")
Back to Journals » International Journal of General Medicine » Volume 14
Variant of TSHR is Not a Frequent Cause of Congenital Hypothyroidism in Chinese Han Patients
Authors Xue P, Yang Y, Yun Q, Cui Y, Yu B , Long W
Received 31 May 2021
Accepted for publication 16 July 2021
Published 3 August 2021 Volume 2021:14 Pages 4135—4143
DOI https://doi.org/10.2147/IJGM.S322726
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Peng Xue,1 Yuqi Yang,2 Qi Yun,1 Yue Cui,1 Bin Yu,2 Wei Long2
1Department of Pediatrics, Changzhou Children’s Hospital, Changzhou, People’s Republic of China; 2Department of Medical Genetics, Affiliated Changzhou Women and Children’s Hospital, Nanjing Medical University, Changzhou, People’s Republic of China
Correspondence: Wei Long
Department of Medical Genetics, Affiliated Changzhou Women and Children’s Hospital, Nanjing Medical University, No. 16, Dingxiang Road, Changzhou, 213000, People’s Republic of China
Fax +86 519 8858 1350
Email [email protected]
Purpose: To screen variants of the thyroid stimulating hormone receptor (TSHR) gene among congenital hypothyroidism (CH) patients.
Patients and Methods: We conducted a genetic screening of the TSHR gene in a cohort of 125 Chinese CH patients. Variants were detected by customized targeted next-generation sequencing.
Results: A total of 11 TSHR missense heterozygous variants were identified in 14 CH patients. Six variants were in the transmembrane domains, four variants were in the leucine-rich repeats and one variant was located in the hinge region of the TSHR protein. p.F525S was the most prevalent variant with an allele frequency of 0.016, followed by p.R450H with an allele frequency of 0.012. The allele frequency of most variants was higher in our cohort than those of other populations.
Conclusion: The prevalence of TSHR variants was 11.2%. Variant p.F525S was the most prevalent variant with an allele frequency of 0.016. The prevalence of TSHR variants was different from other populations.
Keywords: congenital hypothyroidism, thyroid stimulating hormone receptor, variant, prevalence
Introduction
Congenital hypothyroidism (CH) is the most common preventable cause of mental and motor retardation in infants with an incidence of 1:2000 to 1:4000.1 With the development of molecular biotechnology, novel perspectives on the pathogenesis of CH have been reported. To date, numerous studies have reported genetic causes in CH patients, and several lines of evidence support a relevant genetic origin for CH.2–5
According to the causes of the underlying mutated genes, the genetic classification divides CH into two main categories, thyroid dysgenesis and thyroid dyshormonogenesis. The defects of thyroid dysgenesis are classified as agenesis (complete lack of thyroid tissue), ectopy (located in an improper position), hemiagenesis or hypoplasia (severely reduced thyroid size). Thyroid dysgenesis, which accounts for 80–85% of primary CH,6 was reported to result from variants in genes responsible for the development or growth of the thyroid.7 Variants in the paired box gene 8 (PAX8), thyroid transcription factor 1 (TTF1/NKX2-1) gene, thyroid transcription factor 2 (TTF2/FOXE1) gene and NK2 transcription factor related locus 5 (NKX2-5) gene were identified in patients with thyroid dysgenesis. Variants of the thyroid stimulating hormone (TSH) receptor (TSHR) gene have been shown to cause CH due to thyroid hypoplasia8 or TSH resistance.9 Variants in THSR result in a wide range of phenotypes ranging from severe CH to mild euthyroid hyperthyrotropinemia. Mild or severe hypothyroidism, thyroid in situ or apparent athyreosis, and complete or partial TSH resistance were both reported in patients with TSHR mutations.10−12 In many previous studies, TSHR variants were reported mainly in patients with abnormal thyroid morphology because of the poor efficiency of the detection method. However, patients with normal thyroid morphology were not screened for variants in the TSHR gene.12 With the use of next-generation sequencing, variants of TSHR were screened progressively in indiscriminate CH patients. The characteristics of TSHR variant have not been fully established, and little is known about the prevalence of variants among CH patients. More research is needed to clarify the causes and etiology of CH.
In the present study, we performed TSHR gene variant screening in a cohort of nonconsanguineous CH patients, aiming to screen and characterize variants in TSHR.
Materials and Methods
Patients
CH patients included in our study were identified from newborn screening between January 2010 and August 2019. Subclinical CH patients and patients with other congenital diseases were excluded from the present cohort. Informed consent to participate in this study was provided by the participants’ legal guardians, and the study was conducted in accordance with the declaration of Helsinki. A total of 125 non-consanguineous Chinese Han patients were included in our study. The study design and protocol were reviewed and approved by the ethics committee of Changzhou Children’s Hospital and the ethics committee of Changzhou Women and Children’s Hospital affiliated to Nanjing Medical University.
