")
Back to Journals » Drug Design, Development and Therapy » Volume 8
Vemurafenib: an evidence-based review of its clinical utility in the treatment of metastatic melanoma
Authors Swaika A, Crozier JA, Joseph R
Received 14 February 2014
Accepted for publication 25 March 2014
Published 16 June 2014 Volume 2014:8 Pages 775—787
DOI https://doi.org/10.2147/DDDT.S31143
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Abhisek Swaika, Jennifer A Crozier, Richard W Joseph
Department of Hematology and Oncology, Mayo Clinic, Jacksonville, FL, USA
Abstract: The discovery of BRAF mutations in the majority of patients with metastatic melanoma combined with the identification of highly selective BRAF inhibitors have revolutionized the treatment of patients with metastatic melanoma. The first highly specific BRAF inhibitor, vemurafenib, began clinical testing in 2008 and moved towards a rapid approval in 2011. Vemurafenib induced responses in ~50% of patients with metastatic BRAF-mutant melanoma and demonstrated improved overall survival in a randomized Phase III trial. Furthermore, vemurafenib is well-tolerated with a low toxicity profile and rapid onset of action. Finally, vemurafenib is active even in patients with widely metastatic disease. Despite the success of vemurafenib in treating patients with BRAF-mutant metastatic melanoma, most, if not all, patients ultimately develop resistance resulting in disease progression at a median time of ~6 months. Multiple mechanisms of resistance have been described and rationale strategies are underway to combat resistance. This review highlights the development, clinical utility, resistance mechanisms, and future use of vemurafenib both in melanoma and other malignancies. We consulted PubMed, Scopus, MEDLINE, ASCO annual symposium abstracts, and http://clinicaltrials.gov/for the purpose of this review.
Keywords: resistance, vemurafenib, BRAFV600E, immunotherapy
Introduction
In 2002, Davies et al reported the presence of an activating mutation in the gene encoding the protein kinase B-raf (BRAF) in approximately 50% of cutaneous melanomas.1 Over 30 distinct mutations in the BRAF gene have been identified and most localize to the kinase domain; however, the majority of the mutations in BRAF result in a substitution for valine with glutamic acid at the 600 position of the BRAF protein (BRAFV600E).2 BRAF is a member of the rapidly accelerated fibrosarcoma (RAF) family of serine/threonine-specific kinases pathway, and mutation in BRAF leads toward constitutive activation of the mitogen-activated protein kinase (MAPK) pathway and increased cellular proliferation. Subsequent studies have confirmed the presence of BRAF mutations in 40%–50% of cutaneous melanomas.3,4
Vemurafenib, a highly selective inhibitor of mutated BRAF, is extremely active in patients with metastatic melanoma who harbor a mutation in BRAFV600 (including patients with non V600E mutations) by inducing response rates in ~50% of patients and prolonging survival when compared to traditional chemotherapeutic agents.5–7 In addition to excellent clinical activity, vemurafenib is generally well tolerated with the most common side effects being fatigue, arthralgia, and dermatitis. Finally, vemurafenib also holds promise in treating patients with the most lethal form of melanoma: those with active brain metastases.8
Despite the clinical success of vemurafenib, most, if not all, patients eventually develop resistance. Multiple resistance mechanisms have been defined and are generally grouped as either those with reactivation of the MAPK pathway (intrinsic) or those outside of the MAPK pathway (extrinsic). Defining mechanisms of resistance has led to multiple ongoing trials that involve rationale combinations of BRAF-inhibitors with other agents. Finally, in addition to their role in increasing cellular proliferation, BRAF mutations are also associated with the tumor’s ability to evade the immune system, providing the rationale for possible combination therapies involving a BRAF inhibitor with immunostimulatory agents.9
Preclinical data
Defining the pathway
In 2002, Davies et al discovered that ~50% of cutaneous melanomas harbor a mutation in BRAF. A change from valine to glutamic acid at codon 600 (V600E) is the most common mutation (~80%) in BRAF followed by V600K (~10%).1,10 Mutated BRAF leads to constitutive activation of the MAPK pathway, which in turn stimulates growth-factor independent cellular proliferation and drives oncogenic activity with evasion of apoptosis and enhanced invasiveness (Figure 1).2 The MAPK pathway is composed of the rat sarcoma virus homologue (RAS)–RAF–mitogen-activated and extracellular signal-regulated kinase (MEK)–extracellular signal regulated kinase (ERK) signal transduction pathway. The physiological role of this pathway is to link the extra-cellular signals/factors to the nucleus for a regulated expression of genes, which is essential for normal cellular proliferation, differentiation, and survival.11,12 Oncogenic mutations leading to activation of the MAPK pathway are implicated in close to 90% of melanomas, including activating mutations in BRAF (~50%), NRAS (~20%), and other genes in the MAPK pathway.3,13
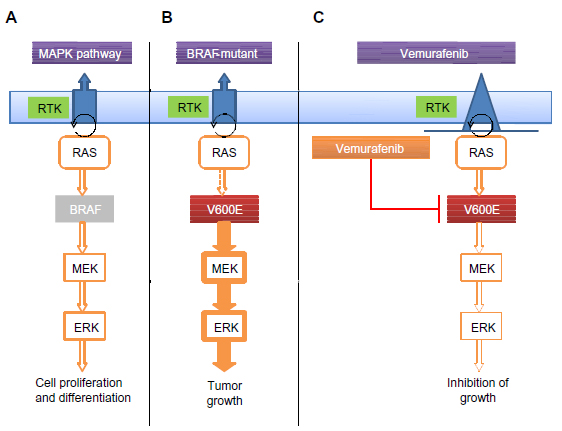 | Figure 1 Activation of the MAPK pathway through a BRAFV600E mutation. |
BRAF-mutated melanomas are more common in younger patients with early but intermittent sun exposure rather than chronic sun-damaged skin.14 BRAF mutations are more common in superficial spreading or nodular melanoma and occur less frequently in mucosal and acral melanoma.15–17 Furthermore, BRAF mutations are not associated with ocular melanoma.18
Previous attempts and lack of success at BRAF inhibition
Soon after the discovery of BRAF mutations in the majority of patients with cutaneous melanomas, preclinical trials involving BRAF inhibitors in melanoma were initiated. Sorafenib, a nonspecific BRAF inhibitor, was unsuccessful at generating meaningful clinical activity in patients with melanoma, secondary to its inability to inhibit mutant BRAF at pharmacologically tolerated doses.19,20 Given this limitation, multiple groups sought to develop a highly selective BRAF inhibitor that would only target mutant BRAF and thus avoid the off-target effects of inhibiting wild type BRAF.
