Back to Journals » Journal of Pain Research » Volume 11
Vaso-occlusive crisis in sickle cell disease: current paradigm on pain management
Authors Uwaezuoke SN, Ayuk AC ![]() , Ndu IK
, Ndu IK ![]() , Eneh CI
, Eneh CI ![]() , Mbanefo NR, Ezenwosu OU
, Mbanefo NR, Ezenwosu OU
Received 28 August 2018
Accepted for publication 12 November 2018
Published 11 December 2018 Volume 2018:11 Pages 3141—3150
DOI https://doi.org/10.2147/JPR.S185582
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Überall
Samuel N Uwaezuoke,1 Adaeze C Ayuk,1 Ikenna K Ndu,2 Chizoma I Eneh,2 Ngozi R Mbanefo,1 Osita U Ezenwosu1
1Department of Pediatrics, College of Medicine, University of Nigeria, Enugu, Nigeria; 2Department of Pediatrics, Enugu State University Teaching Hospital, Enugu, Nigeria
Abstract: This narrative review aims to highlight the current paradigm on pain management in sickle cell vaso-occlusive crisis. It specifically examines the pathophysiologic mechanisms of sickle cell pain as well as the pharmacologic and nonpharmacologic methods of pain management. Recurrent painful episodes constitute the major morbidity in sickle cell disease (SCD). While adolescents and young adults experience mostly acute episodic nociceptive pain, it is now recognized that a significant number of adult patients develop chronic neuropathic and centralized pain. In fact, current evidence points to an age-dependent increase in the frequency of SCD patients with chronic pain.
Management of disease-related pain should be based on its pathophysiologic mechanisms instead of using recommendations from other non-SCD pain syndromes. Pain management in vaso-occlusive crisis is complex and requires multiple interventions such as pharmacologic, nonpharmacologic, and preventive therapeutic interventions. Pharmacologic treatment involves the use of non-opioid and opioid analgesics, and adjuvants – either singly or in combination – depending on the severity of pain. The basic approach is to treat SCD pain symptomatically with escalating doses of non-opioid and opioid analgesics. Given the moderate-to-severe nature of the pain usually experienced in this form of SCD crisis, opioids form the bedrock of pharmacologic treatment. Multimodal analgesia and structured, individualized analgesic regimen appear more effective in achieving better treatment outcomes. Although the current evidence is still limited on the supportive role of cognitive behavioral therapy in pain management, this nonpharmacologic approach is reportedly effective, but needs further exploration as a possible adjunct in analgesia.
Keywords: sickle cell pain, nociceptive pain, non-opioids, opioids, multimodal analgesia, cognitive behavioral therapy
Introduction
Sickle cell disease (SCD) is the most common inherited hemoglobinopathy among the black population worldwide.1–3 The pathologic hallmarks of the disease are vaso-occlusion, chronic hemolysis, and increased erythrocyte adhesiveness to vascular endothelium.4 Ischemic pain from vaso-occlusion is a major clinical feature manifesting with varying degrees of severe, episodic bone pain or abdominal pain. This episodic event is referred to as vaso-occlusive crisis and represents the more frequent type of crises in the disease, with anemic crisis being the other major type comprising hyper-hemolytic, sequestration, and aplastic crises.5
Vaso-occlusive crisis or acute painful crisis constitutes the major morbidity in SCD. Fever appears common even in apparently uncomplicated painful crisis, suggesting that the symptom may be characteristic of the crisis itself and not necessarily a sign of infection.6 In fact, SCD is believed to be a tetrad of pain syndromes, anemia and its sequelae, organ failure (including infection), and comorbid conditions, with pain dominating the clinical picture, and may either be spontaneous or be triggered by the other three components of the tetrad.7 Although the therapeutic approach to this crisis is basically pharmacologic, pain management during each episode has gone beyond the exclusive focus on physical dimension of pain to the incorporation of psychological, social, and behavioral components. A multipronged approach to pain management thus remains crucial to patients’ holistic care. For instance, pain research has shown that cognitive behavioral therapy in SCD is an effective adjunct in the treatment of chronic pain, as it reduces psychological distress and enhances the patient’s confidence and resilience.8 Given the established influence of psychological, sociocultural, and spiritual factors on pain perception, vaso-occlusive crisis may affect the patient in the six fundamental domains of health-related quality of life, comprising physical functioning, psychological functioning, social functioning, role activities, overall life satisfaction, and perception of health status.9
Nevertheless, vaso-occlusive crisis requires immediate and adequate analgesia commensurate with the level of patient-reported pain, according to some recent recommendations.10 Although the pain of vaso-occlusive episodes is nociceptive, neuropathic pain has also been demonstrated in SCD patients aged 7 years and above by using quantitative sensory testing (QST).11 In addition, age-dependent increase in the frequency of SCD patients with chronic pain has been reported, a phenomenon attributed to chronic inflammation, organ damage, and opioid-induced hyperalgesia.12 Thus, despite the use of opioids as the main analgesic therapy, other adjunct pharmacologic agents such as low-dose ketamine (a modulator of opioid tolerance and opioid-induced hyperalgesia) have been recommended for clinical use.13,14 Furthermore, evidence for the efficacy of psychological therapies in SCD-associated pain still appears limited despite renewed interest in this modality of treatment.15
This narrative review aims to appraise the current paradigm on pain management in vaso-occlusive crisis. It specifically discusses the pathophysiologic mechanisms of crisis-related pain, as well as its pharmacologic and nonpharmacologic management.