CH Screening and Diagnosis
The flow path of screening and diagnosis of CH was based on the consensus statement of the Chinese Preventive Medicine Association.13 Briefly, CH screening was performed between 72 h and 7 days after birth. Heel blood of neonates was dropped on filter paper, and dried blood spots were punched for the subsequent TSH test. TSH of newborns’ heel blood (hTSH) was tested first by a time-resolved fluorescence assay. Newborns whose hTSH ranged from 9.0 to 20.0 mIU/L were recalled for a second test of hTSH. Newborns whose hTSH was higher than 20.0 mIU/L at the first test or whose hTSH was higher than 9.0 mIU/L at the second test were recalled again for the test of blood TSH and free thyroxine (FT4) to make a definite diagnosis. Serum TSH and FT4 were determined by electrochemiluminescence assay. The diagnosis of CH was based on elevated TSH levels and decreased FT4 levels. Thyroid morphology was determined using ultrasound scanning.
TSHR Variant Test
The TSHR targeted panel was designed based on the Illumina Sequencing Assay Designer, including entire coding regions and exon-intron boundaries of TSHR(chr14:81421965–81610778). Heel blood of patients was collected, and genomic DNA was extracted using the QIAGEN QIAamp DNA Blood Kit according to the manufacturer’s protocol. Oligonucleotide probes were synthesized and pooled into a custom amplicon tube containing all the probes to generate attempted amplicons. Sample-specific indices were then added to each library by PCR using common primers from the TruSeq Amplicon Index Kit. After a normalization procedure enables simple volumetric pooling of libraries, sequencing was performed on the Illumina MiSeq 2000 system.
Variant Analysis
Illumina Amplicon Viewer was used for variant detection, data analysis, and variant annotation. The impact of variants on the function and structure of TSHR proteins was predicted by in silico tools, including SIFT, Polyphen-2 and MutationTaster. However, synonymous variants were not evaluated. Suspected pathogenic variants were also searched in public databases or previously published studies to interpret the pathogenic variants.
Results
Following the acquisition of consent from guardians, 125 newborns with CH (60 males and 65 females) were enrolled in our study. The average birth weight of the enrolled newborns was 3233 g, while the average gestational age was 38+5 weeks. The average level of hTSH in the newborns screening was 70.88 mIU/L, and the average levels of serum TSH and FT4 at diagnosis were 68.28 mIU/L and 3.97 pmol/L, respectively.
