Back to Journals » Journal of Pain Research » Volume 12
Voltage gated sodium channels as therapeutic targets for chronic pain
Authors Ma RSY, Kayani K ![]() , Whyte-Oshodi D, Whyte-Oshodi A, Nachiappan N
, Whyte-Oshodi D, Whyte-Oshodi A, Nachiappan N ![]() , Gnanarajah S
, Gnanarajah S ![]() , Mohammed R
, Mohammed R ![]()
Received 5 March 2019
Accepted for publication 2 August 2019
Published 9 September 2019 Volume 2019:12 Pages 2709—2722
DOI https://doi.org/10.2147/JPR.S207610
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Überall
Renee Siu Yu Ma,1 Kayani Kayani,1 Danniella Whyte-Oshodi,1 Aiyesha Whyte-Oshodi,2 Nitish Nachiappan,3 Shaene Gnanarajah,1 Raihan Mohammed1
1Department of Medicine, University of Cambridge, Cambridge, UK; 2Faculty of Medicine, University College London, London, UK; 3Faculty of Medicine, Imperial College London, London, UK
Correspondence: Raihan Mohammed
School of Clinical Medicine, University of Cambridge, Hills Road, Cambridge CB2 0SP, UK
Tel +44 787 173 6004
Email [email protected]
Abstract: Being maladaptive and frequently unresponsive to pharmacotherapy, chronic pain presents a major unmet clinical need. While an intact central nervous system is required for conscious pain perception, nociceptor hyperexcitability induced by nerve injury in the peripheral nervous system (PNS) is sufficient and necessary to initiate and maintain neuropathic pain. The genesis and propagation of action potentials is dependent on voltage-gated sodium channels, in particular, Nav1.7, Nav1.8 and Nav1.9. However, nerve injury triggers changes in their distribution, expression and/or biophysical properties, leading to aberrant excitability. Most existing treatment for pain relief acts through non-selective, state-dependent sodium channel blockage and have narrow therapeutic windows. Natural toxins and developing subtype-specific and molecular-specific sodium channel blockers show promise for treatment of neuropathic pain with minimal side effects. New approaches to analgesia include combination therapy and gene therapy. Here, we review how individual sodium channel subtypes contribute to pain, and the attempts made to develop more effective analgesics for the treatment of chronic pain.
Keywords: nociceptors, TTX, neuropathic, electrogenesis, CNS, PNS
Introduction
With a global incidence of 20–25%, chronic pain is a significant health problem that significantly reduces the quality of life and presents a high economic burden.1,2 While both peripheral nervous system (PNS) and central nervous system (CNS) processes underlie the pain experience, PNS changes are necessary and sufficient to initiate and maintain CNS changes in chronic pain states.3,4 This gives reason to focus on controlling pathophysiological changes in the PNS, which is more accessible, and likely to have greater therapeutic impact than targeting the CNS.
Chronic pain that is neuropathic in origin is an important and unmet clinical problem.5 Existing treatments for neuropathic pain deliver inadequate pain relief and/or intolerable side effects necessitating the development of more effective therapeutics.6 Neuropathic pain (a result of somatosensory disease or damage) is distinguished from chronic nociceptive pain (a result of tissue disease or damage in which the nociceptive system is intact) in its underlying mechanisms and the requirement for distinct therapeutics.7 Nociceptor hyperexcitability induced by peripheral nerve injury is an established peripheral mechanism of neuropathic pain.8 Neuropathic pain has diverse etiologies, but hyperexcitability can explain positive clinical symptoms common to many neuropathic pain syndromes such as spontaneous ongoing pain (not stimuli-induced) and evoked pain/hypersensitivity (stimuli-induced), itself comprising allodynia (increased response to otherwise non-noxious stimuli) and hyperalgesia (increased perception of noxious stimuli).8
Voltage gated sodium channels (VGSCs) underlie the transduction and propagation of nociceptive signals and VGSC subtypes are selectively expressed in dorsal root ganglia (DRG) neurons.9
The expression and properties of VGSCs are dramatically altered by nerve injury, implying that the modulation of sodium currents critically contributes to the pathophysiological hyperexcitability that is associated with neuropathic pain states. Nav1.3, Nav1.7, Nav1.8, Nav1.9 are of particular interest due to their preferential distribution in nociceptors. Their importance in pain signaling is demonstrated by animal models of pain and human pain disorders.9 It is desirable but challenging to develop subtype- specific VGSC blockers which can minimize side effects outside the pain axis.
A brief overview of the pain axis
Pain is the unpleasant sensory and emotional experience associated with actual or potential tissue damage.10 The perception of pain begins with signal transduction in nociceptors, which are either slow conducting myelinated C fibers or thinly myelinated Ad fibers. Most nociceptive afferents form glutamatergic synapses onto spinal second-order neurons in the superficial laminae (I and II) in the dorsal horn where integration and processing of sensory inputs occurs. The net output is then carried by several pathways to distinct higher-order brain centers to signal the presence, location and intensity of noxious stimuli.11,12
An overview of VGSCs
VGSCs are hetero-multimeric, typically consisting of an -subunit associated with one or more -subunits.13 Nine mammalian genes (SCN1A-SCN5A and SCN8A- SCN11A) encode nine -subunits NaV1.1-NaV1.9 (henceforth referred as channels). These have distinct electrophysiological properties and characteristic patterns of tissue distribution.9 Conserved transmembrane segments of the -subunit are organized into four homologous domains (I-IV), each with six transmembrane -helices (S1-S6). The -subunit makes up the voltage sensor and channel pore which includes the selectivity filter.14 Most known pharmacological binding sites are located within the-subunit.15 Smaller associated beta subunits (30-40kDa) are encoded by the gene SCN1B- SCN4B and multifunctional. For example, they modulate channel gating properties, facilitate channel stabilization within the plasma membrane and are involved in channel localisation.16,17
VGSCs may be distinguished by their primary structure and kinetic properties.18
VGSCs transit between distinct conformational states in response to changes in membrane potential: resting (closed), activated (open), inactivated (closed), and repriming (a period of recovery from inactivation in which the channel cannot open in response to a depolarization). The inactivated state itself may exist as fast-inactivated (within milliseconds) and slow-inactivated (seconds).19
Pharmacologically, VGSCs may be classified by their sensitivity to the neurotoxin tetrodotoxin (TTX) (Table 1). TTX binds in the channel pore where a single residue determines susceptibility to blockade.9 A serine (Nav1.8, Nav1.9) or cysteine (Nav1.5) confers resistance to M TTX concentrations, while the presence of aromatic residues (like tyrosine in Nav1.7) engages TTX in a cation- interaction that increases the affinity of TTX-channel interaction and confers sensitivity to nM TTX concentrations.14
 |
Table 1 The pharmacological classification of VGSCs according to TTX sensitivity |
Dorsal root ganglia (DRG) neurons express more VGSC subtypes (up to five) than any other neuronal cell type.20 VGSCs are synthesized in the DRG cell body and accumulate at targets including nodes of Ranvier and peripheral terminals via axoplasmic transport mechanisms.21,22 This trafficking is dynamically regulated to ensure the correct complement of VGSCs arrive to confer an appropriate level of excitability.23
While Nav1.1 and Nav1.6 expression is common to CNS and PNS neurons, Nav1.3, Nav1.7, Nav1.8, Nav1.9 are specific to peripheral neurons.20 Nav1.7, Nav1.8 and Nav1.9 are expressed in sensory and myenteric neurons, and Nav1.7 is additionally expressed in sympathetic neurons.14 Immunocytochemical techniques reveal that smaller DRG neurons (likely nociceptors) express both TTX-S and TTX-R channels.24,25 The selective expression of Nav1.7, Nav1.8 and Nav1.9 in functionally identified nociceptors suggests they have evolved a specialized role in pain processing.26–28
Electrogenesis in nociceptors: normal and pathological
It is useful to first consider the processes required for normal nociception. Different modalities of noxious stimuli activate specific thermal-, mechanical- or chemical-sensitive transducer proteins and are converted into a membrane depolarization known as a generator potential. A generator potential exceeding a threshold is amplified by VGSCs to initiate an action potential that is propagated.1 The termination of nociceptors in free nerve endings suggests that sensory transduction is an intrinsic property of the afferent terminal.1
Acute nociceptive pain is a physiological response to external noxious stimuli) that can facilitate survival by warning of impending tissue damage.20 However, pain that is prolonged, magnified or spontaneous is pathological. Hypersensitivity at the site of inflammation is largely a result of sensitization (reduced threshold and increased excitability) of the peripheral terminals of nociceptors mediated by pro-inflammatory mediators.29 Pain may persist depending on the duration and strength of the immune response.
