Back to Journals » Cancer Management and Research » Volume 17

Vitamin K1 Induced Cytotoxic Effects and Transcriptomic Analysis in Jurkat T Lymphocyte Leukemia Cells
Authors Shang Y ![]() , Si S, Qin Y, Li X, Wang H, Ma X
, Si S, Qin Y, Li X, Wang H, Ma X ![]() , Wu Y, Lou X, Song S
, Wu Y, Lou X, Song S ![]()
Received 4 July 2025
Accepted for publication 22 November 2025
Published 17 December 2025 Volume 2025:17 Pages 3171—3183
DOI https://doi.org/10.2147/CMAR.S550692
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Seema Singh
Ying Shang, Shaoyan Si, Yaya Qin, Xinlou Li, Hongjiang Wang, Xiaoyu Ma, Yingying Wu, Xiaotong Lou, Shujun Song
Department of Research, The Ninth Medical Center, Chinese PLA General Hospital, Beijing, People’s Republic of China
Correspondence: Shujun Song, Department of Research, The Ninth Medical Center, Chinese PLA General Hospital, No. 9 Anxiangbeili Street, Chaoyang District, Beijing, 100101, People’s Republic of China, Email [email protected] Xiaotong Lou, Department of Research, The Ninth Medical Center, Chinese PLA General Hospital, No. 9 Anxiangbeili Street, Chaoyang District, Beijing, 100101, People’s Republic of China, Email [email protected]
Purpose: Vitamin K1 (VK1) has been proved to have anticancer properties in various cancer cells. However, little is known about the effects of VK1 on hematologic malignancies. The aim of this study was to evaluate the cytotoxic effects of VK1 on Jurkat T Lymphocyte Leukemia Cells (Jurkat T cells), as well as to investigate the changes in gene expression.
Methods: Jurkat T cells and normal human peripheral blood mononuclear cells (PBMCs) were treated with VK1 at different concentrations, and the cell viabilities were evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Cell apoptosis was detected by Annexin V-FITC cell apoptosis detection kit and the cell cycle was determined by PI staining and flow cytometric analysis. Differentially expressed genes (DEGs) of Jurkat T cells induced by VK1 were analyzed by RNA sequencing. The mRNA expression of HMGCR and HMGCS1 were further verified by real-time RT-qPCR.
Results: VK1 showed an obviously antiproliferative effect on Jurkat T cells but no any effects on normal human PBMCs. VK1 induced Jurkat T cells apoptosis and cell cycle arrest at G0/G1 phase. A total of 21 down-regulated and 34 up-regulated genes were identified by transcriptome analysis. We selected HMGCR and HMGCS1 genes among the significantly up-regulated genes for further RT-qPCR analyzation. It was confirmed that VK1 up-regulated the mRNA expression of HMGCR and HMGCS1.
Conclusion: VK1 has cytotoxic effects and transcriptional regulation of multiple genes on Jurkat T cells. The genes of HMGCR and HMGCS1 related pathways may play roles in this process.
Keywords: vitamin K1, cell proliferation, cell apoptosis, cell cycle, HMGCR, HMGCS1
Introduction
Acute lymphocytic leukemia (ALL) is an aggressive hematological malignancy that affects both children and adults, with incidence peaking between 1 and 4 years of age.1 From 1990 to 2017, the global incidence of ALL increased by 30.81%.2 The 5-year survival rate of ALL varies with age, 85% for children and 50% for adults.3 For patients with relapsed/refractory ALL, the 5-year survival rate is lower than 10%.4 The current therapeutic approaches for ALL typically involve chemotherapy and radiation therapy.5 Although high-dose multi-agent chemotherapy has shown clinical effectiveness, it can also cause some severe side effects, such as myelosuppression.6 Therefore, it is necessary to search for agents that is safe and non-toxicity to improve the management of ALL.
Vitamin K (VK) is a fat-soluble vitamin with a common skeleton of 2-methyl-1,4-naphthoquinone structure, which is modified by different side chains to form various VKs including naturally occurring VK1 (phylloquinone) and VK2 (menaquinone), as well as synthetic forms such as VK3 (menadione), VK4, and VK5. VK1 has a saturated side chain, also known as leaf green quinone, which mainly exists in green leafy vegetables such as broccoli, lettuce, spinach, fermented soy (natto), spring onions, and cabbage naturally.7 VK2 is a compound characterized by partially unsaturated side chains and is commonly produced by various bacteria through the fermentation of meat and dairy products. VK3 is a synthetic vitamin that can be alkylated into VK2 in the liver.
VK is essential in the coagulation process. VK, by serving as a cofactor, plays a vital role in the conversion of glutamic (Glu) residues into γ- Carboxyglutamic acid (Gla) residues, which is important for ensuring proper blood clotting and preventing excessive bleeding.8 In addition to coagulation, VK also plays an active role in maintaining bone and cardiovascular health,9,10 and promoting wound healing.11 In recent years, increasing evidences suggest the cytotoxicity of VKs on different cancer cells. It has been demonstrated that VK1 has anticancer properties across several types of cell lines, including liver, colon, lung, stomach, nasopharynx, breast, pancreatic and oral epidermoid cancers.12–15 VK1 also shows inhibitory effects on Neuroblastoma cells (NBP2).16 In addition, the anticancer effects of VK1 are also observed in combination with other compounds. The combination of VK1 and Lactobacillus rhamnosus GG shows against colon adenocarcinoma cells. The co-treatment with VK1 and Sorafenib induces growth inhibition and apoptosis in pancreatic cancer cells and glioma cells.17,18
The cytotoxic effect of VK1 on cancer cells is due to the promotion of cell cycle arrest and apoptosis through transcription factors such as c-Myc, c-Jun, and c-Fos.19,20 VK1 can trigger apoptosis of liver cancer cells through the phosphorylation of JNK and c-Jun, as well as caspase activation.21 In human colon cancer cells and gastric cancer cells, VK1 induces apoptosis involving mitogen-activated protein kinase (MAPK) pathway.13,14 VK1 starts caspase-dependent apoptosis through MAP kinase pathway and leads to an inhibition of pancreatic cancer cell survival.15
Numerous studies have reported the anticancer effect of VK1 on solid tumors, but little is known about the effects of VK1 on hematological malignancies. Given the urgent need for novel therapeutic agents with better safety profiles in leukemia treatment, exploring the potential of naturally derived compounds like VK1 is of significant value. Furthermore, unlike other vitamin K analogs whose effects on leukemia have been preliminarily explored, the impact of VK1 on T-cell acute lymphoblastic leukemia remains largely uncharacterized. The objectives of this study were to evaluate the cytotoxic effects of VK1 on Jurkat T cells and attempt to understand its underlying mechanism by the observation of VK1 induced DEGs.