Pathophysiologic mechanisms of pain in SCD
Management of pain in SCD should be predicated on its pathophysiologic mechanisms instead of using recommendations from other non-SCD pain syndromes.16 These mechanisms are, therefore, important in understanding the basis for the current pharmacologic interventions. Generally, pain pathways involve the peripheral and central nervous systems. Pain can be nociceptive – in which specific sensory receptors in the visceral and somatic structures are stimulated with preservation of nerve integrity – or neuropathic – in which it arises either from nerve injury involving the peripheral or central nervous systems or from nerve compression.17 Electrical and chemical signals play key roles in transmitting the information, which results in pain. The chemical signals are mediated by prostaglandin E2 (PGE2) – a product of the cyclooxygenase pathway – whose functions partly include elevation of autonomic neurotransmitters and causation of hyperalgesia.18
Molecular mechanisms and opioid metabolism
Recent evidence points to certain molecular mechanisms by which transmission of painful stimuli is modulated based on their frequency and severity. This modulation is achieved through the α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) and N-methyl-d-aspartate (NMDA) receptors. At the beginning, the AMPA channel modulates the transmission of painful stimuli of mild-to-moderate severity. Subsequently, the NMDA channel is activated (after the AMPA channel has attained its membrane-depolarization limit), and progressively facilitates the transmission of painful stimuli resulting in central sensitization and glial activation.19 Interestingly, the refractoriness of painful crisis to increasing opioid doses in some patients (tolerance and opioid-induced hyperalgesia) is also linked to this activation of NMDA receptors, as ketamine (an NMDA-receptor agonist) represents a possible modulator of these side effects in both experimental animals and clinical settings.20 At pharmacogenomics level, the metabolism of each opioid is patient-specific. The metabolism of morphine, hydromorphone, and oxymorphone is characteristically by glucuronidation, while that of all other opioids requires specific cytochrome P450 (CYP) isoenzymes. The activities of each isoenzyme and the metabolites of each opioid differ among patients depending on their genetic features and concurrent environmental variables such as the intake of other medications that may enhance or block the CYP isoenzyme activity.19
Major types of pain in SCD
Sickle cell pain can present typically as acute recurrent painful crises, chronic pain syndromes, and neuropathic pain (Figure 1). However, chronic pain is now a recognized form of pain, especially in adult SCD patients. Interestingly, it has been reported that older SCD patients with chronic pain have higher levels of mast-cell activation markers (serum tryptase and substance P).12 Following degranulation of mast cells, tryptase is released into plasma and can result in inflammation, anaphylaxis, urticaria, and more importantly, neuropathic pain.21 It binds to protease-activated receptor 2, releasing inflammatory mediators and substance P, which induce neurogenic inflammation.21
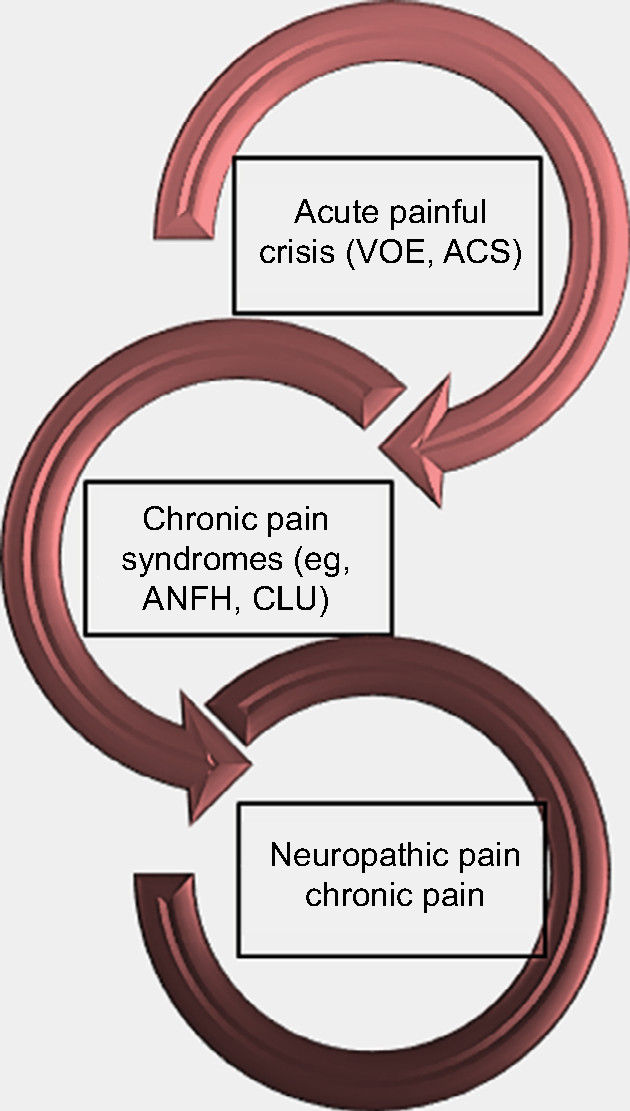 | Figure 1 Forms of presentation of pain in sickle cell disease. Abbreviations: ACS, acute chest syndrome; ANFH, avascular necrosis of the femoral head; CLU, chronic leg ulcers; VOE, vaso-occlusive episodes. |
In SCD patients with acute painful crises, plasma levels of endothelin-1 and PGE2 have been noted to be elevated in comparison with their counterparts with normal hemoglobin genotype.22 As a powerful long-acting mediator of vasoconstriction and inflammation, endothelin-1 may thus play a fundamental role in the ischemia–inflammation cycle, which triggers and maintains the crisis-associated pain.22 In fact, endogenous endothelin peptides play a role in a number of pain-related events such as inflammation-driven pain, skin incision pain, cancer-related pain, and pain of sickle cell crisis, which are all ameliorated by antagonists of endothelin receptors.23 Although crisis-related pain is nociceptive, impairment of autonomic nervous system activity in patients who experienced frequent painful crises in comparison with their crisis-free cohorts has been reported as well.24 This underscores the mediatory action of PGE2 (which increases autonomic neurotransmitters) in the pathophysiology of pain in these patients, and also shows that sickle cell pain can evolve to neuropathic pain. Initially, progressive nociceptive pain from vaso-occlusion and local lesions may evolve over the first two decades of life. Later, in the third and following decades, central neuropathic pain may also develop.25 As previously mentioned, neuropathic pain does occur in pediatric SCD patients.11 Again applying QST, other investigators have corroborated this finding, which was attributed to possible tissue damage following peripheral or central vaso-occlusion in neural tissues.26
In the first form of SCD pain, which is acute painful crisis, there is progression through four phases: prodromal, initial, established, and resolution phases. Every painful crisis is characterized by inflammation and may result in potentially fatal sequelae such as multiorgan failure and sudden death. During crisis, these sequential pathophysiologic events act in synergy: vaso-occlusion, inflammation, and nociception (Figure 2). Thus, acute painful crisis is characterized by nociceptive pain (primarily related to visceral and somatic-tissue injury). The second form of sickle cell pain – the chronic pain syndromes – is associated with avascular necrosis of the femoral head, neuropathic pain, or pain related to persistent low-grade inflammation involving the chest, back, upper and lower extremities, and rarely leg ulcers. But sometimes, some patients who are older and have a history of extremely painful episodes are likely to evolve into a “chronic pain state” as previously reported12; these early signs of disturbed processing of pain may help clinicians who aim to circumvent disease progression.27 Finally, the third form (neuropathic pain) is poorly evaluated in SCD patients but has been demonstrated in transgenic murine models with SCD. Emerging evidence from both human and animal studies, however, suggest that a component of SCD pain may be associated with neuropathic pathways. Studies of molecular and neurobiologic mechanisms that result in and sustain neuropathic pain have opened a new vista on the subject, as the key roles of molecules like protein kinase C and Ca2+/calmodulin-dependent protein kinase II, and their interaction with the NMDA receptors and the transient receptor potential (vanilloid 1 receptor) in the evolution of neuropathic pain have been advanced; these pathways are also implicated in SCD pain.28