Development of and preclinical activity of vemurafenib
One of the first highly selective inhibitors of mutant BRAF was PLX-4720 (Plexxikon, Berkeley, CA, USA).21 PLX-4720 demonstrated marked inhibition of the BRAF mutant cell lines with little impact on BRAF wild type cell lines. Unfortunately, this original formulation could not reach pharmacologic levels in vivo to effectively inhibit BRAFV600. A partnership with F. Hoffmann-La Roche Ltd (Basel, Switzerland) resulted in a reformulation of the agent to PLX-4032 (vemurafenib) that demonstrated acceptable pharmacokinetic properties with an appropriate increase in serum levels with dose escalation.22 PLX-4032 was also highly specific for mutant BRAF including the V600E, V600K, and V600D isoforms, but caused tumor growth in wild type BRAF xenograft models secondary to transactivation of the RAF dimers, enhancing downstream ERK and MEK phosphorylation, thus promoting cellular proliferation and growth.23 Upon identification of a highly active BRAF inhibitor, the pivotal BRAF inhibitors in melanoma (BRIM) clinical trial began.
Clinical activity of vemurafenib
BRIM1 (Phase I)
The Phase 1 trial included patients with advanced solid tumors, with the majority of patients having metastatic melanoma with a BRAFV600E mutation (89%) (Table 1).5 The dose-escalation phase included 55 patients followed by a dose-expansion cohort of 32 patients. Patients with active brain metastases and a poor functional status, as determined by an Eastern Cooperative Oncology Group (ECOG) performance status of greater than 1, were excluded from the study.24 No dose-limiting toxicities were observed at doses lower than 720 mg twice daily; however, grade 1–2 rash, arthralgia, fatigue, nausea, and photosensitivity were relatively common adverse effects. Squamous cell carcinoma (SCC), almost all being of the keratoacanthoma type, was observed as an unexpected adverse effect in ~20% of patients. In the dose escalation cohort, there were 16 patients with melanoma and a BRAF V600E mutation, and eleven of 16 (69%) experienced a response. In the dose-expansion cohort, there were 32 patients with melanoma, all with BRAF V600E mutations, all treated with 960 mg orally twice daily, and with an overall response rate (ORR) of 26/32 (81%). Accelerated responses were noted in several symptomatic patients leading to decreased pain and improving their quality of life. The median progression-free survival (PFS) in the dose-expansion cohort was greater than 7 months with a median survival of 13.8 months.25 In summary, this Phase I trial demonstrated marked clinical activity by generating response rates in >50% of patients and established the recommended Phase II dose of 960 mg orally twice daily.
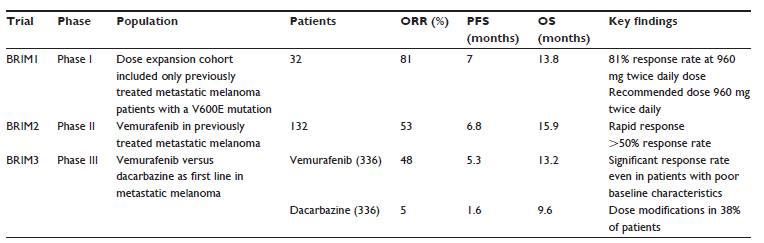 | Table 1 Summary of results from BRIM1, BRIM2, and BRIM3 |
BRIM2 (Phase II)
A multicenter Phase II trial of vemurafenib in previously treated patients with BRAFV600-mutant metastatic melanoma enrolled a total of 132 patients at a dose of 960 mg orally twice daily.6 The primary endpoint was to determine ORR as determined by RECISTv1.1 (PAREXEL International, Waltham, MA, USA). One hundred and twenty-two patients (92%) had the V600E mutation while the remaining ten had V600K mutation. The confirmed ORR was 53%, with 6% of patients achieving a complete response (CR) and 47% attaining a partial response (PR). Primary progression was observed in 14% while 29% of patients had stable disease and 4% had a response that could not be assessed. The subgroup analysis revealed an inferior response (33%) in patients who had an elevated baseline lactate dehydrogenase (LDH) level of greater than 1.5 times normal. Median duration of response was 6.7 months and the median PFS was reported as 6.8 months. The secondary endpoint of overall survival (OS) was 15.9 months (95% confidence interval [CI], 11.6–18.3).
BRIM3 (Phase III)
BRIM3 was the pivotal Phase III study that compared vemurafenib versus dacarbazine in previously untreated metastatic melanoma.7 The co-primary endpoints included OS and PFS, and secondary endpoints included duration of response, time to response, and response rate. Cobas® (Roche Molecular Systems Inc., Branchburg, NJ, USA) testing was performed at one of the five central laboratories worldwide to screen for the BRAF V600E mutation status. Six hundred and seventy five out of 2,107 patients were screened and were randomized to receive either vemurafenib (960 mg orally twice daily) or dacarbazine chemotherapy (1,000 mg/m2 administered intravenously every 3 weeks). Eligibility criteria were similar to the Phase II study and excluded patients with an Eastern Cooperative Oncology Group score of greater than 1 and with active central nervous system metastases. The baseline serum LDH level (normal or elevated) was also included during patient stratification. Tumor assessments were conducted at baseline, week 6, week 12, and subsequently every 9 weeks. RECISTv1.1 was used to assess tumor response. A planned interim analysis by an independent review committee established the accomplishment of the co-primary endpoints. After review of an interim analysis by an independent data and safety monitoring board, crossover was recommended for patients randomized to the dacarbazine arm. The OS at 6 months was reported as 84% (95% CI, 78–89) for the vemurafenib arm as compared to 64% (95% CI, 56–73) for the dacarbazine arm. The median OS (updated), presented by Chapman et al25 at the 2012 American Society of Clinical Oncology (ASCO) annual meetings, was 13.2 months (95% CI, 12.0–15.0) with vemurafenib as compared to 9.6 months (95% CI, 7.9–11.8) for dacarbazine. The hazard ratio for death in vemurafenib group was 0.37 (95% CI, 0.26–0.5; P<0.001) while the hazard ratio for tumor progression was 0.26 (95% CI, 0.20–0.33; P<0.001). The vemurafenib arm had an ORR of 48% with a median PFS of 5.3 months, compared with a 5% response rate and 1.6 months median PFS for dacarbazine. Subgroup analysis revealed a consistent PFS, even with those with elevated baseline LDH levels. The toxicity profile of vemurafenib was similar to the Phase I/II trials with dose modifications/reductions needed in 38% of the patients.
