Back to Journals » OncoTargets and Therapy » Volume 9
Verteporfin, a suppressor of YAP–TEAD complex, presents promising antitumor properties on ovarian cancer
Authors Feng J, Gou J, Jia J, Yi T, Cui T ![]() , Li Z
, Li Z
Received 6 April 2016
Accepted for publication 14 July 2016
Published 29 August 2016 Volume 2016:9 Pages 5371—5381
DOI https://doi.org/10.2147/OTT.S109979
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Juntao Feng,1 Jinhai Gou,1 Jia Jia,1 Tao Yi,2 Tao Cui,1 Zhengyu Li1,2
1Department of Gynecology and Obstetrics, 2Sichuan Key Laboratory of Gynecologic Oncology, West China Second University Hospital, Sichuan University, Chengdu, People’s Republic of China
Abstract: Yes-associated protein (YAP) is a key transcriptional coactivator of Hippo pathway and has been shown to be an oncoprotein in ovarian cancer (OC). Verteporfin (VP), clinically used in photodynamic therapy for neovascular macular degeneration, has been recently proven to be a suppressor of YAP–TEAD complex and has shown potential in anticancer treatment. In this study, we aimed to explore the potential effect of VP in the treatment of OC. Our results showed that VP led to inhibition of proliferation in a time- and dose-dependent manner and to the suppression of migratory and invasive capacities of OC cells. Western blot and real-time polymerase chain reaction demonstrated that VP induced YAP cytoplasmic retention and deregulated inducible YAP and CCNs in OC cells. In vivo, VP exerted a significant effect on tumor growth in OVCAR8 xenograft mice, resulting in tumor nodules with lower average weight and reduced volume of gross ascites. In addition, VP treatment remarkably upregulated cytoplasmic YAP and phosphorylation YAP and downregulated CCN1 and CCN2, but exerted little effect on YAP-upstream components in Hippo pathway. In conclusion, our results suggested that VP may be a promising agent for OC, acting by suppressing YAP–TEAD complex.
Keywords: YAP, CCN2, ovarian cancer, verteporfin, Hippo pathway
Introduction
The Hippo pathway is known to regulate cell proliferation and organ size in both Drosophila and mammals. In mammals, the Hippo pathway consists of a highly conserved kinase cascade, whose upstream proteins SAV1, MST1/2, and LATS1/2 together form a complex to phosphorylate the key transcriptional coactivator Yes-associated protein (YAP), thus facilitating YAP’s retention in the cytoplasm by creating 14-3-3 binding site. Consequently, YAP cannot move to the nucleus where it interacts with the TEAD transcription factors and mediates the expression of the downstream growth-promoting and apoptosis-inhibiting genes such as CCN family.1,2 Over the past decade, YAP has received a surge of interest and been found as a candidate oncogene on the chromosome 11q22 amplicon.3 Recently, several studies have shown a widespread activation/overexpression of the YAP oncoprotein in a variety of human cancers including ovarian cancer (OC).4,5 Studies have demonstrated that upregulated YAP in OC is considered to be a common event in the carcinogenesis process and is associated with poor survival, higher recurrence, and lower sensitivity to platinum-based chemotherapy among patients with OC.6–10 Moreover, YAP has been shown to maintain stemness of OC-initiated cells,11 but such an effect is reversed with the knockdown or downregulation of YAP. These observations indicate that YAP may be a novel potential target for antitumor drug.
We were drawn to a recent study where a screen through Johns Hopkins Drug Library for >3,300 drugs enabled Liu-Chittenden et al to identify a small-molecule verteporfin (VP), capable of disrupting YAP–TEAD interaction without light activation, thus blocking YAP-driven oncogenic growth.12 Lately, VP has shown promising anticancer effect in various malignancies such as breast cancer,13,14 prostate carcinoma,15 pancreatic ductal adenocarcinoma,16 and esophageal cancer,17 especially when combined with standard chemotherapy.
In this study, we aimed to explore the anticancer effect of VP on OC in vitro and in vivo. Our results would provide evidence that VP possesses promising anticancer effects and shows valuable clinical implications for patients with OC.
Material and methods
Cell culture and reagents
The human OC cells OVCAR3 and OVCAR8 were obtained from the American Type Culture Collection (ATCC; LGC Promochem, Manassas, VA, USA). All cell lines were routinely maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA), 1% 100 units/mL penicillin, and 100 units/mL streptomycin. The culture was incubated in a humidified atmosphere containing 5% CO2 at 37°C. Cisplatin (CDDP) was obtained from Haosen Biotechnology, Jiangsu, People’s Republic of China. VP was purchased from Selleck Chemicals (Houston, TX, USA). For in vitro and in vivo study, VP was first dissolved in dimethyl sulfoxide (DMSO; Sigma, St Louis, MO, USA) and further diluted in phosphate-buffered saline (PBS) before use.
MTT assay
Cells were seeded in 96-well plates at a density of 1×104 cells/mL in a final volume of 200 μL and maintained in DMEM medium at 37°C. After different durations of treatment with the desired concentrations of VP, cell viability was assessed by MTT assay as described previously.18
Migration and invasion transwell assays
A 24-well transwell plate (8.0 μm, Corning, NY, USA) was used to determine the migration and invasive capability of each cell line. For the invasion assay, the upper surface of membrane was precoated with Matrigel Basement Membrane Martrix (BD Biosciences, San Jose, CA, USA). A total of 1×105 cells in serum-free medium were seeded into upper chambers treated with the indicated drug. Complete medium with 10% fetal bovine serum was added to the lower chamber as a chemoattractant. After incubation for 18 hours (migration assay) or 36 hours (invasion assay), the invading cells attached to the lower surface were fixed, stained, and counted. A percentage of migratory or invasive cells were calculated from the ratio of attached stained cells to the total number of seeded cells. Experiments were independently performed in triplicate.
Hoechst 33342 staining analysis
The OC cells were plated into 6-well plates, and then treated with vehicle and VP (5, 10, and 20 μM) for 48 hours. Cell apoptosis was analyzed by Hoechst 33258 staining (Apoptosis-Hoechst staining kit; Beyotime Biotechnology, Haimen, People’s Republic of China). Briefly, cells were immersed in 0.5 mL of methanol for 15 minutes, followed by rinsing twice with PBS. Then cells were stained with 1 μg/mL Hoechst 33258 compounds in a dark chamber at room temperature for 10 minutes and again rinsed twice with PBS. Cells were analyzed by fluorescence microscopy. The apoptotic cells are seen as pyknotic and have fragmented nuclei emitting intense fluorescence.