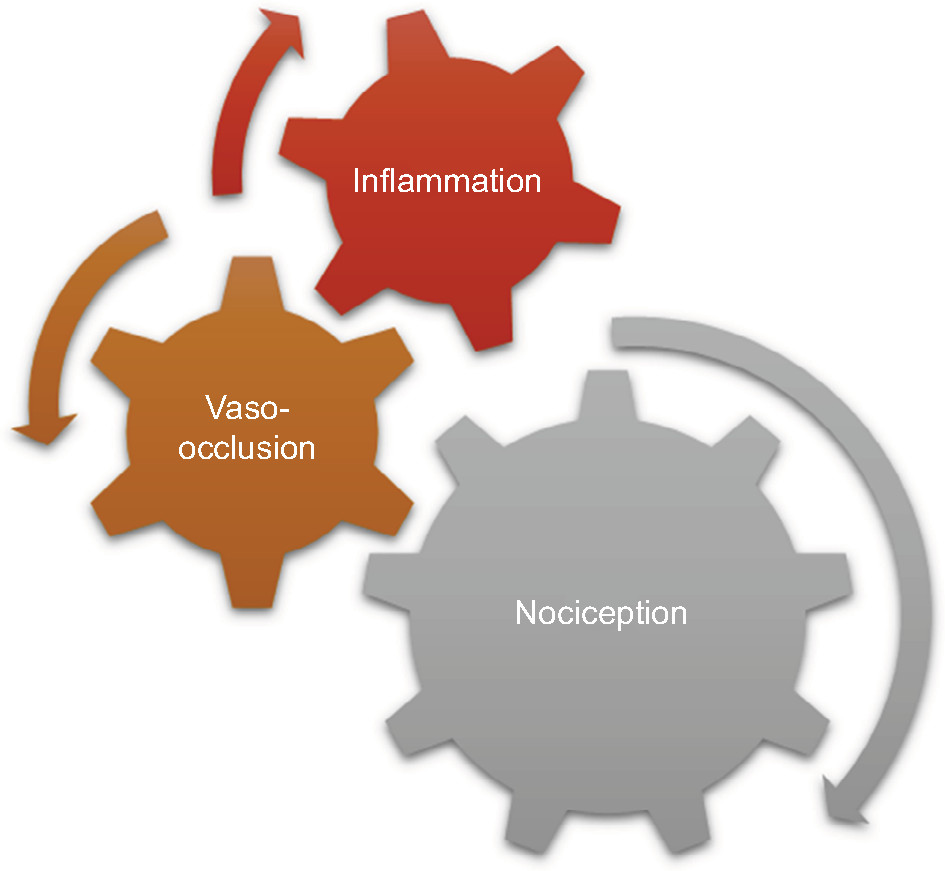 | Figure 2 The pathophysiologic events acting in synergy in acute painful crisis. |
Treatment of pain: the general principles
The treatment of pain essentially involves a sequential approach: pain assessment, pain measurement, and pain management. The major goals of pain assessment are to understand the patient’s experience, the underlying holistic factors, and pathophysiology contributing to the pain, and to obviate the sequelae arising from nontreatment.29 Specifically, pain assessment of the child and adolescent with SCD appears complex and challenging. The limitations of current pain assessment tools have been highlighted in a report, underscoring the need for their improvement.30
The goals of measuring pain include determination of the presence, intensity, duration, and location of pain, as well as treatment efficacy. Pain measurement in children is more complicated than in adults, and involves the use of tools like “FLACC (Face, Legs, Activity, Cry, and Consolability) Scale”, the “Faces Scale”, and the “Visual Analog Scale”.31,32 Finally, pain management comprises pharmacologic and nonpharmacologic methods. The WHO’s three-step analgesic ladder provides the basic concept for the pharmacologic management of pain. The choice of analgesic therapy is determined by the severity, site, and type of pain. In mild pain, step 1 analgesics (non-opioid ± adjuvants) are recommended; in moderate pain, step 2 analgesics (weak opioid ± non-opioid ± adjuvants) should be used; while in severe pain, step 3 analgesics (strong opioid ± non-opioid ± adjuvants) are recommended.33 Non-opioids include paracetamol and nonsteroidal anti-inflammatory drugs (NSAID); weak opioids consist of codeine and tramadol, while morphine and fentanyl are examples of strong opioids. Adjuvants are used depending on the type of pain, and comprise the following: antidepressants such as amitriptyline (for neuropathic pain); anticonvulsants such as carbamazepine, sodium valproate, and gabapentin (for neuropathic pain); antispasmodics like hyoscine butylbromide (for abdominal or renal colic); anxiolytics/muscle relaxants like diazepam (for anxiety-related pain); and corticosteroids such as dexamethasone and prednisolone (for bone pain, neuropathic pain, raised intracranial pressure-induced headache, and edema/inflammation-related pain). Nonpharmacologic methods are meant to be complimentary to pharmacologic methods, and include transcutaneous electrical nerve stimulation, warm or cold compression, and acupuncture. Others consist of emotional support, cognitive methods such as cognitive behavioral therapy, spiritual support, distraction methods, deep breathing, and music therapy.34
The pharmacologic options of pain management in vaso-occlusive crisis
Pain management in vaso-occlusive crisis is equally complex and requires multiple interventions such as pharmacologic, nonpharmacologic, and preventive therapeutic interventions.35–37 For instance, extra fluids are routinely administered as adjunct treatment, irrespective of the patient’s hydration status; this measure aims to retard or stop the sickling process, which helps to achieve pain relief.37 Although this practice has long been advocated, it is largely not evidence-based and may result in circulatory overload. However, given that dehydration is one of the triggers of vaso-occlusive crisis, administration of fluids in this instance may be beneficial in managing crisis-related pain. Other investigators have also reported the possible efficacy of vitamin D replacement in SCD chronic pain because severe vitamin D deficiency has been observed in adolescents with this morbidity.38
Pharmacologic treatment involves the use of non-opioid analgesics, opioid analgesics, and adjuvants – either singly or in combination – depending on the severity of pain. The basic approach remains the symptomatic treatment of pain with escalating doses of non-opioid and opioid analgesics.36 However, using non-opioid analgesics during an acute exacerbation of pain may also limit escalation of opioid doses. In addition, modifying the adhesiveness of sickle cells to inflamed endothelium may potentially reduce vaso-occlusive crisis.39 Nevertheless, careful assessment of painful episodes should precede and follow all the available options for managing this type of crisis. The choice, route, dose, and frequency of administration of opioids should depend on the patient’s presentation. To achieve effective pain treatment, opioids can either be administered on a fixed schedule or by patient-controlled analgesia. Given the moderate-to-severe nature of the pain usually experienced in vaso-occlusive crisis, opioids form the bedrock of pharmacologic treatment.35 However, adult patients who receive opioids more frequently for pain exhibit more disruption of their lives, with reduced activity levels and more pessimistic mood.40 Despite the choice of opioids for acute and chronic pain in SCD, concerns about narcotic addiction continue to pose a barrier to more effective analgesia, although evidence points to lack of addiction.41 The step 2 analgesics (weak opioid ± non-opioid ± adjuvants) or step 3 analgesics (strong opioid ± non-opioid ± adjuvants) are, nonetheless, applicable in most cases of SCD pain. In benchmarking pain outcomes in children with SCD, results revealed three areas to focus on to achieve improvement: enhanced pain assessment