%), and facial paresis (8%).75 Tumor-associated intralabyrinthine hemorrhage may lead to acute hearing loss and vestibulopathy due to endolymphatic hydrops.76 All patients with endolymphatic sac tumors (ELSTs) should undergo diagnostic testing for VHL mutations since endolymphatic sac tumors can be the first manifestation of VHL disease.
In general, early surgical resection is recommended to enhance the possibility of complete resection and avoid hearing loss or vestibular symptoms, which may be irreversible.77 The timing of surgery depends on the severity of symptoms, the slow but variable growth of the tumors, the possibility of injury to the 7th and 8th cranial nerves and possible bilateral occurrence. The role of radiation in the treatment of these tumors remains controversial.78 Description of pharmacological therapies is scarce and is discussed in the section on targeted therapy.
Renal Manifestations
Patients with VHL disease may develop renal cysts and renal cell carcinomas (RCCs).46,79–83 VHL-disease-associated RCCs tend to be low grade and minimally invasive,84 their rate of growth varies widely.85,86 VHL alterations are also observed in 60–70% of sporadic RCCs.87
The goal of therapy is the prevention of metastasis and maintenance of kidney function. Most authors recommend nephron-sparing surgery for carcinomas that exceed 3 cm in size.79,88 Other authors suggest that a diameter of 4 cm is also acceptable.89 The “3 cm rule” takes into account the overall survival and quality of life (considering potential premature renal failure and need for dialysis).
Although the gold standard for treating renal tumors is open and minimally invasive partial nephrectomy, alternative therapies including cryotherapy and radiofrequency ablation are also presently utilized.90 Pharmacological therapy is used for metastatic disease and is discussed in the section on targeted therapy.
Pheochromocytoma
In more than 33% of patients, pheochromocytomas can occur in a familial syndrome such as the VHL syndrome.91,92 The presence of pheochromocytomas defines VHL disease type 2 (A,B,C). More than 26% of VHL patients developed pheochromocytomas in major series.93–96
Few tumors are hormonally inactive with plasma-free metanephrine levels within the normal reference range. The tumors occur bilaterally in up to 39% of patients.93 The tumors can also be found in the sympathetic chain and are then mostly unable to produce and secrete catecholamines.92,97–100 In general, extra-adrenal pheochromocytomas have a higher frequency and potential of malignancy than pheochromocytomas located in the adrenal gland.101 No biomarkers are available that can reliably distinguish a benign from a metastatic pheochromocytoma.
Screening for pheochromocytoma includes measurements of urinary catecholamine metabolites and fractionated metanephrines, as well as of plasma-free metanephrines.94,102,103 The diagnosis can further be established by abdominal computed tomography and/or magnetic resonance imaging.58,105 Given the low frequency of extra-adrenal pheochromocytomas (15%) or metastatic (<5%) pheochromocytomas in patients with VHL disease, selected patients should undergo 123I-MIBG scanning to screen for extra-adrenal or metastatic disease18,101,105 F-FDG, and more recently,67 Ga DOTATATE PET have been shown to offer better test characteristics for assessment and staging of pheochromocytoma/paraganglioma than 106 MIBG scintigraphy107–110 The detection of a pheochromocytoma in patients with VHL syndrome is important to prevent life-threatening hypertensive attacks. Hormonally active pheochromocytomas should generally be treated before surgical treatment of other VHL lesions to prevent intraoperative hypertensive complications and require perioperative blockade.111
It is recommended that pheochromocytoma screening should be started already in childhood at the age of 4 years,58,92,94,96,112 acknowledging that metastatic pheochromocytoma occurs in asymptomatic VHL patients.113
In patients with apparently familial pheochromocytoma there is a 50% chance of VHL disease, whereas the disease is detected in only 10% of patients with sporadic pheochromocytoma.92,114,115 More than 70% of pediatric pheochromocytomas are VHL related.
Treatment of nonmetastatic adrenal pheochromocytoma consists of adrenal cortical sparing (partial) tumor removal, whenever possible.116–119 Minimally invasive adrenal sparing removal of the pheochromocytoma should be performed in most if not all patients.120
This potentially impacts survival as the risk of death from primary adrenal insufficiency may be higher than dying of a recurrent pheochromocytoma. Metastatic pheochromocytoma occurred in 3 patients of a VHL cohort consisting of 273 patients94 and recurrences occurred in 8 of the 273 patients. Bausch and colleagues found a 38% risk of developing a second paraganglioma in pediatric patients diagnosed with pheochromocytoma.121
There is no consensus on whether hormonally inactive pheochromocytomas require treatment. The decision will depend on tumor growth, symptoms, and possible metastasis.106,122 Mutation-adjusted surveillance aims to increase life expectancy.123
Epididymal Cystadenoma
About 50% of male VHL patients develop these non-malignant tumors.124 Epididymal cystadenomas typically arise in the head of the epididymis and contain cystic as well as adenomatous areas and. These lesions are usually asymptomatic. Malignant transformation has not been reported. Obstruction of efferent ductules and spermatic cords may result in fertility problems in rare cases. If necessary, these tumors can be seen on ultrasound.
The female counterparts are cystadenomas of the broad ligaments. They play no major clinical role. “Adnexal papillary cystadenoma” are rare, but may require surgery. They are of probable mesonephric origin.
Pancreatic Manifestations
Pancreatic manifestations in VHL disease include cystadenomas, cysts and pancreatic neuroendocrine tumors.125 A recent series of 48 patients with VHL disease at the Mayo Clinic also revealed 5 patients with branch duct intraductal papillary neoplasm.126
Pancreatic manifestations are observed in 35–70% of VHL patients.55,127,128 It may be difficult to differentiate between benign microcystic adenoma and a pancreatic neuroendocrine tumor. Pancreatic neuroendocrine tumors also reveal a staggering growth pattern similar to hemangioblastoma.129
Besides rare cases of bile duct obstruction or pancreatic insufficiency these tumors are usually asymptomatic. Screening is performed by contrast-enhanced MRI with. Image acquisition has to be performed in the early arterial phase. Pancreatic neuroendocrine tumors are usually solid lesions, whereas microcystic adenomas reveal a multicystic appearance. Microcystic adenomas usually need no treatment, whereas pancreatic neuroendocrine tumors need to be considered for surgery. Mutations in exon 3, especially of codons 161/167 are at enhanced risk for metastatic pancreatic neuroendocrine tumors.130 Most of these lesions are small and slow-growing. In general, VHL-associated pancreatic neuroendocrine tumors have a favorable prognosis compared to sporadic tumors.131 Tumors with a diameter over 2.8 cm should be treated surgically to avoid metastasis according to a recent multicenter study.130 68Ga-DOTATOC PET/CT has better sensitivity in detecting pancreatic NET (size range 4–38 mm) in patients with VHL than 111In-octreoscan.132
Resection of pancreatic tumors may be combined with laparotomy for other VHL associated lesions.133,134 Biliary obstruction with pancreatic insufficiency can be treated by placing biliary stents or replacing pancreatic enzymes. A new option is organ-sparing minimally invasive resection which can be applied to tumors in the tail and the body of the pancreas.135 Chemotherapy is currently under investigation and discussed in the section on targeted therapy.
VHL Screening and Prognosis
A careful family history and screening for other lesions associated with VHL should be performed in all patients with hemangioblastoma, the index tumor of VHL disease. Genetic testing for VHL disease is recommended for all patients with hemangioblastoma.136,137
If a diagnosis of VHL disease is established, patients should undergo an annual screening program (Box 1) for the timely identification of manifestations before irreversible deficits or metastasis occurs. Screening includes periodic contrast-enhanced MRI scans. While CNS gadolinium accumulation will eventually occur in virtually all VHL patients, no pathological effect of this accumulation is known so far.138
The screening programs vary slightly between different centers. A typical program is presented in Box 1. The screening includes several contrast MRI scans. Most centers perform three different scans. Brain, spine and abdomen. Since the discovery of Gadolinium (Gd) depositions in the CNS it is being considered to minimize the use of Gadolinium. These depositions are particularly frequent in VHL patients.138 A shortened 35-mins whole-body MRI protocol with only one Gd injection has been recently suggested139 making VHL screening also quicker and more convenient for patients.
The overall life expectancy of VHL patients used to be limited with a median survival of around 50 years.2,60 However, the introduction of clinical screening enabled timely diagnosis and prophylactic treatment of VHL lesions leading to significantly improved life expectancy.140 Modern management of VHL disease-associated lesions has achieved a life expectancy of additional 10 years.141
Biomarkers in VHL Disease
Timely screening for and preventive treatment of lesions is key to the successful management of VHL patients. Recent developments in oncology are biomarkers including tumor markers, disease activity markers, liquid biopsy and others. We here review the knowledge of systemic biomarkers. Tumor-specific markers, which are detected within the tumor tissue, are not reviewed here. To date, systemic biomarkers do not play a role in the clinical practice of VHL disease, except for plasma and urinary catecholamine metabolites and fractionated metanephrines for pheochromocytoma. However, there are interesting new developments. Certain markers or combinations of markers may be further developed for clinical use in the future and may supplement or even replace other time-consuming screening examinations.
Measuring plasma levels of HIF-dependent molecules appears to be an obvious attempt to monitor disease activity in VHL patients. One group has suggested using plasma VEGF and miRNA210, a hypoxia-inducible micro-RNA, to monitor disease activity in VHL patients with retinal hemangioblastomas. They report that levels of both molecules decreased in all patients under therapy with propranolol.142 Other groups have observed a decrease of serum levels of the HIF-dependent proteins VEGF and also the receptors VEGFR-2 and VEGFR-3 as well as CA9 during therapy with sunitinib and sorafenib in patients with VHL-associated and sporadic RCC.143 Reduction of serum VEGF levels was also detected in mice with xenografted VHL RCCs under propranolol treatment.144
Also, monitoring tumor secretion products may be of interest in VHL disease. VHL patients with pancreatic neuroendocrine tumors had a correlation with plasma vasoactive intestinal peptide and pancreatic polypeptide levels in one study.145
The significance of VHL mutations as a predictive marker in sporadic RCCs is well described. VHL mutations in circulating tumor DNA have been detected by liquid biopsy using a Taqman assay.146
Another study describes the identification of VHL mutations in circulating cancer cells with single-cell genetic analysis.147
Polyglobulia has been described as a paraneoplastic phenomenon in VHL disease. It was reported to occur in CNS hemangioblastomas as a consequence of the secretion of erythropoietin. More recent studies show that this phenomenon is rare, but if present, polyglobulia subsides with the removal of the hemangioblastoma.69 This is consistent with reports of erythropoietic activity in a subset of hemangioblastomas.37 Interestingly, the level of serum erythropoietin and the extent of polyglobulia do not correlate.148 However, the size of the solid tumor component correlates with polyglobulia. Therefore, polyglobulia is a biomarker for tumor growth in a subset of hemangioblastomas. Polycythemia has also been reported in patients with VHL disease and renal cancer149 and pheochromocytoma.150
In general, the knowledge on biomarkers in VHL disease is scarce and this should be addressed in future clinical studies on VHL disease.