Materials and Methods
Cells and Cell Culture
Jurkat T cells were obtained from Academy of Military Medical Sciences, Academy of Military Sciences, Beijing, China. The use of Jurkat T cells was approved by the Ethical Committee of the Ninth Medical Center, Chinese PLA General Hospital. Normal human PBMCs were isolated from the peripheral blood of healthy volunteers according to the method previously described.22 The cells were cultured in RPMI 1640 medium (HyClone, USA) supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS; Gibco BRL, USA), antibiotics (penicillin 100U/mL and streptomycin 100μg/mL; Livning Biotechnology, Beijing, China) at 37 °C under 5% CO2 in air. This study has been approved by the Ethical Committee of the Ninth Medical Center, Chinese PLA General Hospital, Beijing, China (approval number: LL-LCSY-2023-03). The use of Jurkat T cells was also covered under this protocol, and separate approval for the utilization of normal human PBMCs was granted by the same committee.
Cell Viability Measurement
Jurkat T cells and normal human PBMCs were cultured in mediums containing VK1 (Huayang Pharmaceutical, Jiangsu, China) at five different concentrations (4.6 μM, 9.2 μM, 18.3 μM, 27.5 μM and 55 μM) in 96-well plates for 48 h. MTT stock solution (5mg/mL; Sigma Aldrich, USA) was added to the medium at 1/10 of the original culture volume in each well and cells were cultured for additional four hours. After incubation, the supernatant was discarded and DMSO (150μL; Solarbio, Beijing, China) was added to each well. The absorbance was detected at 570nm using microplate reader (Bio-Rad, CA, USA). The relative cell viability was expressed as the ratio of the absorbance of VK1 treated cells against that of the untreated cells.
Apoptotic Analysis
Jurkat T cells (5 × 105–1×106 cells) were cultured in medium containing VK1 (10 μM and 50 μM) for 24 h or 48 h, then washed with PBS. The cells were harvested. The apoptotic cells were examined using Annexin V-FITC Cell Apoptosis Detection kit (Beyotime Biotechnology, Shanghai, China). The apoptotic cells were estimated by Annexin V/ Propidium Iodide (PI) staining. The cells were analyzed by flow cytometer (Navios, Beckman Coulter Inc., Brea, CA, USA) and subsequently classified into four groups, viable (Annexin V− /PI−), early apoptotic (Annexin V+/PI−), late apoptotic (Annexin V+/PI+), and necrotic (Annexin V−/PI+) cells.23 Data were analyzed using Kaluza Analysis 2.1 software (Beckman Coulter, CA, USA).
Cell Cycle Analysis
Jurkat T cells (1 × 106 cells) were seeded in flask and incubated with medium containing VK1 (10 μM and 50 μM) for 24 h or 48 h. The cells were harvested and washed with PBS, then fixed in 100% ethanol at −20 °C for 15min, treated with ribonuclease A (RNaseA) (100 μg/mL) (Sigma Aldrich, USA), and stained with PI (3 μM) (Sigma Aldrich, USA). The DNA content of cells was determined by a flow cytometer (Navios, Beckman Coulter Inc., Brea, CA, USA) and data were analyzed by ModFit LT (version 32, Verity Software House, Topsham, ME, USA).
Transcriptomic Analysis and Protein–Protein Interaction (PPI) Network Analysis
Jurkat T cells (1 × 107 cells) were seeded in flask and incubated with culture medium containing 50 μM VK1 for 15 h. Total RNA was extracted using TRIZOL Reagent (Cat#15596-018, Life technologies, Carlsbad, CA, US) following the standard protocol provided by the manufacturer. The total RNA was further purified using RNAClean XP Kit (Cat A63987, Beckman Coulter, Inc. Kraemer Boulevard Brea, CA, USA) and RNase-Free DNase Set (Cat#79254, QIAGEN, GmBH, Germany) after being quality checked. The purified total RNA was subjected to mRNA isolation, fragmentation, first-strand cDNA synthesis, second-strand cDNA synthesis, end-repair, A-tailing, adapter ligation, and enrichment to construct sequencing libraries. The qualified libraries were applied to the Illumina NovaSeq6000 sequencing platform (Illumina, CA, USA) for PE150 sequencing mode. The DEGs were identified using P ≤ 0.05 and the gene expression fold-change (FC) ≥ 2. The Gene Ontology (http://www.geneontology.org) was used for the GO enrichment analysis of DEGs at three levels: biological process, cellular component, and molecular function. The KEGG pathway analysis (http://www.genome.jp/kegg/) was used to identify the number of DEGs in each pathway. The PPI network was constructed using the STRING database (http://string-db.org) to evaluate the interactive relationship of up-regulated and down-regulated DEGs. Cytoscape software (version 3.9.1) was employed to determine the degree of connectivity and identify hub genes.24
Validation of mRNA Expression of DEGs by Real-Time RT-qPCR
HMGCR and HMGCS1 were selected from DEGs for verification. Jurkat T cells were harvested in logarithmic phase of growth. The cells were seeded in flasks at a density of 1×107 cells per flask and cultured in RPMI 1640 medium containing VK1 (50 μM) for 8 h, 15 h, 24 h and 48 h. Following incubation, the cells were harvested for total RNA extraction using the ArchiPure Total RNA Extration Kit (Microbeads; Rrizol; ROCGENE, Xuzhou, China). cDNA was synthesized using fastKing gDNA Dispeeling RT SuperMix (TIANGEN, Beijing, China). GoTaq®qPCR Master Mix (Promega, Madison, USA) was used for RT-qPCR following the manufacturer’s instructions. Primers were synthesized by Shanghai SANGON Biological Engineering Co. Ltd. (Shanghai, China) and primer sequences listed in Supplementary Table 1. Real-Time RT-qPCR analysis was conducted in SlAN-96S PCR amplification instrument (ZEESAN, Shanghai, China). The procedure was as follows: 95 °C for 2min, followed by 40 cycles of 95 °C for 15sec, 60 °C for 1min. The 2−ΔΔCt method was used to analyze the expression levels of nominated genes normalized to β-actin expression.
Statistical Analysis
IBM SPSS 26.0 software (IBM Corporation, Armonk, NY, USA) and GraphPad Prism 9.0 (GraphPad Software, San Diego, CA, USA) were used for statistical analysis. The values, averaged of at least three repeated experiments, were expressed as mean ± standard deviation (SD). Comparisons between experimental groups were performed using One-way analysis of variance (ANOVA) with Tukey post hoc test for normally distributed data with homogeneous variance, or Dunnett T3 post hoc test for normally distributed data with unequal variance. P-values < 0.05 were considered statistical significance. Graphs were generated using GraphPad Prism 9.0.