A total of 11 TSHR missense heterozygous variants were identified in 14 CH patients. The prevalence of TSHR variants was 11.2% in our unbiased cohort. Among these patients with TSHR variants, we observed the occurrence of thyroid dysgenesis (TD) in 6 patients and goiter in one patient, whereas 6 patients had normal-sized gland-in-situ (GIS), the characteristic of patients are shown in Table 1. Nine of 11 variants were included in the dbSNP (database of SNP) or gnomAD (Genome Aggregation Database, v2.1.1) databases, and two variants, p.S234P and p.C462R, were not included in any variation databases but were reported in previous studies. Of the 14 patients with mutated TSHR, 11 were single missense heterozygous. The remaining three patients harbored multisite heterozygous variants: p.S234P combined with p.R450H, p.C462R combined with p.F525S, and p.G132R combined with p.R450H, respectively.
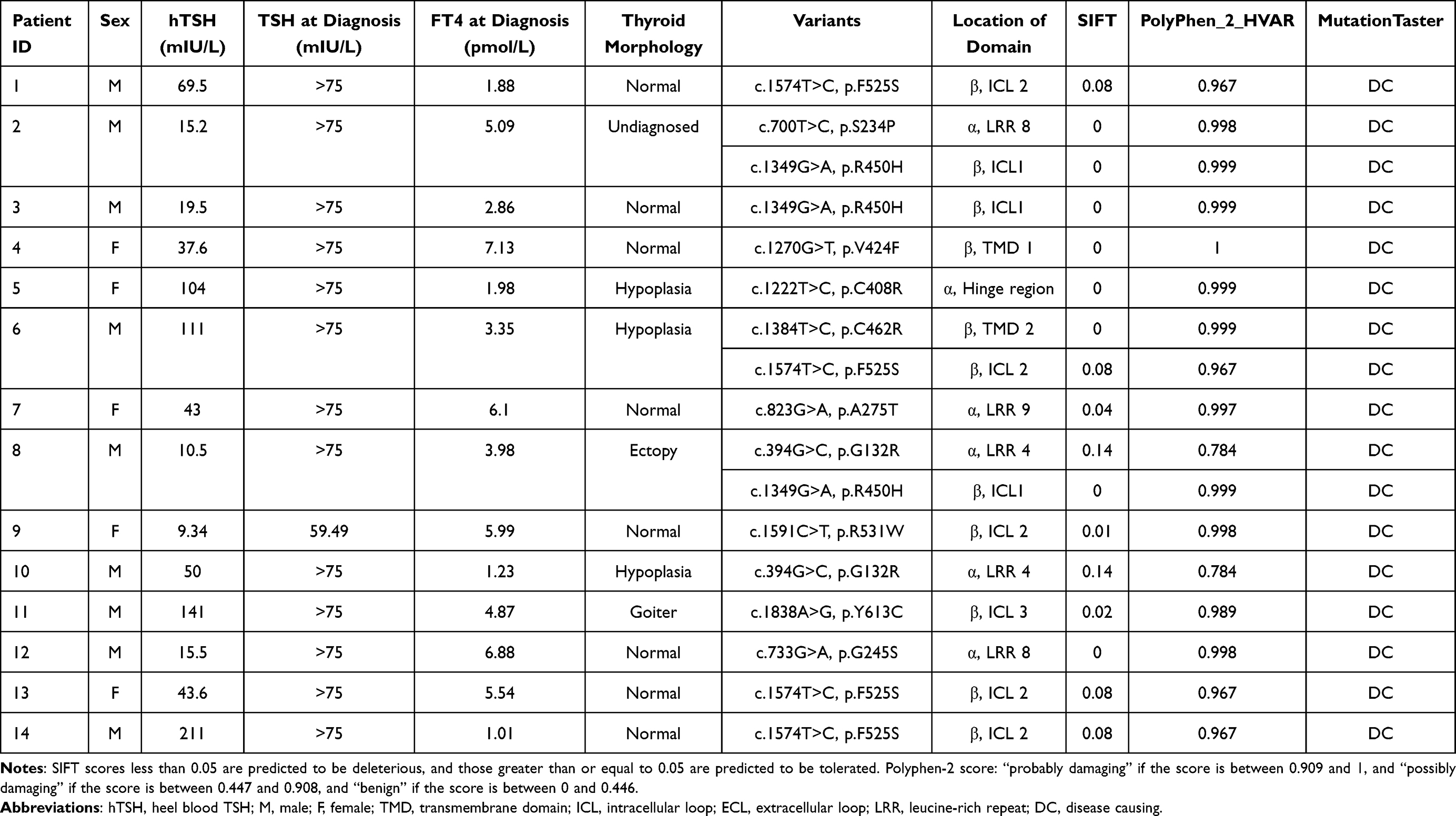 |
Table 1 Characteristics and Damage Prediction of Identified TSHR Variants in 14 Patients |
Amino acids of six variants in the transmembrane domains (TMD) were located in the β subunit of the TSHR protein, including p.V424F, p.R450H, p.C462R, p.F525S, p.R531W and p.Y613C. Four variants were in the leucine-rich repeats (LRRs), including p.G132R, p.S234P, p.G245S and p.A275T. p.C408R was in the hinge region of the TSHR protein. The amino acid locations of the variants are shown in Table 1 and Figure 1.
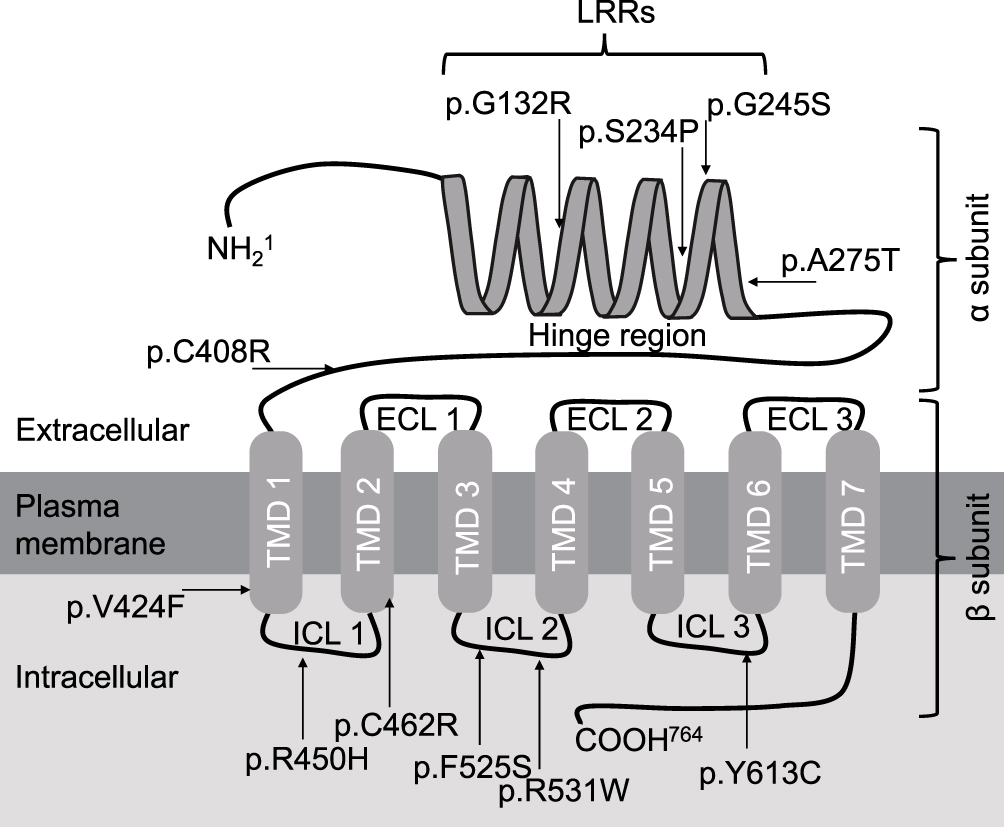 |
Figure 1 Model of TSHR protein structure and localization of variants that identified in the present cohort. Abbreviations: TMD, transmembrane domain; ICL, intracellular loop; ECL, extracellular loop; LRR, leucine-rich repeat. |
The impact of variants on the function and structure of TSHR proteins was predicted by in silico tools. All variants were classified as “disease causing” by MutationTaster, nine of them were classified as “deleterious” by SIFT, and ten of them were classified as “probably damaging” by Polyphen-2 (Table 1). We also analyzed conservation by homology analysis among 11 different species, and most amino acid sequences of variants were located in the highly conserved regions of TSHR (Figure 2).