Tumor models of peritoneal metastases and treatment
The animal study was carried out in strict accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All experimental procedures were approved by the Institutional Animal Care and Use Committee of Sichuan University. All surgery was performed under chloral hydrate anesthesia, and all efforts were made to minimize suffering.
Briefly, about 1×107 cells suspended in 400 μL of serum-free cell DMEM medium were intraperitoneally (IP) injected into female nude mice (BALB/c-nu/nu, 4–6 weeks of age,16–18 g). Fifteen days after tumor injection, the mice were randomly divided into four groups (n=5) and then treated with: 1) 100 mg/kg VP by IP, thrice a week for total 3 weeks as previously described;17 2) 3 mg/kg CDDP via the IP route, once every 4 days for total of five times; and 3) DMSO and PBS at the same concentration and schedule as VP. All mice were sacrificed under chloral hydrate anesthesia at day 21 after the first injection. Tumor nodules in the abdominal cavity were collected and weighed; mice weight and ascites were also evaluated in all groups.
Western blotting
Total protein, cytoplasmic or nuclear protein, from cells and tumor tissues were extracted according to the manufacturer’s instructions (Cwbiotechnology, Beijing, People’s Republic of China). The used primary antibodies were YAP (1:500; Cell Signaling Technology, Danvers, MA, USA, #12395), serine-127 phospho-YAP (pYAP; 1:500; Cell Signaling Technology, #13008), CCN1 (1:500; Santa Cruz Biotechnology, Dallas, TX, USA, #13100), CCN2 (1:500; Santa Cruz Biotechnology, #14939), and GAPDH (1:5,000; Santa Cruz Biotechnology, #48167).
Real-time polymerase chain reaction (RT-PCR) assay
Total RNA was extracted using the miRNeasy Mini Kit (Qiagen, Valencia, CA, USA) according to manufacturer’s instruction. RNA was reverse transcribed using the miScript II Reverse Transcription Kit with miScript HiSpec Buffer (Qiagen). All PCR reactions were carried out in an ABI 7900 HT Sequence Detection System (Applied Biosystems, Foster City, CA, USA) using the miScript SYBR Green Real-Time PCR Kit (Qiagen). After completion of PCR amplification, relative expression of the tested genes was calculated based on the 2-ΔΔCt method. The mRNA level of GAPDH was measured as an internal control. Each sample was run independently in triplicate. The RT-PCR primers used are listed in Table S1.
Immunohistochemical analysis
Immunohistochemistry was used to assess the in vivo effect of VP as described previously. For ki67 (1:100, Santa Cruz Biotechnology, #15402), only nuclear staining was considered as positive. In the case of caspase3 (1:100, Santa Cruz Biotechnology, #65497), cytoplasm staining was considered. At least three randomly selected areas were evaluated.
Statistical analysis
All quantitative data were recorded as mean ± standard deviation. Unpaired t-test was used to compare the significant differences between two groups. Comparisons between multiple groups were done using one-way variance, followed by Dunnett’s test. SPSS 13.0 (SPSS, Chicago, IL, USA) was used for analyses. Statistical significance was defined as P<0.05.
Results
VP inhibits viability of OC cells