and documentation using valid pain tools; more aggressive multimodal management for peak vaso-occlusive pain; and better education and support for pain management at home.42 Multimodal analgesia appears to be the prerequisite for effective pain management in SCD.43 One study on home management of sickle cell pain among pediatric patients suggests that pain relief was substantially better for analgesic combinations than for single analgesics, especially for moderate-to-severe pain.44 Furthermore, opioid tolerance may develop from frequent emergency department (ED) visits and may constitute one of the several barriers to effective pain management; addressing these barriers to pain management for patients in crisis in the emergency setting may improve the quality of the administered pain management.45 Several studies have documented the effectiveness of both unimodal and multimodal analgesia with or without adjuvants in crisis-related pains (Table 1).
 | Table 1 Summary of findings on the different analgesic regimens used in SCD painful crisis Notes: aStrong opioid; badjunct treatment; calso known as heroin (more potent than morphine); dopioid with similar pain-relieving potency to morphine. Abbreviations: ACS, acute chest syndrome; ED, emergency department; LOS, length of stay; PCA, patient-controlled analgesia; SCD, sickle cell disease. |
Use of opioids and adjunct medications
Some authors have reported that a protocol of oral morphine administered to SCD patients in acute painful crisis significantly reduced the number of ED consultations, total number of hours spent in the ED, and the proportion of consultations that subsequently terminated in hospital admissions.46 The authors concluded that the opioid regimen was a useful pharmacologic approach for providing consistent pain relief via the oral route to SCD patients with crisis-related pains. The need for individualized treatment protocols has been highlighted in a review that noted that several patients with SCD are still poorly treated because of nonstandardized and difficult nature of pain management in them.47 Because of its NMDA-receptor-agonist activity as previously mentioned, low-dose ketamine is seen as a useful adjunct medication to opioids in patients with refractory painful crisis. In fact, a recent study has documented the role of a low-dose intravenous ketamine–midazolam combination in the management of severe painful crisis.13 In their retrospective analysis of adult SCD patients with severe painful crisis unresponsive to intravenous morphine and other adjuvant analgesics, the authors documented a significant improvement in pain scores (using the numeric rating scale) after adding the ketamine–midazolam regimen.13 This finding suggests that this regimen may be effective in reducing pain and opioid requirements in SCD patients with severe painful crisis.
Other investigators have further explored the use of intranasal diamorphine in pediatric sickle cell patients with painful crisis. When administered in synergy with either intravenous or oral morphine in an emergency-care setting, there was a rapid improvement in pain scores within 2 hours.48 More importantly, these children reported no adverse reactions, but rated intranasal diamorphine highly as an effective and acceptable analgesic. Intranasal diamorphine was, therefore, recommended for rapid analgesia in children with painful crisis.48 In their adult counterparts, a pain-control program modeled on regimens used in treating chronic cancer pain (intravenous and oral controlled-release morphine) was found to have reduced hospital visits, as the number of admissions for sickle cell pain was reduced by 44%, total inpatient days by 57%, length of hospital stay by 23%, and the number of ED visits by 67%.49 This buttresses the importance of instituting a structured and individualized analgesic regimen in order to achieve optimal outcomes. In fact, individualized pain management plans in emergency settings are effective in delivering high-quality management of vaso-occlusive crisis and are characterized by a high level of patient satisfaction and reduced hospitalizations.50
Another group of investigators have explored the efficacy of nalbuphine hydrochloride (an opioid with similar pain-relieving potency as morphine) in pain control among pediatric sickle cell patients.51 Comparisons were made with those placed on morphine using specific outcome measures. Remarkably, it was noted that patients on nalbuphine hydrochloride were less likely to develop acute chest syndrome and had shorter duration of hospitalizations, even though their use of continuous analgesic infusion with patient-controlled analgesia was seen as a confounder.
Low-molecular-weight heparin as an adjunct therapy
Furthermore, a randomized double-blind clinical trial had evaluated the safety and efficacy of tinzaparin (a low-molecular-weight heparin) in the treatment of acute painful vaso-occlusive crisis.52 In the treatment group, patients received once-daily subcutaneous tinzaparin in combination with supportive care, which included opioid analgesia (morphine) for not more than 7 days, while patients in the control group were exposed to a placebo and similar supportive care for the same duration of time. Interestingly, a statistically significant decrease in the number of days with the highest pain score, total duration of painful crisis, and duration of admission were noted for each comparison of tinzaparin vs placebo. Specifically, the decrease in pain severity was observed to be more abrupt for tinzaparin-treated patients, highlighting the fact that tinzaparin, administered at its appropriate therapeutic regimen, reduced both the severity and duration of acute painful crisis.52 This interesting finding may be related to its antithrombotic activity, which probably helps to ameliorate vaso-occlusion. For instance, it is believed that the degree of adherence of the different blood cells to vascular endothelium correlates well with clinical severity of disease.52 Thus, this tendency of thrombus formation underpins the pharmacologic basis for the potential utility of tinzaparin in vaso-occlusive crisis.