Ectopic activity underlies neuropathic pain
The most common complaint of chronic neuropathic pain patients is spontaneous, ongoing pain, caused by the emergence of sustained discharge at ectopic sites (pacemaker activity). Except for direct CNS injury, pacemaker activity predominantly originates in the PNS, and initiates and maintains “central sensitization”. This is not simply a reduction in threshold but can change the sensory modality of afferents from touch to pain.30,31 Peripheral analgesics aim to prevent ectopic discharges from gaining access to the CNS, which can eliminate both ongoing pain and allodynia.32
Studies show that in DRG neurons, subthreshold membrane potential oscillations are necessary to trigger ectopic repetitive firing. An oscillation sinusoid that crosses threshold evokes the first spike, and depolarizing after-potentials (DAPs) maintain an impulse train.33 It has been showed that the rapid rate of depolarization in the oscillation sinusoid can overcome membrane accommodation to enable spiking.34 The contribution of a few extra millivolts of depolarization is comparatively less important since slow ramp depolarization typical of physiological stimuli did not evoke spikes due to pronounced membrane accommodation. Chronic nerve injury increases the proportion of DRG neurons with subthreshold oscillations and consequently the intensity of ectopic spike discharge.35 Partial Na+ substitution, or bath application of lidocaine or TTX, eliminated oscillations and the associated ectopic discharge while preserving axonal spike propagation.35 This suggests a Na+ conductance sensitive to TTX contributes to oscillations and underlies the molecular pathogenesis of chronic nerve injury.36
Matzner (1992) showed that the threshold for repetitive discharge is distinct to that for evoking a single spike, the former being significantly more sensitive to changes in VGSC density.37 A simulation of increased Na+ conductance predicts a modest reduction in single- spike threshold, facilitating repetitive spiking. The gap between the two thresholds constitutes a “therapeutic window” within which ectopic firing can be suppressed without blocking normal sensory signaling.38 This is exploited by “membrane- stabilizing” drugs that block VGSCs: systemic administration of lidocaine selectively silences ectopia in injured DRG nerves and neuromas without blocking afferent nerve conduction.39
Current evidence suggest that membrane remodeling triggered by nerve injury underlies ectopic activity. Remodeling involves changes in the distribution, expression, and/or biophysical properties of ion channels that alters neuronal excitability. Pathological accumulation of various VGSC subtypes occurs in neuroma endings and patches of demyelination for several reasons.39–41 Altered trafficking disrupts fast axonal transport leading to the local accumulation of channel-loaded transport vesicles; or demyelination that removes myelin-mediated suppression of channel insertion; or axotomy which removes normal downstream targets of distribution all promote redistribution into remaining competent membrane, including sites upstream to injury.23,41 Thus, both permissive and promotional factors generate ectopic pacemaker sites.
Peripheral nerve injury also alters membrane excitability by triggering dysregulated transcription of VGSC genes to produce an abnormal repertoire of VGSCs.42 This is not simply a recapitulation of developmentally expressed VGSC subtypes, since a different set of VGSCs is upregulated post-injury.43 In rat DRG neurons, axotomy upregulates previously undetected Nav1.3 channels and downregulates abundant Nav1.8 and Nav1.9 channels.44–46
This partly due to interrupted access to peripheral sources of neurotrophic factors47 For example, nerve growth factor (NGF) delivery to axotomised DRG neurons upregulates TTX-R currents.45 Therefore, post-injury remodeling of membrane electrical properties in response to neurotrophic factors may be mechanism for aberrant excitability, leading to chronic pain.
Electrophysiological properties of individual VGSCs (Table 2)
Nav1.7 produces a fast -activating and -inactivating, slowly-repriming TTX-S current.49 Nav1.7 displays slow onset of inactivation, a property that permits it to remain available for activation and produce a ramp current in response to small slow depolarizations.50 Thus Nav1.7 acts as a “threshold channel” important in early phases of electrogenesis: it amplifies generator potentials to bring the neuron to the more depolarized firing threshold of Nav1.8.20 It thereby sets the gain in nociceptors where it is co-expressed with Nav1.8.51,52
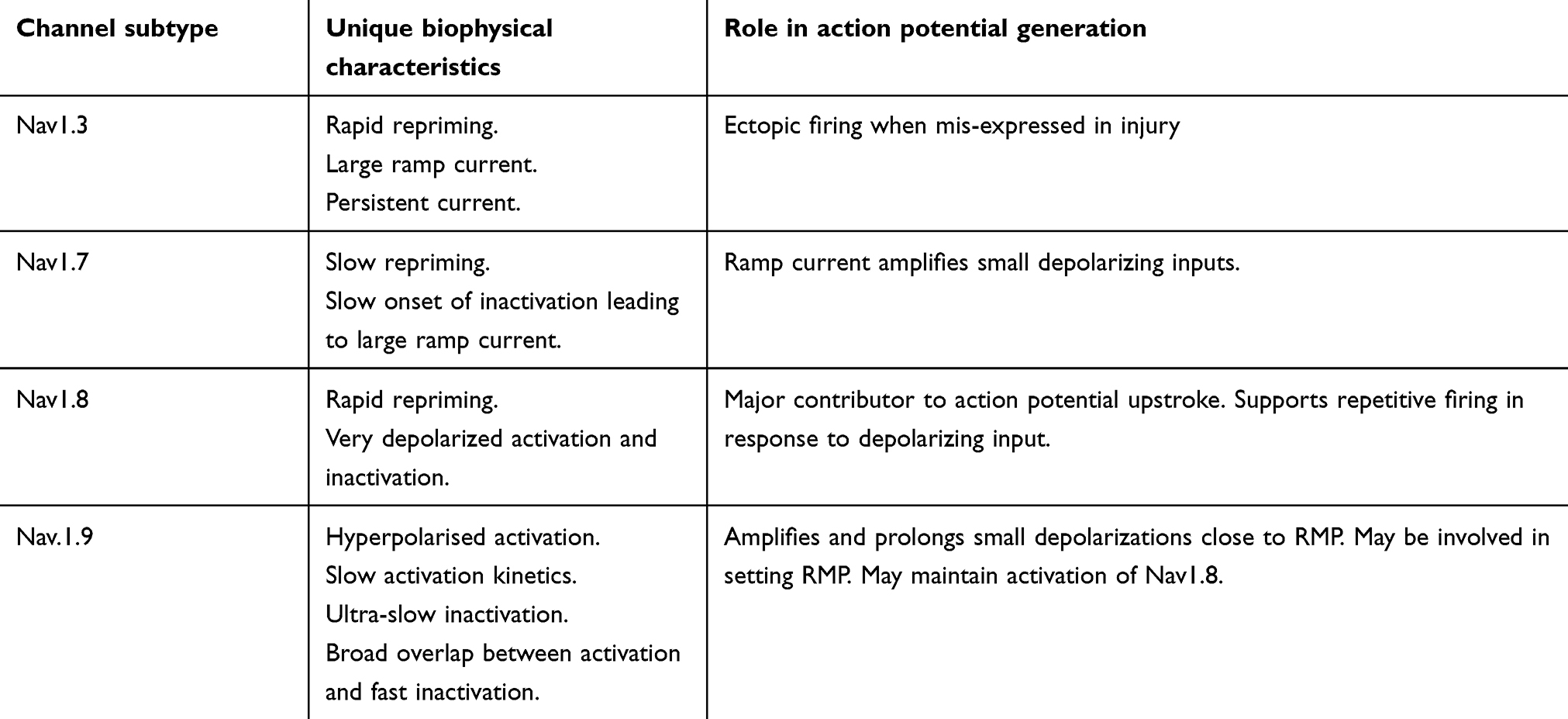 |
Table 2 properties of VGSCs and their role in generating action potentials (AP) |
Nav1.8 produces a slow-inactivating TTX-R current characterized by significantly depolarized activation and inactivation and rapid recovery from fast inactivation.53–55 This enables Nav1.8 to contribute most of the inward current in the action potential upstroke.56 Han et al 2015 demonstrated that human Nav1.8 displays slower inactivation and larger persistent and ramp currents compared to rat Nav1.8, paralleled by longer-lasting action potentials and increased firing frequency.57 Thus Nav1.8 channels play a major role in regulating the firing properties of DRG neurons. It is important to take in consideration the distinct properties of human and rat Nav1.8 channels when extrapolating from rodent pain studies to humans and testing novel blockers for pain treatment.57
Nav1.9 mediates a TTX-R current that is challenging to study due to current instability and poor expression in heterologous systems.58,59 Nav1.9 characteristically activates at hyperpolarized potentials and displays an extremely slow inactivation. The significant overlap of activation and inactivation produces large “window currents” (a wide range of voltages in which a channel may open) around the resting membrane potential (RMP), which are predicted to increase depolarization of the RMP and so can boost weak stimuli.44 The more hyperpolarized activation voltage shown to be around −80 mV in human DRG neurons, indicates that Nav1.9 can be activated by small sub-threshold depolarizations to generate persistent sodium currents.60,61 Knockout studies confirm that Nav1.9 produces the persistent TTX-R current: TTX-R was eliminated in DRG neurons of Nav1.9-knockout mice but restored by expression of recombinant Nav1.9 channels in these neurons.62 The extremely slow kinetics of Nav1.9 suggest that it minimally contributes to the action potential upstroke. Instead, current evidence supports its role as a threshold channel, through contributing a Na+ conductance that regulates the RMP and prolongs the depolarizing response to subthreshold stimuli, lowering the threshold for single action potentials and supporting repetitive firing.63 Computer simulations show that Nav1.9 conductance depolarizes the cell resting potential even at channel densities as low as 20% of the density estimated to be present in DRG neurons, supporting this conclusion.64
Contribution of individual VGSCs to pain
Nav1.3
Nav1.3 expression in rat embryonic neural tissues is lost within a few days after birth.43 Initial interest in Nav1.3 was due to strong evidence for its re- expression in primary-, secondary-, and third-order neurons following peripheral nerve and after chronic constriction injury (CCI) of the sciatic nerve in adult rats, while other channel subtypes are downregulated.44,60,64,65 Nav1.3 upregulation is paralleled by the emergence of a rapidly-repriming TTX-S current.36 Re-expression and the property of fast recovery from inactivation suggests the role of Nav1.3 in sustaining higher-than-normal frequency firing in chronic pain conditions.48 This is compatible with the observation of Nav1.3 accumulation within the transected axon tips of both rat and painful human neuromas, as well as the reversal of neuropathic pain behavior by intrathecal administration of oligonucleotides (ODN) against Nav1.3 mRNA.44,64,66,67
However, the specificity of antisense studies is not absolute and using different ODNs failed to replicate the finding.68 Moreover, global or DRG-specific knock- out of Nav1.3 with no genetic compensation, does not impair pain behavior after nerve injury.69 The increase in Na+ currents contributed by upregulated Nav1.3 appears to be insignificant, and injury-induced hyperexcitability is likely to be mediated by other VGSC subtypes.20 It appears that Nav1.3 is neither necessary nor sufficient to drive neuropathic pain, ruling it out as an effective analgesic target and ATP.79,80
Nav1.7
Animal studies of inflammatory pain indicate a role for Nav1.7 in acquired channelopathies. Peripheral tissue inflammation significantly increases TTX-S current density in DRG neurons, paralleled by increased Nav1.7 transcript and protein levels. The increase in Nav1.7 is more robust than that of Nav1.3, the other TTX-S channel upregulated under these conditions.81 NGF, an important inflammatory can also increase Nav1.7 expression.82,83 These findings suggest that upregulation of Nav1.7 leading to neuronal hyperexcitability is important in inflammatory pain signaling.52