To evaluate the antitumor effects of VP on OC, MTT assay was performed to examine its effect on the viability of two human OC cell lines OVCAR3 and OVCAR8. As shown in Figure 1A, after treatment with 2.5–20 μM VP for 24, 48, 72, and 96 hours, respectively, cell viability was decreased in a dose- and time-dependent manner in OVCAR3 and OVCAR8 cells. Approximately 20 μM VP reduced growth in these two cell lines by over 50%. The 72 hours half maximal inhibitory concentration (IC50) was 10.55 and 17.92 μM, respectively. Hoechst staining analysis of apoptosis in both cell lines showed no significant differences after treatment with VP compared to no treatment (see Figure S1).
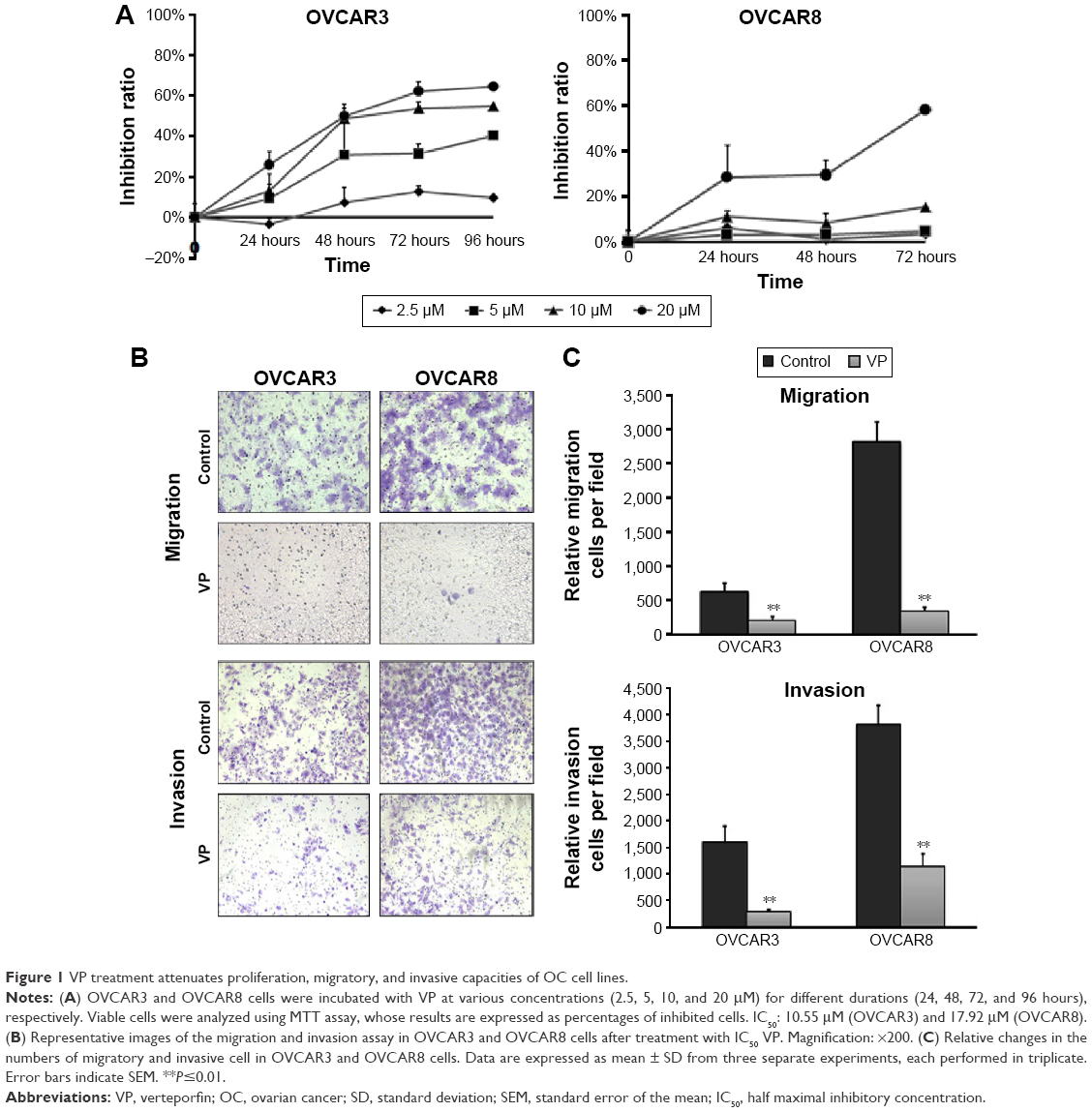 | Figure 1 VP treatment attenuates proliferation, migratory, and invasive capacities of OC cell lines. |
VP inhibits the migratory/invasive capacity of OC cells
Transwell assay was performed to evaluate the potential effects of VP on the migratory and invasive capacities of cells. OVCAR3 and OVCAR8 cells in the upper chamber were incubated with VP of IC50 dose for 18 (migration assay) and 36 hours (invasion assay), respectively. As shown in Figure 1B and C, both the migration and invasion ability of these three cell lines were hindered by VP. In OVCAR3, the number of migratory and invasive cells was 214±56 and 288±47, compared with 630±125 and 1,594±313 (P<0.05) in control groups, respectively. As for OVCAR8, the numbers stood at 346±68 and 1,149±241 for migratory and invasive cells, respectively, in contrast to 2,815±295 and 3,811±372 (P<0.05) in control group.
VP induces YAP cytoplasmic retention and deregulates inducible YAP and CCNs in OC cells
We next investigated whether YAP and its direct target genes cysteine-rich protein 61 (CYR61/CCN1) and connective tissue growth factor (CTGF/CCN2)19,20 change in response to VP treatment in these two OC cells. Similar effects were observed – that YAP cytoplasmic retention and Ser127 phosphorylation could be significantly induced after 10 μM VP incubation for 120 minutes (Figure 2A). CCN1 and CCN2 also showed notable downregulation at both gene transcription and translation levels in a time-dependent manner in OVCAR8 cells (Figure 2B and C). Furthermore, we induced the expression of total YAP and its targeted genes by adding S1P (sphingosine-1-phosphate, 0.3 μM) to OVCAR8 cells.21 Interestingly, we observed that the increase in the expression of total YAP, CCN1, and CCN2 was abrogated after treatment with VP (10 μM) for 180 minutes in comparison to untreated groups (Figure 2D).
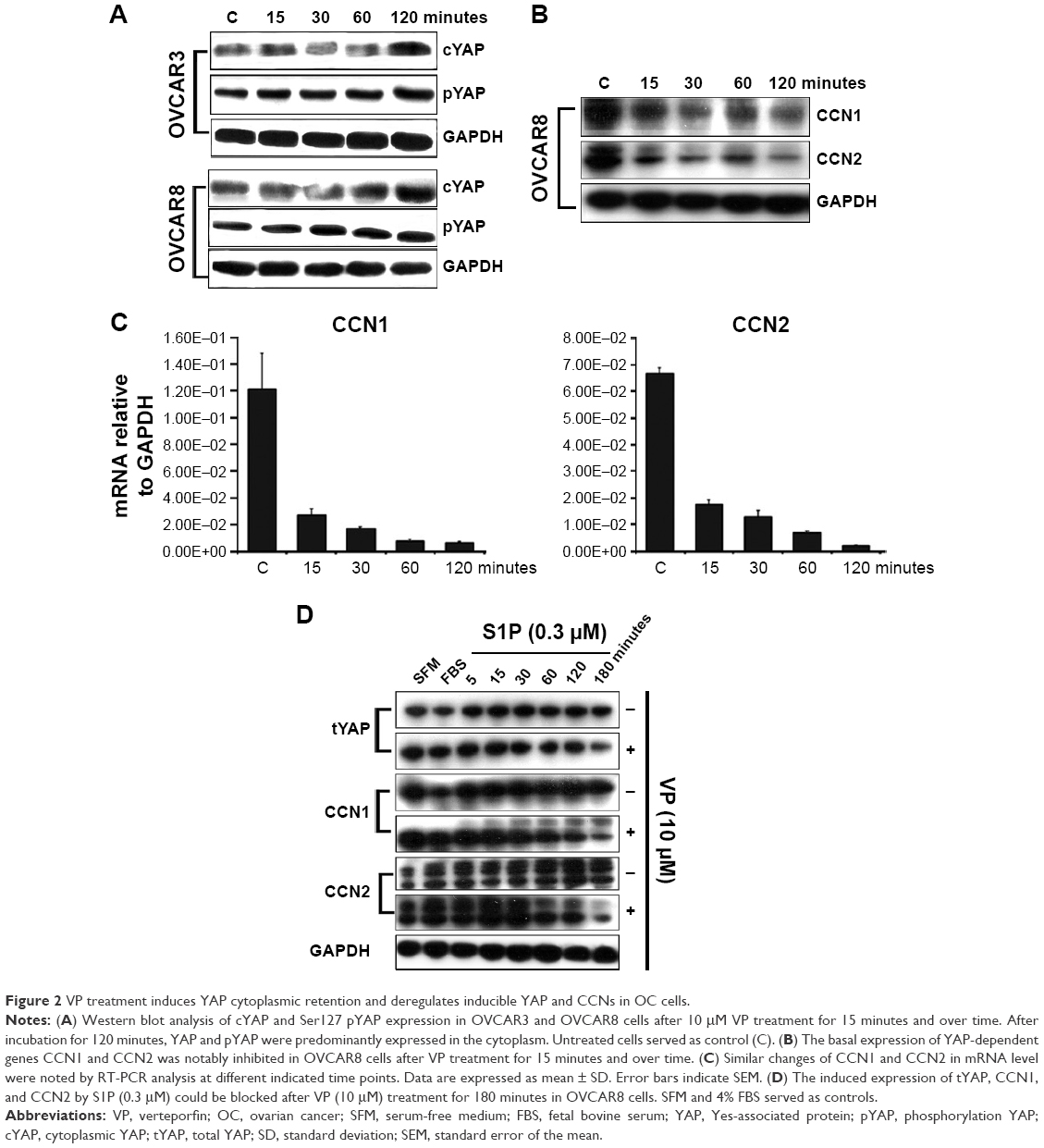 | Figure 2 VP treatment induces YAP cytoplasmic retention and deregulates inducible YAP and CCNs in OC cells. |
VP exerts little effect on YAP-upstream components in Hippo pathway in vitro
To determine whether VP affects YAP-upstream factors, the mRNA level of MST1/2 and LATS1/2 in OVCAR8 cells was evaluated after exposure to different concentrations (0, 1.25, 2.5, 5, and 10 μM) of VP for 2 hours. As shown in Figure 3, little change of these factors was observed. In contrast, CCN1 and CCN2 experienced a significant downregulation in a dose-dependent manner, of which CCN2 presented much sensitivity to VP.