Use in adjuvant setting
The substantial clinical benefit vemurafenib provided in the metastatic setting has prompted further studies of using BRAF inhibitors in the adjuvant or neoadjuvant setting (BRIM8, NCT01667419). Currently, there are multiple ongoing studies (Table 3) using BRAF inhibitors or the combination of BRAF and MEK inhibitors in the adjuvant setting in patients with stage IIIb and IIIc BRAF-mutant melanoma who are at high risk of developing disease recurrence. While the use of BRAF inhibitors in the adjuvant setting certainly has good rationale, there are concerns that the use of BRAF inhibitors in the adjuvant setting could lead to the development of BRAF-resistance tumors sooner, and thereby limit future therapeutic options in the metastatic setting. Other concerns for the use of BRAF inhibitors in the adjuvant setting include management of toxicities as well as the unknown duration of treatment with these agents.
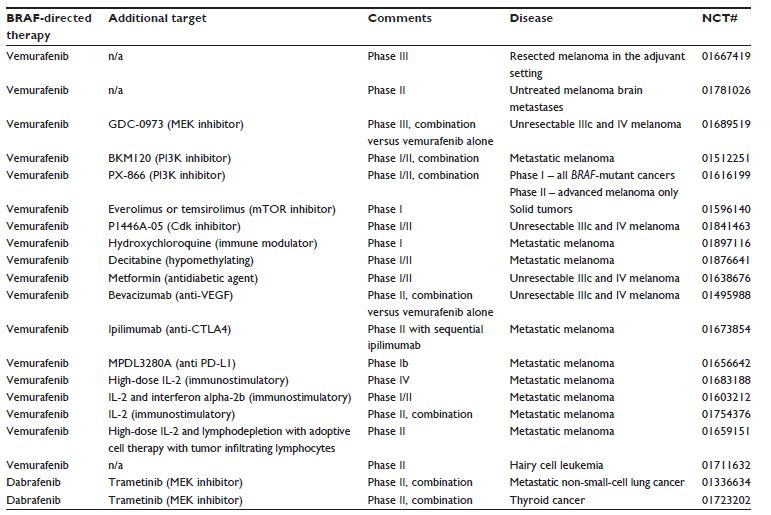 | Table 3 Active trials with BRAF inhibitors referenced in the manuscript |
Activity in brain metastases
Patients with active brain metastases were excluded from the initial pivotal studies (BRIM2 and BRIM3). Drummer et al recently published the results of an open-label trial in 24 BRAFV600-mutant metastatic melanoma patients with nonresectable previously treated brain metastases.8 Median PFS was reported as 3.9 months (95% CI, 3.0–5.5) and median survival as 5.3 months (95% CI, 3.9–6.6). Approximately half of the patients had meaningful intracranial tumor regression, with other signs of improvement such as improved performance status and decreased need for systemic corticosteroids. A Phase II study assessing the clinical efficacy and safety of vemurafenib as a neoadjuvant treatment for untreated brain metastases (nonresectable, not amenable to stereotactic-radiosurgery, or more than 4 lesions) in BRAFV600-mutant melanoma is underway (NCT01781026).
Activity in non-V600E BRAF-mutant metastatic melanoma
Both BRIM2 and BRIM3 focused mainly on patients with BRAFV600E mutations, largely excluding patients with V600K. The trials utilized the now US Food and Drug Administration-approved companion diagnostic, Cobas® 4800 BRAFV600 Mutation test (Roche Molecular Systems Inc.), to identify the BRAFV600E mutation in patients with metastatic melanoma. The discordant or non-BRAF V600E results underwent Sanger sequencing and massively parallel pyrosequencing to detect non-V600E mutations. In the BRIM2 study, four of the ten patients with V600K mutations attained PR.6 The results of BRIM3 based on V600E and V600K were also recently published.26 In the 598 patients (91%) who had V600E, the median OS was 13.3 months in the vemurafenib arm versus 10.0 months in the dacarbazine arm. In the 57 patients (9%) who harbored a V600K mutation, the median OS was 14.5 months in the vemurafenib arm and 7.6 months in the dacarbazine arm. The authors appropriately conclude that vemurafenib improves the survival in patients with metastatic melanoma who harbor a BRAFV600E or V600K mutation.
Pharmacologic data and pharmacokinetics
The recommended dose of vemurafenib is 960 mg orally twice daily. The inhibitory concentration at 50% for V600E was calculated at 31 nmol/L with an estimated half-life of 50 hours, and median time to peak plasma concentration of ~4 hours.7 Vemurafenib is highly protein bound and biliary excretion of the unchanged compound appears to be the primary route of drug elimination. The study evaluating the effects of food on the bioavailability of the drug proved that a rich fatty meal increased the drug concentration without affecting its mean terminal half-life.27 Vemurafenib inhibits the CYP1A2 enzyme while inducing the CYP3A4 enzyme.
In terms of pharmacokinetics, vemurafenib is extremely fast acting. In the Phase I study of vemurafenib, biopsies performed at day 1 and day 14 revealed robust inhibition of the downstream marker of BRAF activation and phosphorylated ERK as well as significant decrease in metabolic activity as measured by Fludeoxyglucose (FDG)-Positron emission tomography (PET).5
Management of toxicity
Vemurafenib is fairly well tolerated with a very manageable side effect profile. The most common adverse effects include arthralgia (60%) and fatigue (~35%–50%). These are mostly grade 1 or 2 side effects as per the Common Terminology Criteria for Adverse Events (CTCAE) and are typically managed conservatively.28 An outline of the most common toxicities reported in BRIM2 and BRIM3 are presented in Table 2.