This is supported by both knock-down (KD) and knock-out (KO) mice studies. Global Nav1.7 deletion is neonatal lethal. Conditional Nav1.7 KO in Nav1.8-positive nociceptors leads to loss of acute and inflammatory pain, and selective Nav1.7 KD attenuated inflammatory hyperalgesia.70,71 Nav1.7 KO in nociceptors preserved neuropathic pain behavior of mice, yet a class of benzazepinone Nav1.7 blockers reversed tactile allodynia in a rat model of neuropathic pain.71,84 Perhaps a contribution of Nav1.7 to neuropathic pain lies in a very limited population of Nav1.8-negative DRG neurons.48
SCN9A located in chromosome (2q31-32) encodes Nav1.7. Genetic studies that directly link SCN9A mutations to three inherited human pain syndromes strongly implicates Nav1.7 in pain-signaling.85 Dominantly-inherited gain-of-function Nav1.7 mutations lead to severe pain in inherited erythromelalgia (IEM), characterized by recurrent episodes of bilateral burning pain, erythema and mild swelling in the extremities triggered by mild warmth or physical exertion; and in paroxysmal extreme pain disorder (PEPD), a visceral pain condition characterized by paroxysms of rectal, ocular or mandibular burning pain that may be induced by bowel movement or probing of perianal areas.86–89 Pain in these syndromes is either evoked by mild stimuli or spontaneous, closely resembling neuropathic pain symptoms. In contrast, recessive loss-of-function mutations lead to truncated non-functional Nav1.7 channels and congenital insensitivity to pain (CIP), where patients are otherwise normal apart from the severe loss of pain perception and anosmia.52,90,91 This provides genetic validation for Nav1.7 to be a promising analgesic target with minimal side effects.
Ten Nav1.7 missense mutations have been identified in early- and delayed-onset IEM, and eight mutations in PEPD.86 Biophysical characterization of these mutations reveals that IEM mutations in Nav1.7 affect channel regions involved in activation, while PEPD mutations affect regions regulating fast-inactivation.13 Documented IEM mutations in Nav1.7 all significantly hyperpolarize activation voltage-dependency; some slow deactivation (transition from an open to closed state) and increase the ramp response to small slow depolarizations.20 In contrast, PEPD mutations do not affect activation but depolarize fast inactivation voltage-dependency and may make inactivation incomplete resulting in a persistent current.20 Therefore, although the mechanisms for increased Na+ current through mutant Nav1.7 channels differ, lowered activation in IEM and impaired inactivation in PEPD both drive DRG neuron hyperexcitability, consistent with warm-evoked pain in IEM and normal bowel movement evoked pain in PEPD patients. This provides an understanding how Nav1.7 contributes to pain pathophysiology at a molecular level.
Whole cell current-clamp studies confirmed that IEM Nav1.7 mutations lead to neuronal hyperexcitability. Wild-type or mutant Nav1.7 (L858H, F1449A, A863P) were transfected into DRG neurons and the effects of mutations on firing properties were investigated.92–94 As predicted, IEM mutants lower the threshold for single action potentials and increase firing frequency in response to supra- threshold stimuli. The PEPD mutant (M1627K) increases DRG firing frequency.19 Therefore, hyperexcitbailty of nociceptive DRG neurons induced by mutant Nav1.7 channels can explain pain associated with IEM and PEPD.
Interestingly, the Nav1.7 mutant (L858H) produces functionally opposing phenotypes of hyperexcitability in DRG and hypo-excitability in sympathetic (superior cervical ganglion, SCG) neurons. This may be explained by the selective presence of Nav1.8 in sensory but not sympathetic neurons. Introduction of Nav1.8 into SCG rescues SCG firing properties.92 Sympathetic neuron hypoexcitability can explain attenuated cutaneous vasoconstriction and skin flushing observed in IEM and PEPD patients, although it is unclear why there is no global sympathetic dysfunction. While highlighting the role of Nav1.8 in supporting neuron hyperexcitbailty (further discussed in 6.3.1), this example illustrates that VGSCs do not act in isolation to alter neuronal excitability. The impact of an ion channel mutation on neuronal excitability is not necessarily predictable solely based on changes in the mutated channel; likewise, the predicted effects of drugs targeting a particular VGSC may not translate to functional effects. The ensemble of ion channels present in the cell must be considered.
IEM is generally refractory to pharmacotherapy. VGSC blockers like lidocaine or mexiletine are largely ineffective, a case of early onset IEM (N395K) suggesting this results from reduced drug affinity of the mutant channel.52,86,95 By contrast, PEPD symptoms are well controlled by the anti-convulsant VGSC blocker carbamazepine, a use-dependent inhibitor that preferentially binds and stabilizes the inactivated state.19 Hence counteracting impaired inactivation of PEPD mutants can account for drug efficacy. Carbamazepine is not expected to be effective for IEM patients since most IEM mutations do not alter channel inactivation. These findings demonstrate how drug efficacy depends on the effect of the underlying genetic mutations on channel function.
Nav1.8
A role for Nav1.8 in initiating and maintaining inflammatory pain is well documented in animal studies.96 Nav1.8-null mice have impaired NGF- and carrageenan- induced thermal hyperalgesia.77,97 Visceral inflammatory pain responses were impaired after capsaicin, a model in which hyperalgesia and pain are maintained by ongoing activity due to sensitization on initial application, consistent with Nav1.8 expression in all DRG neurons innervating the colon.98,99 This supports the essential role of Nav1.8 in mediating spontaneous activity in sensitized nociceptors.78 The role for Nav1.8 in inflammatory pain is further supported by the upregulation of Nav1.8 in DRG neurons in rats after direct treatment with inflammatory mediators.81,100–102 Nav1.8 anti-sense treatment demonstrated Nav1.8 TTX-R channels to be involved in afferent nerve sensitization after chemical irritation of the rat bladder, suggesting them to represent a new target to treat visceral inflammatory pain.76
The role of Nav1.8 in neuropathic pain is less understood. Normal neuropathic pain behavior is observed in Nav1.8 KO mice as well as in double Nav1.7/Nav1.8 KO.71,97 This argues that Nav1.8 does not contribute to neuropathic pain. Nav1.8 knock-down by antisense ODN or siRNA attenuated mechanical allodynia and hyperalgesia in animal models of chronic pain, implicating a functional role for Nav1.8 at least in the expression of experimental neuropathic pain.72,74 However, this was not immediately obvious since peripheral nerve injury downregulated Nav1.8 mRNA, protein and associated TTX-R currents in injured axons.46,60 Later, functional Nav1.8 channels were observed to be redistributed in uninjured afferents, possibly in response to inflammatory cytokines like NGF produced during Wallerian degeneration.75,103 Therefore, an injury-induced redistribution of Nav1.8 to uninjured axons leading to hyperexcitability provides a plausible explanation for how Nav1.8 contributes neuropathic pain in animal models. Blocking Nav1.8 pharmacologically or the processes underlying redistribution may selectively eliminate neuropathic pain behavior.75
In chronic neuropathic pain patients, pre-synthesized channel proteins translocate and accumulate in sites proximal to injury and in neuromas after an initial reduction in Nav1.8 expression, leading to ectopic firing and persistent hypersensitivity.66,104,105 This supports Nav1.8 as a useful target to treat chronic local hypersensitivity
More convincing data comes from gain-of-function mutations in SCN10A encoding Nav1.8, identified in human patients with painful neuropathies.106–108 Current clamp showed that mutations (L554P, A1304T), others reduce current threshold and increase firing frequency in response to supra-threshold stimuli depolarize resting potential (A1304) or induce spontaneous firing of small DRG neurons including nociceptors (L554P). The first three changes would lower threshold, or increase the intensity of evoked pain, while the latter change would contribute to spontaneous pain. Subtle changes in channel biophysics caused by Nav1.8 mutations markedly alter neuronal excitability, highlighting the importance of even small changes in human Nav1.8 channel properties for pain signaling.106
Nav1.9
Animal studies support a role for Nav1.9 in inflammatory pain. Nav1.9-null mice have greatly impaired or absent inflammatory hyperalgesia in response to inflammatory mediators.79,80,109 Second messengers like prostaglandin E2 acting through a G protein-coupled pathway, increases Nav1.9 current density in DRG neurons in vitro; whilst treatment with IL-1 increases persistent TTX-R that is associated with Nav1.9 in a p38 mitogen-activated protein kinase (MAPK)-dependent manner.102,110 These findings support the notion that various inflammatory mediators potentiate Nav1.9 currents to maintain inflammation-induced hyperalgesia. Thus Nav1.9 is an attractive target to develop analgesics for inflammatory disorders.