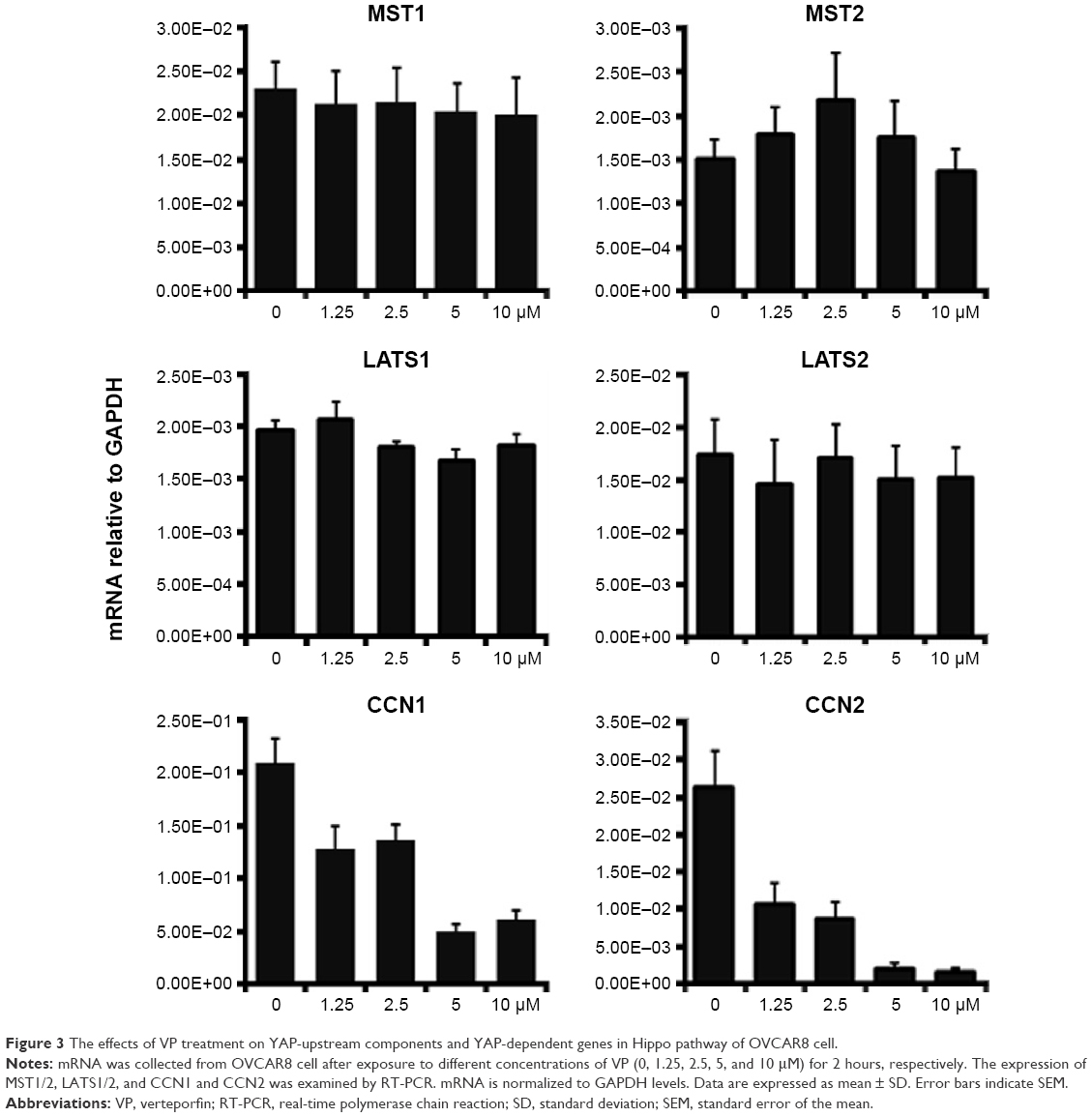 | Figure 3 The effects of VP treatment on YAP-upstream components and YAP-dependent genes in Hippo pathway of OVCAR8 cell. |
VP inhibits OC xenograft in vivo
To further explore the mechanisms of the antitumor effects of VP, we established OVCAR8 IP metastasis mice models. We found that treatment with VP caused a moderate but significant decrease in the tumor growth in comparison with PBS and DMSO groups, but the effect was less prominent than that in CDDP group (Figure 4A). The average tumor nodule weight in PBS, DMSO, CDDP, and VP groups weighed (g, mean ± standard deviation) 1.68±0.34, 1.55±0.28, 0.38±0.11, and 1.16±0.17, respectively. Besides, treatment with VP led to a remarkable and significant decrease in the gross ascites volume in comparison with PBS and DMSO groups (150 μL vs 4,300 and 3,200 μL; Figure 4B). Further immunohistochemistry staining analysis indicated VP diminished cell proliferation by inhibiting expression of Ki67 (Figure 4C), while apoptosis index caspase3 showed no significant change (Figure S2). These findings were in agreement with our in vitro results. In addition, VP-treated tumors showed a markedly increased expression of cytoplasmic YAP and pYAP as well (Figure 4D). Moreover, during the therapeutic session, the bodyweight of the mice from VP-treated xenograft model kept increasing (Table S2).