Results
The Effect of VK1 on the Cell Viability
The Jurkat T cell viability was compared between VK1 (4.6μM, 9.2μM, 18.3 μM, 27.5 μM, and 55 μM) treated cells to the control cells (0μM). Similarly, in the normal human PBMCs, the comparison of the cell viability was made between VK1 (4.6μM, 9.2μM, 18.3 μM, 27.5 μM, and 55 μM) treated cells to the control cells (0μM). The proliferation of Jurkat T cells was significantly inhibited by VK1 treatment (Figure 1). The viability of the cells decreased to 59.5%, 37% and 24.4% in cells treated with VK1 at concentrations of 18.3 μM, 27.5 μM and 55 μM (P<0.01), respectively. The inhibitory effect of VK1 was in a dose-dependent manner. The IC50 of VK1 was 18.58 μM. The antiproliferative effects of VK1 were not observed in normal human PBMCs (Figure 1). These results are summarized in Figure 1. Together, these data clearly demonstrate that VK1 exerts a potent and dose-dependent antiproliferative effect on Jurkat T leukemia cells, with a calculated IC50 of 18.58 μM, while exhibiting no cytotoxicity against normal human PBMCs. This establishes the selective anti-leukemic potential of VK1.
 |
Figure 1 VK1 exhibits a selective antiproliferative effect on Jurkat T cells. Cell viability of Jurkat T cells (●) and normal human peripheral blood mononuclear cells (PBMCs, ■) was assessed by MTT assay after 48 h of treatment with various concentrations of VK1. Data are presented as mean ± SD (n≥3). **P < 0.01 compared with the untreated control group of Jurkat T cells. |
The Effect of VK1 on the Cell Apoptosis
To determine if the suppression of cell viability by VK1 was driven by the induction of programmed cell death, we analyzed apoptosis using Annexin V-FITC/PI staining followed by flow cytometry. Jurkat T cells were treated with VK1 (10 μM and 50 μM) for 24 and 48 hours. Figure 2a presents representative dot plots from this analysis, where the lower right quadrant (Annexin V+/PI-) represents cells in early apoptosis, and the upper right quadrant (Annexin V+/PI+) represents cells in late apoptosis or necrosis.
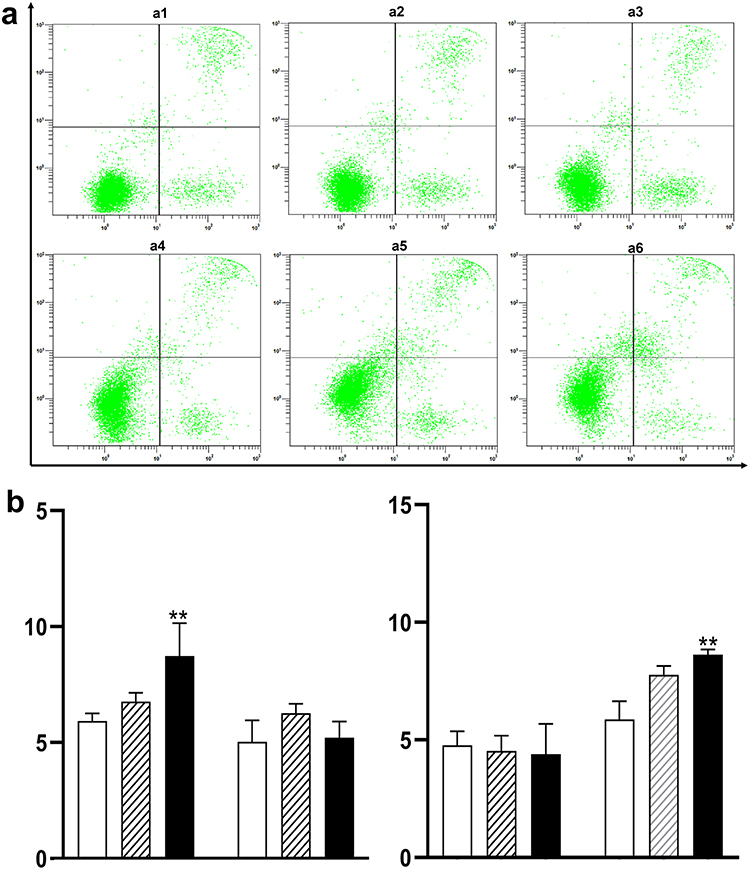 |
Figure 2 VK1 induces apoptosis in Jurkat T cells in a time- and dose-dependent manner. (a) Representative flow cytometry dot plots of Annexin V-FITC/PI staining. The X-axis represents Annexin V-FITC fluorescence intensity, and the Y-axis represents Propidium Iodide (PI) fluorescence intensity. The lower left quadrant (Annexin V−/PI−) represents viable cells, the lower right quadrant (Annexin V⁺/PI−) represents early apoptotic cells, and the upper right quadrant (Annexin V⁺/PI⁺) represents late apoptotic/necrotic cells. Panels represent: a1, Control, 24 h; a2, VK1 10 µM, 24 h; a3, VK1 50 µM, 24 h; a4, Control, 48 h; a5, VK1 10 µM, 48 h; a6, VK1 50 µM, 48 h. (b) Quantitative analysis of apoptotic cells after 24 h and 48 h of treatment. The left cluster of bars in each panel corresponds to early apoptosis, and the right cluster to late apoptosis. Data are presented as mean ± SD (n≥3). Groups are represented as follows: open bars (□), control; hatched bars (////), 10 µM VK1; solid bars (■), 50 µM VK1. **P < 0.01 compared with the untreated control group at the corresponding time point. |
The quantitative analysis of these results is summarized in Figure 2b. After 24 hours of treatment, a significant induction of early apoptosis was observed. Specifically, the population of early apoptotic cells increased by 2.8% in the 50 μM VK1 treatment group compared to the control (P < 0.01). By 48 hours, the primary mode of cell death shifted to late apoptosis, with the 50 μM VK1 treatment leading to a significant 2.77% increase in late apoptotic cells (P < 0.01). This temporal progression from early to late apoptosis indicates that VK1 triggers a cascade of apoptotic events in Jurkat T cells over time. These findings demonstrate that the induction of apoptosis is a key mechanistic component of the cytotoxic effects exerted by VK1.
The Effect of VK1 on the Cell Cycle
Further, we investigated whether the changes of cell cycle involving the inhibitory effect of VK1 on cell growth. Cells were treated with VK1 for 24 h or 48 h, and the results are showed in Figure 3.
 |
Figure 3 VK1 induces cell cycle arrest at G0/G1 phase in Jurkat T cells.(a) Representative flow cytometry histograms of cell cycle distribution analyzed by Propidium Iodide (PI) staining. The X-axis represents DNA content, and the Y-axis represents cell count. Panels represent: A1, Control, 24 h; A2, VK1 10 µM, 24 h; A3, VK1 50 µM, 24 h; A4, Control, 48 h; A5, VK1 10 µM, 48 h; A6, VK1 50 µM, 48 h. The peaks from left to right correspond to cells in G0/G1, S, and G2/M phases of the cell cycle, respectively. (b) Quantitative analysis of cell cycle distribution after 24 h and 48 h of treatment. The Y-axis represents the percentage of cells in each phase. The left set of bars corresponds to the 24 h time point, and the right set to the 48 h time point. Data are presented as mean ± SD (n≥3). Groups are represented as follows: open bars (□), control; hatched bars (////), 10 µM VK1; solid bars (■), 50 µM VK1. *P < 0.05, **P < 0.01 compared with the untreated control group at the corresponding time point. |
The percentage of cells in G0/G1 phase increased significantly after 24 h treatment with VK1 (P < 0.01). Compared with the control group, the average value of cell population in G0/G1 phase increased by 5.2% and 9.48% in cells treated with VK1 at concentrations of 10 μM and 50 μM, respectively. Meanwhile, the percentage of cells in S phase and G2/M phase declined in VK1 treated cells. After 48 h treatment with VK1, the cell population in G0/G1 phase showed a similar change as 24 h treatment. Hence, VK1 (10 μM and 50 μM) induced cell cycle arrest at G0/G1 phase at two selected time points. The flow cytometric profiles and quantitative analysis are presented in Figure 3a and b, respectively. The significant increase in the G0/G1 population, accompanied by a concomitant decrease in S and G2/M phases, indicates that VK1 effectively triggers a cell cycle arrest at the G1/S checkpoint in Jurkat T cells. This arrest provides a mechanistic explanation for the observed inhibition of cell proliferation.