The correlation of Nav1.9 activity with neuropathic pain is uncertain in animal models. Nav1.9 expression is downregulated in injured neurons without a significant change in neighboring uninjured neurons.36,45,111 Antisense ODN-mediated Nav1.9 KD did not ameliorate neuropathic pain.73 Nav1.9 KO mice show impaired somatic inflammatory pain behavior, but unaltered neuropathic pain.79,80 However, orofacial neuropathic pain (characteristic of trigeminal neuralgia) produced by constriction of the infraorbital nerve in mice is dependent on Nav1.9.112 Moreover, Nav1.9 levels increase in large-diameter neurons of diabetic rats but are unaltered in small DRG neurons suggesting a contribution of Nav1.9 to diabetic neuropathy pain.113 Thus current evidence indicates a role for Nav1.9 in inflammatory, diabetic neuropathy and orofacial neuropathic pain.
Seven different mutations in SCN11A encoding Nav1.9 channels identified in peripheral neuropathy patients confirm Nav1.9 involvement in neuropathic pain. For example, the missense mutations (I381 and L1158P) reduce current threshold and increase firing frequency in response to supra-threshold stimuli, leading to hyperexcitability.114
Implications for therapeutic approaches for neuropathic pain
Small molecule pharmacotherapy
Currently, most VGSC blockers clinically used to alleviate pain are non-selective as they bind to highly conserved residues in the pore domain. Blockade is often state-dependent. The local anesthetic lidocaine more readily accesses channels in the open state and exhibits highest affinity for fast-inactivated state.115 These underlie its use-dependence (blockage increases with firing frequency), thereby limiting hyperexcitability. Systemic lidocaine is shown to be effective in a variety of neuropathic pain states, providing long-term relief with minimal side effects if infusion is limited to five mg/kg/hour.116 Topical lidocaine (5%) is effective for post-herpetic neuralgia (PHN) in which pain is triggered at a specific dermatome.117 Other clinically effective agents include anticonvulsants carbamazepine and lamotrigine for human immunodeficiency virus-associated neuropathic pain, the anti-dysrhythmic mexelentine and tricyclic antidepressants (also block neuronal VGSCs).118–121 Despite their uses, the lack of selectivity of current analgesics to VGSC subtypes lead to narrow therapeutic windows and limited efficacy.
It is desirable for drugs to selectively block ectopic hyperexcitability while preserving physiological nerve conduction. Lacosamide is an effective anticonvulsant that is also a promising analgesic. It effectively reduces pain-associated behavior with minimal adverse effects in animal models of neuropathic pain, and is successful as monotherapy for diabetic neuropathic pain.122–124 It acts in a novel mechanism by selectively enhancing slow inactivation.125 Whole-cell patch-clamp electrophysiology showed that at a holding potential of −80 mV, Lacosamide at clinically relevant concentrations (10–70 M) effectively reduces Nav1.7 and Nav1.8 currents and to a lesser extent Nav1.3.95 It only enhances the voltage-dependence of steady state inactivation, contrasting with carbamazepine and lidocaine that enhance steady- state fast-inactivation. Moreover, Lacosamide demonstrated a greater ability to selectively block inactivated rather than resting VGSCs. This suggests a greater ability to inhibit chronically depolarized neurons while sparing those with normal RMP, which predicts a better safety profile.95
Molecularly-selective blockers
One strategy of improving selectivity is engineering agents that bind poorly conserved regions outside the pore. The term “molecularly-selective” implies inhibition is independent of channel state.126 PF-05089771 (Pfizer) is an aryl sulfonamide compound with 1000-fold selectivity for Nav1.7 over Nav1.5 and Nav1.8, stabilizing Nav1.7 in a non- conductive state.127 It is currently in Phase II trials (NCT02215252) to treat diabetic peripheral neuropathy pain. Current data shows that sulfonamides present a principal class used to develop Nav1.7 inhibitors.126
Nav1.8-specific blockers
A-803467 is a potent and highly selective Nav1.8 blocker, exhibiting up to 1000-fold greater potency for Nav1.8 blockage than other VGSC subtypes; blockage is voltage-dependent without significant frequency-dependence.128 A-803467 effectively suppressed spontaneous and electrically evoked firing in rat DRG neurons, and dose- dependently reduced nociception in experimental pain model. A-803467 was found to be most effective in reducing pain in models of neuropathic and inflammatory pain. It is interesting that the analgesic profile of A-8034687 is consistent with the pattern of anti- nociceptive effects of antisense OGN treatment. The fact that analgesic effects are not equivalent across all pain models suggests that Nav1.8 channels may differentially mediate certain forms of nociceptive processing, or that other VGSC subtypes contribute to nociception in specific pain states. Furthermore, A-803467 via spinal or systemic administration can attenuate both spontaneous and mechanically evoked firing of wide dynamic range neurons in the dorsal horn of nerve-injured rats.129 Although formulation of A-803467 suitable for human use is challenging due to poor bioavailability, its identification proves that subtype-specific VGSC blockers can be synthesized.
Toxins
Natural neurotoxins are highly potent, non-selective VGSCs blockers with therapeutic potential. TTX shows little selectivity for TTX-S channels in the nM range but have up to
100-fold reduced affinity to cardiac Nav1.5 channels.58 Despite lack of selectivity, a phase III trial although underpowered, indicates that subcutaneous TTX (TEC-006) may provide clinically meaningful analgesia for persistent refractory cancer pain.130 Although selectivity and systemic toxicity of toxins constrains clinical use, they are promising scaffolds for more specific inhibitors.
Peptide -conotoxins of marine cone snails, GIIIA and GIIB, block the rat skeletal muscle Nav1.4 by binding neurotoxin site 1.131 It is desirable to develop - conotoxin derivatives targeting neuronal VGSC subtypes. MrVIB is a synthetic -conotoxin showing significant analgesic activity in animal models of pain due to a 10-fold higher affinity for Nav1.8 than other VGSC subtypes.132 MrVIB therefore provides a basis for development of Nav1.8-selective blockers that will have greater therapeutic index than non-selective blockers like lidocaine.
Peptide tarantula toxins bind and impede the movement of VGSC voltage sensors, thereby reducing the peak of Na+ conductance.133 Some are subtype-selective. ProTx-II shows up to 50-fold greater selectivity for Nav1.7 than Nav1.5 channels, whilst Huwentoxin-I and -IV have virtually no effects on muscle VGSCs but potently inhibit neuronal TTX-S channels particularly Nav1.7.133–136 Developing analgesics using large peptide toxins is advantageous since their interaction with multiple residues increases subtype-specificity. Moreover, their charge prevents them from crossing the blood-brain barrier (BBB), limiting effects to the periphery. However, peptide toxins typically have poor bioavailability on oral administration limiting their clinical utility.12
Combination therapy
The link of Nav1.7 to human pain disorders has energized a focus on Nav1.7 as a logical analgesic target that in theory, should have minimal side effects. Potent specific antagonists have been tested in humans but with limited success in replicating a CIP phenotype.137 Surprisingly, an increased selectivity of inhibitors for Nav1.7 is associated with reduced analgesic potency. An explanation is provided by opioid-mediated analgesia that seems to account for most of the CIP phenotype. The major role of opioids is supported by analgesia in Nav1.7-null mutant mice and humans shown to be reversible by naloxone (opioid antagonist). Loss of Nav1.7 expression is linked to upregulation of Penk (precursor of met-enkephalin). High levels (0.5 M) of TTX can produce complete Nav1.7 block in wild type DRG neurons that also leads to opioid upregulation; but TTX at five times the IC50 for Nav1.7 could prevent enhanced encephalin expression. This suggests that recapitulation of the CIP phenotype requires a 100% Nav1.7 block, an unrealistic pharmacological goal.138 Thus, combining specific Nav1.7 antagonist with opioid or enkephalinase blocker should provide an alternative strategy to produce analgesia.