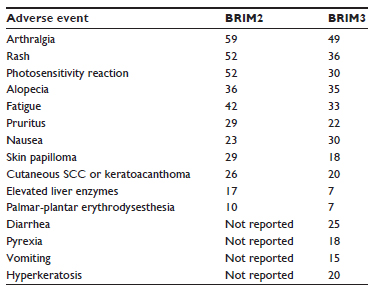 | Table 2 Adverse events of any grade reported in at least 5% of patients treated with vemurafenib |
Dermatologic toxicities
The most common toxicities from BRAF inhibitors are dermatologic. Cutaneous SCC occurs in a high percentage (20%–25%) of all treated patients and usually develops within a few weeks (average 8 weeks) of starting treatment. They are mostly of the keratoacanthoma type and may occur as a single or multiple dome-shaped lesions.7,29 The SCCs have a high proliferative index but seem to be localized, with no cases of metastatic SCC reported yet. Advanced age (≥65 years) and chronic sun exposure are predisposing factors. These lesions only require simple excision and do not warrant any dose reductions or interruptions even though they are considered as grade 3 adverse effects as per the CTCAE.30 Accelerated growth of previously quiescent keratinocytes containing RAS mutations and/or a paradoxical activation of the MAPK signaling pathway plays a role in development of these cutaneous SCCs.31 Interestingly, the incidence of cutaneous SCC is markedly reduced when BRAF inhibition is combined with MEK inhibition. A second common dermatologic toxicity for patients treated with vemurafenib is photosensitivity, with a reported rate of 12% of grade 2/3 in the BRIM3 trial; however, this can largely be avoided using skin block and sun avoidance. Stevens–Johnson syndrome is a rare but reported event while on vemurafenib, but is subsequently resolved after discontinuation of the drug. Other skin toxicities include alopecia, pruritus, acneiform eruptions, painful panniculitis, xerosis, and hypo- or hyperpigmentation. Most of these are managed conservatively and do not require any dose reductions/interruptions.30 Regular meticulous dermatology follow-up is mandatory for all patients on treatment with vemurafenib. Finally, severe cutaneous toxicities have been reported in two patients who started vemurafenib after progressing on anti-PD1 (programmed death 1) agents, emphasizing the point that unexpected toxicities of BRAF inhibitors should be carefully monitored.32
Additional toxicities
Elevated liver enzymes were documented in close to 20% of treated patients. Most of these were less than grade 3 and resolved with temporary interruption of treatment. Grade 1 and 2 toxicity generally does not require any dose adjustment. Grade 3 toxicity prompts treatment interruption with dose reduction to 720 mg twice daily with further reduction of dose to 480 mg twice daily at the second incident after reinitiation. Grade 4 toxicity warrants treatment interruption and dose reduction to 480 mg twice daily. The treatment dose of vemurafenib is never lowered to less than 480 mg twice daily.
Prolongation of the QTc interval and cardiac arrhythmias were reported in 8% of patients. The treatment is held with QTc >500 ms with reinitiation at a reduced dose and only if the QTc is lower than 500 ms. If the QTc is persistently greater than 500 ms, or it is constantly greater than 60 ms above the baseline electrocardiogram (EKG), the drug is permanently discontinued.
Mechanism of resistance
Primary resistance
Primary resistance to vemurafenib occurs in approximately 5%–10% of patients treated with vemurafenib, with multiple possible mechanisms reported. One possible explanation of primary resistance is the intratumor heterogeneity of varying proportions of wild type and mutant BRAF subclones within an individual tumor.23,33 Determination of the actual proportion of the mutant BRAF cells in the tumor might become essential to predict an initial/sustained response. More recently, Girotti et al demonstrated that activation of the epidermal growth factor (EGF) pathway through over-expression of the EGF receptors (EGFR) plays a role in both intrinsic and acquired resistance to BRAF inhibitors.34 The use of EGFR inhibitors and Dasatinib, a broad-specificity tyrosine kinase inhibitor, reverses the inhibition and also inhibits the proliferation and invasion of the resistant melanoma cells.32 BRAF-mutant melanoma cell lines with increased cyclin D1 expression also exhibited primary resistance to BRAF inhibitors.35 This outcome was amplified when the melanoma cells concomitantly overexpressed both cyclin D1 and cyclin-dependent kinase 4. Tumor microenvironment also plays an important role in such native resistance. The reactivation of the MAPK and the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K/AKT) signaling pathway due to the stromal cell secretion of hepatocyte growth factor (HGF) also confers resistance to BRAF inhibitor therapy. Straussman et al exhibited a reversal of this resistance by combining a BRAF inhibitor with HGF and/or HGF receptor (MET) inhibitor.36
Secondary resistance
Unlike primary resistance, the development of secondary resistance to vemurafenib and all BRAF inhibitors is nearly universal at a median time of 6–7 months. Several mechanisms of resistance have been proposed and in general can be divided into reactivation of the MAPK pathway (intrinsic pathway) or activation of the PI3K/AKT/mTOR (mammalian target of rapamycin) pathway (extrinsic). Below we review both the intrinsic and extrinsic mechanisms of resistance, which are summarized in Figure 2.