Transcriptomic and PPI Network Analysis
DEGs
Cells were treated with VK1 (50 μM) for 15 h, and the transcriptomic changes of the cells were analyzed by RNA-seq. The gene expression profiles were shown by volcano plot and heat map in Figure 4. Fifty-five DEGs (P<0.05 and FC>2) were identified in VK1 treated cells, including 21 down-regulated genes and 34 up-regulated genes (Figure 4a and b). The overall landscape of these transcriptional changes is visualized in the volcano plot (Figure 4a), which highlights the significantly up- and down-regulated genes, and the heatmap (Figure 4b), which shows the consistent expression pattern of these DEGs across replicates.
 |
Figure 4 Transcriptomic profiling of Jurkat T cells in response to VK1 treatment. (a) Volcano plot of DEGs. The X-axis represents the log2 fold-change (log2FC) in gene expression between VK1-treated and control cells. The Y-axis represents the statistical significance of the change as -log10 of the adjusted p-value (q-value). The two vertical dashed lines indicate the thresholds for |log2FC| > 1 (corresponding to a twofold change). The horizontal dashed line indicates the significance threshold of q < 0.05. Each point represents a single gene, colored based on its expression and significance: red points, significantly up-regulated DEGs; blue points, significantly down-regulated DEGs; gray points, genes with no significant change. (b) Heatmap of the 55 identified DEGs. Rows represent individual genes. Columns represent biological replicates: the first three columns correspond to the three VK1-treated samples (VK-3, VK-2, VK-1), and the last two columns correspond to the two control samples (C2, C1). There are only two sets of samples in the control group since the outlier group C3 was eliminated through principal components analysis (PCA). This result was further confirmed by RT-qPCR. The color scale (Z-score) indicates the relative expression level of each gene across samples, from down-regulated (blue) to up-regulated (red). (c) GO enrichment analysis of DEGs. The bubble chart displays the top 30 significantly enriched GO terms. The X-axis represents the Rich Factor. The Y-axis lists the enriched GO terms. The size of each bubble is proportional to the number of DEGs mapped to the specific GO term. The color of the bubbles represents the GO domain: biological process (red), cellular component (green), and molecular function (blue). The color intensity of the bubbles corresponds to the statistical significance (-log10(q-value)), with more intense colors indicating higher significance. (d) KEGG pathway enrichment analysis of DEGs. The bar chart displays the top 30 significantly enriched KEGG pathways. The X-axis represents the Rich Factor. The Y-axis lists the enriched pathways. The length of each bar corresponds to the Rich Factor value for that pathway. The color of the bars corresponds to the statistical significance (-log10(q-value)), with more intense colors indicating higher significance. The number of DEGs mapped to each pathway increases with the color intensity of the bar. (e) PPI network of the DEGs. Nodes represent proteins, and edges (gray lines) represent predicted or known interactions. The core nodes with the highest connectivity scores, including HMGCR and HMGCS1, are centrally located within the network. |
GO Enrichment and KEGG Analysis
The GO enrichment of DEGs was analyzed for three aspects of biological process, cellular component and molecular function. The biological processes involved DEGs are mainly concentrated in cholesterol biosynthetic, cholesterol metabolic, lipid biosynthetic and steroid metabolic process, etc. (Figure 4c).
KEGG pathway enrichment analysis revealed that the top 30 pathway DEGs were significantly enriched in metabolic pathways, steroid biosynthesis, terpenoid backbone biosynthesis, apoptosis, AMPK signaling pathway, p53 signaling pathway, cholesterol metabolism, bile secretion and other pathways (Figure 4d). The functional implications of these DEGs are summarized in Figure 4c and d. The GO and KEGG enrichment analyses reveal that VK1 treatment profoundly disrupts metabolic processes, particularly cholesterol and steroid biosynthesis, and engages key oncogenic pathways including apoptosis and p53 signaling, thereby providing a molecular basis for the phenotypic effects we observed.
PPI Network Analysis
It is widely recognized that genes carry out their biological functions through interactions with other genes, rather than functioning independently. Thus, PPI network analysis of the 55 DEGs was performed in order to determine any significant gene associations. It was found that HMGCR (log2FC=1.38, P=2.00E-21) showed the highest number of interactions with other DEGs, as well as HMGCS1 (log2FC=1.50, P=4.18E-10), LDLR (log2FC=1.01, P=2.89E-09), SQLE (log2FC=1.04, P=1.11E-09), and INSIG1 (log2FC=1.53, P=3.92E-15). Consequently, we selected HMGCR and HMGCS1 in up-regulated DEGs for further verification (Figure 4e). The PPI network (Figure 4e) identified HMGCR and HMGCS1 as central hub genes among the up-regulated DEGs, indicating their pivotal role in the cellular response to VK1 and guiding our selection for further validation.
Verification of the DEGs, HMGCR and HMGCS1
The DEGs, HMGCR and HMGCS1, were further verified by RT-qPCR. Cells were treated with VK1 (50 μM) for 8 h, 15 h, 24 h and 48 h, and total RNA was isolated for analyzation (Figure 5). Compared with the control group, the mRNA expression of HMGCR was up-regulated by VK1 at 15, 24, 48 hours treatment with the peak observed at 15 h time-point (P < 0.01). The up-regulation of HMGCS1 mRNA expression induced by VK1 was observed at 8 h (P < 0.01). VK1 had a time-dependent impact on the mRNA expression of HMGCR and HMGCS1. The time-dependent mRNA expression profiles of HMGCR and HMGCS1 are shown in Figure 5. The successful verification of these key mevalonate pathway genes by RT-qPCR not only confirms the reliability of our transcriptomic data but also strengthens the hypothesis that VK1 exerts its anti-leukemic effects by perturbing cholesterol metabolic homeostasis.