 |
Figure 2 Multiple sequence alignment of TSHR protein among 11 different species. |
We reviewed the literatures and analyzed the various detection rate of TSHR variants among different populations in 23 studies,10,14–35 the results are shown in Table 2. The prevalence of variants among different populations was also analyzed. In the present cohort, p.F525S was the most prevalent variant with an allele frequency of 0.016, subsequent p.R450H with an allele frequency of 0.012. The allele frequency of all nine variants was higher in our cohort than the allele frequency of the global population. However, the allele frequencies of six variants (p.G132R, p.C408R, p.V424F, p.R450H, p.F525S and p.R531W) were higher than those in East Asia (Table 3).
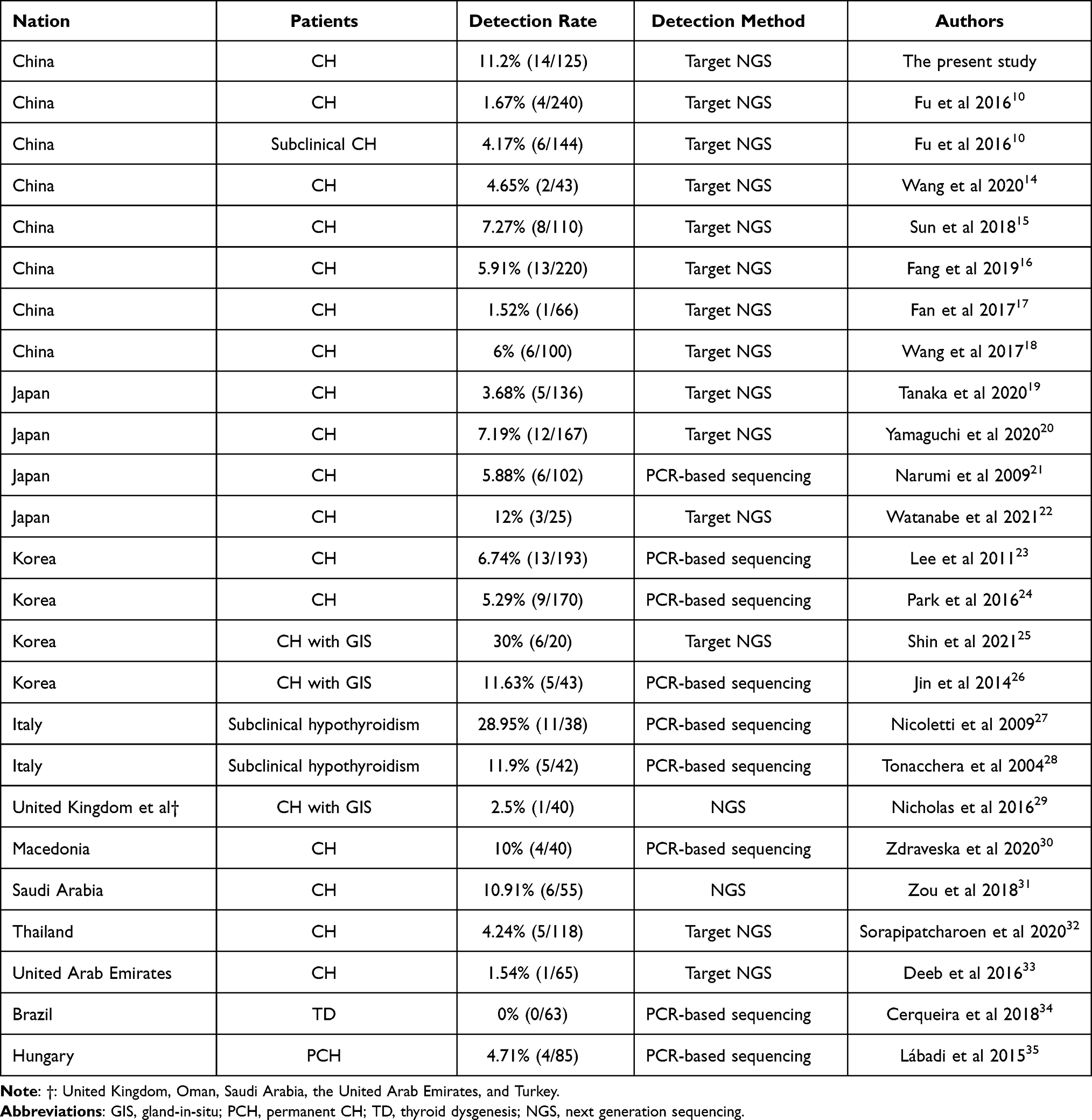 |