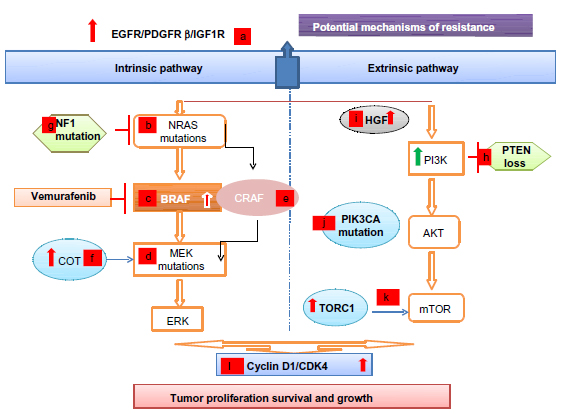 | Figure 2 Resistance to BRAF inhibitors: intrinsic and extrinsic mechanisms. |
Intrinsic pathways
Extracellular
Tumors that develop resistance to BRAF inhibitors have been shown to upregulate extracellular receptors including EGFR, platelet derived growth factor receptor beta, and the insulin-like growth factor receptor 1 expression.34,37–40
Intracellular
Activating mutations in RAS (NRAS or Kirsten rat sarcoma viral oncogene homolog KRAS) and MEK ([mitogen-activated and extracellular signal-regulated kinase kinase 1] MEK1) present in the resistant tumor but not in the pretreatment tumor specimen have been reported.41–43 While additional mutations in BRAF at the time of resistance are rare, the presence of BRAF amplification and splice variants has been demonstrated in the setting of acquired resistance.41,44 Most recently, Van Allen et al performed a whole-exome sequencing of BRAF-mutant melanoma tumor samples, pre- and posttreatment with a BRAF inhibitor, and identified dysregulation of genes encoding transcription factors of the MAPK pathway (dual specificity mitogen-activated protein kinase kinase 2 [MAP2K2], microphthalmia-associated transcription factor [MITF]) as well as confirmed previously known genetic mutations leading to the MAPK reactivation.45
Downstream pathways
The increased expression of cancer Osaka thyroid, a MAPK, leads to ERK activation through MEK, independent of BRAF signaling, promoting resistance to BRAF inhibitors.46 Elevated cellular-rapidly accelerated fibrosarcoma (CRAF) levels can lead to a shift of tumor cells’ dependency from BRAF to CRAF.47,48 The heat shock protein 90 inhibitors can overcome resistance to BRAF inhibitors by promoting the degradation of CRAF and by other potential mechanisms, showing promise in improvement of PFS in these resistant clones.49 Finally, the loss of function of the tumor suppressor neurofibromin, that inhibits RAS activity, has also been associated with resistance to BRAF and MEK inhibitors.50
Extrinsic pathway
In addition to reactivation of the MAPK pathway, resistance to vemurafenib is also associated with activation of pathways outside of the MAPK pathway including activation of the PI3K/AKT/mTOR pathway through several described mechanisms. First, the loss of the tumor suppressor, phosphatase and tensin homologue (PTEN), is associated with resistance to BRAF inhibitors.51 Recently, it was shown that patients with PTEN loss treated with a BRAF inhibitor had a decreased PFS as compared to patients without loss of PTEN.52 Second, gain-of-function phosphatidylinositol-4, 5-bisphosphate 3-kinase, catalytic subunit alpha mutations have been reported in melanomas resistant to BRAF inhibitors.53 Third, higher pretreatment levels of target of rapamycin complex 1, a downstream component of the AKT-PI3K pathway are associated with resistance to BRAF inhibitors. Corcoran et al demonstrated that patients with lower phosphorylated S6 had a shorter PFS when treated with a BRAF inhibitor with preservation of target of rapamycin complex 1 activity.54 Finally, activation of the AKT-PI3K pathway can also be induced through the tumor stromal cells’ secretion of HGF.36 It should also be noted that resistance mechanisms to BRAF inhibitors can vary even within the same patient, which presents an even more challenging clinical situation.41
Drug dependency
Another model of resistance to BRAF inhibitors is at least partially driven by dependence on the actual inhibitor. Das Thakur et al generated xenografts resistant to vemurafenib by dosing mice with subtherapeutic levels of vemurafenib.55 Interestingly, growth of these cells in vitro was dependent upon media containing ~50 nM of vemurafenib, and that cessation of the drug led to decreased cellular proliferation. The authors concluded that altered dosing of vemurafenib may forestall the emergence of resistance; however, this strategy has yet to be tried in humans.
Future use of BRAF inhibitors
As outlined above, defining mechanisms of resistance to BRAF inhibitors has inspired multiple clinical trials combining BRAF inhibitors with additional agents. Below we review some of these trials and we summarize these trials in Table 3.
Combining BRAF and MEK inhibitors
One of the first studies to address resistance to BRAF inhibitors was the combination of the BRAF inhibitor dabrafenib with a selective MEK inhibitor, trametinib, as presented by Flaherty et al in a pivotal Phase I and II trial.56 The primary endpoints of this trial were PFS, incidence of SCC, and overall response, while the OS and pharmacokinetic activity constituted the secondary endpoints. In the maximum tolerated dose, the combination of dabrafenib and trametinib generated a median PFS of 11 months compared to only 5.8 months in the single agent dabrafenib arm (hazard ratio for progression or death, 0.39; 95% CI, 0.25–0.62; P<0.001). ORR in the combination was 76%, as compared with 54% with monotherapy (P=0.03). The incidence of SCC was only 7% with the combination regimen as compared to 19% with dabrafenib alone. The combination regimen did require more frequent dose reductions and interruptions, mainly due to increased incidence of pyrexia (71% versus 26%), though it was rarely grade 3 or 4. Neutropenia was the most common grade 3 or 4 adverse effect in the combination regimen, with one incidence of febrile neutropenia. Given the success of this approach, in January 2014, the US Food and Drug Administration approved the combination of dabrafenib and trametinib for patients with BRAF-mutant metastatic melanoma. A separate trial combining vemurafenib with another MEK inhibitor (GDC-0973) has also demonstrated promising early activity with the expected final results of the coBRIM study, a Phase III study comparing a MEK inhibitor (GDC-0973) in combination with vemurafenib versus vemurafenib alone, anticipated in 2016.
Combination of BRAF inhibitors with PI3K/AKT/mTOR inhibitors
Vemurafenib is also being currently investigated in various clinical trials in combination with other small molecular inhibitors targeting the PI3K/AKT/mTOR pathway: BKM-120, a PI3K inhibitor (NCT01512251); PX-866, which is an oral irreversible pan-isoform inhibitor of the PI-3-kinase/PTEN/AKT signaling pathway (NCT01616199); and mTOR inhibitors everolimus or temsirolimus (NCT01596140).57
Additional combinations
Given the multiple reported mechanisms of resistance to BRAF inhibitors, there is an increasing number of ongoing clinical trials investigating various combination regimens of vemurafenib with other molecularly targeted drugs of the MAPK and the PI3K/AKT/mTOR pathway to overcome the aforementioned resistance mechanisms.
The combination of an oral cyclin dependent kinase (CDK) inhibitor with vemurafenib is similarly being studied (NCT01841463). Novel approaches, such as a combination regimen of hydroxychloroquine with vemurafenib (NCT01897116), decitabine with vemurafenib (NCT01876641), and metformin and vemurafenib (NCT01638676) are also being scrutinized in Phase I trials.
Increased angiogenesis through tumor expression of the vascular endothelial growth factor is associated with a worse clinical outcome.58,59 A randomized Phase II trial combining vemurafenib with a monoclonal antibody against vascular endothelial growth factor bevacizumab (NCT01495988) is ongoing.