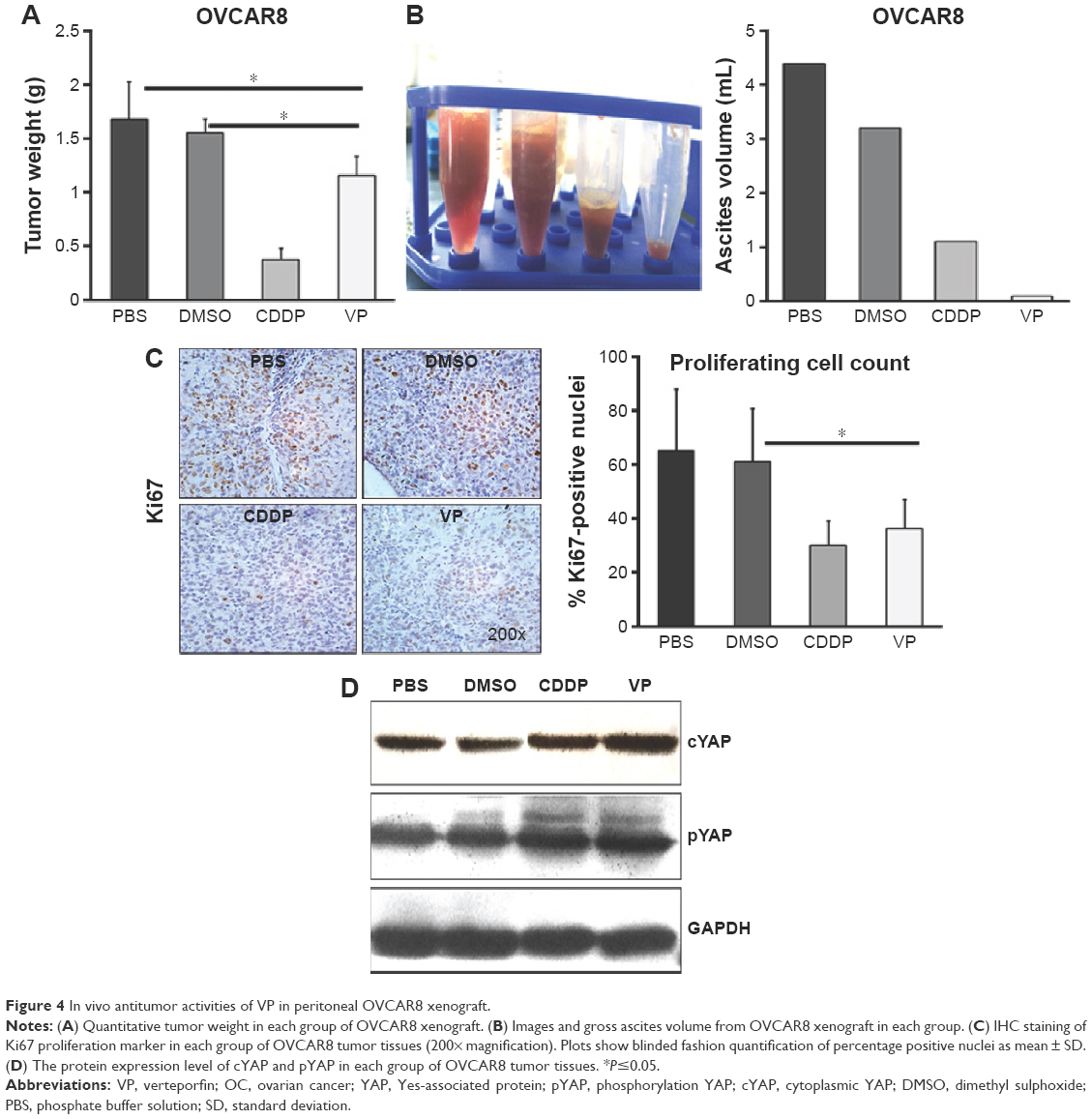 | Figure 4 In vivo antitumor activities of VP in peritoneal OVCAR8 xenograft. |
VP exerts little effect on YAP-upstream components in Hippo pathway in vivo
We next investigated changes in YAP-upstream components and YAP-dependent genes in OVCAR8 tumor tissue after VP treatment via RT-PCR analysis (Figure S3). Significant downregulation of CCN1 and CCN2 was noted. However, MST1/2 and LATS1/2 revealed little effect from VP.
Discussion
Hippo pathway is a newly discovered and evolutionarily conserved signaling network in mammals that controls cell fate and organ size. However, a handful of studies have led to the discovery of tumorigenesis role of Hippo pathway over the past decade.22,23 Deregulation of the Hippo pathway contributes to a number of cellular processes associated with cancer progression, such as increased cellular proliferation, inhibited apoptosis, and deregulated cellular differentiation.24 YAP deregulation is the best understood mechanism. As noted in the “Introduction” section, YAP overexpression occurs in a broad range of human cancers, including OC. A research group found that YAP plays an important role in regulating ovarian adult granulosa cell tumor cell proliferation, migration, and steroidogenesis.25 They also found that YAP interacts with epidermal growth factor receptor signaling pathway to regulate growth of ovarian surface epithelial cells and OC cells.26 Most recently, the same group came to a conclusion that YAP may be involved in the initiation and progression of fallopian tube epithelia-derived cancer.27 These evidence suggest that YAP may be a therapeutic target in OC.
VP, a newly identified YAP inhibitor,12 has shown encouraging results in the treatment of human cancers. However, few of these studies focus on OC. In this study, we provided evidence that pharmaceutical inhibition of YAP by VP is feasible for OC therapy, and both the in vitro and in vivo results verified the expected Hippo pathway changes. In other words, VP treatment could prevent YAP from accumulating in nucleus, block YAP’s transcriptional coactivator activity, decrease the downstream factors that govern cell migration and proliferation such as CCN1 (CYR61) and CCN2 (CTGF),28,29 and ultimately, suppress the progression of OC cells.