Benefits of hydroxyurea
Another treatment paradigm for acute painful crisis has been the use of hydroxyurea. For several years now, it has been observed that the clinical trajectory of sickle cell patients can be ameliorated by this cytotoxic drug. The clinical improvement in some of the patients is thought to be related to its induction of fetal hemoglobin (Hb F) production. However, its possible mechanisms of action have further been explored, and these include reduction of red blood cell adhesion receptor expression in young SCD subjects and in vitro induction of changes in endothelial cells, affecting the degree of adherence of different blood cells, as well as modulating the determinants of intercellular and endothelial adhesion such as soluble vascular adhesion molecule-1 (sVCAM-1) and myeloperoxidase levels.53 Decreasing sVCAM-1 and myeloperoxidase levels suggests a reduction in the erythrocyte–endothelial interactions and in neutrophil activity, respectively; the latter outcome may help to reduce the propagation phase of a vaso-occlusive crisis. Other proven mechanisms of action of hydroxyurea include its reduction of hypercoagulability markers in SCD such as von Willebrand’s factor and factor VIII,54 as well as nitric oxide (NO) depletion, which occurs in disease-associated hemolytic anemia.55 In SCD pathology, hemolytic anemia is associated with decreased bioavailability of NO, which results in the impairment of vasodilation among other effects.56 Although findings from the study of hydroxyurea in SCD indicate its beneficial effects in shortening the duration of crisis-related admissions and reducing the net dose of opioids,57,58 there have been concerns about its safety profile in pediatric sickle cell patients. Nevertheless, there is compelling evidence to support its use in patients as young as 9 months, given its reported ability to reduce the frequency of vaso-occlusive crises and acute chest syndrome with little or no adverse reactions.59 In fact, results from a protocol suggest minimal genotoxicity or carcinogenicity with long-term hydroxyurea exposure.60 A recent review further lends credence to its safety and efficacy in both pediatric and adult patients as there was no reported increase in the incidence of leukemia and teratogenicity.61 In a longitudinal study of a cohort of sickle cell children in the United States, a group of researchers compared the number of vaso-occlusive pain episodes (including acute chest syndrome/pneumonia episodes) and treatment expenditure in those treated with hydroxyurea and those not treated with it, during a period of 2–3 years.62 Remarkably, treatment with hydroxyurea was associated with a significant reduction in vaso-occlusive pain episodes, hospital admissions, and cumulative expenditure for care.62 A multicenter study among adult patients also reported that hydroxyurea usage resulted in significant reduction in daily patient-rated pain intensity, analgesic use, and utilization, findings that are in tandem with previously documented reports about its ameliorating effects on vaso-occlusive crisis.63 Better still, the extent of pain relief in these patients was found to be directly correlated with the size of Hb F therapeutic response to hydroxyurea. More importantly, some authors have advocated that an increased utilization of hydroxyurea by pediatric and adult patients would most likely reduce their emergency-department visits for pain management.64
Role of inhaled nitric oxide
Inhaled nitric oxide may serve as a potential medication in the treatment of vaso-occlusive crisis. For instance, the microvascular responses to vaso-occlusive events are dependent on oxidative stress and endothelial cell dysfunction; reactive oxygen- and nitrogen-induced events contributing to SCD-associated vasculopathy are driven by changes in substrate and cofactor availability for endothelial-cell nitric oxide synthase, highlighting the emerging role of reactive oxygen and nitrogen species in disease pathogenesis.65 Despite the evidence about the efficacy of inhaled nitric oxide in murine SCD models and in few reported human studies, a report indicates that the use of inhaled nitric oxide vs inhaled nitrogen placebo among hospitalized SCD patients with vaso-occlusive crisis did not improve time to resolution of crisis.66 In addition, there were no significant differences in duration of hospitalization, visual analog pain-scale scores, cumulative opioid usage, and rate of acute chest syndrome between the treatment and placebo groups.66
Treatment outcome of a multimodal analgesia
In a recent study of adult sickle cell patients who received two different types of multimodal analgesia, namely ketorolac (NSAID)+ tramadol (weak opioid) regimen as well as ketorolac + tramadol + fentanyl buccal tablet (strong opioid) regimen (during two separate severe episodes of vaso-occlusive crises), the time-weighted sum of pain intensity differences in the first 24 hours of acute pain management was higher with the latter treatment regimen.67 Moreover, the pain-intensity difference, the total pain relief, and the time-weighted sum of anxiety in the first 24 hours were more ameliorated with the ketorolac + tramadol + fentanyl regimen than with the ketorolac + tramadol regimen, suggesting that fentanyl may be a potent analgesic in early management of acute pain under emergency settings.67 Although ketorolac is a first-generation NSAID used for short-term management of moderate-to-severe pain,68 it is obvious that further improvement in pain relief is achieved when used as an adjuvant to opioid medications, as was shown in this study.
Magnesium sulfate as an adjuvant drug
Novel adjuvant drugs have also been shown to be effective in modulating the outcome of vaso-occlusive crisis when used in conjunction with the conventional pain-management protocols. For instance, a group of investigators studied the effect of intravenous magnesium sulfate on the duration of hospitalization for children admitted with vaso-occlusive crisis.69 Two sequential treatment protocols of intravenous magnesium sulfate were added to standard inpatient management comprising intravenous fluids, opioids, and NSAIDs. Interestingly, intravenous magnesium sulfate apparently reduced the duration of hospitalization for these children in crisis.69 Although magnesium sulfate had been found effective in severe asthma attacks70 and in reducing systolic blood pressure in eclampsia of pregnancy,71 its potentially beneficial use in SCD vaso-occlusive crisis may be related to the vasodilatory action in blood pressure reduction.