Development of the next generation RAF inhibitors (PLX7904) has elucidated promise in this patient population. They seem to block the survival and growth of vemurafenib resistant cells by annulling the effect of mutant BRAF splice variants. They also do not have paradoxical effects on wild type BRAF while effectively inhibiting mutant BRAFV600E melanoma cells.60
Relationship of BRAF mutations and BRAF inhibitors with immunotherapy
The clinical responses to immune stimulation, occasional spontaneous regression, and the presence of tumor infiltrating lymphocytes have categorized melanoma as one of the most immunogenic tumors.61 Boni et al demonstrated that targeted BRAF inhibition in melanoma cells can lead to enhanced T-cell recognition by upregulating the expression of melanocyte differentiation antigens, thus validating the benefit of combining targeted therapy with immunotherapy.62 There is also evidence that BRAF inhibition shifts the balance of the tumor microenvironment to a pro-inflammatory state by increasing intratumoral cytotoxic T-cells.63 These lymphocytes retain their function and viability even after being exposed to high concentrations of the BRAF inhibitor, thereby endorsing the combination therapy.64,65 Most recently, Callahan et al proposed that paradoxical activation of ERK signaling in T-cells could be the reason for the improved antitumor activity of the combination of RAF inhibitors with cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) blockade.66
The improved understanding of tumor immunology and immune escape phenomena led to the development of ipilimumab, which is a fully human Immunoglobulin G1 monoclonal antibody that blocks the CTLA-4. CTLA-4 is an immune checkpoint molecule that functions as a negative costimulatory molecule for the T-cell.67,68 Ipilimumab increases the T-cell activity of the tumor infiltrating lymphocytes, which forms the basis of its antitumor activity.69
In view of this anticipated clinical benefit with the combination regimen, Ribas et al conducted a Phase I clinical trial with concomitant administration of vemurafenib and ipilimumab.70 The primary goal was to assess clinical safety and an administration schedule of the combination regimen. All patients had BRAFV600 mutant metastatic melanoma and were naive to any treatment with a BRAF/MEK inhibitor or any CTLA-4/programmed death (PD-1) blocking antibodies. Dose limiting grade 3 hepatotoxicity (elevations in aminotransferase levels) developed in four out of six patients at the 960 mg dose and in two of the four patients treated with the 720 mg dose of twice daily vemurafenib in combination with ipilimumab (3 mg/kg dose). The study was discontinued prematurely and the patients’ liver enzymes recovered with the discontinuation of the drug or with steroid treatment. Currently, a Phase II study evaluating the safety and benefit of sequential treatment with vemurafenib and ipilimumab is ongoing (NCT01673854).
The PD-1 receptor is another immune checkpoint that functions as an inhibitory receptor of T-cells.71,72 Monoclonal antibodies that block PD-1 or its ligand (PD-L1) have demonstrated excellent clinical activity in patients with metastatic melanoma, and there appears to be an association between tumor expression of PD-L1 and degree of benefit from these antibody therapies.73–75 Preliminary data suggests that BRAF inhibition can augment tumor expression of PD-L1,77 and a current Phase 1b clinical trial (NCT01656642) is exploring the clinical utility of the combination of vemurafenib with MPDL3280A (anti-PD-L1).
Immunotherapy with interleukin-2 (IL-2) has historically been the preferred treatment in melanoma patients with an excellent performance status by generating long lasting responses in a minority of patients. Whether activation of the MAPK pathway influences response to IL-2 is uncertain; however, there is some preliminary data to suggest that patients with an NRAS mutation may have a higher response rate to high dose IL-2.78 The rationale of increased tumor cell antigen expression with enhanced T-cell recognition by the BRAF inhibitors has inspired the clinical trials investigating the combination treatment of vemurafenib with high dose IL-2 (PROCLIVITY, NCT01683188, NCT01603212), and aldesleukin (NCT01754376).62 Concomitant blockade of interleukin-1 may also have synergistic effects with other immunotherapeutic options in BRAF wild-type melanoma, treated with a BRAF inhibitor.79
In preclinical melanoma models, BRAF inhibition has proven to paradoxically activate the MAPK signaling in cells with wild-type BRAF. In addition, BRAF inhibition has been shown to increase the in vivo activity of adoptively transferred lymphocytes through increased intratumoral cytokine secretion without affecting their ability to traffic to the tumor, leading to enhanced antitumor activity as compared to BRAF inhibitor alone.65,80 Attempts to isolate, expand, and infuse tumor infiltrating lymphocytes for the treatment of cancer is termed adoptive cell therapy (ACT). ACT with tumor infiltrating lymphocytes’ infusion and nonmyeloablative lymphodepletion with cytotoxic chemotherapy have previously shown clinical benefit in patients with metastatic melanoma.81–84 The combination of ACT with vemurafenib is also being investigated in a clinical trial (NCT01659151).
Use of vemurafenib and BRAF inhibitors in malignancies other than melanoma
In addition to melanoma, multiple other malignancies harbor mutations in BRAF. Close to 100% of hairy cell leukemias harbor a BRAF mutation, and preliminary reports demonstrate BRAF inhibition results in durable responses.85–87 In addition, non-small cell cancers harbor BRAF mutations in ~1% of tumors, and dramatic responses to vemurafenib have been characterized.88,89 BRAF mutations are also present in 50% of papillary thyroid tumors, and inhibition with vemurafenib has shown activity in patients with metastatic papillary thyroid cancer.90,91 On the other hand, vemurafenib and other BRAF inhibitors do not generate responses in the 10% of colorectal cancer patients who harbor BRAF mutations, likely secondary to activation of alternate pathways such as the EGFR signaling pathway.37,92,93 In summary, BRAF mutations occur in multiple malignancies outside of melanoma; however, the presence of BRAF mutation does not always equate with clinical efficacy with BRAF inhibitors and each disease state requires further study.
Conclusion
The combined discovery of BRAF mutations present in the majority of patients with metastatic melanoma, as well as the development of selective inhibitors of mutated BRAF, have revolutionized the treatment of metastatic melanoma. The next step is to further understand the baseline tumor biology, individual mechanisms of resistance, and the impact of BRAF mutations on the immune microenvironment. Ongoing and future clinical trials will translate these efforts with rationally designed drug combinations that will lead to more potent and durable treatment options.
Disclosure
The authors report no conflicts of interest in this work.