Gene therapy
Gene therapy enabling cell-type specific inhibition of neuronal excitability is a potential strategy, but technical problems present a major challenge. Control of gene expression through a drug-dependent regulation system maintains appropriate levels of gene products within the therapeutic window. Examples are adeno-associated virus (AAV)-mediated gene delivery, but irreversible gene silencing and the lack of neuron-specificity are potential problems;the Tet-on system is limited by an immune response to components of the viral delivery system.137,139
Conclusion
Peripheral nerve block has been long used to treat pain conditions through inhibition of VGSCs. Animal and human studies have validated Nav1.7, Nav1.8 and Nav1.9 as attractive targets for pain therapeutics. These three VGSC subtypes play central roles in rendering nociceptors hyperexcitable, a fundamental mechanism leading to neuropathic pain. Despite a detailed characterisation of the underlying mechanisms leading to hyperexcitability, development of effective therapeutics has not progressed remarkably compared to other areas of medicine. Knowledge of the diverse mechanisms underlying different types of pain is still limited. A significant challenge are the many factors complicating data interpretation. In animal studies, differences in animal species and sex, and inter-strain genetic differences between rats and mice in which most KD and KO studies are performed respectively may explain conflicting findings; multiple splice isoforms of VGSC subtypes may have differential contributions to hyperexcitability; the off-targets effects of antisense treatment may account for mismatches between KO and KD studies. The fact that none of the channels function in isolation adds further complexity.140
VGSC blockers that target aberrant activity in nociceptors and are weakly brain penetrant have distinct advantages over currently available broad-spectrum blockers in treating pain, such as circumvention of CNS side effects like ataxia and sedation. One example is cyclopentane dicarboxamide (CDA54) exhibiting 33-fold lower brain than plasma concentrations and effectively reduced neuropathic pain in two different animal nerve injury models.109 CDA54 further demonstrates that blocking peripheral VGSCs is sufficient for analgesic efficacy. Despite the great advances in whole genome sequencing, genetic manipulation in mice is invaluable in providing mechanistic insight that enable drug design.137 Nav1.7 is currently the most promising target for alleviating chronic pain. Combination therapy has been shown to be effective in animal models but requires confirmation in humans. Given that effective pain management is a majorly unmet clinical need, the pursuit for better pain therapeutics is hugely rewarding.
Author contributions
All authors contributed to data analysis, drafting or revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Gold MS, Gebhart GF. Nociceptor sensitization in pain pathogenesis. Nat Med. 2010;16(11):1248–1257. doi:10.1038/nm.2235
2. McCarberg BH, Billington R. Consequences of neuropathic pain: quality-of-life issues and associated costs. Am J Manag Care. 2006;12(9 Suppl):S263–S268. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16774458. Accessed August 20, 2019.
3. Staud R, Nagel S, Robinson ME, Price DD. Enhanced central pain processing of fibromyalgia patients is maintained by muscle afferent input: a randomized, double-blind, placebo-controlled study. Pain. 2009;145(1–2):96–104. doi:10.1016/j.pain.2009.05.020
4. Verne GN, Robinson ME, Vase L, Price DD. Reversal of visceral and cutaneous hyperalgesia by local rectal anesthesia in irritable bowel syndrome (IBS) patients. Pain. 2003;105(1–2):223–230. doi:10.1016/S0304-3959(03)00210-0
5. Bouhassira D, Lantéri-Minet M, Attal N, Laurent B, Touboul C. Prevalence of chronic pain with neuropathic characteristics in the general population. Pain. 2008;136(3):380–387. doi:10.1016/j.pain.2007.08.013
6. Rice ASC, Hill RG. New treatments for neuropathic pain. Annu Rev Med. 2006;57(1):535–551. doi:10.1146/annurev.med.57.121304.131324
7. Baron R. Mechanisms of disease: neuropathic pain - A clinical perspective. Nat Clin Pract Neurol. 2006;2(2):95–106. doi:10.1038/ncpneuro0113
8. Devor M. Neuropathic pain: what do we do with all these theories? Acta Anaesthesiol Scand. 2001;45(9):1121–1127. doi:10.1034/j.1399-6576.2001.450912.x
9. Dib-Hajj SD, Waxman SG. Diversity of composition and function of sodium channels in peripheral sensory neurons. Pain. 2015;156(12):2406–2407. doi:10.1097/j.pain.0000000000000353
10. Merskey H NB. IASP Terminology.
11. Kuner R. Central mechanisms of pathological pain. Nat Med. 2010;16(11):1258–1266. doi:10.1038/nm.2231
12. Dib-Hajj SD, Binshtok AM, Cummins TR, Jarvis MF, Samad T, Zimmermann K. Voltage-gated sodium channels in pain states: role in pathophysiology and targets for treatment. Brain Res Rev. 2009;60(1):65–83. doi:10.1016/j.brainresrev.2008.12.005
13. Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26(1):13–25. Available from: https://www.cell.com/neuron/pdf/S0896-6273(00)81133-2.pdf. Accessed August 20, 2019.
14. Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International union of pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev. 2005;57(4):411–425. doi:10.1124/pr.57.4.5
15. Sula A, Booker J, Ng LCT, Naylor CE, Decaen PG, Wallace BA. The complete structure of an activated open sodium channel. Nat Commun. 2017;8:1–9. doi:10.1038/ncomms14205
16. Namadurai S, Balasuriya D, Rajappa R, et al. Crystal structure and molecular imaging of the Na V channel β3 subunit indicates a trimeric assembly. J Biol Chem. 2014;289(15):10797–10811. doi:10.1074/jbc.M113.527994
17. Brackenbury WJ, Isom LL. Voltage-gated Na + channels: potential for β subunits as therapeutic targets. Expert Opin Ther Targets. 2008;12(9):1191–1203. doi:10.1517/14728222.12.9.1191
18. Catterall WA. Cellular and molecular biology of voltage-gated sodium channels. Physiol Rev. 1992;72(suppl_4):S15–S48. doi:10.1152/physrev.1992.72.suppl_4.S15
19. Dib-Hajj SD, Estacion M, Jarecki BW, et al. Paroxysmal extreme pain disorder M1627K mutation in human Na v 1.7 renders DRG neurons hyperexcitable. Mol Pain. 2008;4:
20. Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. Sodium channels in normal and pathological pain. Annu Rev Neurosci. 2010;33(1):325–347. doi:10.1146/annurev-neuro-060909-153234
21. Lombet A, Laduron P, Mourre C, Jacomet Y, Lazdunski M. Axonal transport of the voltage-dependent Na+ channel protein identified by its tetrodotoxin binding site in rat sciatic nerves. Brain Res. 1985;345(1):153–158. doi:10.1016/0006-8993(85)90846-7
22. Wonderlin WF, French RJ. Ion channels in transit: voltage-gated Na and K channels in axoplasmic organelles of the squid Loligo pealei. Proc Natl Acad Sci. 1991;88(10):4391–4395. doi:10.1073/pnas.88.10.4391
23. Leterrier C, Brachet A, Fache MP, Dargent B. Voltage-gated sodium channel organization in neurons: protein interactions and trafficking pathways. Neurosci Lett. 2010;486(2):92–100. doi:10.1016/j.neulet.2010.08.079
24. Amaya F, Decosterd I, Samad TA, et al. Diversity of expression of the sensory neuron-specific TTX-resistant voltage-gated sodium ion channels SNS and SNS2. Mol Cell Neurosci. 2000;15(4):331–342. doi:10.1006/mcne.1999.0828
25. Fjell J, Hjelmström P, Hormuzdiar W, et al. Localization of the tetrodotoxin-resistant sodium channel NaN in nociceptors. Neuroreport. 2000;11(1):199–202. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10683857. Accessed August 20, 2019.
26. Fang X, Djouhri L, Black JA, Dib-Hajj SD, Waxman SG, Lawson SN. The presence and role of the tetrodotoxin-resistant sodium channel Nav1.9 (NaN) in nociceptive primary afferent neurons. J Neurosci. 2002;22(17):7425–7433. Available from: http://www.jneurosci.org/content/22/17/7425.abstract. Accessed August 20, 2019.
27. Djouhri L, Fang X, Okuse K, Wood JN, Berry CM, Lawson SN. The TTX-resistant sodium channel Nav 1.8 (SNS/PN3): expression and correlation with membrane properties in rat nociceptive primary afferent neurons. J Physiol. 2003;550(3):739–752. doi:10.1113/jphysiol.2003.042127
28. Djouhri L, Newton R, Levinson SR, Berry CM, Carruthers B, Lawson SN. Sensory and electrophysiological properties of guinea-pig sensory neurones expressing Nav 1.7 (PN1) Na+ channel α subunit protein. J Physiol. 2003;546(2):565–576. doi:10.1113/jphysiol.2002.026559
29. Lai J, Porreca F, Hunter JC, Gold MS. Voltage-gated sodium channels and hyperalgesia. Annu Rev Pharmacol Toxicol. 2004;44(1):371–397. doi:10.1146/annurev.pharmtox.44.101802.121627
30. Woolf CJ. Evidence for a central component of post-injury pain hypersensitivity. Nature. 1983;306(5944):686–688.Available from: http://www.ncbi.nlm.nih.gov/pubmed/6656869. Accessed August 20, 2019.