 |
Figure 5 VK1 upregulates the mRNA expression of HMGCR and HMGCS1 in a time-dependent manner. Relative mRNA expression levels of HMGCR (left panel) and HMGCS1 (right panel) in Jurkat T cells treated with 50 µM VK1 for the indicated times (8, 15, 24, and 48 hours), as determined by RT-qPCR. Gene expression was normalized to β-actin and is presented relative to the control group (0 h). The Y-axis represents the relative mRNA expression level. Within each cluster of two bars at a given time point, the left, open bar (□) represents the control group and the right, solid black bar (■) represents the VK1-treated group. Data are presented as mean ± SD (n≥3). **P < 0.01 compared with the untreated control group at the corresponding time point. |
Discussion
VK, an essential nutrient commonly linked to the clotting cascade, has been shown to possess anticancer properties as well. In recent years, the antitumor effects of VK have been observed in vivo and in vitro. A study has demonstrated that VK2 significantly inhibits the tumor growth in sarcoma-180 tumor-bearing mice.25 In a clinical trial, VK3 supplementation results in a reduction in tumor volume and an increase in average survival time for 17% in patients with late-stage hepatocellular carcinoma (HCC).26 A survey study found a negative correlation between the intake of VK1 and dihydrovitamin K1, not VK2, in the diet and the risk of pancreatic cancer.27 An in vitro study has shown that VK2 has a dose-dependent inhibitory effect on cell growth in multiple lung carcinoma cell lines.28 In human colon cancer cell lines, VK1 inhibits cell proliferation, induces cell apoptosis and leads to cell cycle arrest.14 It has been proposed that VK may play a potential role in inhibiting the growth of solid tumors by actively inducing cell cycle arrest, differentiation, and apoptosis.29,30 However, few studies about VK on hematological malignancies have been reported.
It has been reported that VK3 and VK5 can inhibit leukemia cell proliferation and induce the cell apoptosis.31 VK2 has shown to induce cell apoptosis and autophagy in leukemia cell lines and primary cultured leukemia cells.32–34 In this study, we demonstrated that VK1 significantly inhibited the proliferation of Jurkat T cells while it did not cause any inhibition on normal human PBMCs. The concentrations of VK1 used in this study are consistent with those employed in prior in vitro investigations of its anticancer properties,13,18 ensuring pharmacological relevance for mechanistic exploration. Additionally, VK1 promoted cell apoptosis and induced cell cycle arrest at G0/G1 phase. These results suggest that VK1 has potential cytotoxic effects on Jurkat T cells, a type of hematological malignancy cell line35 has cytotoxic effects of VK1, VK2, and VK3 against human T lymphocytic leukemia cell lines including Jurkat T cells. Their results showed that VK2 and VK3, not VK1, reduced cell viabilities of Jurkat T cells at the concentration of 0.1μM, 1μM, 10μM and 100 μM. Their results about VK1 are contrary to ours. The inconsistent results may attribute to the different experimental conditions or methods. Collectively, it appears that VK has anticancer effects in hematological malignancies.
Almost all reported mechanisms about VK on cancer cells focus on solid tumors. The anticancer effects of VK3 is mainly through oxidative stress and aromatization, while VK1 and VK2 are mainly through non-oxidative stress mechanisms.36 VK promotes cancer cell apoptosis by altering intracellular calcium homeostasis and activating pro-apoptotic factors such as c-Jun N-terminal kinases (JNKs), Fas-dependent and -independent signaling pathways, and nuclear factor kappa-B (NF-κB).37 VK2 inhibits cell growth by suppressing NF-κB binding site-dependent cyclin D1 promoter activity in HCC cells.29 However, little is known about how VK exerts anticancer effects on hematologic malignancies.
In our study, a total of 21 down-regulated and 34 up-regulated genes were identified by transcriptomic analysis. These VK1 regulated genes involved various cell functions, including metabolic pathways, steroid biosynthesis, terpenoid backbone biosynthesis, apoptosis, AMPK signaling pathway, p53 signaling pathway, cholesteric metabolism, bile secretion, etc. These results suggest that the cytotoxic effects of VK1 on Jurkat T cells may be related to multiple signaling pathways. Our KEGG pathway analysis provides a potential molecular basis for the cytotoxic phenotypes we observed. The enrichment of DEGs in the “Apoptosis” and “p53 signaling pathway” (Figure 4d) is highly consistent with our flow cytometric results showing induction of both early and late apoptosis (Figure 2). Furthermore, the “p53 signaling pathway” is a well-known regulator of the G1/S cell cycle checkpoint. The engagement of this pathway offers a plausible explanation for the G0/G1 phase arrest induced by VK1 (Figure 3), as it can lead to the inhibition of cyclin-dependent kinases required for S phase entry. Thus, the transcriptomic data robustly support our functional findings that VK1 induces apoptosis and cell cycle arrest in Jurkat cells.
While the precise entry mechanism of VK1 into Jurkat cells was not directly investigated in this study, it is noteworthy that the transcriptomic profile did not show significant enrichment in known vitamin K transport pathways. As a fat-soluble vitamin, VK1 may passively diffuse across the plasma membrane or utilize specific transporters, a process that could differ between leukemic cells and their normal counterparts, potentially explaining the selective toxicity we observed. This represents an important area for future investigation. Additionally, while previous reports on VK1 in solid tumors have emphasized roles for MAPK and NF-κB pathways, our KEGG analysis did not identify these pathways as among the most significantly enriched. Instead, our data point towards a pronounced impact on metabolic pathways, particularly cholesterol and steroid biosynthesis. This suggests that the anticancer mechanism of VK1 may be cell context-dependent, and in Jurkat T leukemia cells, it may pivot towards disrupting metabolic homeostasis. This notion of context-dependent mechanisms is supported by recent comprehensive reviews, which highlight the diverse and complex roles of Vitamin K beyond coagulation, including its involvement in various cell fate decisions and signaling pathways across different cancer types.38
We selected HMGCR and HMGCS1 genes from the significantly up-regulated DEGs for RT-qPCR analysis. The results confirmed that the mRNA expression of HMGCR and HMGCS1 were up-regulated by VK1. The study implicates that HMGCR and HMGCS1 related pathway could be involved in the anticancer effects of VK1 on hematologic malignancies. HMGCR and HMGCS1 are known to be key enzymes in mevalonate (MVA) pathway.39 The MVA pathway plays central roles in cholesterol biosynthesis. Importantly, a recent study demonstrated that the inhibition of HMGCS1 by metformin effectively suppresses tumorigenesis, providing direct experimental evidence for targeting this enzyme in cancer therapy.39
In addition, recent study has shown that abnormalities in MVA pathway could promote malignant transformation.40 The MVA pathway utilizes Acetyl-CoA to synthesize sterols that play a crucial role in the growth and progression of tumors.41 Acetyl-CoA undergoes catalysis by HMGCS, leading to the synthesis of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA). This compound is then reduced to MVA through the action of HMGCR. Subsequently, cholesterol is produced via a series of enzymatic reactions.42 HMGCS1 is located upstream of HMGCR and is required in the catalytic synthesis of HMG-CoA, which serves as the substrate of HMGCR. HMGCR and HMGCS1 have been identified in various types of cancers. In vivo tumorigenesis experiments demonstrate that HMGCR and HMGCS1 are critical for primary tumorigenesis, angiogenesis, and cell survival of breast cancer cells, while HMGCR is also essential for the extravasation and subsequent engraftment of breast cancer cells in the lung parenchyma.43 The HMGCR expression level is positively correlated with good prognosis in patients with breast cancer.44 Statins, powerful inhibitors of HMGCR,45 can induce specific apoptosis in hematological malignancies when it is used in the treatment of hypercholesterolemia.46,47 This apoptotic induction effect of statins is attributed to direct inhibition of HMGCR in tumor cells.46 Pandyra found that statin administration increased the expression of HMGCR and HMGCS1 in multiple myeloma and acute myelogenous leukemia (AML) cell lines. Dipyridamole, as an antiplatelet agent, has been found to enhance the anticancer activity of statins through reducing the feedback response that up-regulates HMGCS1 and HMGCR.46 Therefore, the activation of HMGCR and HMGCS1 may play a role for the anticancer effects of VK in some types of cancer cells.