Targeted Therapy in VHL Disease
Targeted cancer therapy aims to block specific molecular pathways of cancer cells. Targeted drugs can be subdivided into compounds that inhibit the growth of the primary tumor, prevent invasion in nearby tissue; or drugs that block angiogenesis, metastasis and consequent development of secondary tumors. Because most VHL related tumors are benign, or only carry a low rate of metastasis, targeted VHL therapy mainly focuses on inhibiting primary tumor growth and angiogenesis.
Up to date, the incomplete understanding of VHL tumorigenesis has thwarted the development of a targeted therapy against the disease. Furthermore, VHL tumors are known for their saltatory growth pattern with quiescent phases, which complicates the distinction between response to treatment and natural behavior. Considerable effort has been spent on pharmacological trials, harvesting mixed results. There are no systematic reviews on pharmacological trials in VHL disease. We have reviewed more than 50 studies or case reports and summarized the findings here. The studies are categorized by the main action of their respective drug (Figure 6).
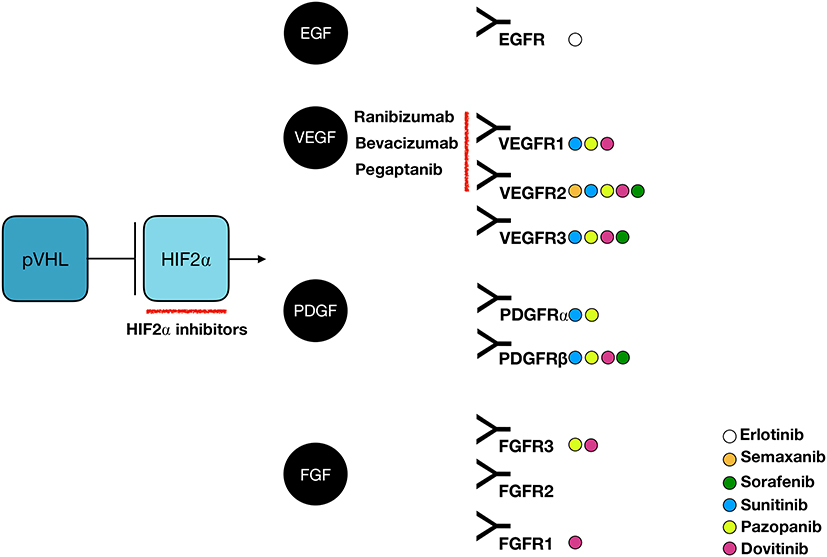 |
Figure 6 Molecular targeting in VHL disease: action mechanisms. Abbreviations: EGF, epidermal growth factor; EGFR, epidermal growth factor receptor; FGF, fibroblast growth factor; FGFR, fibroblast growth factor receptor; HIF, hypoxia-inducible factor; PDGF, platelet-derived growth factor; PDGFR, platelet-derived growth factor receptor; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor. |
Monoclonal Antibodies
Because VHL tumor vascularization (and consequently also tumor growth) is largely dependent on VEGF, many VHL treatment trials have focused on blocking the VEGF-pathway.
Intravitreal injection of anti-VEGF monoclonal antibodies has been widely used as therapy for neovascular age-related macular degeneration. It is now being repurposed for retinal hemangioblastomas in VHL disease.151
Bevacizumab is a monoclonal antibody that prevents interaction between VEGF-A (isoforms 206, 189, 165, 121 and 110) and its receptors (VEGFR1/2). Research groups have reported moderate effects of intravitreal bevacizumab on retinal hemangioblastomas.
Ach et al reported a VHL patient who experienced growth stabilization of retinal hemangioblastomas after several years of intravitreal bevacizumab injections.152
Tano et al treated two VHL patients with intravitreal bevacizumab in combination with vitrectomy, to stabilize a proliferative vitreoretinopathy caused by retinal hemangioblastomas. Even though exudation was suppressed, hemangioblastoma volumes remained stable.153 Hrisomalos et al reported a similar response in one VHL patient, after a regimen of 12 bevacizumab injections over 60 months.154 Tsai et al published contradictory results: photodynamic therapy in combination with intravitreal bevacizumab and corticosteroids reduced the volume of multiple retinal hemangioblastomas in one VHL patient, but exudation was not affected. According to Tsai et al, corticosteroids seem to be more effective in reducing exudation than bevacizumab.155 Walker et al described a VHL patient with rebound neovascularization after intravitreal bevacizumab injection, again without effect on tumor volume.156
Also, the systemic use of bevacizumab has been tested. Wackernagel et al reported the active development of retinal hemangioblastomas during systemic bevacizumab treatment in a VHL patient. Side effects of systemic use were paresthesias and nail petechiae.157
To conclude; laser photocoagulation, cryotherapy, photodynamic therapy and pars plana vitrectomy seem to remain the gold standard for the treatment of retinal VHL hemangioblastomas. Bevacizumab may be considered as adjunctive therapy to stabilize exudation caused by multiple treatments, and to halt neovascularization.
Ranibizumab is another monoclonal antibody that prevents interaction between VEGF-A (isoforms 165, 121, 110 and theoretically 206 and 189) and its receptors (VEGFR1/2). After three intravitreal ranibizumab injections, Michels et al saw stabilization and decreased exudation of a retinal hemangioblastoma in a VHL patient.158 Wong et al conducted a prospective case series including 5 VHL patients with advanced retinal hemangioblastomas, who followed a 24-week regimen of 7 intravitreal ranibizumab injections. Three subjects noticed transient vision improvement, which was however followed by progressive tumor growth and macular exudation. In one patient, macular exudate even increased immediately. One patient experienced prolonged vision improvement and a decrease of macular exudate after 10 injections, with stabilization of tumor volume.159
Pegaptanib is a monoclonal antibody that selectively binds the VEGF-A 165 isoform (which is most predominant in hemangioblastomas) and thereby prevents interaction with its receptors (VEGFR1/2). Dahr et al published a pilot case series including 5 VHL patients with retinal hemangioblastomas that were ineligible for conventional treatment. After six intravitreal pegaptanib injections, macular exudation decreased in two patients, and one of them experienced improved visual acuity. Three patients had adverse events that necessitated treatment withdrawal; such as retinal detachment after laser treatment, retinal detachment caused by exudation, and progressive macular edema. Pegaptanib did not reduce retinal hemangioblastoma volumes and was therefore considered as a palliative treatment modality only.160
We conclude that monoclonal antibodies directed against VEGF are mainly applied as adjunctive treatment to reduce macular exudation; in VHL patients with advanced juxtapapillary or peripheral retinal hemangioblastomas, which are ineligible for conventional treatment. However, because of mixed results, larger prospective randomized trials may be needed to evaluate their efficacy and safety.
Tyrosine Kinase Inhibitors
Tyrosine kinase inhibitors are targeted drugs that block cellular signal transduction cascades. As for monoclonal antibodies, pharmacologic VHL trials have mainly focused on the VEGF tumor pathway.
Semaxanib (SU5416) is a small molecule tyrosine kinase inhibitor of VEGFR2 and stem cell factor receptor c-kit. It has been widely used as a chemotherapeutic for liver metastases of colon cancer and other malignancies. To date semaxanib is not licensed for human use outside clinical trials. Its application in VHL disease has been tested in case reports and small series.
In 2002, Aiello et al reported a VHL patient with an optic nerve head hemangioblastoma, who was treated with 10 cycles of systemic semaxanib. Fundoscopic findings did not change; however, a positive correlation was found between semaxinib dose and visual acuity. Interestingly, Aiello et al reported no change in tumor volumes of the patient’s synchronous cerebellar, spinal and pancreatic lesions.161
Later, other semaxanib trials have followed. Richard et al conducted a toxicity assessment study on 3 VHL patients. Semaxanib caused secondary polycythemia, without effect on hemangioblastoma volumes.162
Girmens et al reported the decrease in macular exudate around a retinal hemangioblastoma, in a VHL patient who was treated with semaxanib for 7 months. However, volume and vision remained stable.163
In 2004, Madhusudan et al conducted a Phase I/Phase II study with 4-week cycles of 8 semaxanib injections, including 6 VHL patients. In one patient, brain/spinal hemangioblastomas and pancreatic cyst volumes remained stable after 22.4 months of ongoing treatment and after 41.4 months of follow-up. However, upper motor neuron symptoms decreased. Another patient with retinal/brain/spinal hemangioblastomas and renal/pancreatic/epididymal cysts only experienced complete regression of a sole retinal lesion (which was also treated with laser coagulation). The patient had improved strength and sexual function after 10 months of treatment. Tumor volumes remained stable on imaging. Clinical response in the other four patients was not described.164
A similar improvement of upper motor neuron symptoms caused by semaxanib, without effect on tumor volume, was described by Schuch et al in a VHL patient with multiple spinal hemangioblastomas.165
To our great interest, Jennens et al reported a VHL patient with the complete metabolic and radiologic response of a metastatic renal cell carcinoma, after 11 systemic semaxanib injections.166
Sunitinib is a first-generation tyrosine kinase inhibitor of multiple receptors (including VEGFR1/2/3, and PDGFR αand β). It is by far the most investigated targeted therapy in VHL disease. However, side effects often lead to disruption of treatment: fatigue, nausea, vomiting, diarrhea, mucositis, hematologic disturbances, nausea, hypertension and hand-foot neuropathy.167–179
Jimenez et al used oral sunitinib to treat a VHL patient with a metastatic pelvic pheochromocytoma (positively staining for VEGF and PDGF beta on immunohistochemistry). The pheochromocytoma started shrinking after 6 months. Interestingly, also the patient’s renal (staining positive for VEGF) and pancreatic tumor volumes decreased.167
Other researchers published similar results in VHL patients undergoing several years of sunitinib treatment: Ali et al (1 patient),169 Wang et al (1 patient),170 Kobayashi et al (1 patient),172 Oudard et al (6 patients),173 Kim et al (4 patients),174 Yuan et al (3 patients),176 Tsimafeyeu et al (1 patient),178 Babinska et al (1 patient)179 and Nuñez et al (3 patients).177 Results range from disease stability to partial response in (metastatic) renal cell carcinomas, in some (metastatic) pancreatic neuroendocrine tumors and in one pheochromocytoma.