Strategies to improve pain management
Finally, the modality of analgesic administration is equally important as it can influence treatment outcomes. For example, some authors developed a protocol to determine whether commencement of patient-controlled analgesia in a pediatric ED would reduce the length of time between opioid bolus doses and initiation of patient-controlled analgesia as compared with standard in-patient initiation of patient-controlled analgesia.72 They also compared patient satisfaction and inpatient length of stay for the two groups. Notably, the authors reported that this protocol not only reduced the time of initiating patient-controlled analgesia in the emergency setting but was also associated with patient preference.72 Generally, the use of patient-controlled analgesia has positive outcomes for patients in both pain management and satisfaction.73 It is a method that entails administering parenteral analgesics (especially opioids) through a microprocessor-controlled infusion pump used by patients once pain is felt by them.74 Patient-controlled analgesia is predicated on the patient’s ability to assess the pain intensity and the analgesic potency. At the beginning of pain, the patient activates the dosing system and receives a doctor-programmed analgesic dose, which is followed by activation of a protection system to circumvent overdose.74
Furthermore, a report shows that opioid delivery in nebulized form can be an effective modality in the management of acute chest pain in patients with SCD.75 It has, therefore, been recommended as a preferable alternative in patients with difficult venous access and may more specifically target chest pain.75 Interestingly, it has been observed that the type of health care personnel administering analgesia in sickle cell patients may also affect treatment outcomes. For instance, a single-center retrospective review of pain-management patterns and outcomes in adult patients admitted for vaso-occlusive crisis revealed that patients managed by hospitalists had shorter duration of admission, and did not have a significant increase in readmission rates when compared with those managed by hematologists.76 Similarly, nursing knowledge has been reported as fundamental to the reduction of stigma and in the improvement of pain management in SCD, as nurses play a key role in the introduction of evidence-based practice within the clinical setting.77 A group of researchers also assessed whether a clinical pathway improves the acute management of crisis pain in the pediatric ED. Interesting findings of the study include significant improvements of time interval to first analgesic use from 74 to 42 minutes and to first opioid use from 94 to 46 minutes, and significant increase in the percentage of patients who received ketorolac from 57% to 82%, suggesting that the use of a clinical pathway for vaso-occlusive crisis in this setting can improve important parameters of pain management.78
Nonpharmacologic options of pain management
Recurrent and frequent episodes of vaso-occlusive crisis may be associated with psychosocial problems such as low self-esteem, anxiety, depression, dissatisfaction with body image, poor school performance, social isolation, reduced participation in normal activities of daily living, as well as poor peer and family relationships.79 Psychologic interventions for painful crisis in SCD patients should, therefore, complement current pharmacologic options. However, a systematic review that was conducted to examine if the coping abilities of SCD patients were improved following psychologic interventions showed limited evidence for their efficacy.15 One of these interventions that have received empirical support is the cognitive behavioral therapy. It has been advocated that sickle cell patients need to have more information about their illness as well as better access to psychosocial interventions.80 Cognitive behavioral therapy is a psychosocial intervention that aims to improve mental health.81 It focuses on challenging and changing the patients’ cognitive distortions and behaviors concerning the disease, and on developing personal coping strategies that target solving any current psychosocial problems. Cognitive behavioral therapy can thus serve as an effective adjunct in managing chronic pain in SCD as it may help to boost patients’ resilience or develop coping strategies.8 Despite paucity of evidence-based role for psychologic therapies in SCD pain management, further studies are advocated to establish the efficacy of these nonpharmacologic options.
Conclusion
The pharmacologic options for pain management in sickle cell vaso-occlusive crisis are effective in both pediatric and adult patients. Non-opioids and opioids still form the mainstay of analgesic therapy for acute and chronic pain of the disease. Multimodal analgesia and structured, individualized analgesic regimen appear more effective in achieving better treatment outcomes. Novel analgesics and adjuvant drugs are still emerging and are proving equally effective in pain relief. A disease modifier like hydroxyurea may be the “turning point” for reducing pain-related morbidity in children and adults with SCD. Although current evidence is still limited on the supportive role of cognitive behavioral therapy in pain management, this nonpharmacologic approach is reportedly effective, but needs further exploration as a possible adjunct in providing analgesia.
Acknowledgment
The authors acknowledge the invaluable information obtained from the handbook Beating Pain: A Pocket Guide for Pain Management in Africa,82 during the preparation of the manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Serjeant GR. Sickle-cell disease. Lancet. 1997;350(9079):725–730. | ||
Roberts I, de Montalembert M. Sickle cell disease as a paradigm of immigration hematology: new challenges for hematologists in Europe. Haematologica. 2007;92(7):865–871. | ||
Bonds DR. Three decades of innovation in the management of sickle cell disease: the road to understanding the sickle cell disease clinical phenotype. Blood Rev. 2005;19(2):99–110. | ||
Okpala I. Leukocyte adhesion and the pathophysiology of sickle cell disease. Curr Opin Hematol. 2006;13(1):40–44. | ||
Nkrumah FK, Neequaye JE, Ankra-Badu G. Bone marrow in sickle cell anaemia at time of anaemic crisis. Arch Dis Child. 1984;59(6):561–565. | ||