To our knowledge, the present study is the first time to report the evidence about the metabolic-related genes, HMGCR and HMGCS1, may participate in the inhibitory effects of VK1 on Jurkat T cells. The central role of HMGCR and HMGCS1 in our transcriptomic dataset is particularly intriguing. Beyond their canonical function in cholesterol synthesis, the mevalonate pathway outputs, such as geranylgeranyl pyrophosphate (GGPP), are essential for the prenylation and thus proper membrane localization and function of small GTPases (eg, Ras, Rho). These proteins are critical downstream regulators of both cell proliferation and survival signaling. Therefore, the VK1-induced upregulation of HMGCR/HMGCS1 could represent a compensatory feedback response to a deeper disruption of the mevalonate pathway. This disruption, by limiting GGPP availability, would impair the function of these GTPases, ultimately leading to the cell cycle arrest and apoptosis we observed. This hypothesis aligns with the known mechanism of statins, which directly inhibit HMGCR and can induce similar cytotoxic effects in leukemia cells.
However, studies have also shown that HMGCR and HMGCS1 promote cancer progression in some types of cancers. It has been reported that the overexpression of HMGCR promotes cell growth and migration in gastric cancer cells. While, knocking down the expression of HMGCR inhibits the cell growth, migration, and tumorigenesis.46 Ashida found that overexpression of HMGCS1 or HMGCR in prostate cancer cells promotes cell growth, but is accompanied by low invasiveness. Their explanation is that HMGCR and HMGCS1 are required for the growth of prostate cancer cells until they progress to aggressive disease, after which their expression is down-regulated during invasion or metastasis.48 Previous studies have shown that HMGCR expression is negatively correlated with the aggressiveness of breast cancer.44,49 The discrepancies between studies may be due to the dual properties of cancer-related genes. The critical role of the mevalonate pathway in oncogenesis is increasingly recognized beyond cholesterol synthesis. Recent work has shown that this pathway is vital for primary tumor growth and metastasis in breast cancer,50 and its interaction with key tumor suppressors like p53 can modulate the response to therapies such as statins.40,51 This evolving understanding underscores the pathway’s complexity and context-dependency. Numerous studies have revealed the dual nature of some cancers-related genes, showing their ability to either promote or suppress the development of cancers. This duality is contingent upon various factors such as the specific malignant tumors, tissue type, cancer stage, gene dosage, and their intricate interactions with other contributors in the process of carcinogenesis.52,53 The discovery of phosphatase and tensin homolog (PTEN) in the metabolic pathways associated with HMGCR and HMGCS1 provides strong evidence to support this viewpoint. PTEN, as a traditional tumor suppressor, has been extensively investigated in various types of malignancies.54 Currently, PTEN has been found to be an agent with dual roles in tumorigenesis. The loss of PTEN protein expression has been found to be associated with decreased survival rates in diffuse large B-cell lymphoma (DLBCL) patients with excessive activity of a serine threonine kinase, AKT. Conversely, in patients with normal AKT activation, high levels of PTEN expression are associated with poorer survival rates. It can be seen that PTEN has shown different impacts on the survival outcomes of DLBCL depending on the activity of AKT.55 Disabled-2 (DAB2) and YAP/TAZ, which are widely recognized as tumor suppressors, have also been shown to have the nature of pro-tumorigenesis.56,57 It remains unclear whether HMGCR and HMGCS1 also possess dual properties as other dual nature genes in tumorigenesis. Further studies are necessary to gain a better understanding about roles played by HMGCR and HMGCS1 in the anticancer activities of VK1.
Our study provides the first transcriptomic evidence that VK1 exerts cytotoxic effects in Jurkat T lymphocytic leukemia cells primarily by perturbing metabolic pathways and inducing cell cycle arrest and apoptosis. The distinct transcriptional response observed, centered on genes like HMGCR and HMGCS1, highlights a potentially novel mechanism of action for VK1 in hematological malignancies, differing from its established roles in solid tumors. Future studies employing genetic knockdown (eg, siRNA) of key identified genes like HMGCR and HMGCS1, combined with metabolic flux analysis and rescue experiments, will be crucial to validate their essential role in VK1-induced cytotoxicity and to elucidate the precise molecular cascade initiated by VK1 in leukemia cells. Furthermore, future investigations should also aim to validate the observed gene expression changes at the protein level (eg, by Western Blot) and to elucidate the precise mechanism of VK1 cellular uptake, which may underpin its selective toxicity.
Conclusion
In conclusion, by integrating cellular phenotyping with unbiased transcriptomics, our study provides a comprehensive framework for understanding the anti-leukemic action of VK1. We have demonstrated that VK1 inhibits proliferation, induces apoptosis, and causes G0/G1 cell cycle arrest in Jurkat T cells. Crucially, the transcriptomic data not only corroborate these phenotypes by revealing the activation of relevant pathways but also uncover a previously underappreciated metabolic vulnerability-the mevalonate pathway-as a central and potentially novel target of VK1 in hematological malignancies. This work successfully achieves our stated objective of elucidating the underlying mechanism of VK1 and positions it within the broader research context of targeting cancer metabolism for therapeutic benefit.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding authors (S. Song and X. Lou) on reasonable request. The datasets generated and/or analyzed during the current study are available in the Gene Expression Omnibus repository, [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?&acc=GSE237219].
Ethics Statement
The present study was approved by the Ethics Committee of the Ninth Medical Center, Chinese PLA General Hospital (approval number: LL-LCSY-2023-03). The use of Jurkat T cells was also approved by the same committee. Written informed consent was obtained from all study participants.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work. Ying Shang and Shaoyan Si are Co-first authors.
Funding
Funding information is not applicable.
Disclosure
The authors declare that they have no competing interests.
References
1. Malard F, Mohty M. Acute lymphoblastic leukaemia. Lancet. 2020;395(10230):1146–1162. doi:10.1016/S0140-6736(19)33018-1
2. Yi M, Zhou LH, Li AP, Luo SX, Wu KM. Global burden and trend of acute lymphoblastic leukemia from 1990 to 2017. Aging. 2020;12(22):22869–22891. doi:10.18632/aging.103982
3. Hunger SP, Lu X, Devidas M, et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: a report from the children’s oncology group. J Clin Oncol. 2012;30(14):1663–1669. doi:10.1200/JCO.2011.37.8018
4. Gökbuget N, Dombret H, Ribera JM, et al. International reference analysis of outcomes in adults with B-precursor Ph-negative relapsed/refractory acute lymphoblastic leukemia. Haematologica. 2016;101(12):1524–1533. doi:10.3324/haematol.2016.144311
5. Zhang XM, Chen X, Liu J, et al. Knockdown of WISP1 inhibit proliferation and induce apoptosis in ALL Jurkat cells. Int J Clin Exp Pathol. 2015;8(11):15489–15496.