Tsimafeyeu et al describe complete regression of a metastatic renal cell carcinoma in a VHL patient, after 11 months of sunitinib treatment.178
Jonasch et al conducted a major pilot trial, treating 15 VHL patients with sunitinib. After four cycles, response was absent in hemangioblastomas and partial in 33% of renal cell carcinomas. Rebound growth in both tumor groups occurred after 48 months. According to Jonasch et al, the higher concentrations of VEGFR2 in renal cell carcinomas compared to hemangioblastomas may explain differences in treatment sensitivity. Pancreatic NETs and cysts, retinal hemangioblastomas and renal cysts remained stable. Two patients with retinal hemangioblastomas experienced increased ocular discomfort, a finding also reported by Oudard.168,173
In a retrospective analysis of 14 VHL patients, Roma et al reported partial response to sunitinib in 64% of different VHL tumors (progressive renal cell carcinomas; and pancreatic, adrenal and ovarian lesions), yielding a progression-free survival of 71.4% after 2 years. Hemangioblastomas remained stable.171
In conclusion, sunitinib response seems to be dependent on several individual patient characteristics; such as overall VHL disease severity, performance status, age and tumor VEGFR status. Sunitinib is mainly used as a palliative treatment modality for temporary symptom control of renal cell carcinomas and pancreatic neuroendocrine tumors. Complete remission of renal cell carcinomas has been described. Sunitinib does not seem to affect cerebellar and retinal hemangioblastomas. Transient shrinking of medullary hemangioblastomas, with neurologic symptom improvement, was described by Oudard.173
Similar to sunitinib, pazopanib is a first-generation inhibitor of multiple tyrosine kinases (including VEGFR 1/2/3; FGFR3 and PDGFR αand β). It has been approved for the treatment of advanced renal cell cancer and advanced soft tissue sarcoma.
In 2012, Kim et al reported a VHL patient with multifocal renal cell carcinoma and multiple cerebellar hemangioblastomas that were all progressive under sunitinib treatment. Subsequent pazopanib treatment improved bulbar symptoms and ambulation. On imaging, cerebellar hemangioblastoma volume decreased, and renal cell carcinoma growth rate slowed.180
Also remarkable is the report of Migliorini et al in 2015, about a tetraplegic VHL patient with a non-resectable spinal hemangioblastoma, who regained ambulation after pazopanib treatment. The tumor seemed to be reshaped on MRI.181
Taylor et al described a VHL patient with the radiologic shrinking of two spinal hemangioblastomas, after pazopanib treatment. Neurologic symptoms did not change, nor did the tumor volume of other hemangioblastomas and one renal cell carcinoma.182
In 2018, Jonasch et al conducted a single-arm nonrandomized phase II study with pazopanib on 30 VHL patients. After a median follow-up of 12 months, the radiologic objective response rate was 52% in renal cell carcinomas (59 tumors; 2 complete responses, 29 partial responses, 28 stable diseases), 53% in pancreatic serous cystadenomas (17 tumors; no complete responses, 9 partial responses, 8 stable diseases), and 4% in central nervous system hemangioblastomas (49 tumors; no complete responses, 2 partial responses, 47 stable diseases, 2 patients with intratumoral hemorrhage). None of the 30 patients developed new VHL related lesions or progressive disease during pazopanib treatment. Interestingly, the type of VHL mutation did not correlate with disease response.183
Finally, Salim described the 9-month stability of a VHL patient with retinal hemangioblastomas treated with pazopanib184.
Reported side effects of pazopanib are fatigue, diarrhea, increase of transaminases, oral mucositis, neutropenia, alopecia/hair depigmentation, hypertension, gastritis and appendicitis.180,181,183
In conclusion, pazopanib seems to have a larger therapeutic effect on renal cell carcinomas, compared to sunitinib. However, study results are inconsistent and side effects are not to be neglected.
Erlotinib is a tyrosine kinase inhibitor of EGFR, indicated for the treatment of metastatic non-small cell lung carcinoma and metastatic pancreatic cancer. In 2001, Rogers et al reported progressive disease under oral erlotinib in a young VHL patient with multiple cerebellar and spinal hemangioblastomas.185
Dovitinib is a tyrosine kinase inhibitor of multiple receptors (including VEGFR1/2/3, PDGFRβ and FGFR1/3). It is currently under investigation for refractory multiple myeloma and metastatic renal cell carcinoma. In 2018, Pilié et al conducted a pilot study of dovitinib in 6 VHL patients. All patients discontinued treatment due to noncompliance or side effects (fatigue, vomiting, severe rash, transaminitis, neutropenia, mucositis and dyspnea). The best response was a stable disease of central nervous system hemangioblastomas.186
Sorafenib is a multiple tyrosine kinase inhibitor of VEGFR2/3 and PDGFR β, among other receptors. In 2017, Choi et al report partial response to sorafenib of multiple small renal cell carcinomas and pancreatic cysts in 2 VHL patients. Retinal and spinal hemangioblastomas did not respond. Manageable diarrhea was reported as a side effect.187
Biological Response Modifiers
Biological response modifiers are drugs that boost or modulate the host’s autologous immune response to neoplasia. Some of these modifiers have been tested in vitro and in small VHL patient groups: roquinimex, thalidomide, IFN-α-2a, HIF2α inhibitors, octreotide, clarithromycin and immunotherapy.
Roquinimex is a derivative quinoline that exerts its anti-neoplastic potential by blocking angiogenesis and decreasing TNF-alpha synthesis. Second, by stimulating NK cell and macrophage activity, it also has immunostimulant properties. In 1999, roquinimex caused a shrinking of von-Hippel-Lindau paraganglioma xenografts in mice.188 No further studies have followed.
Thalidomide acts as an immunosuppressive and anti-angiogenic agent, by modulating the actions of several cytokines. It was withdrawn from the market due to its teratogenic side effects. However, in 2004, Tan et al used thalidomide together with rofecoxib to control post-radiation inflammatory reaction in a VHL patient with a cerebral hemangioblastoma.189 In 2009, Sardi et al describe the growth stabilization of two progressive spinal hemangioblastomas, in a VHL patient treated with thalidomide during a follow-up period of 37 months.190
IFN-α-2a binds to type 1 interferon receptors. It has been previously applied as an antiviral agent in chronic hepatitis C and as an antineoplastic agent in several types of leukemia. In 2011, Niemela et al treated 3 VHL patients with recombinant IFN-α-2a. They reported no significant shrinking of retinal, cerebellar nor spinal hemangioblastomas. Also, pancreatic and renal cysts remained stable. A mild diminishment of blood flow was noticed in retinal hemangioblastomas, potentially by inhibiting VEGF secretion.191
Following VHL inactivation, HIF2α is constitutively upregulated in sporadic and VHL-associated renal cell carcinomas, stimulating tumor growth by activating multiple downstream targets such as VEGF, PDGF, EGFR, TGFα, FGF, GLUT1, Epo, NO-synthase and cyclin D. Therefore, a strong rationale exists for directly inhibiting HIF2α, instead of VEGF alone.
In 2015, Metelo et al reported that pharmacological HIF2α mRNA inhibition in zebrafish improved VHL phenotype (decreasing erythrocytosis and blood vessel formation in the retina, brain, liver and kidney). Further preclinical pharmacologic moderations are needed before its implementation in clinical studies.192 A remaining question is whether HIF2α inhibitors also influence VHL-associated hemangioblastomas, or (extra-adrenal) paragangliomas, which can be either HIF-driven or non-HIF driven.193
In 2017, Sizdahkhani et al demonstrated the presence of somatostatin receptors (1, 2a, 4 and 5) in VHL related hemangioblastomas. They discovered apoptosis of cultured VHL hemangioblastoma cells that were treated with the somatostatin analogue octreotide. Octreotide also caused volume and symptom reduction of an inoperable suprasellar hemangioblastoma in one VHL patient.194
Also in 2017, O’toole et al used octreotide in a VHL patient with multiple pancreatic neuroendocrine tumors, yielding partial response in all tumors.195
Yaghobi Joybari et al reported that octreotide/everolimus caused symptom improvement in a VHL patient with a metastatic pancreatic neuroendocrine carcinoma. Radiological follow-up data were not available.196
Clarithromycin, an antibiotic, has been repurposed as an anti-cancer agent. By modulating cytokine biology and VEGF expression, it has inhibitory effects on inflammation and angiogenesis. In 2019, Ma et al used clarithromycin to treat a VHL patient with a progressive ELST tumor. After 3 months, the ELST tumor remained stable on CT imaging. Pancreatic and renal lesions shrunk or remained stable. Some renal lesions showed progressive growth. No side-effects were observed.197
Only little data are available on immunotherapy in VHL disease. In 1993, Weidmann et al established in vitro sensitization of lymphocytes to autologous renal cell carcinoma tumor cells in two VHL patients. Combined with the injection of interleukin-2, the sensitized lymphocytes caused volume reduction of pulmonary metastasis, without affecting renal cell carcinoma volume.198
Miscellaneous
Based on its previous success rates with infantile hemangiomas, in 2015 Albiñana et al investigated the effect of propranolol on hemangioblastomas in VHL disease. Acting as a nonselective β1 and β2-adrenergic antagonist, propranolol caused apoptosis and inhibition of downstream HIF targets in hemangioblastoma cell cultures (such as VEGF and FGF). However, its exact mechanism of action was unknown.199 Two years later, the same research group used propranolol to treat 7 VHL patients with retinal hemangioblastomas. There was no effect on tumor volume. Resorption of tumor exudate was seen in two patients.142 Another two years later, Albiñana et al applied a highly specific β2-adrenergic receptor blocker (ICI-118,551), which exhibited similar effects on hemangioblastoma cells in vitro.200
In 2018, Shepard et al conducted a similar experiment with propranolol on in vitro VHL hemangioblastoma and renal cell carcinoma cells. Both tumor cell types responded with signs of apoptosis. Propranolol also reduced tumor volume of renal cell carcinoma xenografts in mice. Afterwards, Shepard et al retrospectively analyzed the effect of propranolol on 66 hemangioblastomas in 3 VHL patients who used to receive propranolol for other clinical reasons. Median growth rate of hemangioblastomas was reduced, but did not stop, during a 2.1-year median follow-up.144
Up to date, there are no reports on alkylating agents nor antimetabolites used in VHL patients. However, VHL mutation status is an important predictor for response to alkylating agents in sporadic renal cell carcinomas.201
Halofuginone, a plant derivate, decreases gene expression of collagen type 1 and matrix metalloproteinases; thereby thwarting VEGF-regulated angiogenesis. In 2003, Gross et al discovered that halofuginone led to angiogenic inhibition of a VHL pheochromocytoma xenograft in mice. There was no tumor regression. The authors suggested the future investigation of its applicability as a chemotherapeutic in the early stages of highly vascular VHL tumors.202
Little is known about radiopharmacological treatment. In 1995, Pujol et al report near-complete regression of a metastatic pheochromocytoma in a VHL patient treated with Iodine-131 metaiodobenzylguanidine ([131I]MIBG).203
Conclusion
Even though randomized prospective trials are needed, intravitreal bevacizumab injections may be considered for refractory and exudative retinal hemangioblastomas. The effects and safety profile of ranibizumab and pegaptanib are less well studied, neither are the effects of bevacizumab on other VHL-related tumors.