References
Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. | |
Wan PT, Garnett MJ, Roe SM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–867. | |
Jakob JA, Bassett RL Jr, Ng CS, et al. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer. 2012;118:4014–4023. | |
Bucheit AD, Syklawer E, Jakob JA, et al. Clinical characteristics and outcomes with specific BRAF and NRAS mutations in patients with metastatic melanoma. Cancer. 2013;119:3821–3829. | |
Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819. | |
Sosman JA, Kim KB, Schuchter L, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–714. | |
Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. | |
Dummer R, Goldinger SM, Turtschi CP, et al. Vemurafenib in patients with BRAF(V600) mutation-positive melanoma with symptomatic brain metastases: Final results of an open-label pilot study. Eur J Cancer. 2014;50:611–621. | |
Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med. 2006;203:1651–1656. | |
Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–2147. | |
Raman M, Chen W, Cobb MH. Differential regulation and properties of MAPKs. Oncogene. 2007;26:3100–3112. | |
Garnett MJ, Marais R. Guilty as charged: B-RAF is a human oncogene. Cancer Cell. 2004;6:313–319. | |
Omholt K, Platz A, Kanter L, Ringborg U, Hansson J. NRAS and BRAF mutations arise early during melanoma pathogenesis and are preserved throughout tumor progression. Clin Cancer Res. 2003;9:6483–6488. | |
Viros A, Fridlyand J, Bauer J, et al. Improving melanoma classification by integrating genetic and morphologic features. PLoS Med. 2008;5:e120. | |
Platz A, Egyhazi S, Ringborg U, Hansson J. Human cutaneous melanoma; a review of NRAS and BRAF mutation frequencies in relation to histogenetic subclass and body site. Mol Oncol. 2008;1:395–405. | |
Greaves WO, Verma S, Patel KP, et al. Frequency and spectrum of BRAF mutations in a retrospective, single-institution study of 1112 cases of melanoma. J Mol Diagn. 2013;15:220–226. | |
Zebary A, Omholt K, Vassilaki I, et al. KIT, NRAS, BRAF and PTEN mutations in a sample of Swedish patients with acral lentiginous melanoma. J Dermatol Sci. 2013;72:284–289. | |
Rimoldi D, Salvi S, Liénard D, et al. Lack of BRAF mutations in uveal melanoma. Cancer Res. 2003;63:5712–5715. | |
Eisen T, Ahmad T, Flaherty KT, et al. Sorafenib in advanced melanoma: a Phase II randomised discontinuation trial analysis. Br J Cancer. 2006;95:581–586. | |
Hauschild A, Agarwala SS, Trefzer U, et al. Results of a phase III, randomized, placebo-controlled study of sorafenib in combination with carboplatin and paclitaxel as second-line treatment in patients with unresectable stage III or stage IV melanoma. J Clin Oncol. 2009;27:2823–2830. | |
Tsai J, Lee JT, Wang W, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A. 2008;105:3041–3046. | |
Bollag G, Hirth P, Tsai J, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010;467:596–599. | |
Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. | |
Oken, MM, Creech RH, Tormey DC et al. Toxicity and Response Criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol. 1982;5:649–655. | |
Chapman PB, Hauschild A, Robert C, et al. Updated overall survival (OS) results for BRIM-3, a phase III randomized, open-label, multicenter trial comparing BRAF inhibitor vemurafenib (vem) with dacarbazine (DTIC) in previously untreated patients with BRAF(V600E)-mutated melanoma. Journal of Clinical Oncology. 2012;30:8502. | |
McArthur GA, Chapman PB, Robert C, et al. Safety and efficacy of vemurafenib in BRAFV600E and BRAFV600K mutation-positive melanoma (BRIM-3):extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014;15:323–332. | |
Ribas A, Zhang W, Chang I, et al. The effects of a high-fat meal on single-dose vemurafenib pharmacokinetics. J Clin Pharmacol. 2014;54:368–374. | |
National Cancer Institute. National Cancer Institute Common Terminology Criteria for Adverse Events, NCI-CTCAE v.4 (2010). Available: http://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_5x7.pdf. Accessed February 5th, 2014. | |
Anforth RM, Blumetti TC, Kefford RF, et al. Cutaneous manifestations of dabrafenib (GSK2118436):a selective inhibitor of mutant BRAF in patients with metastatic melanoma. Br J Dermatol. 2012;167:1153–1160. | |
Robert C, Sibaud V, Mateus C, Cherpelis BS. Advances in the management of cutaneous toxicities of targeted therapies. Semin Oncol. 2012;39:227–240. | |
Flaherty KT, Robert C, Hersey P, et al. Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107–114. | |
Johnson DB, Wallender EK, Cohen DN, et al. Severe cutaneous and neurologic toxicity in melanoma patients during vemurafenib administration following anti-PD-1 therapy. Cancer Immunol Res. 2013;1:373. | |
Yancovitz M, Litterman A, Yoon J, et al. Intra- and inter-tumor heterogeneity of BRAF(V600E) mutations in primary and metastatic melanoma. PLoS One. 2012;7:e29336. | |
Girotti MR, Pedersen M, Sanchez-Laorden B, et al. Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov. 2013;3:158–167. | |
Smalley KS, Lioni M, Dalla Palma M, et al. Increased cyclin D1 expression can mediate BRAF inhibitor resistance in BRAF V600E-mutated melanomas. Mol Cancer Ther. 2008;7:2876–2883. | |
Straussman R, Morikawa T, Shee K, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487:500–504. | |
Prahallad A, Sun C, Huang S, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–103. | |
Corcoran RB, Ebi H, Turke AB, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2:227–235. | |
Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. | |
Villanueva J, Vultur A, Lee JT, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–695. | |
Romano E, Pradervand S, Paillusson A, et al. Identification of multiple mechanisms of resistance to vemurafenib in a patient with BRAFV600E-mutated cutaneous melanoma successfully rechallenged after progression. Clin Cancer Res. 2013;19:5749–5757. | |
Emery CM, Vijayendran KG, Zipser MC, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci U S A. 2009;106:20411–20416. | |
Wagle N, Emery C, Berger MF, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29:3085–3096. | |
Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4:80–93. | |
Van Allen EM, Wagle N, Sucker A, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014;4:94–109. | |
Johannessen CM, Boehm JS, Kim SY, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–972. | |