Brodowska et al30 reported that VP affects growth inhibition, apoptosis, and G0/G1-phase cell cycle arrest in human retinoblastoma cells in vitro. From our ovarian cell lines study, it can be seen that the viability of OVCAR3 and OVCAR8 cells were affected by VP treatment, which was indicated by our MTT analysis, while apoptosis analysis using Hoechst staining did not demonstrate significant effect of VP in comparison with nontreatment group. The in vivo analysis for detecting xenograft proliferation and apoptosis showed a concordant result that no significant differences in activated caspase3 apoptotic index were noted, but the proliferation index Ki67 differed significantly. These results suggest that tumor inhibition role of VP is associated with inhibition of proliferation rather than induction of apoptosis, which can be well explained by the physiological activity of Hippo pathway in cell proliferation. Furthermore, we found that VP could hinder the migratory and invasive capacity of OC cells. These results were consistent with observations by Hua et al27 who also showed that VP eliminates YAP activation-induced proliferation and migration of fallopian tube secretory epithelial cells. These lines of results may be supported by Cai et al31 who found that, in OC cells, YAP acts via LPA3/G13/RhoA/ROCK/protein phosphatase1A (PP1)/dpYAP, which results in secretion of epidermal growth factor receptor ligand amphiregulin and epidermal growth factor receptor–dependent cell migration.31
We also employed a preclinical xenograft mouse model of OC to investigate the in vivo efficacy of VP on OVCAR8 cells. In accordance with our in vitro findings, the results showed that VP could cause a remarkable effect on production of ascites and a moderate effect on tumor growth of OC in vivo. Recent studies found that better anticancer treatment effect could be achieved in VP when in combination with chemoagents.16,32 Additionally, knockdown of YAP could sensitize OC cells to various cancer therapeutic agents in vitro.33 Thus, we hypothesized that pharmacologic inhibition of YAP by VP may maximize therapeutic effects as well when combined with other anticancer drugs in the treatment of OC in vivo. More extensive studies are needed in the near future.
Furthermore, on the basis of our observations, there was no apparent decrease in body weight of the VP-treated animals, which indirectly indicates low or no toxicity of VP. This might be explained as a result of two aspects:
- The direct inhibition of YAP–TEAD may avoid possible side effects resulting from modulated upstream components involved in multiple other cellular pathways besides Hippo pathway, such as Wnt/β-catenin, Shh, BMP/TGFβ, and GPCR signaling.34 This is supported by our RT-PCR studies. VP treatment induced few mRNA changes of the YAP-upstream components of Hippo pathway both in vitro and in vivo.
- As previous work has reported that YAP is dispensable for the normal growth and homeostasis of some tissues,35 VP’s effect of YAP inhibition minimized toxicity on healthy tissue. These findings carry critical implications for potential use of VP as a safe and efficient agent for OC.
Despite the promising results we achieved in this study, there are still some limitations that should be acknowledged. First, although nude mice xenografts are considered to be generally used models in cancer research,36 they are not ideal to mimic the human disease due to the immunodeficient environment. Second, VP has a low half-life (t1/2 ~3 hours) because of rapid excretion in the feces, which may not be suitable for long-term treatments.37 Further investigation about the real efficacy of VP, its pharmacokinetics, or the appropriate administration route should be done.