6. Kim ES, Putnam JB, Komaki R, et al. Phase II study of a multidisciplinary approach with induction chemotherapy, followed by surgical resection, radiation therapy, and consolidation chemotherapy for unresectable malignant thymomas: final report. Lung Cancer. 2004;44(3):369–379. doi:10.1016/j.lungcan.2003.12.010
7. Ivanova D, Zhelev Z, Getsov P, et al. Vitamin K: redox-modulation, prevention of mitochondrial dysfunction and anticancer effect. Redox Biol. 2018;16:352–358. doi:10.1016/j.redox.2018.03.013
8. Rodríguez C R-O, Díaz CM. Vitamin K and bone health: a review on the effects of vitamin k deficiency and supplementation and the effect of non-vitamin k antagonist oral anticoagulants on different bone parameters. J Osteoporosis. 2019;2019:2069176. doi:10.1155/2019/2069176
9. Evenepoel P, Claes K, Meijers B, et al. Poor vitamin k status is associated with low bone mineral density and increased fracture risk in end‐stage renal disease. J Bone Miner Res. 2019;34(2):262–269. doi:10.1002/jbmr.3608
10. Popa D-S, Bigman G, Rusu ME. The role of vitamin k in humans: implication in aging and age-associated diseases. Antioxidants. 2021;10(4):566. doi:10.3390/antiox10040566
11. Tang SQ, Ruan Z, Ma AX, Wang D, Kou JS. Effect of vitamin K on wound healing: a systematic review and meta-analysis based on preclinical studies. Front Pharmacol. 2022;13.
12. Wu FY, Liao WC, Chang HM. Comparison of antitumor activity of vitamins K1, K2 and K3 on human tumor cells by two (MTT and SRB) cell viability assays. Life Sci. 1993;52(22):1797–1804. doi:10.1016/0024-3205(93)90469-J
13. Linsalata M, Orlando A, Tutino V, Notarnicola M, D’Attoma B, Russo F. Inhibitory effect of vitamin K1 on growth and polyamine biosynthesis of human gastric and colon carcinoma cell lines. Int J Oncol. 2015;47(2):773–781. doi:10.3892/ijo.2015.3033
14. Orlando A, Linsalata M, Tutino V, D’Attoma B, Notarnicola M, Russo F. Vitamin K1 exerts antiproliferative effects and induces apoptosis in three differently graded human colon cancer cell lines. Biomed Res. Int. 2015;2015:1–15. doi:10.1155/2015/296721
15. Showalter SL, Wang Z, Costantino CL, et al. Naturally occurring K vitamins inhibit pancreatic cancer cell survival through a caspase-dependent pathway. J Gastroenterol Hepatol. 2010;25(4):738–744. doi:10.1111/j.1440-1746.2009.06085.x
16. Prasad KN, Edwards-Prasad J, Sakamoto A. Vitamin K3 (menadione) inhibits the growth of mammalian tumor cells in culture. Life Sci. 1981;29(13):1387–1392. doi:10.1016/0024-3205(81)90683-4
17. Wei G, Wang M, Carr BI. Sorafenib combined vitamin K induces apoptosis in human pancreatic cancer cell lines through RAF/MEK/ERK and c-Jun NH2-terminal kinase pathways. J Cell Physiol. 2010;224(1):112–119. doi:10.1002/jcp.22099
18. Du W, Zhou JR, Wang DL, Gong K, Zhang QJ. Vitamin K1 enhances sorafenib-induced growth inhibition and apoptosis of human malignant glioma cells by blocking the Raf/MEK/ERK pathway. World J Surg Oncol. 2012;10:1–7. doi:10.1186/1477-7819-10-60
19. Dasari S, Ali SM, Zheng G, et al. Vitamin K and its analogs: potential avenues for prostate cancer management. Oncotarget. 2017;8(34):57782–57799. doi:10.18632/oncotarget.17997
20. Chen A, Li J, Shen N, Huang H, Hang Q. Vitamin K: new insights related to senescence and cancer metastasis. Biochim Biophys Acta Rev on Cancer. 2024;1879(2):189057. doi:10.1016/j.bbcan.2023.189057
21. Carr BI, Wang Z, Wang MF, Wei G. Differential Effects of Vitamin K1 on AFP and DCP Levels in Patients with Unresectable HCC and in HCC Cell Lines. Dig Dis Sci. 2011;56(6):1876–1883. doi:10.1007/s10620-010-1521-x
22. Xu W, Meng K, Wu H, et al. Vitamin K2 immunosuppressive effect on pediatric patients with atopic dermatitis. Pediatr Int. 2019;61(12):1188–1195. doi:10.1111/ped.14014
23. Xu W, Wang X, Tu Y, et al. Tetrandrine and cepharanthine induce apoptosis through caspase cascade regulation, cell cycle arrest, MAPK activation and PI3K/Akt/mTOR signal modification in glucocorticoid resistant human leukemia Jurkat T cells. Chem. Biol. Interact. 2019;310:108726. doi:10.1016/j.cbi.2019.108726
24. Lou W, Liu J, Ding B, Xu L, Fan W. Identification of chemoresistance-associated miRNAs in breast cancer. Cancer Manag Res. 2018;10:4747–4757. doi:10.2147/CMAR.S172722
25. Shi J, Zhou S, Kang L, et al. Evaluation of the antitumor effects of vitamin K 2 (menaquinone-7) nanoemulsions modified with sialic acid-cholesterol conjugate. Drug Delivery Transl Res. 2018;8(1):1–11. doi:10.1007/s13346-017-0424-1
26. Sarin SK, Kumar M, Garg S, Hissar S, Pandey C, Sharma BC. High dose vitamin K3 infusion in advanced hepatocellular carcinoma. J Gastroenterol Hepatol. 2006;21(9):1478–1482. doi:10.1111/j.1440-1746.2006.04383.x
27. Yu DW, Li QJ, Cheng L, et al. Dietary vitamin k intake and the risk of pancreatic cancer: a prospective study of 101,695 American Adults. Am J Epidemiol. 2021;190(10):2029–2041. doi:10.1093/aje/kwab131
28. Yoshida T, Miyazawa K, Kasuga I, et al. Apoptosis induction of vitamin K2 in lung carcinoma cell lines: the possibility of vitamin K2 therapy for lung cancer. Int J Oncol. 2003;23(3):627–632.
29. Ozaki I, Zhang H, Mizuta T, et al. Menatetrenone, a vitamin k2 analogue, inhibits hepatocellular carcinoma cell growth by suppressing cyclin d1 expression through inhibition of nuclear factor κb activation. Clin Cancer Res. 2007;13(7):2236–2245. doi:10.1158/1078-0432.CCR-06-2308
30. Mizuta T, Ozaki I. Hepatocellular carcinoma and vitamin K. Vitamin hormon. 2008;78:435–442.
31. Nakaoka E, Tanaka S, Onda K, Sugiyama K, Hirano T. Effects of vitamin k3 and k5 on daunorubicin-resistant human t lymphoblastoid leukemia cells. Anticancer Res. 2015;35(11):6041–6048.