Table 2 Detection Rate of TSHR Variants Among Different Populations |
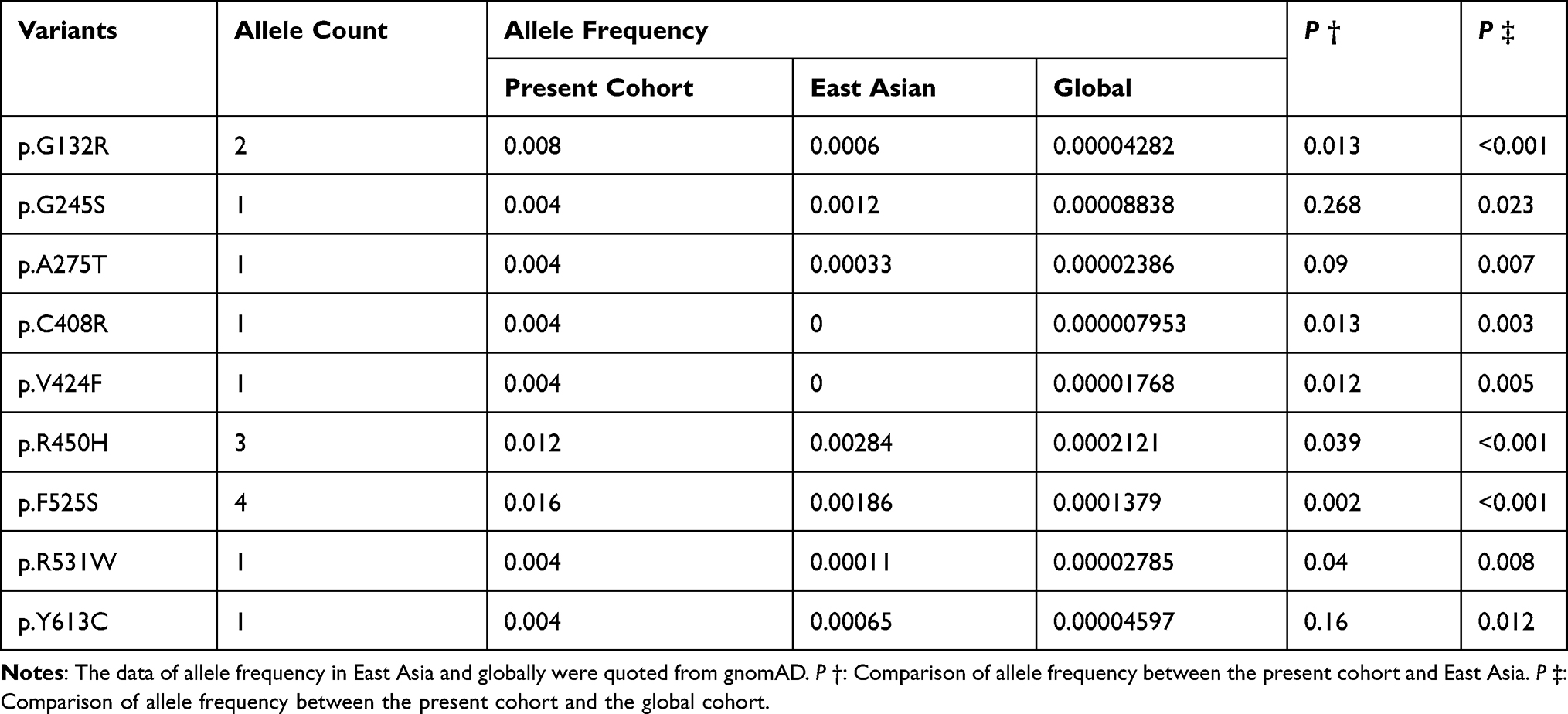 |
Table 3 Allele Frequency of Identified Variants Among Different Populations |
Discussion
In the present study, we studied TSHR variants by targeted sequencing. Targeted sequencing uses oligonucleotide probes designed to target and capture regions of interest, followed by next-generation sequencing. This method enables researchers to analyze genetic variation in specific genomic regions. It also reduces sequencing costs and turnaround time compared to broader approaches such as whole-genome sequencing. In addition, ultradeep sequencing of PCR products allows efficient variant identification and characterization. Eleven missense variants were identified in a cohort of 125 patients with CH, and all the variants were heterozygous.