Regarding monoclonal antibodies, larger randomized trials including control groups are needed to differentiate therapeutic stabilization from natural tumor behavior. Semaxanib causes symptomatic improvement in some central nervous system hemangioblastomas, without affecting tumor volume. We believe that the same temporary relief may be expected from corticosteroids. The effects of sunitinib and pazopanib are potentially depending on different factors such as VEGFR activity, and range from disease stability to partial response in (metastatic) renal cell carcinomas and (metastatic) pancreatic neuroendocrine tumors. Complete regression of one metastatic renal cell carcinoma has been described for both semaxanib and sunitinib. The effect of pazopanib on renal cell carcinomas seems slightly greater. Sunitinib causes disease stability to partial response in some pheochromocytomas. Sunitinib and pazopanib do not seem to affect hemangioblastoma tumor volumes, but some patients experienced symptomatic improvement after treatment with pazopanib. Sunitinib and pazopanib have severe side effects.
The in vitro effect of propranolol on hemangioblastoma and renal cell carcinoma cell cultures seems promising. Again, larger randomized trials including control groups are needed to differentiate therapeutic stabilization due to propranolol, from natural tumor behavior.
Agents such as sorafenib, thalidomide, HIF2α, octreotide and immunotherapy may seem promising, but more preclinical and larger patient studies are needed to assess their efficacy and safety profile.
Acknowledgments
We dedicate this article to Professor emeritus Hartmut P.H. Neumann, who has spent his career in the advancement in diagnosis and treatment of patients suffering from VHL disease.
Author Contributions
All authors contributed to literature analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
Prof. Dr. Christian A Koch received royalties for book and editorship from Springer and Elsevier. He also received honorarium as a consultant for DOC events. The authors report no other conflicts of interest in this work.
References
1. Maher ER, Iselius L, Yates JR, et al. Von Hippel-Lindau disease: a genetic study. J Med Genet. 1991;28(7):443–447. doi:10.1136/jmg.28.7.443
2. Neumann HP. Clustering of features and genetics of von Hippel-Lindau syndrome [letter; comment]. Lancet. 1991;338(8761):258. doi:10.1016/0140-6736(91)90401-A
3. Maher ER, Yates JR, Harries R, et al. Clinical features and natural history of von Hippel-Lindau disease [see comments]. Q J Med. 1990;77(283):1151–1163.
4. von Hippel E. Ueber eine seltene Erkrankung der Netzhaut. Graefes Arch Opthalmol. 1904;59:83–106.
5. Lindau A. Studien uber Kleinhirnzysten. Bau, Pathogenese und Beziehung zur Angiomatosis retinae. Acta Path Et Microbiol Scand. 1926;Suppl 1:1–126.
6. Lindau A. Zur Frage der Angiomatosis retinae und ihrer Hirnkomplikation. Acta Ophthalmol. 1927;4:193–226.
7. Zbar B, Kishida T, Chen F, et al. Germline mutations in the Von Hippel-Lindau disease (VHL) gene in families from North America, Europe, and Japan. Hum Mutat. 1996;8(4):348–357.
8. Couvalard A, Hammel P, Komminoth P, et al. Von Hippel-Lindau Syndrome. In: Lloyd RV, Osamura RY, Klöppel G, Rosai J, editors. WHO Classification of Tumors of Endocrine Organs,
9. Latif F, Tory K, Gnarra J, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260(5112):1317–1320.
10. Nordstrom-O’Brien M, van der Luijt RB, van Rooijen E, et al. Genetic analysis of von Hippel-Lindau disease. Hum Mutat. 2010;31(5):521–537. doi:10.1002/humu.21219
11. Kaelin WG
12. Pause A, Lee S, Worrell RA, et al. The von Hippel-Lindau tumor-suppressor gene product forms a stable complex with human CUL-2, a member of the Cdc53 family of proteins. Proc Natl Acad Sci U S A. 1997;94(6):2156–2161. doi:10.1073/pnas.94.6.2156
13. Lonergan KM, Iliopoulos O, Ohh M, et al. Regulation of hypoxia-inducible mRNAs by the von Hippel-Lindau tumor suppressor protein requires binding to complexes containing elongins B/C and Cul2. Mol Cell Biol. 1998;18(2):732–741. doi:10.1128/MCB.18.2.732
14. Lisztwan J, Imbert G, Wirbelauer C, Gstaiger M, Krek W. The von Hippel-Lindau tumor suppressor protein is a component of an E3 ubiquitin-protein ligase activity. Genes Dev. 1999;13(14):1822–1833. doi:10.1101/gad.13.14.1822
15. Iliopoulos O, Kibel A, Gray S, Kaelin WG
16. Semenza GL. HIF-1 and mechanisms of hypoxia sensing. Curr Opin Cell Biol. 2001;13(2):167–171. doi:10.1016/S0955-0674(00)00194-0
17. Iyer NV, Kotch LE, Agani F, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1alpha. Genes Dev. 1998;12(2):149–162. doi:10.1101/gad.12.2.149
18. Vortmeyer AO, Tran MG, Zeng W, et al. Evolution of VHL tumourigenesis in nerve root tissue. J Pathol. 2006;210(3):374–382. doi:10.1002/path.2062
19. Shively SB, Falke EA, Li J, et al. Developmentally arrested structures preceding cerebellar tumors in von Hippel–Lindau disease. Mod Pathol. 2011;24(8):1023–1030. doi:10.1038/modpathol.2011.61
20. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3(10):721–732. doi:10.1038/nrc1187
21. Iliopoulos O, Levy AP, Jiang C, Kaelin WG
22. Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399(6733):271–275.
23. Krieg M, Haas R, Brauch H, Acker T, Flamme I, Plate KH. Up-regulation of hypoxia-inducible factors HIF-1alpha and HIF-2alpha under normoxic conditions in renal carcinoma cells by von Hippel-Lindau tumor suppressor gene loss of function. Oncogene. 2000;19(48):5435–5443.
24. Kaelin WG
25. Frew IJ, Krek W. Multitasking by pVHL in tumour suppression. Curr Opin Cell Biol. 2007;19(6):685–690.
26. Young AP, Schlisio S, Minamishima YA, et al. VHL loss actuates a HIF-independent senescence programme mediated by Rb and p400. Nat Cell Biol. 2008;10(3):361–369.
27. Glasker S, Lonser RR, Tran MG, et al. Effects of VHL deficiency on endolymphatic duct and sac. Cancer Res. 2005;65(23):10847–10853.
28. Glasker S, Li J, Xia JB, et al. Hemangioblastomas share protein expression with embryonal hemangioblast progenitor cell. Cancer Res. 2006;66(8):4167–4172.
29. Vortmeyer AO, Yuan Q, Lee YS, Zhuang Z, Oldfield EH. Developmental effects of von Hippel-Lindau gene deficiency. Ann Neurol. 2004;55(5):721–728.
30. Vortmeyer AO, Gnarra JR, Emmert-Buck MR, et al. von Hippel-Lindau gene deletion detected in the stromal cell component of a cerebellar hemangioblastoma associated with von Hippel-Lindau disease. Hum Pathol. 1997;28(5):540–543.
31. Lee JY, Dong SM, Park WS, et al. Loss of heterozygosity and somatic mutations of the VHL tumor suppressor gene in sporadic cerebellar hemangioblastomas. Cancer Res. 1998;58(3):504–508.
32. Wizigmann-Voos S, Breier G, Risau W, Plate KH. Up-regulation of vascular endothelial growth factor and its receptors in von Hippel-Lindau disease-associated and sporadic hemangioblastomas. Cancer Res. 1995;55(6):1358–1364.
33. Bohling T, Hatva E, Kujala M, Claesson-Welsh L, Alitalo K, Haltia M. Expression of growth factors and growth factor receptors in capillary hemangioblastoma. J Neuropathol Exp Neurol. 1996;55(5):522–527.
34. Hasselblatt M, Jeibmann A, Gerss J, et al. Cellular and reticular variants of haemangioblastoma revisited: a clinicopathologic study of 88 cases. Neuropathol Appl Neurobiol. 2005;31(6):618–622.
35. Shively SB, Beltaifa S, Gehrs B, et al. Protracted haemangioblastic proliferation and differentiation in von Hippel-Lindau disease. J Pathol. 2008;216(4):514–520.