In conclusion, our results suggest that treatment with VP could effectively reduce proliferation and inhibit the growth of OC cells in vitro and in vivo by direct interruption of the YAP–TEAD complex. In future work, the efficacy of VP in combating OC warrants in-depth study.
Acknowledgments
We thank Lisa Tsang for her help in correcting possible grammatical or spelling errors of this manuscript. This work was supported by the National Natural Science Foundation of China (Grant numbers: 81170592 and 81101991) and Sichuan Youth Foundation of Science of Technology (Grant number: 2015JQ0026).
Disclosure
The authors report no conflicts of interest in this work.
References
Santucci M, Vignudelli T, Ferrari S, et al. The hippo pathway and YAP/TAZ-TEAD protein-protein interaction as targets for regenerative medicine and cancer treatment. J Med Chem. 2015;58(12):4857–4873. | ||
Zhao B, Tumaneng K, Guan KL. The Hippo pathway in organ size control, tissue regseneration and stem cell self-renewal. Nat Cell Biol. 2011;13(8):877–883. | ||
Overholtzer M, Zhang J, Smolen GA, et al. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci U S A. 2006;103(33):12405–12410. | ||
Barron DA, Kagey JD. The role of the Hippo pathway in human disease and tumorigenesis. Clin Transl Med. 2014;3:25. | ||
Deel MD, Li JJ, Crose LE, Linardic CM. A review: molecular aberrations within hippo signaling in bone and soft-tissue sarcomas. Front Oncol. 2015;5:190. | ||
Hall CA, Wang R, Miao J, et al. Hippo pathway effector Yap is an ovarian cancer oncogene. Cancer Res. 2010;70(21):8517–8525. | ||
Steinhardt AA, Gayyed MF, Klein AP, et al. Expression of Yes-associated protein in common solid tumors. Hum Pathol. 2008;39(11):1582–1589. | ||
Zhang X, George J, Deb S, et al. The Hippo pathway transcriptional co-activator, YAP, is an ovarian cancer oncogene. Oncogene. 2011;30(25):2810–2822. | ||
Xia Y, Chang T, Wang Y, et al. YAP promotes ovarian cancer cell tumorigenesis and is indicative of a poor prognosis for ovarian cancer patients. PLoS One. 2014;9(3):e91770. | ||
Jeong W, Kim SB, Sohn BH, et al. Activation of YAP1 is associated with poor prognosis and response to taxanes in ovarian cancer. Anticancer Res. 2014;34(2):811–817. | ||
Xia Y, Zhang YL, Yu C, Chang T, Fan HY. YAP/TEAD co-activator regulated pluripotency and chemoresistance in ovarian cancer initiated cells. PLoS One. 2014;9(11):e109575. | ||
Liu-Chittenden Y, Huang B, Shim JS, et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012;26(12):1300–1305. | ||
Chen Q, Zhang N, Gray RS, et al. A temporal requirement for Hippo signaling in mammary gland differentiation, growth, and tumorigenesis. Genes Dev. 2014;28(5):432–437. | ||
Lin C, Pelissier FA, Zhang H, et al. Microenvironment rigidity modulates responses to the HER2 receptor tyrosine kinase inhibitor lapatinib via YAP and TAZ transcription factors. Mol Biol Cell. 2015;26(22):3946–3953. | ||
Jiang N, Hjorth-Jensen K, Hekmat O, et al. In vivo quantitative phosphoproteomic profiling identifies novel regulators of castration-resistant prostate cancer growth. Oncogene. 2015;34(21):2764–2776. | ||
Donohue E, Thomas A, Maurer N, et al. The autophagy inhibitor verteporfin moderately enhances the antitumor activity of gemcitabine in a pancreatic ductal adenocarcinoma model. J Cancer. 2013;4(7):585–596. | ||
Song S, Ajani JA, Honjo S, et al. Hippo coactivator YAP1 upregulates SOX9 and endows esophageal cancer cells with stem-like properties. Cancer Res. 2014;74(15):4170–4182. | ||
Li Z, Min W, Gou J. Knockdown of cyclophilin A reverses paclitaxel resistance in human endometrial cancer cells via suppression of MAPK kinase pathways. Cancer Chemother Pharmacol. 2013;72(5):1001–1011. | ||