Montagut C, Sharma SV, Shioda T, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68:4853–4861. | |
Heidorn SJ, Milagre C, Whittaker S, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–221. | |
Paraiso KH, Haarberg HE, Wood E, et al. The HSP90 inhibitor XL888 overcomes BRAF inhibitor resistance mediated through diverse mechanisms. Clin Cancer Res. 2012;18:2502–2514. | |
Whittaker SR, Theurillat JP, Van Allen E, et al. A genome-scale RNA interference screen implicates NF1 loss in resistance to RAF inhibition. Cancer Discov. 2013;3:350–362. | |
Paraiso KH, Xiang Y, Rebecca VW, et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011;71:2750–2760. | |
Nathanson KL, Martin AM, Wubbenhorst B, et al. Tumor genetic analyses of patients with metastatic melanoma treated with the BRAF inhibitor dabrafenib (GSK2118436). Clin Cancer Res. 2013;19:4868–4878. | |
Falchook GS, Trent JC, Heinrich MC, et al. BRAF mutant gastrointestinal stromal tumor: first report of regression with BRAF inhibitor dabrafenib (GSK2118436) and whole exomic sequencing for analysis of acquired resistance. Oncotarget. 2013;4:310–315. | |
Corcoran RB, Rothenberg SM, Hata AN, et al. TORC1 suppression predicts responsiveness to RAF and MEK inhibition in BRAF-mutant melanoma. Sci Transl Med. 2013;5:196ra98. | |
Das Thakur M, Salangsang F, Landman AS, et al. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494:251–255. | |
Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–1703. | |
Faber AC, Coffee EM, Costa C, et al. mTOR inhibition specifically sensitizes colorectal cancers with KRAS or BRAF mutations to BCL-2/BCL-XL inhibition by suppressing MCL-1. Cancer Discov. 2014;4:42–52. | |
Sharma A, Trivedi NR, Zimmerman MA, Tuveson DA, Smith CD, Robertson GP. Mutant V599EB-Raf regulates growth and vascular development of malignant melanoma tumors. Cancer Res. 2005;65:2412–2421. | |
Graells J, Vinyals A, Figueras A, et al. Overproduction of VEGF concomitantly expressed with its receptors promotes growth and survival of melanoma cells through MAPK and PI3K signaling. J Invest Dermatol. 2004;123:1151–1161. | |
Basile KJ, Le K, Hartsough EJ, Aplin AE. Inhibition of mutant BRAF splice variant signaling by next generation, selective RAF inhibitors. Pigment Cell Melanoma Res. Epub January 15, 2014. | |
Finn L, Markovic SN, Joseph RW. Therapy for metastatic melanoma: the past, present, and future. BMC Med. 2012;10:23. | |
Boni A, Cogdill AP, Dang P, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010;70:5213–5219. | |
Wilmott JS, Long GV, Howle JR, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res. 2012;18:1386–1394. | |
Comin-Anduix B, Chodon T, Sazegar H, et al. The oncogenic BRAF kinase inhibitor PLX4032/RG7204 does not affect the viability or function of human lymphocytes across a wide range of concentrations. Clin Cancer Res. 2010;16:6040–6048. | |
Liu C, Peng W, Xu C, et al. BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin Cancer Res. 2013;19:393–403. | |
Callahan MK, Masters G, Pratilas CA, et al. Paradoxical activation of T cells via augmented ERK signaling mediated by a RAF inhibitor. Cancer Immunol Res. 2014;2(1). | |
Melero I, Hervas-Stubbs S, Glennie M, Pardoll DM, Chen L. Immunostimulatory monoclonal antibodies for cancer therapy. Nat Rev Cancer. 2007;7:95–106. | |
Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. | |
Robert C, Thomas L, Bondarenko I, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517–2526. | |
Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368:1365–1366. | |
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–264. | |
Okazaki T, Chikuma S, Iwai Y, Fagarasan S, Honjo T. A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat Immunol. 2013;14:1212–1218. | |
Brahmer JR, Drake CG, Wollner I, et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol. 2010;28:3167–3175. | |
Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455–2465. | |
Hamid O, Robert C, Daud A, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–144. | |
Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. | |
Taube JM, Anders RA, Young GD, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med. 2012;4:127ra37. | |
Joseph RW, Sullivan RJ, Harrell R, et al. Correlation of NRAS mutations with clinical response to high-dose IL-2 in patients with advanced melanoma. J Immunother. 2012;35:66–72. | |
Khalili JS, Liu S, Rodríguez-Cruz TG, et al. Oncogenic BRAF(V600E) promotes stromal cell-mediated immunosuppression via induction of interleukin-1 in melanoma. Clin Cancer Res. 2012;18:5329–5340. | |
Koya RC, Mok S, Otte N, et al. BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer Res. 2012;72:3928–3937. | |
Rosenberg SA, Yannelli JR, Yang JC, et al. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J Natl Cancer Inst. 1994;86:1159–1166. | |
Rosenberg SA, Dudley ME. Cancer regression in patients with metastatic melanoma after the transfer of autologous antitumor lymphocytes. Proc Natl Acad Sci U S A. 2004;101 Suppl 2:14639–14645. | |
Dudley ME, Wunderlich JR, Yang JC, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–2357. | |
Rosenberg SA, Yang JC, Sherry RM, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550–4557. | |
Tiacci E, Trifonov V, Schiavoni G. BRAF mutations in hairy-cell leukemia. N Engl J Med. 2011;364:2305–2315. | |
Samuel J, Macip S, Dyer MJ. Efficacy of vemurafenib in hairy-cell leukemia. N Engl J Med. 2014;370:286–288. | |
Munoz J, Schlette E, Kurzrock R. Rapid response to vemurafenib in a heavily pretreated patient with hairy cell leukemia and a BRAF mutation. J Clin Oncol. 2013;31:e351–e352. | |
Peters S, Michielin O, Zimmermann S. Dramatic response induced by vemurafenib in a BRAF V600E-mutated lung adenocarcinoma. J Clin Oncol. 2013;31:e341–e344. | |
Gautschi O, Pauli C, Strobel K, et al. A patient with BRAF V600E lung adenocarcinoma responding to vemurafenib. J Thorac Oncol. 2012;7:e23–e24. | |
Kim KB, Cabanillas ME, Lazar AJ, et al. Clinical responses to vemurafenib in patients with metastatic papillary thyroid cancer harboring BRAF(V600E) mutation. Thyroid. 2013;23:1277–1283. | |
Cohen Y, Xing M, Mambo E, et al. BRAF mutation in papillary thyroid carcinoma. J Natl Cancer Inst. 2003;95:625–627. | |
Kopetz SJD, Chan E, Hecht JR, et al. PLX4032 in metastatic colorectal cancer patients with mutant BRAF tumors. J Clin Oncol. 2010;28(15) (suppl;abstr 3534). | |
Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.