36. Stein AA, Schilp AO, Whitfield RD. The histogenesis of hemangioblastoma of the brain. J Neurosurg. 1960;17:751.
37. Vortmeyer AO, Frank S, Jeong SY, et al. Developmental arrest of angioblastic lineage initiates tumorigenesis in von Hippel-Lindau disease. Cancer Res. 2003;63(21):7051–7055.
38. Park DM, Zhuang Z, Chen L, et al. von Hippel-Lindau disease-associated hemangioblastomas are derived from embryologic multipotent cells. PLoS Med. 2007;4(2):e60.
39. Vortmeyer AO, Tran M, Zeng W, et al. Evolution of VHL tumorigenesis in nerve root tissue. J Pathol. 2006;210(3):374–382.
40. Chan CC, Vortmeyer AO, Chew EY, et al. VHL gene deletion and enhanced VEGF gene expression detected in the stromal cells of retinal angioma. Arch Ophthalmol. 1999;117(5):625–630.
41. Vortmeyer AO, Chan CC, Chew EY, et al. Morphologic and genetic analysis of retinal angioma associated with massive gliosis in a patient with von Hippel-Lindau disease. Graefes Arch Clin Exp Ophthalmol. 1999;237(6):513–517.
42. Heffner DK. Low-grade adenocarcinoma of probable endolymphatic sac origin A clinicopathologic study of 20 cases. Cancer. 1989;64(11):2292–2302.
43. Choyke PL, Glenn GM, Walther MM, et al. The natural history of renal lesions in von Hippel-Lindau disease: a serial CT study in 28 patients. Am J Roentgenol. 1992;159:1229–1234.
44. Solomon D, Schwartz A. Renal pathology in von Hippel-Lindau disease. Hum Pathol. 1988;19(9):1072–1079.
45. Walther MM, Lubensky IA, Venzon D, Zbar B, Linehan WM. Prevalence of microscopic lesions in grossly normal renal parenchyma from patients with von Hippel-Lindau disease, sporadic renal cell carcinoma and no renal disease: clinical implications. J Urol. 1995;154(6):
46. Lubensky IA, Gnarra JR, Bertheau P, Walther MM, Linehan WM, Zhuang Z. Allelic deletions of the VHL gene detected in multiple microscopic clear cell renal lesions in von Hippel-Lindau disease patients. Am J Pathol. 1996;149(6):2089–2094.
47. Mandriota SJ, Turner KJ, Davies DR, et al. HIF activation identifies early lesions in VHL kidneys: evidence for site-specific tumor suppressor function in the nephron. Cancer Cell. 2002;1(5):459–468.
48. Glasker S, Tran MG, Shively SB, et al. Epididymal cystadenomas and epithelial tumorlets: effects of VHL deficiency on human epididymis. J Pathol. 2006;210(1):32–41.
49. Mehta GU, Shively SB, Duong H, et al. Progression of epididymal maldevelopment into hamartoma-like neoplasia in VHL disease. Neoplasia. 2008;10(10):1146–1153.
50. Odrzywolski KJ, Mukhopadhyay S. Papillary cystadenoma of the epididymis. Arch Pathol Lab Med. 2010;134(4):630–633.
51. Shen T, Zhuang Z, Gersell DJ, Tavassoli FA. Allelic deletion of VHL gene detected in papillary tumors of the broad ligament, epididymis, and retroperitoneum in von Hippel-Lindau disease patients. Int J Surg Pathol. 2000;8(3):207–212.
52. Compagno J, Oertel JE. Microcystic adenomas of the pancreas (glycogen-rich cystadenomas): a clinicopathologic study of 34 cases. Am J Clin Pathol. 1978;69(3):289–298.
53. Vortmeyer AO, Lubensky IA, Fogt F, Linehan WM, Khettry U, Zhuang Z. Allelic deletion and mutation of the von Hippel-Lindau (VHL) tumor suppressor gene in pancreatic microcystic adenomas. Am J Pathol. 1997;151(4):951–956.
54. Mohr VH, Vortmeyer AO, Zhuang Z, et al. Histopathology and molecular genetics of multiple cysts and microcystic (serous) adenomas of the pancreas in von Hippel-Lindau patients. Am J Pathol. 1615;157(5):1615–1621.
55. Lubensky IA, Pack S, Ault D, et al. Multiple neuroendocrine tumors of the pancreas in von Hippel-Lindau disease patients: histopathological and molecular genetic analysis. Am J Pathol. 1998;153(1):223–231.
56. Wilson RA, Ibanez ML. A comparative study of 14 cases of familial and nonfamilial pheochromocytomas. Hum Pathol. 1978;9(2):181–188.
57. Koch CA, Mauro D, Walther MM, et al. Pheochromocytoma in von Hippel-Lindau disease: distinct histopathologic phenotype compared to pheochromocytoma in multiple endocrine neoplasia type 2. Endocr Pathol. 2002;13(1):17–27.
58. Lonser RR, Glenn GM, Walther M, et al. von Hippel-Lindau disease. Lancet. 2003;361(9374):2059–2067.
59. Maher ER, Kaelin WG
60. Lamiell JM, Salazar FG, Hsia YE. von Hippel-Lindau disease affecting 43 members of a single kindred. Medicine. 1989;68(1):1–29.
61. Friedrich CA. Von Hippel-Lindau syndrome. A pleomorphic condition. Cancer. 1999;86(11 Suppl):2478–2482.
62. Singh AD, Nouri M, Shields CL, Shields JA, Perez N. Treatment of retinal capillary hemangioma. Ophthalmology. 2002;109(10):1799–1806.
63. Schmidt D, Natt E, Neumann HP. Long-term results of laser treatment for retinal angiomatosis in von Hippel-Lindau disease. Eur J Med Res. 2000;5(2):47–58.
64. Gaudric A, Krivosic V, Duguid G, Massin P, Giraud S, Richard S. Vitreoretinal surgery for severe retinal capillary hemangiomas in von hippel-lindau disease. Ophthalmology. 2011;118(1):142–149.
65. Chew EY. Ocular manifestations of von Hippel-Lindau disease: clinical and genetic investigations. Trans Am Ophthalmol Soc. 2005;103:495–511.
66. Wanebo J, Lonser R, Glenn G, Oldfield E. The natural history of hemangioblastomas of the central nervous system in patients with von Hippel-Lindau disease. J Neurosurg. 2003;98(1):82–94.
67. Lonser RR, Butman JA, Huntoon K, et al. Prospective natural history study of central nervous system hemangioblastomas in von Hippel-Lindau disease. J Neurosurg. 2014;120(5):1055–1062.
68. Lonser RR, Vortmeyer AO, Butman JA, et al. Edema is a precursor to central nervous system peritumoral cyst formation. Ann Neurol. 2005;58(3):392–399.
69. Glasker S, Kruger MT, Klingler JH, et al. Hemangioblastomas and neurogenic polyglobulia. Neurosurgery. 2013;72(6):
70. Weil R, Lonser R, DeVroom H, Wanebo J, Oldfield E. Surgical management of brainstem hemangioblastomas in patients with von Hippel-Lindau disease. J Neurosurg. 2003;98(1):95–105.
71. van Velthoven V, Reinacher PC, Klisch J, Neumann HP, Glasker S. Treatment of intramedullary hemangioblastomas, with special attention to von Hippel-Lindau disease. Neurosurgery. 2003;53(6):1306–1313.
72. Hwang SW, Malek AM, Schapiro R, Wu JK. Intraoperative use of indocyanine green fluorescence videography for resection of a spinal cord hemangioblastoma. Neurosurgery. 2010;67(3 Suppl Operative):
73. Benedetto N, Aquila F, Vannozzi R. Use of near-infrared indocyanine videoangiography and flow 800 in the resectioning of a spinal cord haemangioblastoma. Br J Neurosurg. 2013;27(6):847–849.
74. Asthagiri AR, Mehta GU, Zach L, et al. Prospective evaluation of radiosurgery for hemangioblastomas in von Hippel-Lindau disease. Neuro Oncol. 2010;12(1):80–86.
75. Manski TJ, Heffner DK, Glenn GM, et al. Endolymphatic sac tumors. A source of morbid hearing loss in von Hippel-Lindau disease. JAMA. 1997;277(18):1461–1466.
76. Butman JA, Kim HJ, Baggenstos M, et al. Mechanisms of morbid hearing loss associated with tumors of the endolymphatic sac in von Hippel-Lindau disease. JAMA. 2007;298(1):41–48.
77. Hojo M, Arakawa Y, Funaki T, et al. Usefulness of tumor blood flow imaging by intraoperative indocyanine green videoangiography in hemangioblastoma surgery. World Neurosurg. 2014;82(3–4):e495–501.
78. Hao S, Li D, Ma G, Yang J, Wang G. Application of intraoperative indocyanine green videoangiography for resection of spinal cord hemangioblastoma: advantages and limitations. J Clin Neurosci. 2013;20(9):1269–1275.
79. Neumann HP, Riegler P, Huber W, et al. The challenge of kidney lesions in von Hippel-Lindau disease. [Review] [39 refs]. Contrib Nephrol. 2001;136:193–207.
80. Richard S, Chauveau D, Chretien Y, et al. Renal lesions and pheochromocytoma in von Hippel-Lindau disease. Adv Nephrol Necker Hosp. 1994;23:1–27.
81. Grubb RL
82. Kaelin WG
83. Linehan WM, Walther MM, Zbar B. The genetic basis of cancer of the kidney. J Urol. 2003;170(6 Pt 1):2163–2172.
84. Poston CD, Jaffe GS, Lubensky IA, et al. Characterization of the renal pathology of a familial form of renal cell carcinoma associated with von Hippel-Lindau disease: clinical and molecular genetic implications. J Urol. 1995;153(1):22–26.
85. Walther MM, Choyke PL, Glenn G, et al. Renal cancer in families with hereditary renal cancer: prospective analysis of a tumor size threshold for renal parenchymal sparing surgery. J Urol. 1999;161(5):1475–1479.
86. Jilg CA, Neumann HP, Glasker S, et al. Growth kinetics in Von Hippel-Lindau-associated renal cell carcinoma. Urol Int.