32. Yaguchi M, Miyazawa K, Katagiri T, et al. Vitamin K2 and its derivatives induce apoptosis in leukemia cells and enhance the effect of all-trans retinoic acid. Leukemia. 1997;11(6):779–787. doi:10.1038/sj.leu.2400667
33. Yaguchi M, Miyazawa K, Otawa M, et al. Vitamin K2 selectively induces apoptosis of blastic cells in myelodysplastic syndrome: flow cytometric detection of apoptotic cells using APO2.7 monoclonal antibody. Leukemia. 1998;12(9):1392–1397. doi:10.1038/sj.leu.2401143
34. Yokoyama T, Miyazawa K, Naito M, et al. Vitamin K2 induces autophagy and apoptosis simultaneously in leukemia cells. Autophagy. 2008;4(5):629–640. doi:10.4161/auto.5941
35. Xu WC, Wu HG, Chen SH, et al. Cytotoxic effects of vitamins K1, K2, and K3 against human T lymphoblastoid leukemia cells through apoptosis induction and cell cycle arrest. Chem. Biol. Drug Des. 2020;96(4):1134–1147. doi:10.1111/cbdd.13696
36. Lamson DW, Plaza SM. The anticancer effects of vitamin K. Alternative Med Rev J Clin Ther. 2003;8(3):303–318.
37. Caricchio R, Kovalenko D, Kaufmann WK, Cohen PL. Apoptosis provoked by the oxidative stress inducer menadione (Vitamin K(3)) is mediated by the Fas/Fas ligand system. Clin immunol. 1999;93(1):65–74. doi:10.1006/clim.1999.4757
38. Sadler RA, Shoveller AK, Shandilya UK, et al. Beyond the coagulation cascade: vitamin k and its multifaceted impact on human and domesticated animal health. Curr Issues Mol Bio. 2024;46(7):7001–7031. doi:10.3390/cimb46070418
39. Chen Y, Li M, Yang Y, Lu Y, Li X. Antidiabetic drug metformin suppresses tumorigenesis through inhibition of mevalonate pathway enzyme HMGCS1. J Biol Chem. 2022;298(12):102678. doi:10.1016/j.jbc.2022.102678
40. Pereira M, Matuszewska K, Glogova A, Petrik J. Mutant p53, the mevalonate pathway and the tumor microenvironment regulate tumor response to statin therapy. Cancers. 2022;14(14):3500. doi:10.3390/cancers14143500
41. Mullen PJ, Yu R, Longo J, Archer MC, Penn LZ. The interplay between cell signalling and the mevalonate pathway in cancer. Nat Rev Cancer. 2016;16(11):718–731. doi:10.1038/nrc.2016.76
42. Sharpe LJ, Brown AJ. Controlling cholesterol synthesis beyond 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR). J Biol Chem. 2013;288(26):18707–18715. doi:10.1074/jbc.R113.479808
43. Conde J, Fernández-Pisonero I, Lorenzo-Martín LF, et al. The mevalonate pathway contributes to breast primary tumorigenesis and lung metastasis. Mol Oncol. 2025;19(1):56–80. doi:10.1002/1878-0261.13716
44. Borgquist S, Djerbi S, Pontén F, et al. HMG-CoA reductase expression in breast cancer is associated with a less aggressive phenotype and influenced by anthropometric factors. Int J Cancer. 2008;123(5):1146–1153. doi:10.1002/ijc.23597
45. Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi:10.1038/343425a0
46. Pandyra A, Mullen PJ, Kalkat M, et al. Immediate utility of two approved agents to target both the metabolic mevalonate pathway and its restorative feedback loop. Cancer Res. 2014;74(17):4772–4782. doi:10.1158/0008-5472.CAN-14-0130
47. Williams AB, Li L, Nguyen B, Brown P, Levis M, Small D. Fluvastatin inhibits FLT3 glycosylation in human and murine cells and prolongs survival of mice with FLT3/ITD leukemia. Blood. 2012;120(15):3069–3079. doi:10.1182/blood-2012-01-403493
48. Ashida S, Kawada C, Inoue K. Stromal regulation of prostate cancer cell growth by mevalonate pathway enzymes HMGCS1 and HMGCR. Oncol Lett. 2017;14(6):6533–6542. doi:10.3892/ol.2017.7025
49. Gustbée E, Tryggvadottir H, Markkula A, et al. Tumor-specific expression of HMG-CoA reductase in a population-based cohort of breast cancer patients. BMC Clin Pathol. 2015;15:8. doi:10.1186/s12907-015-0008-2
50. Göransson S, Olofsson H, Johansson HJ, et al. Mechanical control of breast cancer malignancy by promotion of mevalonate pathway enzyme synthesis. Matrix Biol. 2025;140:1–15. doi:10.1016/j.matbio.2025.05.005
51. Juarez D, Buono R, Matulis SM, et al. Statin-induced mitochondrial priming sensitizes multiple myeloma cells to BCL2 and MCL-1 Inhibitors. Cancer Res Commun. 2023;3(12):2497–2509. doi:10.1158/2767-9764.CRC-23-0350
52. Zhang J, Chen YH, Lu Q. Pro-oncogenic and anti-oncogenic pathways: opportunities and challenges of cancer therapy. Future Oncol. 2010;6(4):587–603. doi:10.2217/fon.10.15
53. Qiu MJ, Zhang L, Fang XF, et al. Research on the circadian clock gene HNF4a in different malignant tumors. Int J Med Sci. 2021;18(6):1339–1347. doi:10.7150/ijms.49997
54. Cetintas VB, Batada NN. Is there a causal link between PTEN deficient tumors and immunosuppressive tumor microenvironment? J Transl Med. 2020;18(1):1–11. doi:10.1186/s12967-020-02219-w
55. Wang X, Cao X, Sun R, et al. Clinical Significance of PTEN Deletion, Mutation, and Loss of PTEN expression in de novo diffuse large b-cell lymphoma. Neoplasia. 2018;20(6):574–593. doi:10.1016/j.neo.2018.03.002
56. Price ZK, Lokman NA, Yoshihara M, Kajiyama H, Oehler MK, Ricciardelli C. Disabled-2 (DAB2): a key regulator of anti- and pro-tumorigenic pathways. Int J Mol Sci. 2022;24(1):696. doi:10.3390/ijms24010696
57. Pearson JD, Huang K, Pacal M, et al. Binary pan-cancer classes with distinct vulnerabilities defined by pro- or anti-cancer YAP/TEAD activity. Cancer Cell. 2021;39(8):1115–1134. doi:10.1016/j.ccell.2021.06.016
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
AC067930.4/miR-375 Axis Regulates the Malignant Progression of Colon Adenocarcinoma Through the Cuproptosis-Related Gene SLC6A6
Hou B, Liu Q, Li Z
Cancer Management and Research 2026, 18:585644
Published Date: 27 May 2026