TSHR, which is located on the surface of thyroid follicular cells, is a member of the G-protein-coupled receptor superfamily. Variants in TSHR play a key role in the main regulatory cAMP and Gq/phospholipase C cascade pathways, which mediate most effects of hormone synthesis in the thyroid gland, including iodide uptake, expression of thyroid genes, biosynthesis of thyroid hormone, TPO activity, thyroid H2O2 generating system, endocytosis, proteolysis and hormone release.36 Loss-of-function variants in the TSHR gene are expected to cause uncompensated TSH resistance.37,38 Most of these variants lead to misfolding of the protein, affecting the signaling pathway.39 The TSHR ectodomain, consisting mainly of 9 LRRs and an N-terminal tail, forms the binding domain for TSH.40,41 The 7 TMDs are joined intracellularly by connecting loops that interact with G proteins when the receptor is activated.42 According to the function of the domains, variants in LRRs will result in decreased binding activity of TSH and subsequently lead to a reduction in cAMP production activities.21,43 However, in vitro experiments confirmed that the TMD variant p.R450H not only leads to a reduction in cAMP production activities but also results in a decreased activity of TSH-binding.21 Pathogenesis may be more complicated even contrary to the in silico analysis.44
A various prevalence of TSHR variants was reported among the different populations. The prevalence of TSHR variants was 11.2% in our cohort, which was higher than that in other Chinese population studies.10,45 In contrast to the variants in DUOX2 or DUOXA2,17 THSR variants are not considered a major cause of CH in the Chinese population.15 p.F525S was the most frequent variant in our cohort. Previous studies identified the variant among populations of Korea23,46,47 and China,48 indicating that p.F525S is a high allele frequency variant among East Asia. p.R450H was also identified as a high-frequency THSR variant in East Asia. It has been reported that p.R450H account for approximately 70% of TSHR variants in Japanese CH patients.21 In Taiwanese CH patients, the frequency of homozygous p.R450H was 1.4% and that of heterozygous p.R450H was 5.6%.49 p.R450H was the second most frequent variant, with a rate of 0.024 (3/125) in our cohort. While the variant rate of p.R450H was 0.0026 (1/384) in a previous study in Guangxi Zhuang Autonomous Region of China, and the rate was lower than that in our cohort. This demonstrated that the variant spectrum of TSHR is different among different populations. Other variants, p.G132R,18 p.G245S,23 p.V424F,50 p.R531W51 and p.Y613C,10 were reported and are related to CH.
In the present cohort, the thyroid morphology was normal in most patients, although ectopy and hypoplasia were observed. It reported that phenotypic variability is a characteristic in patients with TSHR gene mutations, ranging from severe CH to only mild elevations of TSH in the absence of signs and symptoms of hypothyroidism.52 In addition, the correlation between phenotype and genotype remains unclear. Previous studies indicated that heterozygous TSHR mutations have been associated with mildly elevated TSH levels, and biallelic mutations in the TSHR gene result in mild or moderate hypothyroidism with high TSH concentrations, or severe hypothyroidism with a hypoplastic thyroid gland or athyreosis.53 However, studies have also reported that heterozygous mutations were identified in patients with athyreosis,18 and the monoallelic TSHR mutations were recognized as a pathologic role of CH.54 Further studies are required to clarify the molecular etiology and genotype-phenotype correlation in CH with TSHR mutations.
Conclusion
In the present study, we conducted a variant screening of the TSHR gene by customized targeted next-generation sequencing. The prevalence of TSHR variants was 11.2% in our cohort, and the prevalence of TSHR variants was different from other populations. The identification of variants could contribute to the accurate diagnosis and classification of defects, and it also helps to further understand the gene variant spectrum and genetic pathogenesis of CH.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant number 81903400), Changzhou Key Laboratory of High-tech Research (grant number CM20193009), Changzhou Social Development Science and Technology Support Project (grant number CE20205035), Young Talent Development Plan of Changzhou Health Commission (grant number CZQM2020096).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Rastogi MV, LaFranchi SH. Congenital hypothyroidism. Orphanet J Rare Dis. 2010;5(1):17.
2. Park S, Chatterjee V. Genetics of congenital hypothyroidism. J Med Genet. 2005;42(5):379–389.
3. Cherella CE, Wassner AJ. Congenital hypothyroidism: insights into pathogenesis and treatment. Int J Pediatr Endocrinol. 2017;2017(1):11.