87. Kim BJ, Kim JH, Kim HS, Zang DY. Prognostic and predictive value of VHL gene alteration in renal cell carcinoma: a meta-analysis and review. Oncotarget. 2017;8(8):13979–13985.
88. Walther MM, Choyke PL, Weiss G, et al. Parenchymal sparing surgery in patients with hereditary renal cell carcinoma. J Urol. 1995;153(3 Pt 2):913–916.
89. Jilg CA, Neumann HP, Glasker S, et al. Nephron sparing surgery in von Hippel-Lindau associated renal cell carcinoma; clinicopathological long-term follow-up. Fam Cancer. 2012.
90. Allasia M, Soria F, Battaglia A, et al. Radiofrequency ablation for renal cancer in von hippel-lindau syndrome patients: a prospective cohort analysis. Clin Genitourin Cancer. 2017.
91. Jafri M, Maher ER. The genetics of phaeochromocytoma: using clinical features to guide genetic testing. Eur J Endocrinol. 2012;166(2):151–158.
92. Neumann HP, Bausch B, McWhinney SR, et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;346(19):1459–1466.
93. Walther MM, Reiter R, Keiser HR, et al. Clinical and genetic characterization of pheochromocytoma in von Hippel-Lindau families: comparison with sporadic pheochromocytoma gives insight into natural history of pheochromocytoma. J Urol. 1999;162(3 Pt 1):659–664.
94. Aufforth RD, Ramakant P, Sadowski SM, et al. Pheochromocytoma screening initiation and frequency in von Hippel-Lindau syndrome. J Clin Endocrinol Metab. 2015;100(12):4498–4504.
95. Li SR, Nicholson KJ, McCoy KL, Carty SE, Yip L. Clinical and biochemical features of pheochromocytoma characteristic of Von Hippel-Lindau syndrome. World J Surg. 2020;44(2):570–577.
96. Fagundes GFC, Petenuci J, Lourenco DM
97. Welander J, Soderkvist P, Gimm O. Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr Relat Cancer. 2011;18(6):R253–276.
98. Papaspyrou K, Mewes T, Rossmann H, et al. Head and neck paragangliomas: report of 175 patients (1989-2010). Head Neck. 2012;34(5):632–637.
99. Gaal J, van Nederveen FH, Erlic Z, et al. Parasympathetic paragangliomas are part of the Von Hippel-Lindau syndrome. J Clin Endocrinol Metab. 2009;94(11):4367–4371. doi:10.1210/jc.2009-1479
100. Boedeker CC, Erlic Z, Richard S, et al. Head and neck paragangliomas in von Hippel-Lindau disease and multiple endocrine neoplasia type 2. J Clin Endocrinol Metab. 2009;94(6):1938–1944. doi:10.1210/jc.2009-0354
101. Bhatia KSS, Ismail MM, Sahdev A, et al. 123 I-metaiodobenzylguanidine (MIBG) scintigraphy for the detection of adrenal and extra-adrenal phaeochromocytomas: CT and MRI correlation. Clin Endocrinol (Oxf). 2008;69(2):181–188. doi:10.1111/j.1365-2265.2008.03256.x
102. Eisenhofer G, Lenders JW, Timmers H, et al. Measurements of plasma methoxytyramine, normetanephrine, and metanephrine as discriminators of different hereditary forms of pheochromocytoma. Clin Chem. 2011;57(3):411–420. doi:10.1373/clinchem.2010.153320
103. Eisenhofer G, Lenders JWM, Linehan WM, Walther MM, Goldstein DS, Keiser HR. Plasma normetanephrine and metanephrine for detecting pheochromocytoma in von Hippel–Lindau disease and multiple endocrine neoplasia type 2. N Engl J Med. 1999;340(24):1872–1879. doi:10.1056/NEJM199906173402404
104. Neumann HPH, Young WF
105. Koch CA. Should 123I-MIBG scintigraphy be part of the workup for pheochromocytomas? Nat Clin Pract Endocrinol Metab. 2009;5(2):76–77. doi:10.1038/ncpendmet1047
106. Ayala-Ramirez M, Feng L, Johnson MM, et al. Clinical risk factors for malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas: primary tumor size and primary tumor location as prognostic indicators. J Clin Endocrinol Metab. 2011;96(3):717–725. doi:10.1210/jc.2010-1946
107. Timmers HJLM, Chen CC, Carrasquillo JA, et al. Comparison of 18F-fluoro-L-DOPA, 18F-fluoro-deoxyglucose, and 18F-fluorodopamine PET and 123I-MIBG scintigraphy in the localization of pheochromocytoma and paraganglioma. J Clin Endocrinol Metab. 2009;94(12):4757–4767. doi:10.1210/jc.2009-1248
108. Timmers HJLM, Chen CC, Carrasquillo JA, et al. Staging and functional characterization of pheochromocytoma and paraganglioma by 18F-fluorodeoxyglucose (18F-FDG) positron emission tomography. JNCI. 2012;104(9):700–708. doi:10.1093/jnci/djs188
109. Nockel P, El Lakis M, Gaitanidis A, et al. Preoperative 18F-FDG PET/CT in pheochromocytomas and paragangliomas allows for precision surgery. Ann Surg. 2019;269(4):741–747. doi:10.1097/SLA.0000000000002671
110. Chang CA, Pattison DA, Tothill RW, et al. 68Ga-DOTATATE and 18F-FDG PET/CT in paraganglioma and Pheochromocytoma: utility, patterns and heterogeneity. Cancer Imaging. 2016;16(1):22. doi:10.1186/s40644-016-0084-2
111. Waingankar N, Bratslavsky G, Jimenez C, Russo P, Kutikov A. Pheochromocytoma in urologic practice. Eur Urol Focus. 2016;1(3):231–240. doi:10.1016/j.euf.2015.09.011
112. Weise M, Merke DP, Pacak K, Walther MM, Eisenhofer G. Utility of plasma free metanephrines for detecting childhood pheochromocytoma. J Clin Endocrinol Metab. 2002;87(5):1955–1960. doi:10.1210/jcem.87.5.8446
113. Colvin A, Saltzman AF, Walker J, Bruny J, Cost NG. Metastatic pheochromocytoma in an asymptomatic 12-year-old with von Hippel-Lindau Disease. Urology. 2018;119:140–142. doi:10.1016/j.urology.2017.12.007
114. Gergics P, Patocs A, Toth M, et al. Germline VHL gene mutations in Hungarian families with von Hippel–Lindau disease and patients with apparently sporadic unilateral pheochromocytomas. Eur J Endocrinol. 2009;161(3):495–502. doi:10.1530/EJE-09-0399
115. Iacobone M, Schiavi F, Bottussi M, et al. Is genetic screening indicated in apparently sporadic pheochromocytomas and paragangliomas? Surgery. 2011;150(6):1194–1201. doi:10.1016/j.surg.2011.09.024
116. Brunt LM, Lairmore TC, Doherty GM, Quasebarth MA, DeBenedetti M, Moley JF. Adrenalectomy for familial pheochromocytoma in the laparoscopic era. Ann Surg. 2002;235(5):711–713. doi:10.1097/00000658-200205000-00014
117. Neumann HP, Reincke M, Bender BU, Elsner R, Janetschek G. Preserved adrenocortical function after laparoscopic bilateral adrenal sparing surgery for hereditary pheochromocytoma. J Clin Endocrinol Metab. 1999;84(8):2608–2610. doi:10.1210/jcem.84.8.5872
118. Walz MK, Peitgen K, Neumann HPH, Janssen OE, Philipp T, Mann K. Endoscopic treatment of solitary, bilateral, multiple, and recurrent pheochromocytomas and paragangliomas. World J Surg. 2002;26(8):1005–1012. doi:10.1007/s00268-002-6632-x
119. Neumann HPH, Tsoy U, Bancos I, et al. Comparison of pheochromocytoma-specific morbidity and mortality among adults with bilateral pheochromocytomas undergoing total adrenalectomy vs cortical-sparing adrenalectomy. JAMA Netw Open. 2019;2(8):e198898. doi:10.1001/jamanetworkopen.2019.8898
120. Dickson PV, Jimenez C, Chisholm GB, et al. Posterior retroperitoneoscopic adrenalectomy: a contemporary American experience. J Am Coll Surg. 2011;212(4):657–659. doi:10.1016/j.jamcollsurg.2010.12.023
121. Bausch B, Wellner U, Bausch D, et al. Long-term prognosis of patients with pediatric pheochromocytoma. Endocr Relat Cancer. 2013;21(1):17–25. doi:10.1530/ERC-13-0415
122. Singer J, Koch CA, Kassahun W, et al. A patient with a large recurrent pheochromocytoma demonstrating the pitfalls of diagnosis. Nat Rev Endocrinol. 2011;7(12):749–755. doi:10.1038/nrendo.2011.132
123. Nolting N, Ullrich U, Pietzsch P, et al. Current management of pheochromocytoma/paraganglioma: a guide for the practicing clinician in the era of precision medicine. Cancers. 2019;11(10):1505. doi:10.3390/cancers11101505
124. Choyke PL, Glenn GM, Wagner JP, et al. Epididymal cystadenomas in von Hippel-Lindau disease. Urology. 1997;49(6):926–931. doi:10.1016/S0090-4295(97)00074-5
125. Park TY, Lee SK, Park J-S, et al. Clinical features of pancreatic involvement in von Hippel–Lindau disease: a retrospective study of 55 cases in a single center. Scand J Gastroenterol. 2015;50(3):360–367. doi:10.3109/00365521.2014.992364
126. Sharma A, Mukewar S, Vege SS. Clinical profile of pancreatic cystic lesions in von Hippel-Lindau disease: a series of 48 patients seen at a tertiary institution. Pancreas. 2017;46(7):948–952. doi:10.1097/MPA.0000000000000875
127. Neumann HPH, Dinkel E, Brambs H, et al. Pancreatic lesions in the von Hippel-Lindau syndrome. Gastroenterology. 1991;101(2):465–471. doi:10.1016/0016-5085(91)90026-H
128. Hough DM, Stephens DH, Johnson CD, Binkovitz LA. Pancreatic lesions in von Hippel-Lindau disease: prevalence, clinical significance, and CT findings. AJR Am J Roentgenol. 1994;162(5):1091–1094. doi:10.2214/ajr.162.5.8165988
129. Weisbrod AB, Kitano M, Thomas F, et al. Assessment of tumor growth in pancreatic neuroendocrine tumors in von Hippel Lindau syndrome. J Am Coll Surg. 2014;218(2):163–169. doi:10.1016/j.jamcollsurg.2013.10.025
130. Krauss T, Ferrara AM, Links TP, et al. Preventive medicine of von Hippel–Lindau disease-associated pancreatic neuroendocrine tumors. Endocr Relat Cancer. 2018;25(9):783–793. doi:10.1530/ERC-18-0100
131. de Mestier L, Gaujoux S, Cros J, et al. Long-term prognosis of resected pancreatic neuroendocrine tumors in von hippel-lindau disease is favorable and not influenced by small tumors left in place. Ann Surg. 2014.