31. Campbell JN, Raja SN, Meyer RA, Mackinnon SE. Myelinated afferents signal the hyperalgesia associated with nerve injury. Pain. 1988;32(1):89–94. doi:10.1016/0304-3959(88)90027-9
32. Gracely RH, Lynch SA, Bennett GJ. Painful neuropathy: altered central processing maintained dynamically by peripheral input. Pain. 1992;51(2):175–194. doi:10.1016/0304-3959(92)90259-E
33. Amir R, Michaelis M, Devor M. Burst discharge in primary sensory neurons: triggered by subthreshold oscillations, maintained by depolarizing afterpotentials. J Neurosci. 2002;22(3):1187–1198. doi:10.1523/JNEUROSCI.22-03-01187.2002
34. Kovalsky Y, Amir R, Devor M. Subthreshold oscillations facilitate neuropathic spike discharge by overcoming membrane accommodation. Exp Neurol. 2008;210(1):194–206. doi:10.1016/j.expneurol.2007.10.018
35. Amir R, Michaelis M, Devor M. Membrane potential oscillations in dorsal root ganglion neurons: role in normal electrogenesis and neuropathic pain. J Neurosci. 1999;19(19):8589–8596. Available from: http://www.ncbi.nlm.nih.gov/pubmed/10493758. Accessed August 20, 2019.
36. Cummins TR, Waxman SG. Downregulation of tetrodotoxin-resistant sodium currents and upregulation of a rapidly repriming tetrodotoxin-sensitive sodium current in small spinal sensory neurons after nerve injury. J Neurosci. 1997;17(10):3503–3514. doi:10.1523/jneurosci.3386-11.2011
37. Matzner O, Devor M. Na+conductance and the threshold for repetitive neuronal firing. Brain Res. 1992;597(1):92–98. doi:10.1016/0006-8993(92)91509-D
38. McMahon. Wall and Melzack’s Textbook of Pain. Elsevier/Saunders; 2013.
39. Devor M, Wall PD, Catalan N. Systemic lidocaine silences ectopic neuroma and DRG discharge without blocking nerve conduction. Pain. 1992;48(2):261–268. doi:10.1016/0304-3959(92)90067-L
40. Coggan JS, Prescott SA, Bartol TM, Sejnowski TJ. Imbalance of ionic conductances contributes to diverse symptoms of demyelination. Proc Natl Acad Sci. 2010;107(48):20602–20609. doi:10.1073/pnas.1013798107
41. Black JA, Felts P, Smith KJ, Kocsis JD, Waxman SG. Distribution of sodium channels in chronically demyelinated spinal cord axons: immuno-ultrastructural localization and electrophysiological observations. Brain Res. 1991;544(1):59–70. doi:10.1016/0006-8993(91)90885-Y
42. Boucher TJ, Okuse K, Bennett DL, Munson JB, Wood JN, McMahon SB. Potent analgesic effects of GDNF in neuropathic pain states. Science. 2000;290(5489):124–127. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11021795. Accessed August 20, 2019.
43. Waxman SG, Kocsis JD, Black JA. Type III sodium channel mRNA is expressed in embryonic but not adult spinal sensory neurons, and is reexpressed following axotomy. J Neurophysiol. 1994;72(1):466–470. doi:10.1152/jn.1994.72.1.466
44. Black JA, Cummins TR, Plumpton C, et al. Upregulation of a silent sodium channel after peripheral, but not central, nerve injury in DRG neurons. J Neurophysiol. 1999;82(5):2776–2785. doi:10.1152/jn.1999.82.5.2776
45. Dib-Hajj SD, Black JA, Cummins TR, Kenney AM, Kocsis JD, Waxman SG. Rescue of α-SNS sodium channel expression in small dorsal root ganglion neurons after axotomy by nerve growth factor in vivo. J Neurophysiol. 1998;79(5):2668–2676. doi:10.1152/jn.1998.79.5.2668
46. Sleeper AA, Cummins TR, Dib-Hajj SD, et al. Changes in expression of two tetrodotoxin-resistant sodium channels and their currents in dorsal root ganglion neurons after sciatic nerve injury but not rhizotomy. J Neurosci. 2000;20(19): 7279–7289. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11007885. Accessed August 20, 2019.
47. Waxman SG. Transcriptional channelopathies: an emerging class of disorders. Nat Rev Neurosci. 2001;2(9):652–659. doi:10.1038/35090026
48. Momin A, Wood JN. Sensory neuron voltage-gated sodium channels as analgesic drug targets. Curr Opin Neurobiol. 2008;18(4):383–388. doi:10.1016/j.conb.2008.08.017
49. Klugbauer N, Lacinova L, Flockerzi V, Hofmann F. Structure and functional expression of a new member of the tetrodotoxin-sensitive voltage-activated sodium channel family from human neuroendocrine cells. Embo J. 1995;14(6):1084–1090. Available from: http://www.ncbi.nlm.nih.gov/pubmed/7720699%0Ahttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC398185. Accessed August 20, 2019.
50. Cummins TR, Howe JR, Waxman SG. Slow closed-state inactivation: a novel mechanism underlying ramp currents in cells expressing the hNE/PN1 sodium channel. J Neurosci. 1998;18(23):9607–9619. doi:10.1523/JNEUROSCI.18-23-09607.1998
51. Waxman SG. A channel sets the gain on pain. Nature. 2006;444(7121):831–832. doi:10.1038/444831a
52. Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. From genes to pain: Nav1.7 and human pain disorders. Trends Neurosci. 2007;30(11):555–563. doi:10.1016/j.tins.2007.08.004
53. Akopian AN, Sivilotti L, Wood JN. A tetrodotoxi n-resistant voltage-gated sodium channel expressed by sensory neurons. Nature. 1996;379(6562):257–262. doi:10.1038/379257a0
54. Sangameswaran L, Fish LM, Koch BD, et al. A novel tetrodotoxin-sensitive, voltage-gated sodium channel expressed in rat and human dorsal root ganglia. J Biol Chem. 1997;272(23):14805–14809. doi:10.1074/jbc.272.23.14805
55. Elliott AA, Elliott JR. Characterization of TTX-sensitive and TTX-resistant sodium currents in small cells from adult rat dorsal root ganglia. J Physiol. 1993;463:39–56. Available from:: http://www.ncbi.nlm.nih.gov/pubmed/8246189. Accessed August 20, 2019.
56. Renganathan M, Cummins TR, Waxman SG. Contribution of Na v 1.8 sodium channels to action potential electrogenesis in DRG neurons. J Neurophysiol. 2017;86(2):629–640. doi:10.1152/jn.2001.86.2.629
57. Han C, Estacion M, Huang J, et al. Human Na(v)1.8: enhanced persistent and ramp currents contribute to distinct firing properties of human DRG neurons. J Neurophysiol. 2015;113(9):3172–3185. doi:10.1152/jn.00113.2015
58. Leffler A, Herzog RI, Dib-Hajj SD, Waxman SG, Cummins TR. Pharmacological properties of neuronal TTX-resistant sodium channels and the role of a critical serine pore residue. Pflugers Arch Eur J Physiol. 2005;451(3):454–463. doi:10.1007/s00424-005-1463-x
59. Goral RO, Leipold E, Nematian-Ardestani E, Heinemann SH. Heterologous expression of NaV1.9 chimeras in various cell systems. Pflugers Arch Eur J Physiol. 2015;467(12):2423–2435. doi:10.1007/s00424-015-1709-1
60. Dib-Hajj SD, Fjell J, Cummins TR, et al. Plasticity of sodium channel expression in DRG neurons in the chronic constriction injury model of neuropathic pain. Pain. 1999;83(3):591–600. doi:10.1016/S0304-3959(99)00169-4
61. Coste B, Osorio N, Padilla F, Crest M, Delmas P. Gating and modulation of presumptive NaV1.9 channels in enteric and spinal sensory neurons. Mol Cell Neurosci. 2004;26(1):123–134. doi:10.1016/j.mcn.2004.01.015
62. Östman JAR, Nassar MA, Wood JN, Baker MD. GTP up-regulated persistent Na + current and enhanced nociceptor excitability require Na V 1.9. J Physiol. 2008;586(4):1077–1087. doi:10.1113/jphysiol.2007.147942
63. Baker MD, Chandra SY, Ding Y, Waxman SG, Wood JN. GTP-induced tetrodotoxin-resistant Na+current regulates excitability in mouse and rat small diameter sensory neurones. J Physiol. 2003;548(2):373–382. doi:10.1113/jphysiol.2003.039131
64. Hains BC. Altered sodium channel expression in second-order spinal sensory neurons contributes to pain after peripheral nerve injury. J Neurosci. 2004;24(20):4832–4839. doi:10.1523/jneurosci.0300-04.2004
65. Zhao P, Waxman SG, Hains BC. Sodium channel expression in the ventral posterolateral nucleus of the thalamus after peripheral nerve injury. Mol Pain. 2006;2:
66. Black JA, Nikolajsen L, Kroner K, Jensen TS, Waxman SG. Multiple sodium channel isoforms and mitogen-activated protein kinases are present in painful human neuromas. Ann Neurol. 2008;64(6):644–653. doi:10.1002/ana.21527
67. Hains BC, Klein JP, Saab CY, Craner MJ, Black JA, Waxman SG. Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J Neurosci. 2003;23(26):8881–8892. Available from: http://www.ncbi.nlm.nih.gov/pubmed/14523090. Accessed August 20, 2019.