Serjeant GR, Ceulaer CD, Lethbridge R, Morris J, Singhal A, Thomas PW. The painful crisis of homozygous sickle cell disease: clinical features. Br J Haematol. 1994;87(3):586–591. | ||
Ballas SK. Pain management of sickle cell disease. Hematol Oncol Clin North Am. 2005;19(5):785–802. | ||
Thomas V. Cognitive behavioural therapy in pain management for sickle cell disease. Int J Palliat Nurs. 2000;6(9):434–442. | ||
Berzon R, Hays RD, Shumaker SA. International use, application and performance of health-related quality of life instruments. Qual Life Res. 1993;2(6):367–368. | ||
Yawn BP, John-Sowah J. Management of sickle cell disease: recommendations from the 2014 expert panel report. Am Fam Physician. 2015;92(12):1069–1076. | ||
Brandow AM, Panepinto JA. Clinical interpretation of quantitative sensory testing as a measure of pain sensitivity in patients with sickle cell disease. J Pediatr Hematol Oncol. 2016;38(4):288–293. | ||
Pope M, Albo C, Kidwell KM. Evolution of chronic pain in sickle-cell disease. Blood. 2016;128(22):1297. | ||
Tawfic QA, Faris AS, Kausalya R. The role of a low-dose ketamine-midazolam regimen in the management of severe painful crisis in patients with sickle cell disease. J Pain Symptom Manage. 2014;47(2):334–340. | ||
Neri CM, Pestieau SR, Darbari DS. Low-dose ketamine as a potential adjuvant therapy for painful vaso-occlusive crises in sickle cell disease. Paediatr Anaesth. 2013;23(8):684–689. | ||
Anie KA, Green J, Cochrane Cystic Fibrosis and Genetic Disorders Group. Psychological therapies for sickle cell disease and pain. Cochrane Database Syst Rev. 2015;32(7):CD001916. | ||
Ballas SK, Gupta K, Adams-Graves P. Sickle cell pain: a critical reappraisal. Blood. 2012;120(18):3647–3656. | ||
Watson M, Lucas C, Hoy A, Wells J. Oxford Handbook of Palliative Care. 2nd ed. Oxford, UK: Oxford University Press; 2009. | ||
Rang HP. Pharmacology. 5th ed. Edinburgh: Churchill Livingstone; 2003:234. | ||
Ballas SK. Pathophysiology and principles of management of the many faces of the acute vaso-occlusive crisis in patients with sickle cell disease. Eur J Haematol. 2015;95(2):113–123. | ||
Jennings CA, Bobb BT, Noreika DM, Coyne PJ. Oral ketamine for sickle cell crisis pain refractory to opioids. J Pain Palliat Care Pharmacother. 2013;27(2):150–154. | ||
Kuei N, Patel N, Xu H. Characteristics and potential biomarkers for chronic pain in patients with sickle cell disease. Blood. 2015;126(23):986. | ||
Graido-Gonzalez E, Doherty J, Bergreen E. Plasma endothelin-1, cytokine, and prostaglandin E2 levels in sickle cell disease and acute vaso-occlusive sickle crisis. Blood. 1988;92(7):2551–2555. | ||
Khodorova A, Montmayeur JP, Strichartz G. Endothelin receptors and pain. J Pain. 2009;10(1):4–28. | ||
Nebor D, Bowers A, Hardy-Dessources MD, et al. Frequency of pain crises in sickle cell anemia and its relationship with the sympatho-vagal balance, blood viscosity and inflammation. Haematologica. 2011;96(11):1589–1594. | ||
Smith WR, Scherer M. Sickle-cell pain: advances in epidemiology and etiology. Hematology Am Soc Hematol Educ Program. 2010;2010(1):409–415. | ||
Jacob E, Chan VW, Hodge C, Zeltzer L, Zurakowski D, Sethna NF. Sensory and thermal quantitative testing in children with sickle cell disease. J Pediatr Hematol Oncol. 2015;37(3):185–189. | ||
Hollins M, Stonerock GL, Kisaalita NR, Jones S, Orringer E, Gil KM. Detecting the emergence of chronic pain in sickle cell disease. J Pain Symptom Manage. 2012;43(6):1082–1093. | ||
Wang ZJ, Wilkie DJ, Molokie R. Neurobiological mechanisms of pain in sickle cell disease. Hematology Am Soc Hematol Educ Program. 2010;2010:403–408. | ||
Portenoy RK. Treatment of cancer pain. Lancet. 2011;377(9784):2236–2247. | ||
Schiavenato M, Alvarez O. Pain assessment during a vaso-occlusive crisis in the pediatric and adolescent patient: rethinking practice. J Pediatr Oncol Nurs. 2013;30(5):242–248. | ||
Hicks CL, von Baeyer CL, Spafford PA, van Korlaar I, Goodenough B. The Faces Pain Scale-Revised: toward a common metric in pediatric pain measurement. Pain. 2001;93(2):173–183. | ||
Merkel S, Voepel-Lewis T, Shayevitz JR, Malviya S. The FLACC: A behavioral scale for scoring postoperative pain in young children. Pediatr Nursing. 1997;23(3):293–297. | ||
World Health Organization. Cancer for Pain Relief with a Guide to Opioid Availability. Geneva, Switzerland: World Health Organization; 1996. | ||
Pujol LA, Monti DA. Managing cancer pain with nonpharmacologic and complementary therapies. J Am Osteopath Assoc. 2007;107(12 Suppl 7):ES15–ES21. | ||
Ballas SK. Management of sickle pain. Curr Opin Hematol. 1997;4(2):104–111. | ||
Richard RE. The management of sickle cell pain. Curr Pain Headache Rep. 2009;13(4):295–297. | ||
Okomo U, Meremikwu MM. Fluid replacement therapy for acute episodes of pain in people with sickle cell disease. Cochrane Database Syst Rev. 2012;6(6):CD005406. | ||
Osunkwo I, Tangpricha V, James-Herry A. Profound vitamin D deficiency in adolescents with sickle cell chronic pain. J Investig Med. 2010;58(2):482. | ||
Quinlan J, Carter K. Acute pain management in patients with persistent pain. Curr Opin Support Palliat Care. 2012;6(2):188–193. | ||
Anie KA, Steptoe A. Pain, mood and opioid medication use in sickle cell disease. Hematol J. 2003;4(1):71–73. | ||
Payne R. Pain management in sickle cell anemia. Anesthesiol Clin North America. 1997;15(2):305–318. | ||
Vijenthira A, Stinson J, Friedman J, et al. Benchmarking pain outcomes for children with sickle cell disease hospitalized in a tertiary referral pediatric hospital. Pain Res Manag. 2012;17(4):291–296. | ||
Yaster M, Kost-Byerly S, Maxwell LG. The management of pain in sickle cell disease. Pediatr Clin North Am. 2000;47(3):699–710. | ||
Dampier C, Ely E, Brodecki D, O’Neal P, O’Neal P. Home management of pain in sickle cell disease: a daily diary study in children and adolescents. J Pediatr Hematol Oncol. 2002;24(8):643–647. | ||
Sawyer J. Sickle cell pain management meets technology: Everybody wins. Acute Pain. 2005;7(1):1–3. | ||
Conti C, Tso E, Browne B. Oral morphine protocol for sickle cell crisis pain. Md Med J. 1996;45(1):33–35. | ||
Niscola P, Sorrentino F, Scaramucci L, de Fabritiis P, Cianciulli P. Pain syndromes in sickle cell disease: an update. Pain Med. 2009;10(3):470–480. | ||