132. Prasad V, Tiling N, Denecke T, Brenner W, Plockinger U. Potential role of 68Ga-DOTATOC PET/CT in screening for pancreatic neuroendocrine tumour in patients with von Hippel-Lindau disease. Eur J Nucl Med Mol Imaging. 2016;43(11):2014–2020. doi:10.1007/s00259-016-3421-6
133. Libutti SK, Choyke PL, Bartlett DL, et al. Pancreatic neuroendocrine tumors associated with von Hippel Lindau disease: diagnostic and management recommendations. Surgery. 1998;124(6):1153–1159. doi:10.1067/msy.1998.91823
134. Libutti SK, Choyke PL, Alexander HR, et al. Clinical and genetic analysis of patients with pancreatic neuroendocrine tumors associated with von Hippel-Lindau disease. Surgery. 2000;128(6):1022–1028. doi:10.1067/msy.2000.110239
135. von Ducker L, Walz MK, Voss C, Arnold G, Eng C, Neumann HPH. Laparoscopic organ-sparing resection of von Hippel-Lindau disease-associated pancreatic neuroendocrine tumors. World J Surg. 2011;35(3):563–567. doi:10.1007/s00268-010-0878-5
136. Glasker S, Bender BU, Apel TW, et al. The impact of molecular genetic analysis of the VHL gene in patients with haemangioblastomas of the central nervous system. J Neurol Neurosurg Psychiatry. 1999;67(6):758–762. doi:10.1136/jnnp.67.6.758
137. Woodward ER, Wall K, Forsyth J, Macdonald F, Maher ER. VHL mutation analysis in patients with isolated central nervous system haemangioblastoma. Brain. 2007;130(3):836–842. doi:10.1093/brain/awl362
138. Vergauwen E, Vanbinst A M, Brussaard C, et al. Central nervous system gadolinium accumulation in patients undergoing periodical contrast MRI screening for hereditary tumor syndromes. Hered Cancer Clin Pract. 2018;16(1):2. doi:10.1186/s13053-017-0084-7
139. Vanbinst A M, Brussaard C, Vergauwen E, et al. A focused 35-minute whole body MRI screening protocol for patients with von Hippel-Lindau disease. Hered Cancer Clin Pract. 2019;17(1):22. doi:10.1186/s13053-019-0121-9
140. Singh AD, Shields CL, Shields JA. von Hippel–Lindau disease. Surv Ophthalmol. 2001;46(2):117–142. doi:10.1016/S0039-6257(01)00245-4
141. Wilding A, Ingham SL, Lalloo F, et al. Life expectancy in hereditary cancer predisposing diseases: an observational study. J Med Genet. 2012;49(4):264–269. doi:10.1136/jmedgenet-2011-100562
142. Albinana V, Escribano RMJ, Soler I, et al. Repurposing propranolol as a drug for the treatment of retinal haemangioblastomas in von Hippel-Lindau disease. Orphanet J Rare Dis. 2017;12(1):122. doi:10.1186/s13023-017-0664-7
143. George S, Bukowski RM. Biomarkers in clear cell renal cell carcinoma. Expert Rev Anticancer Ther. 2007;7(12):1737–1747. doi:10.1586/14737140.7.12.1737
144. Shepard MJ, Bugarini A, Edwards NA, et al. Repurposing propranolol as an antitumor agent in von Hippel-Lindau disease. J Neurosurg Oct. 2018;1–9.
145. Tirosh A, Papadakis GZ, Millo C, et al. Association between neuroendocrine tumors biomarkers and primary tumor site and disease type based on total 68Ga-DOTATATE-Avid tumor volume measurements. Eur J Endocrinol. 2017;176(5):575–582. doi:10.1530/EJE-16-1079
146. Corro C, Hejhal T, Poyet C, et al. Detecting circulating tumor DNA in renal cancer: an open challenge. Exp Mol Pathol. 2017;102(2):255–261. doi:10.1016/j.yexmp.2017.02.009
147. Broncy L, Njima BB, Mejean A, et al. Single-cell genetic analysis validates cytopathological identification of circulating cancer cells in patients with clear cell renal cell carcinoma. Oncotarget. 2018;9(28):20058–20074. doi:10.18632/oncotarget.25102
148. Kruger MT, Klingler J H, Jilg C, et al. Polyglobulia in patients with hemangioblastomas is related to tumor size but not to serum erythropoietin. Hered Cancer Clin Pract. 2018;16(1):15. doi:10.1186/s13053-018-0097-x
149. Da Silva JL, Lacombe C, Bruneval P, et al. Tumor cells are the site of erythropoietin synthesis in human renal cancers associated with polycythemia. Blood. 1990;75(3):577–582. doi:10.1182/blood.V75.3.577.577
150. Capodimonti S, Teofili L, Martini M, et al. Von hippel-lindau disease and erythrocytosis. J clin oncol. 2012;30(13):e137–139. doi:10.1200/JCO.2011.38.6797
151. Rosenfeld PJ, Brown DM, Heier JS, et al. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. 2006;355(14):1419–1431. doi:10.1056/NEJMoa054481
152. Ach T, Thiemeyer D, Hoeh AE, Schaal KB, Dithmar S. Intravitreal bevacizumab for retinal capillary haemangioma: longterm results. Acta Ophthalmol. 2009;88(4):e137–138. doi:10.1111/j.1755-3768.2009.01594.x
153. Tano R, Kakurai K, Sakurai T, Fujiwara R, Mano T, Maeno T. Intravitreal bevacizumab (avastin) combined with vitrectomy for recurrences of proliferative vitreoretinopathy in Von Hippel-Lindau disease. Acta Ophthalmol. 2012;90(2):e157–158. doi:10.1111/j.1755-3768.2011.02108.x
154. Hrisomalos FN, Maturi RK, Pata V. Long-term use of intravitreal bevacizumab (avastin) for the treatment of von hippel-lindau associated retinal hemangioblastomas. Open Ophthalmol J. 2010;4(1):66–69. doi:10.2174/1874364101004010066
155. Tsai F Y, Lau L I, Chen S J, Lee F L, Lee S M. Persistent exudative retinal detachment after photodynamic therapy and intravitreal bevacizumab injection for multiple retinal capillary hemangiomas in a patient with von Hippel–Lindau disease. J Chin Med Assoc. 2014;77(1):52–56. doi:10.1016/j.jcma.2013.09.001
156. Walker RA, Colleaux KM. Preretinal neovascularization after bevacizumab injections in a patient with von Hippel-Lindau syndrome. Can J Ophthalmol. 2009;44(5):608–609. doi:10.3129/i09-128
157. Wackernagel W, Lackner E M, Pilz S, Mayer C, Stepan V. von Hippel-Lindau disease: treatment of retinal haemangioblastomas by targeted therapy with systemic bevacizumab. Acta Ophthalmol. 2010;88(7):e271–272. doi:10.1111/j.1755-3768.2009.01611.x
158. Michels S, Messmer E, Sutter F, Kurz-Levin MM. Intravitreale anti-VEGF-Therapie von retinalen kapillären Hämangioblastomen bei von-Hippel-Lindau-Erkrankung. Klin Monbl Augenheilkd. 2008;225(4):292–294. doi:10.1055/s-2008-1027175
159. Wong WT, Liang KJ, Hammel K, Coleman HR, Chew EY. Intravitreal ranibizumab therapy for retinal capillary hemangioblastoma related to von Hippel-Lindau disease. Ophthalmology. 2008;115(11):1957–1964. doi:10.1016/j.ophtha.2008.04.033
160. Dahr SS, Cusick M, Rodriguez-Coleman H, et al. Intravitreal anti-vascular endothelial growth factor therapy with pegaptanib for advanced von Hippel-Lindau disease of the retina. Retina. 2007;27(2):150–158. doi:10.1097/IAE.0b013e318030a290
161. Aiello LP, et al. Rapid and durable recovery of visual function in a patient with von hippel-lindau syndrome after systemic therapy with vascular endothelial growth factor receptor inhibitor su5416. Ophthalmology. 2002;109(9):1745–1751. doi:10.1016/S0161-6420(02)01159-4
162. Richard S, Croisille L, Yvart J, et al. Paradoxical secondary polycythemia in von Hippel-Lindau patients treated with anti–vascular endothelial growth factor receptor therapy. Blood. 2002;99(10):3851–3853. doi:10.1182/blood.V99.10.3851
163. Girmens J F, Erginay A, Massin P, Scigalla P, Gaudric A, Richard S. Treatment of von Hippel-Lindau retinal hemangioblastoma by the vascular endothelial growth factor receptor inhibitor SU5416 is more effective for associated macular edema than for hemangioblastomas. Am J Ophthalmol. 2003;136(1):194–196. doi:10.1016/S0002-9394(03)00101-6
164. Madhusudan S, et al. Antiangiogenic therapy for von Hippel-Lindau disease. JAMA. 2004;291(8):943–944. doi:10.1001/jama.291.8.943
165. Schuch G, de Wit M, Holtje J, et al. Difficult diagnostic cases. J Clin Oncol. 2005;23(15):3624–3626. doi:10.1200/JCO.2005.01.184
166. Jennens RR, Rosenthal MA, Lindeman GJ, Michael M. Complete radiological and metabolic response of metastatic renal cell carcinoma to SU5416 (semaxanib) in a patient with probable von Hippel-Lindau syndrome. Urol Oncol. 2004;22(3):193–196. doi:10.1016/j.urolonc.2004.01.011
167. Jimenez C, Cabanillas ME, Santarpia L, et al. Use of the tyrosine kinase inhibitor sunitinib in a patient with von Hippel-Lindau disease: targeting angiogenic factors in pheochromocytoma and other von Hippel-Lindau disease-related tumors. J Clin Endocrinol Metab. 2009;94(2):386–391. doi:10.1210/jc.2008-1972
168. Jonasch E, McCutcheon IE, Waguespack SG, et al. Pilot trial of sunitinib therapy in patients with von Hippel–Lindau disease. Ann Oncol. 2011;22(12):2661–2666. doi:10.1093/annonc/mdr011
169. Ali T, Kandil D, Piperdi B. Long-term disease control with sunitinib in a patient with metastatic pancreatic neuroendocrine tumor (NET) associated with Von Hippel-Lindau syndrome (VHL). Pancreas. 2012;41(3):492–493. doi:10.1097/MPA.0b013e31822a645e
170. Wang W, Jiang CY, Wang HW. Use of sunitinib in a 30-year-old woman with pancreatic neuroendocrine tumors associated with Von Hippel-Lindau syndrome. J Clin Gastroenterol. 2015;49(1):89–90. doi:10.1097/MCG.0000000000000160
171. Roma A, Maruzzo M, Basso U, et al. First-Line sunitinib in patients with renal cell carcinoma (RCC) in von Hippel–Lindau (VHL) disease: clinical outcome and patterns of radiological response. Fam Cancer. 2015;14(2):309–316. doi:10.1007/s10689-014-9771-y
172. Kobayashi A, Takahashi M, Imai H, et al. Attainment of a long-term favorable outcome by sunitinib treatment for pancreatic neuroendocrine tumor and renal cell carcinoma associated with von hippel-lindau disease. Intern Med. 2016;55(6):629–634. doi:10.2169/internalmedicine.55.5796
173. Oudard S, Elaidi R, Brizard M, et al. Sunitinib for the treatment of benign and malignant neoplasms from von Hippel-Lindau disease: A single-arm, prospective phase II clinical study from the PREDIR group. Oncotarget. 2016;7(51):85306–85317. doi:10.18632/oncotarget.13301
174. Kim HC, Lee JS, Kim SH, So HS, Woo CY, Lee JL. Sunitinib treatment for metastatic renal cell carcinoma in patients with von hippel-lindau disease. Cancer Res Treat. 2013;45(4):349–353. doi:10.4143/crt.2013.45.4.349
175. Knickelbein JE, Jacobs-El N, Wong WT, et al. Systemic sunitinib malate treatment for advanced juxtapapillary retinal hemangioblastomas associated with von Hippel-Lindau disease. Ophthalmol Retina. 2017;1(3):181–187. doi:10.1016/j.oret.2016.10.007
176. Yuan G, Liu Q, Tong D, et al. A retrospective case study of sunitinib treatment in three patients with Von Hippel-Lindau disease. Cancer Biol Ther Jun. 2018;1–20.