68. Lindia JA, Köhler MG, Martin WJ, Abbadie C. Relationship between sodium channel NaV1.3 expression and neuropathic pain behavior in rats. Pain. 2005;117(1–2):145–153. doi:10.1016/j.pain.2005.05.027
69. Nassar MA, Baker MD, Levato A, et al. Nerve injury induces robust allodynia and ectopic discharges in Na v 1.3 null mutant mice. Mol Pain. 2006;2:
70. Yeomans DC, Levinson SR, Peters MC, et al. Decrease in inflammatory hyperalgesia by herpes vector-mediated knockdown of Na v 1.7 sodium channels in primary afferents. Hum Gene Ther. 2005;16(2):271–277. doi:10.1089/hum.2005.16.271
71. Nassar MA, Levato A, Stirling LC, Wood JN. Neuropathic pain develops normally in mice lacking both Na v 1.7 and Na v 1.8. Mol Pain. 2005;1:
72. Lai J, Gold MS, Kim CS, et al. Inhibition of neuropathic pain by decreased expression of the tetrodotoxin-resistant sodium channel, NaV1.8. Pain. 2002;95(1–2):143–152. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11790477. Accessed August 20, 2019.
73. Porreca F, Lai J, Bian D, et al. A comparison of the potential role of the tetrodotoxin-insensitive sodium channels, PN3/SNS and NaN/SNS2, in rat models of chronic pain. Proc Natl Acad Sci. 1999;96(14):7640–7644. doi:10.1073/pnas.96.14.7640
74. Joshi SK, Mikusa JP, Hernandez G, et al. Involvement of the TTX-resistant sodium channel Nav 1.8 in inflammatory and neuropathic, but not post-operative, pain states. Pain. 2006;123(1–2):75–82. doi:10.1016/j.pain.2006.02.011
75. Gold MS, Weinreich D, Kim C-S, et al. Redistribution of Na(V)1.8 in uninjured axons enables neuropathic pain. J Neurosci. 2003;23(1):158–166.
76. Yoshimura N, Seki S, Novakovic SD, et al. The involvement of the tetrodotoxin-resistant sodium channel Na v 1.8 (PN3/SNS) in a rat model of visceral pain. J Neurosci. 2001;21(21):8690–8696. doi:10.1523/JNEUROSCI.21-21-08690.2001
77. Akopian AN, Okuse K, Souslova V, England S, Ogata N, Wood JN. Trans-splicing of a voltage-gated sodium channel is regulated by nerve growth factor. FEBS Lett. 1999;445(1):177–182. doi:10.1016/S0014-5793(99)00126-X
78. Laird JMA, Souslova V, Wood JN, Cervero F. Deficits in visceral pain and referred hyperalgesia in Nav1.8 (SNS/PN3)-null mice. J Neurosci. 2002;22(19):8352–8356. doi:10.1523/JNEUROSCI.22-19-08352.2002
79. Priest BT, Liberator P, Kaczorowski GJ, et al. Contribution of the tetrodotoxin-resistant voltage-gated sodium channel NaV1.9 to sensory transmission and nociceptive behavior. Proc Natl Acad Sci. 2005;102(26):9382–9387. doi:10.1073/pnas.0501549102
80. Amaya F, Wang H, Costigan M, et al. The voltage-gated sodium channel Na(v)1.9 is an effector of peripheral inflammatory pain hypersensitivity. J Neurosci. 2006;26(50):12852–12860. doi:10.1523/JNEUROSCI.4015-06.2006
81. Black JA, Liu S, Tanaka M, Cummins TR, Waxman SG. Changes in the expression of tetrodotoxin-sensitive sodium channels within dorsal root ganglia neurons in inflammatory pain. Pain. 2004;108(3):237–247. doi:10.1016/j.pain.2003.12.035
82. Toledo-Aral JJ, Brehm P, Halegoua S, Mandel G. A single pulse of nerve growth factor triggers long-term neuronal excitability through sodium channel gene induction. Neuron. 1995;14(3):607–611. doi:10.1016/0896-6273(95)90317-8
83. Gould HJ, Gould TN, England JD, Paul D, Liu ZP, Levinson SR. A possible role for nerve growth factor in the augmentation of sodium channels in models of chronic pain. Brain Res. 2000;854(1–2):19–29. doi:10.1016/S0006-8993(99)02216-7
84. Hoyt SB, London C, Gorin D, et al. Discovery of a novel class of benzazepinone Nav1.7 blockers: potential treatments for neuropathic pain. Bioorganic Med Chem Lett. 2007;17(16):4630–4634. doi:10.1016/j.bmcl.2007.05.076
85. Drenth JPH, Finley WH, Breedveld GJ, et al. The primary erythermalgia–susceptibility gene is located on chromosome 2q31-32. Am J Hum Genet. 2001;68(5):1277–1282. doi:10.1086/320107
86. Drenth JPH, Waxman SG. Mutations in sodium-channel gene. J Clin Invest. 2007;117(12):3603–3610. doi:10.1172/JCI33297.type
87. Hisama FM, Dib-Hajj SD, Waxman SG SCN9A-related inherited erythromelalgia. GeneReviews®. 2006. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20301342.
88. Novella SP, Hisama FM, Dib-Hajj SD, Waxman SG. A case of inherited erythromelalgia. Nat Clin Pract Neurol. 2007;3(4):229–234. doi:10.1038/ncpneuro0425
89. Fertleman CR, Baker MD, Parker KA, et al. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron. 2006;52(5):767–774. doi:10.1016/j.neuron.2006.10.006
90. Cox JJ, Reimann F, Nicholas AK, et al. An SCN9A channelopathy causes congenital inability to experience pain. Nature. 2006;444(7121):894–898. doi:10.1038/nature05413
91. Goldberg Y, MacFarlane J, MacDonald M, et al. Loss-of-function mutations in the Nav1.7 gene underlie congenital indifference to pain in multiple human populations. Clin Genet. 2007;71(4):311–319. doi:10.1111/j.1399-0004.2007.00790.x
92. Rush AM, Dib-Hajj SD, Liu S, Cummins TR, Black JA, Waxman SG. A single sodium channel mutation produces hyper- or hypoexcitability in different types of neurons. Proc Natl Acad Sci. 2006;103(21):8245–8250. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1472458%5C&tool=pmcentrez%5C&rendertype=abstract http://www.pnas.org/cgi/doi/10.1073/pnas.0602813103%5Cnpapers2://publication/doi/10.1073/pnas.0602813103. Accessed August 20, 2019.
93. Dib-Hajj SD, Rush AM, Cummins TR, et al. Gain-of-function mutation in Nav1.7 in familial erythromelalgia induces bursting of sensory neurons. Brain. 2005;128(8):1847–1854. doi:10.1093/brain/awh514
94. Harty TP, Dib-Hajj SD, Tyrrell L, et al. Nav1.7 mutant A863P in erythromelalgia: effects of altered activation and steady-state inactivation on excitability of nociceptive dorsal root ganglion neurons. J Neurosci. 2006;26(48):12566–12575. doi:10.1523/JNEUROSCI.3424-06.2006
95. Sheets PL, Jackson JO, Waxman SG, Dib-hajj SD, Cummins TR. A Nav 1.7 channel mutation associated with hereditary erythromelalgia contributes to neuronal hyperexcitability and displays reduced lidocaine sensitivity. J Physiol. 2007;581(3):1019–1031. doi:10.1113/jphysiol.2006.127027
96. Gold MS. Tetrodotoxin-resistant Na+ currents and inflammatory hyperalgesia. Proc Natl Acad Sci. 1999;96(14):7645–7649. doi:10.1073/pnas.96.14.7645
97. Kerr BJ, Souslova V, McMahon SB, Wood JN. A role for the TTX-resistant sodium channel Nav 1.8 in NGF-induced hyperalgesia, but not neuropathic pain. Neuroreport. 2001;12(14):3077–3080. Available from: http://www.ncbi.nlm.nih.gov/pubmed/11568640. Accessed August 20, 2019.
98. Schmelz M, Schmid R, Handwerker HO, Torebjörk HE. Encoding of burning pain from capsaicin-treated human skin in two categories of unmyelinated nerve fibres. Brain. 2000;123 Pt 3:560–571. Available from:: http://www.ncbi.nlm.nih.gov/pubmed/10686178. Accessed August 20, 2019.