4. Persani L, Rurale G, de Filippis T, Galazzi E, Muzza M, Fugazzola L. Genetics and management of congenital hypothyroidism. Best Pract Res Clin Endocrinol Metab. 2018;32(4):387–396.
5. Watanabe D, Yagasaki H, Ishii S, Mitsui Y, Nakane T, Inukai T. A novel c.1391_1428delinsT mutation in TSHR as a cause of familial congenital hypothyroidism with delayed onset. Pediatr Neonatol. 2019;61(1):114–116.
6. Rastogi MV, LaFranchi SH. Congenital hypothyroidism. Orphanet J Rare Dis. 2010;5:17.
7. Abu-Khudir R, Larrivee-Vanier S, Wasserman JD, Deladoey J. Disorders of thyroid morphogenesis. Best Pract Res Clin Endocrinol Metab. 2017;31(2):143–159.
8. Nettore IC, Cacace V, De Fusco C, Colao A, Macchia PE. The molecular causes of thyroid dysgenesis: a systematic review. J Endocrinol Invest. 2013;36(8):654–664.
9. Grasberger H, Refetoff S. Resistance to thyrotropin. Best Pract Res Clin Endocrinol Metab. 2017;31(2):183–194.
10. Fu C, Wang J, Luo S, et al. Next-generation sequencing analysis of TSHR in 384 Chinese subclinical congenital hypothyroidism (CH) and CH patients. Clin Chim Acta. 2016;462:127–132.
11. Schoenmakers N, Chatterjee VK. Thyroid gland: TSHR mutations and subclinical congenital hypothyroidism. Nat Rev Endocrinol. 2015;11(5):258–259.
12. Cassio A, Nicoletti A, Rizzello A, Zazzetta E, Bal M, Baldazzi L. Current loss-of-function mutations in the thyrotropin receptor gene: when to investigate, clinical effects, and treatment. J Clin Res Pediatr Endocrinol. 2013;5(Suppl 1):29–39.
13. Of Endocrinologic SG [Consensus statement on the diagnosis and management of congenital hypothyroidism]. Zhonghua Er Ke Za Zhi. 2011;49(6):421–424.
14. Wang H, Kong X, Pei Y, et al. Mutation spectrum analysis of 29 causative genes in 43 Chinese patients with congenital hypothyroidism. Mol Med Rep. 2020.
15. Sun F, Zhang JX, Yang CY, et al. The genetic characteristics of congenital hypothyroidism in China by comprehensive screening of 21 candidate genes. Eur J Endocrinol. 2018;178(6):623–633.
16. Fang Y, Sun F, Zhang RJ, et al. Mutation screening of the TSHR gene in 220 Chinese patients with congenital hypothyroidism. Clin Chim Acta. 2019;497:147–152.
17. Fan X, Fu C, Shen Y, et al. Next-generation sequencing analysis of twelve known causative genes in congenital hypothyroidism. Clin Chim Acta. 2017;468:76–80.
18. Wang F, Liu C, Jia X, et al. Next-generation sequencing of NKX2.1, FOXE1, PAX8, NKX2.5, and TSHR in 100 Chinese patients with congenital hypothyroidism and athyreosis. Clin Chim Acta. 2017;470:36–41.
19. Tanaka T, Aoyama K, Suzuki A, Saitoh S, Mizuno H. Clinical and genetic investigation of 136 Japanese patients with congenital hypothyroidism. J Pediatr Endocrinol Metab. 2020;33(6):691–701.
20. Yamaguchi T, Nakamura A, Nakayama K, et al. Targeted next-generation sequencing for congenital hypothyroidism with positive neonatal TSH screening. J Clin Endocrinol Metab. 2020;105(8):e2825–e2833.
21. Narumi S, Muroya K, Abe Y, et al. TSHR mutations as a cause of congenital hypothyroidism in Japan: a population-based genetic epidemiology study. J Clin Endocrinol Metab. 2009;94(4):1317–1323.
22. Watanabe D, Yagasaki H, Narusawa H, et al. Screening of frequent variants associated with congenital hypothyroidism: a comparison with next generation sequencing. Endocr J. 2021:EJ21–0353.
23. Lee ST, Lee DH, Kim JY, et al. Molecular screening of the TSH receptor (TSHR) and thyroid peroxidase (TPO) genes in Korean patients with nonsyndromic congenital hypothyroidism. Clin Endocrinol (Oxf). 2011;75(5):715–721.
24. Park KJ, Park HK, Kim YJ, et al. DUOX2 mutations are frequently associated with congenital hypothyroidism in the Korean Population. Ann Lab Med. 2016;36(2):145–153.
25. Shin JH, Kim HY, Kim YM, et al. Genetic evaluation of congenital hypothyroidism with gland in situ using targeted exome sequencing. Ann Clin Lab Sci. 2021;51(1):73–81.
26. Jin HY, Heo SH, Kim YM, et al. High frequency of DUOX2 mutations in transient or permanent congenital hypothyroidism with eutopic thyroid glands. Horm Res Paediatr. 2014;82(4):252–260.