Telfer P, Criddle J, Sandell J, Davies F, Morrison I, Challands J. Intranasal diamorphine for acute sickle cell pain. Arch Dis Child. 2009;94(12):979–980. | ||
Brookoff D, Polomano R. Treating sickle cell pain like cancer pain. Ann Intern Med. 1992;116(5):364–368. | ||
Krishnamurti L, Smith-Packard B, Gupta A, Campbell M, Gunawardena S, Saladino R. Impact of individualized pain plan on the emergency management of children with sickle cell disease. Pediatr Blood Cancer. 2014;61(10):1747–1753. | ||
Buchanan ID, Woodward M, Reed GW. Opioid selection during sickle cell pain crisis and its impact on the development of acute chest syndrome. Pediatr Blood Cancer. 2005;45(5):716–724. | ||
Qari MH, Aljaouni SK, Alardawi MS, et al. Reduction of painful vaso-occlusive crisis of sickle cell anaemia by tinzaparin in a double-blind randomized trial. Thromb Haemost. 2007;98(2):392–396. | ||
Saleh AW, Hillen HF, Duits AJ. Levels of endothelial, neutrophil and platelet-specific factors in sickle cell anemia patients during hydroxyurea therapy. Acta Haematol. 1999;102(1):31–37. | ||
Ballas SK, Bauserman RL, McCarthy WF, et al. Hydroxyurea and acute painful crises in sickle cell anemia: effects on hospital length of stay and opioid utilization during hospitalization, outpatient acute care contacts, and at home. J Pain Symptom Manage. 2010;40(6):870–882. | ||
Colella MP, De Paula EV, Conran N, et al. Hydroxyurea is associated with reductions in hypercoagulability markers in sickle cell anemia. J Thromb Haemost. 2012;10(9):1967–1970. | ||
Gladwin MT, Shelhamer JH, Ognibene FP, et al. Nitric oxide donor properties of hydroxyurea in patients with sickle cell disease. Br J Haematol. 2002;116(2):436–444. | ||
Sparkenbaugh E, Pawlinski R. Interplay between coagulation and vascular inflammation in sickle cell disease. Br J Haematol. 2013;162(1):3–14. | ||
Smith WR, Ballas SK, McCarthy WF, et al. The association between hydroxyurea treatment and pain intensity, analgesic use, and utilization in ambulatory sickle cell anemia patients. Pain Med. 2011;12(5):697–705. | ||
Yawn BP, John-Sowah J. Management of sickle cell disease: recommendations from the 2014 expert panel report. Am Fam Physician. 2015;92(12):1069–1076. | ||
McGann PT, Ware RE. Hydroxyurea for sickle cell anemia: what have we learned and what questions still remain? Curr Opin Hematol. 2011;18(3):158–165. | ||
Cannas G, Poutrel S, Thomas X. Hydroxycarbamine: from an old drug used in malignant hemopathies to a current standard in sickle cell disease. Mediterr J Hematol Infect Dis. 2017;9(1):e2017015. | ||
Stallworth JR, Jerrell JM, Tripathi A. Cost-effectiveness of hydroxyurea in reducing the frequency of pain episodes and hospitalization in pediatric sickle cell disease. Am J Hematol. 2010;85(10):795–797. | ||
Smith WR, Ballas SK, Mccarthy WF, et al. The association between hydroxyurea treatment and pain intensity, analgesic use, and utilization in ambulatory sickle cell anemia patients. Pain Med. 2011;12(5):697–705. | ||
Inoue S, Khan I, Mushtaq R, et al. Pain management trend of vaso-occulsive crisis (VOC) at a community hospital emergency department (ED) for patients with sickle cell disease. Ann Hematol. 2016;95(2):221–225. | ||
Wood KC, Granger DN. Sickle cell disease: role of reactive oxygen and nitrogen metabolites. Clin Exp Pharmacol Physiol. 2007;34(9):926–932. | ||
Gladwin MT, Kato GJ, Weiner D, et al. Nitric oxide for inhalation in the acute treatment of sickle cell pain crisis: a randomized controlled trial. JAMA. 2011;305(9):893–902. | ||
de Franceschi L, Mura P, Schweiger V, et al. Fentanyl buccal tablet: a new breakthrough pain medication in early management of severe vaso-occlusive crisis in sickle cell disease. Pain Pract. 2016;16(6):680–687. | ||
Mallinson TE. A review of ketorolac as a prehospital analgesic. J Paramed Pract. 2017;9(12):522–526. | ||
Brousseau DC, Scott JP, Hillery CA, Panepinto JA. The effect of magnesium on length of stay for pediatric sickle cell pain crisis. Acad Emerg Med. 2004;11(9):968–972. | ||
Powell C, Kolamunnage-Dona R, Lowe J, et al. Magnesium sulphate in acute severe asthma in children (MAGNETIC): a randomised, placebo-controlled trial. Lancet Respir Med. 2013;1(4):301–308. | ||
Dudley L, Henderson-Smart DJ, Walker GJ, Chou D. Magnesium sulphate versus diazepam for eclampsia. Cochrane Database Syst Rev. 2010;12:CD000127. | ||
Melzer-Lange MD, Walsh-Kelly CM, Lea G, Hillery CA, Scott JP. Patient-controlled analgesia for sickle cell pain crisis in a pediatric emergency department. Pediatr Emerg Care. 2004;20(1):2–4. | ||
Myers M, Eckes EJ. A novel approach to pain management in persons with sickle cell disease. Medsurg Nurs. 2012;21(5):293–298. | ||
Zubrzycki M, Liebold A, Skrabal C, et al. Assessment and pathophysiology of pain in cardiac surgery. J Pain Res. 2018;11:1599–1611. | ||
Ballas SK, Viscusi ER, Epstein KR. Management of acute chest wall sickle cell pain with nebulized morphine. Am J Hematol. 2004;76(2):190–191. | ||
Shah N, Rollins M, Landi D, Shah R, Bae J, de Castro LM. Differences in pain management between hematologists and hospitalists caring for patients with sickle cell disease hospitalized for vasoocclusive crisis. Clin J Pain. 2014;30(3):266–268. | ||
Wilson BH, Nelson J. Sickle cell disease pain management in adolescents: a literature review. Pain Manag Nurs. 2015;16(2):146–151. | ||
Ender KL, Krajewski JA, Babineau J, et al. Use of a clinical pathway to improve the acute management of vaso-occlusive crisis pain in pediatric sickle cell disease. Pediatr Blood Cancer. 2014;61(4):693–696. | ||
Jacob E. The pain experience of patients with sickle cell anemia. Pain Manag Nurs. 2001;2(3):74–83. | ||
Edwards LY, Edwards CL. Psychosocial treatments in pain management of sickle cell disease. J Natl Med Assoc. 2010;102(11):1084–1094. | ||
Field TA, Beeson ET, Jones LK. The New ABCs: a practitioner’s guide to neuroscience-informed cognitive-behavior therapy. J Ment Health Couns. 2015;37(3):206–220. | ||
African Palliative Care Association. Beating pain: a Pocket-guide for Pain Management in Africa’. APCA: Kampala, Uganda; 2010. Available from: http://apps.who.int/medicinedocs/documents/s19114en/s19114en.pdf. Accessed December 5, 2018. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.