99. Gold MS, Zhang L, Wrigley DL, Traub RJ. Prostaglandin E 2 modulates TTX-R I Na in rat colonic sensory neurons. J Neurophysiol. 2002;88(3):1512–1522. doi:10.1152/jn.2002.88.3.1512
100. Gold MS, Levine JD. DAMGO inhibits prostaglandin E2-induced potentiation of a TTX-resistant Na+ current in rat sensory neurons in vitro. Neurosci Lett. 1996;212(2):83–86. doi:10.1016/0304-3940(96)12791-9
101. Jin X. Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor. J Neurosci. 2006;26(1):246–255. doi:10.1523/jneurosci.3858-05.2006
102. Binshtok AM, Wang H, Zimmermann K, et al. Nociceptors are interleukin-1beta sensors. J Neurosci. 2008;28(52):14062–14073. doi:10.1523/JNEUROSCI.3795-08.2008
103. Wu G, Ringkamp M, Hartke TV, et al. Early onset of spontaneous activity in uninjured C-fiber nociceptors after injury to neighboring nerve fibers. J Neurosci. 2001;21(8):RC140. doi:10.1523/JNEUROSCI.21-08-j0002.2001
104. Coward K, Plumpton C, Facer P, et al. Immunolocalization of SNS/PN3 and NaN/SNS2 sodium channels in human pain states. Pain. 2000;85(1–2):41–50. doi:10.1016/S0304-3959(99)00251-1
105. Yiangou Y, Birch R, Sangameswaran L, Eglen R, Anand P. SNS/PN3 and SNS2/NaN sodium channel-like immunoreactivity in human adult and neonate injured sensory nerves. FEBS Lett. 2000;467(2–3):249–252. doi:10.1016/S0014-5793(00)01166-2
106. Faber CG, Lauria G, Merkies ISJ, et al. Gain-of-function Nav1.8 mutations in painful neuropathy. Proc Natl Acad Sci U S A. 2012;109(47):19444–19449. doi:10.1073/pnas.1216080109
107. Huang J, Yang Y, Zhao P, et al. Small-fiber neuropathy Nav1.8 mutation shifts activation to hyperpolarized potentials and increases excitability of dorsal root ganglion neurons. J Neurosci. 2013;33(35):14087–14097. doi:10.1523/JNEUROSCI.2710-13.2013
108. Han C, Vasylyev D, Macala LJ, et al. The G1662S NaV1.8 mutation in small fibre neuropathy: impaired inactivation underlying DRG neuron hyperexcitability. J Neurol Neurosurg Psychiatry. 2014;85(5):499–505. doi:10.1136/jnnp-2013-306095
109. Brochu RM. Block of peripheral nerve sodium channels selectively inhibits features of neuropathic pain in rats. Mol Pharmacol. 2005;69(3):823–832. doi:10.1124/mol.105.018127
110. Rush AM, Waxman SG. PGE2increases the tetrodotoxin-resistant Nav1.9 sodium current in mouse DRG neurons via G-proteins. Brain Res. 2004;1023(2):264–271. doi:10.1016/j.brainres.2004.07.042
111. Decosterd I, Ji RR, Abdi S, Tate S, Woolf CJ. The pattern of expression of the voltage-gated sodium channels Nav1.8 and Nav1.9 does not change in uninjured primary sensory neurons in experimental neuropathic pain models. Pain. 2002;96(3):269–277. doi:10.1016/S0304-3959(01)00456-0
112. Lulz AP, Kopach O, Santana-Varela S, Wood JN. The role of Na v 1.9 channel in the development of neuropathic orofacial pain associated with trigeminal neuralgia. Mol Pain. 2015;11:
113. Black JA, Waxman SG, Craner MJ, Klein JP, Renganathan M. Changes of sodium channel expression in experimental painful diabetic neuropathy. Ann Neurol. 2002;52(6):786–792. doi:10.1002/ana.10364
114. Huang J, Han C, Estacion M, et al. Gain-of-function mutations in sodium channel NaV1.9 in painful neuropathy. Brain. 2014;137(6):1627–1642. doi:10.1093/brain/awu079
115. Catterall WA. Molecular mechanisms of gating and drug block of sodium channels. Novartis Found Symp. 2002;241:
116. Bach FW, Jensen TS, Kastrup J, Stigsby B, Dejgård A. The effect of intravenous lidocaine on nociceptive processing in diabetic neuropathy. Pain. 1990;40(1):29–34. doi:10.1016/0304-3959(90)91047-M
117. Dworkin RH, O’Connor AB, Backonja M, et al. Pharmacologic management of neuropathic pain: evidence-based recommendations. Pain. 2007;132(3):237–251. doi:10.1016/j.pain.2007.08.033
118. Mcquay H, Carroll D, Jadad AR, Wiffen P, Moore A. Anticonvulsant drugs for management of pain: a systematic review. Bmj. 1995;311(7012):1047. doi:10.1136/bmj.311.7012.1047
119. Simpson DM, McArthur JC, Olney R, et al. Lamotrigine for HIV-associated painful sensory neuropathies: a placebo-controlled trial. Neurology. 2003;60(9):1508–1514. doi:10.1212/01.WNL.0000063304.88470.D9
120. Chabal C, Jacobson L, Mariano A, Chaney E, Britell CW. The use of oral mexiletine for the treatment of pain after peripheral nerve injury. Anesthesiology. 1992;76(4):513–517. Available from: http://www.ncbi.nlm.nih.gov/pubmed/1312797. Accessed August 20, 2019.
121. Pancrazio JJ, Kamatchi GL, Roscoe AK, Lynch C. Inhibition of neuronal Na+ channels by antidepressant drugs. J Pharmacol Exp Ther. 1998;284(1):208–214. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9435180. Accessed August 20, 2019.
122. Beyreuther B, Callizot N, Stöhr T. Antinociceptive efficacy of lacosamide in a rat model for painful diabetic neuropathy. Eur J Pharmacol. 2006;539(1–2):64–70. doi:10.1016/j.ejphar.2006.04.009
123. Beyreuther BK, Freitag J, Heers C, Krebsfänger N, Scharfenecker U, Stöhr T. Lacosamide: a review of preclinical properties. CNS Drug Rev. 2007;13(1):21–42. doi:10.1111/j.1527-3458.2007.00001.x
124. Doty P, Rudd GD, Stoehr T, Thomas D. Lacosamide. Neurotherapeutics. 2007;4(January):145–148. doi:10.1016/j.nurt.2006.10.002
125. Errington AC, Stohr T, Heers C, Lees G. The investigational anticonvulsant lacosamide selectively enhances slow inactivation of voltage-gated sodium channels. Mol Pharmacol. 2007;73(1):157–169. doi:10.1124/mol.107.039867
126. Sun S, Cohen CJ, Dehnhardt CM. Inhibitors of voltage-gated sodium channel Nav1.7: patent applications since 2010. Pharm Pat Anal. 2014;3(5):509–521. doi:10.4155/ppa.14.39
127. Theile JW, Fuller MD, Chapman ML. The selective Nav1.7 inhibitor, PF-05089771, interacts equivalently with fast and slow inactivated Nav1.7 channels. Mol Pharmacol. 2016;90(5):540–548. doi:10.1124/mol.116.105437
128. Jarvis MF, Honore P, Shieh -C-C, et al. A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc Natl Acad Sci U S A. 2007;104(20):8520–8525. doi:10.1073/pnas.0611364104
129. McGaraughty S, Chu K, Scanio M. Nav1. 8 sodium channel blocker, A-803467, attenuates spinal neuronal activity in neuropathic rats. Pharmacol. 2008;324(3):1204–1211. doi:10.1124/jpet.107.134148.1990
130. Hagen NA, Cantin L, Constant J, et al. Tetrodotoxin for moderate to severe cancer-related pain: a multicentre, randomized, double-blind, placebo-controlled, parallel-design trial. Pain Res Manag. 2017;2017:1–7. doi:10.1155/2017/7212713
131. Stephan MM, Potts JF, Agnew WS. The microI skeletal muscle sodium channel: mutation E403Q eliminates sensitivity to tetrodotoxin but not to mu-conotoxins GIIIA and GIIIB. J Membr Biol. 1994;137(1):1–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/7911843. Accessed August 20, 2019.
132. Ekberg J, Wood JN, Adams DJ, et al. O-conotoxin MrVIB selectively blocks Nav1.8 sensory neuron specific sodium channels and chronic pain behavior without motor deficits. Proc Natl Acad Sci. 2006;103(45):17030–17035. doi:10.1073/pnas.0601819103
133. Xiao Y, Bingham J-P, Zhu W, Moczydlowski E, Liang S, Cummins TR. Tarantula huwentoxin-IV inhibits neuronal sodium channels by binding to receptor site 4 and trapping the domain ii voltage sensor in the closed configuration. J Biol Chem. 2008;283(40):27300–27313. doi:10.1074/jbc.M708447200
134. Middleton RE, Warren VA, Kraus RL, et al. Two tarantula peptides inhibit activation of multiple sodium channels. Biochemistry. 2002;41(50):14734–14747. doi:10.1021/bi026546a
135. Smith JJ, Cummins TR, Alphy S, Blumenthal KM. Molecular Interactions of the Gating Modifier Toxin ProTx-II with Na v 1.5. J Biol Chem. 2007;282(17):12687–12697. doi:10.1074/jbc.m610462200
136. Peng K, Shu Q, Liu Z, Liang S. Function and solution structure of huwentoxin-IV, a potent neuronal tetrodotoxin (TTX)-sensitive sodium channel antagonist from Chinese bird spider Selenocosmia huwena. J Biol Chem. 2002;277(49):47564–47571. doi:10.1074/jbc.M204063200
137. Emery EC, Luiz AP, Wood JN. Na v 1.7 and other voltage-gated sodium channels as drug targets for pain relief. Expert Opin Ther Targets. 2016;20(8):975–983. doi:10.1517/14728222.2016.1162295
138. Minett MS, Pereira V, Sikandar S, et al. Endogenous opioids contribute to insensitivity to pain in humans and mice lacking sodium channel Nav1.7. Nat Commun. 2015;6(1):8967. doi:10.1038/ncomms9967
139. Le Guiner C, Stieger K, Toromanoff A, et al. Transgene regulation using the tetracycline-inducible TetR-KRAB system after AAV-mediated gene transfer in rodents and nonhuman primates. Lewin AS, ed. PLoS One. 2014;9(9):e102538. doi:10.1371/journal.pone.0102538
140. Mogil JS. Animal models of pain: progress and challenges. Nat Rev Neurosci. 2009;10(4):283–294. doi:10.1038/nrn2606
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.