Lai D, Ho KC, Hao Y, Yang X. Taxol resistance in breast cancer cells is mediated by the hippo pathway component TAZ and its downstream transcriptional targets Cyr61 and CTGF. Cancer Res. 2011;71(7):2728–2738. | ||
Reddy P, Deguchi M, Cheng Y, Hsueh AJ. Actin cytoskeleton regulates Hippo signaling. PLoS One. 2013;8(9):e73763. | ||
Miller E, Yang J, DeRan M, et al. Identification of serum-derived sphingosine-1-phosphate as a small molecule regulator of YAP. Chem Biol. 2012;19(8):955–962. | ||
Lau AN, Curtis SJ, Fillmore CM, et al. Tumor-propagating cells and Yap/Taz activity contribute to lung tumor progression and metastasis. EMBO J. 2014;33(5):468–481. | ||
Lu L, Li Y, Kim SM, et al. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc Natl Acad Sci U S A. 2010;107(4):1437–1442. | ||
Barron DA, Kagey JD. The role of the Hippo pathway in human disease and tumorigenesis. Clin Transl Med. 2014;3:25. | ||
Fu D, Lv X, Hua G, et al. YAP regulates cell proliferation, migration, and steroidogenesis in adult granulosa cell tumors. Endocr Relat Cancer. 2014;21(2):297–310. | ||
He C, Lv X, Hua G, et al. YAP forms autocrine loops with the ERBB pathway to regulate ovarian cancer initiation and progression. Oncogene. 2015;34(50):6040–6054. | ||
Hua G, Lv X, He C, et al. YAP induces high-grade serous carcinoma in fallopian tube secretory epithelial cells. Oncogene. 2016;35(17):2247–2265. | ||
Zhao B, Ye X, Yu J, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008;22(14):1962–1971. | ||
Young N, Pearl DK, Van Brocklyn JR. Sphingosine-1-phosphate regulates glioblastoma cell invasiveness through the urokinase plasminogen activator system and CCN1/Cyr61. Mol Cancer Res. 2009;7(1):23–32. | ||
Brodowska K, Al-Moujahed A, Marmalidou A, et al. The clinically used photosensitizer Verteporfin (VP) inhibits YAP-TEAD and human retinoblastoma cell growth in vitro without light activation. Exp Eye Res. 2014;124:67–73. | ||
Cai H, Xu Y. The role of LPA and YAP signaling in long-term migration of human ovarian cancer cells. Cell Commun Signal. 2013;11(1):31. | ||
Ciamporcero E, Shen H, Ramakrishnan S, et al. YAP activation protects urothelial cell carcinoma from treatment-induced DNA damage. Oncogene. 2016;35(12):1541–1553. | ||
Huang JM, Nagatomo I, Suzuki E, et al. YAP modifies cancer cell sensitivity to EGFR and survivin inhibitors and is negatively regulated by the non-receptor type protein tyrosine phosphatase 14. Oncogene. 2013;32(17):2220–2229. | ||
Kim M, Jho EH. Cross-talk between Wnt/beta-catenin and Hippo signaling pathways: a brief review. BMB Rep. 2014;47(10):540–545. | ||
Cai J, Zhang N, Zheng Y, de Wilde RF, Maitra A, Pan D. The Hippo signaling pathway restricts the oncogenic potential of an intestinal regeneration program. Genes Dev. 2010;24(21):2383–2388. | ||
Shultz LD, Goodwin N, Ishikawa F, Hosur V, Lyons BL, Greiner DL. Human cancer growth and therapy in immunodeficient mouse models. Cold Spring Harb Protoc. 2014;2014(7):694–708. | ||
Gibault F, Corvaisier M, Bailly F, Huet G, Melnyk P, Cotelle P. Non-photoinduced biological properties of verteporfin. Curr Med Chem. 2016;23(11):1171–1184. |
Supplementary materials
 | Table S1 Primer sequences for RT-PCR |
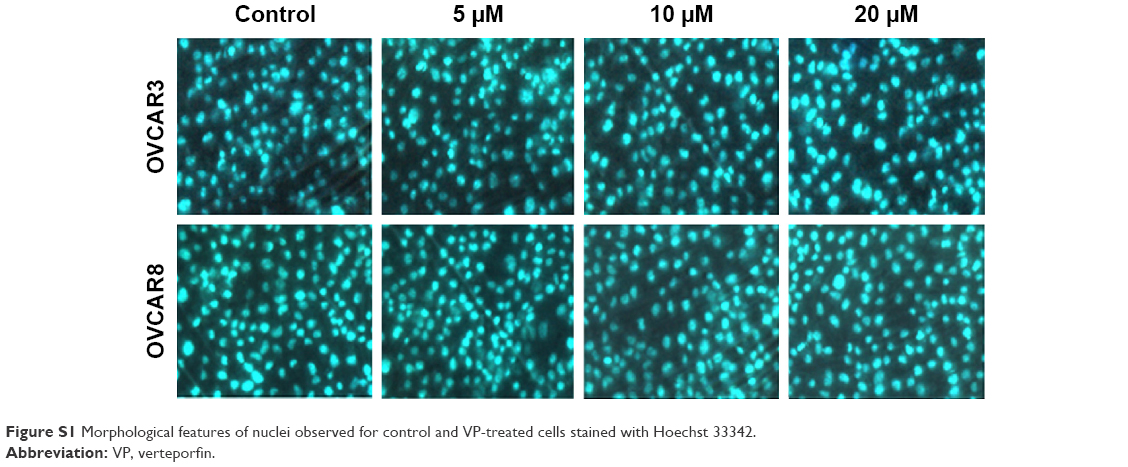 | Figure S1 Morphological features of nuclei observed for control and VP-treated cells stained with Hoechst 33342. |
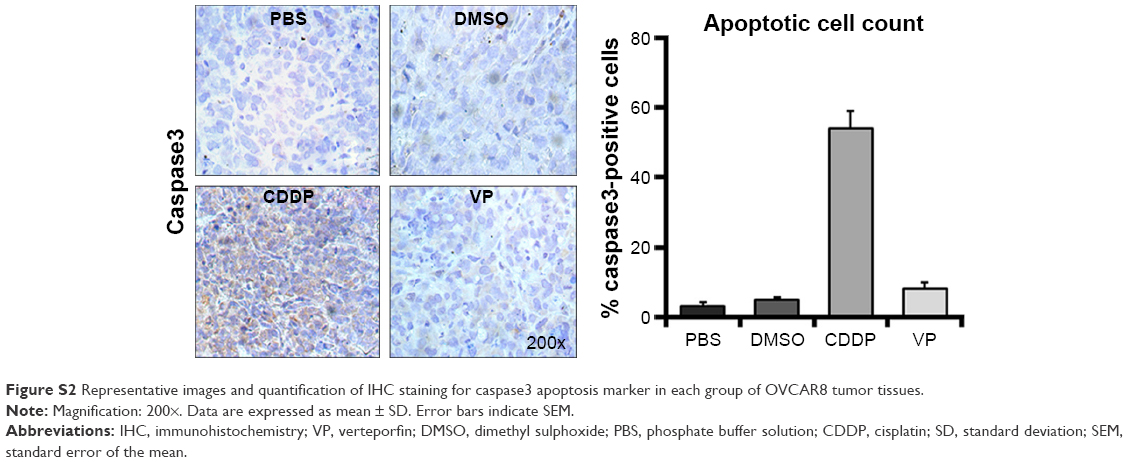 | Figure S2 Representative images and quantification of IHC staining for caspase3 apoptosis marker in each group of OVCAR8 tumor tissues. |
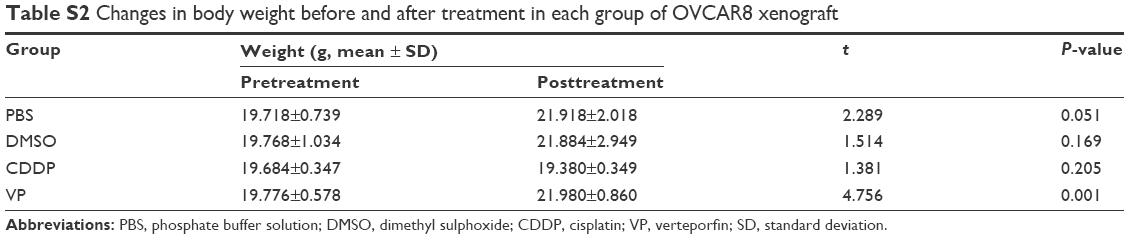 | Table S2 Changes in body weight before and after treatment in each group of OVCAR8 xenograft |
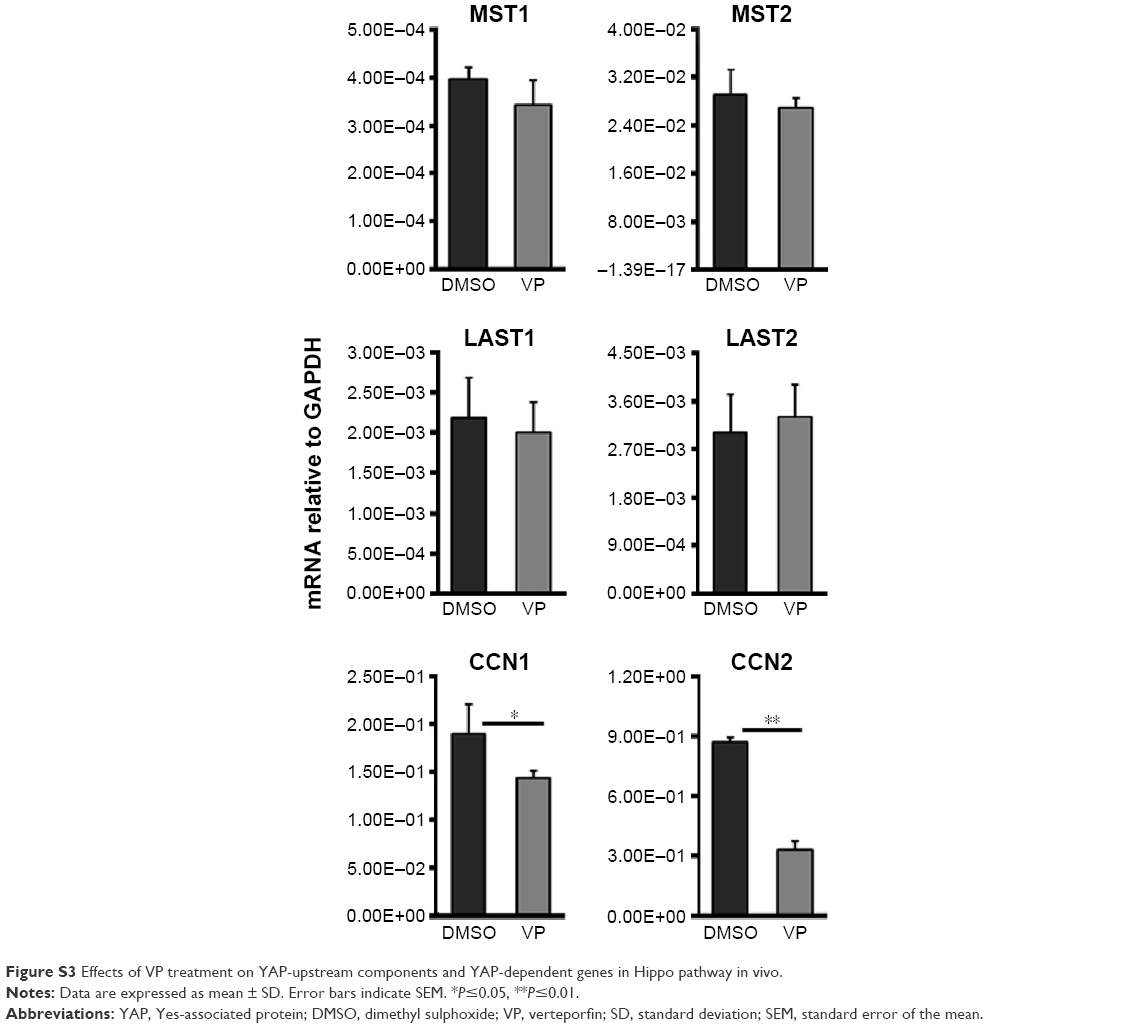 | Figure S3 Effects of VP treatment on YAP-upstream components and YAP-dependent genes in Hippo pathway in vivo. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.