177. Nunez JE, Donadio M, Filho DR, et al. The efficacy of everolimus and sunitinib in patients with sporadic or germline mutated metastatic pancreatic neuroendocrine tumors. J Gastrointest Oncol. 2019;10(4):645–651. doi:10.21037/jgo.2019.01.33
178. Tsimafeyeu I. Sunitinib treatment of metastatic renal cell carcinoma in von Hippel-Lindau disease. J Cancer Res Ther. 2015;11(4):920–922. doi:10.4103/0973-1482.162120
179. Babinska A, Studniarek M, Swiatkowska-Stodulska R, Sworczak K. Sunitinib treatment for multifocal renal cell carcinoma (RCC) and pancreatic neuroendocrine tumor (NET) in patient with Von Hippel-Lindau disease. case report. Neuro Endocrinol Lett. 2015;36(6):517–520.
180. Kim BYS, Jonasch E, McCutcheon IE. Pazopanib therapy for cerebellar hemangioblastomas in von Hippel–Lindau disease. Target Oncol. 2012;7(2):145–149. doi:10.1007/s11523-012-0214-0
181. Migliorini D, Haller S, Merkler D, et al. Recurrent multiple CNS hemangioblastomas with VHL disease treated with pazopanib: a case report and literature review. CNS Oncology. 2015;4(6):387–392. doi:10.2217/cns.15.22
182. Taylor DG, Ilyas A, Mehta GU, et al. Variable response of CNS hemangioblastomas to pazopanib in a single patient with von Hippel-Lindau disease: case report. J Clin Neurosci. 2018;50:154–156. doi:10.1016/j.jocn.2018.01.040
183. Jonasch E, McCutcheon IE, Gombos DS, et al. Pazopanib in patients with von Hippel-Lindau disease: a single-arm, single-centre, Phase 2 trial. Lancet Oncol. 2018;19(10):1351–1359. doi:10.1016/S1470-2045(18)30487-X
184. Salim DK. Continuous angiogenesis inhibition in the treatment for von Hippel–Lindau-related hemangioblastomas of retina and spinal cord. J Oncol Pharm Pract. 2019;25(8):2049–2051. doi:10.1177/1078155219827635
185. Rogers LR, LoRusso P, Nadler P, Malik G, Shields A, Kaelin W. Erlotinib therapy for central nervous system hemangioblastomatosis associated with von Hippel-Lindau disease: a case report. J Neurooncol. 2011;101(2):307–310. doi:10.1007/s11060-010-0244-3
186. Pilie P, Hasanov E, Matin SF, et al. Pilot study of dovitinib in patients with von Hippel-Lindau disease. Oncotarget. 2018;9(34):23390–23395. doi:10.18632/oncotarget.25171
187. Choi KH, Yu YD, Kang MH, Park DS. Sorafenib treatment for recurrent stage T1 bilateral renal cell carcinoma in patients with Von Hippel- Lindau disease: A case report and literature review. Can Urol Assoc J. 2015;9(9–10):E651–653. doi:10.5489/cuaj.2863
188. Gross DJ, Reibstein I, Weiss L, et al. The antiangiogenic agent linomide inhibits the growth rate of von Hippel-Lindau paraganglioma xenografts to mice. Clin Cancer Res. 1999;5(11):3669–3675.
189. Tan DS, Evanson J, Plowman PN, Chew SL. Post-radiation inflammatory reaction controlled with thalidomide and rofecoxib. Clin Oncol. 2004;16(8):585–586. doi:10.1016/j.clon.2004.09.005
190. Sardi I, Sanzo M, Giordano F, et al. Monotherapy with thalidomide for treatment of spinal cord hemangioblastomas in a patient with von Hippel-Lindau disease. Pediatr Blood Cancer. 2009;53(3):464–467. doi:10.1002/pbc.22065
191. Niemela M, Lemeta S, Sainio M, et al. Hemangioblastomas of the retina: impact of von Hippel-Lindau disease. Invest Ophthalmol Vis Sci. 2000;41(7):1909–1915.
192. Metelo AM, Noonan HR, Li X, et al. Pharmacological HIF2α inhibition improves VHL disease–associated phenotypes in zebrafish model. J Clin Invest. 2015;125(5):1987–1997. doi:10.1172/JCI73665
193. Tarade D, Ohh M. The HIF and other quandaries in VHL disease. Oncogene. 2018;37(2):139–147. doi:10.1038/onc.2017.338
194. Sizdahkhani S, Feldman MJ, Piazza MG, et al. Somatostatin receptor expression on von Hippel-Lindau-associated hemangioblastomas offers novel therapeutic target. Sci Rep. 2017;7(1):40822. doi:10.1038/srep40822
195. OʼToole SM, Drake WM. Response to somatostatin analog therapy in a patient with von Hippel-Lindau disease and multiple pancreatic neuroendocrine tumors. Pancreas. 2017;46(7):e57. doi:10.1097/MPA.0000000000000865
196. Yaghobi Joybari A, Azadeh P. Von Hippel-Lindau disease with multi-organ involvement: a case report and 8-year clinical course with follow-up. Am J Case Rep. 2017;18:1220–1224. doi:10.12659/AJCR.907356
197. Ma X, Jing Y, Liu Y, Yu L. Effect of clarithromycin in von Hippel-Lindau syndrome: a case report. J Int Med Res. 2019;47(2):973–981. doi:10.1177/0300060518792368
198. Weidmann E, Logan TF, Yasumura S, Kirkwood JM, Trucco M, Whiteside TL. Evidence for oligoclonal T-cell response in a metastasis of renal cell carcinoma responding to vaccination with autologous tumor cells and transfer of in vitro-sensitized vaccine-draining lymph node lymphocytes. Cancer Res. 1993;53(20):4745–4749.
199. Albinana V. Villar Gomez de Las Heras K, Serrano-Heras G, et al. Propranolol reduces viability and induces apoptosis in hemangioblastoma cells from von Hippel-Lindau patients. Orphanet J Rare Dis. 2015;10(1):118.
200. Cuesta AM, Albinana V, Gallardo-Vara E, et al. The beta2-adrenergic receptor antagonist ICI-118,551 blocks the constitutively activated HIF signalling in hemangioblastomas from von Hippel-Lindau disease. Sci Rep. 2019;9(1):10062.
201. Verbiest A, Lambrechts D, Van Brussel T, et al. Polymorphisms in the Von Hippel-Lindau gene are associated with overall survival in metastatic clear-cell renal-cell carcinoma patients treated with VEGFR tyrosine kinase inhibitors. Clin Genitourin Cancer. 2018;16(4):266–273.
202. Gross DJ, Reibstein I, Weiss L, et al. Treatment with halofuginone results in marked growth inhibition of a von Hippel-Lindau pheochromocytoma in vivo. Clin Cancer Res. 2003;9(10 Pt 1):3788–3793.
203. Pujol P, Bringer J, Faurous P, Jaffiol C. Metastatic phaeochromocytoma with a long-term response after iodine-131 metaiodobenzylguanidine therapy. Eur J Nucl Med. 1995;22(4):382–384.
204. Maher ER. Von Hippel-Lindau disease. Curr Mol Med. 2004;4(8):833–842.
205. Glasker S, Smith J, Raffeld M, Li J, Oldfield EH, Vortmeyer AO. VHL-deficient vasculogenesis in hemangioblastoma. Exp Mol Pathol. 2014;9(2):162–167.
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.