27. Nicoletti A, Bal M, De Marco G, et al. Thyrotropin-stimulating hormone receptor gene analysis in pediatric patients with non-autoimmune subclinical hypothyroidism. J Clin Endocrinol Metab. 2009;94(11):4187–4194.
28. Tonacchera M, Perri A, De Marco G, et al. Low prevalence of thyrotropin receptor mutations in a large series of subjects with sporadic and familial nonautoimmune subclinical hypothyroidism. J Clin Endocrinol Metab. 2004;89(11):5787–5793.
29. Nicholas AK, Serra EG, Cangul H, et al. Comprehensive screening of eight known causative genes in congenital hypothyroidism with gland-in-situ. J Clin Endocrinol Metab. 2016;101(12):4521–4531.
30. Zdraveska N, Kocova M, Nicholas AK, Anastasovska V, Schoenmakers N. Genetics of gland-in-situ or hypoplastic congenital hypothyroidism in macedonia. Front Endocrinol (Lausanne). 2020;11:413.
31. Zou M, Alzahrani AS, Al-Odaib A, et al. Molecular analysis of congenital hypothyroidism in Saudi Arabia: SLC26A7 mutation is a novel defect in thyroid dyshormonogenesis. J Clin Endocrinol Metab. 2018;103(5):1889–1898.
32. Sorapipatcharoen K, Tim-Aroon T, Mahachoklertwattana P, et al. DUOX2 variants are a frequent cause of congenital primary hypothyroidism in Thai patients. Endocr Connect. 2020;9(11):1121–1134.
33. Deeb A, Elkadry I, Attia S, Al Suwaidi H, Obaid L, Schoenmakers NA. Biochemical, radiological, and genetic characterization of congenital hypothyroidism in Abu Dhabi, United Arab Emirates. J Pediatr Endocrinol Metab. 2016;29(7):801–806.
34. Cerqueira TLO, Ramos YR, Strappa GB, et al. Mutation screening in the genes PAX-8, NKX2-5, TSH-R, HES-1 in cohort of 63 Brazilian children with thyroid dysgenesis. Arch Endocrinol Metab. 2018;62(4):466–471.
35. Lábadi Á, Grassi ES, Gellén B, et al. Loss-of-function variants in a Hungarian cohort reveal structural insights on TSH receptor maturation and signaling. J Clin Endocrinol Metab. 2015;100(7):E1039–1045.
36. Vassart G, Dumont JE. The thyrotropin receptor and the regulation of thyrocyte function and growth. Endocr Rev. 1992;13(3):596–611.
37. Sunthornthepvarakul T, Gottschalk ME, Hayashi Y, Refetoff S. Brief report: resistance to thyrotropin caused by mutations in the thyrotropin-receptor gene. N Engl J Med. 1995;332(3):155–160.
38. Van Sande J, Parma J, Tonacchera M, Swillens S, Dumont J, Vassart G. Somatic and germline mutations of the TSH receptor gene in thyroid diseases. J Clin Endocrinol Metab. 1995;80(9):2577–2585.
39. Targovnik HM, Scheps KG, Rivolta CM. Defects in protein folding in congenital hypothyroidism. Mol Cell Endocrinol. 2019;110638.
40. Da Costa CR, Johnstone AP. Production of the thyrotrophin receptor extracellular domain as a glycosylphosphatidylinositol-anchored membrane protein and its interaction with thyrotrophin and autoantibodies. J Biol Chem. 1998;273(19):11874–11880.
41. Davies TF, Ando T, Lin RY, Tomer Y, Latif R. Thyrotropin receptor-associated diseases: from adenomata to Graves disease. J Clin Invest. 2005;115(8):1972–1983.
42. Chazenbalk GD, Nagayama Y, Russo D, Wadsworth HL, Rapoport B. Functional analysis of the cytoplasmic domains of the human thyrotropin receptor by site-directed mutagenesis. J Biol Chem. 1990;265(34):20970–20975.
43. Sanders J, Bolton J, Sanders P, et al. Effects of TSH receptor mutations on binding and biological activity of monoclonal antibodies and TSH. Thyroid. 2006;16(12):1195–1206.
44. Cerqueira TL, Carre A, Chevrier L, et al. Functional characterization of the novel sequence variant p.S304R in the hinge region of TSHR in a congenital hypothyroidism patients and analogy with other formerly known mutations of this gene portion. J Pediatr Endocrinol Metab. 2015;28(7–8):777–784.
45. Ma SG, Fang PH, Hong B, Yu WN. The R450H mutation and D727E polymorphism of the thyrotropin receptor gene in a Chinese child with congenital hypothyroidism. J Pediatr Endocrinol Metab. 2010;23(12):1339–1344.
46. Park K-J, Park H-K, Kim Y-J, et al. DUOX2 mutations are frequently associated with congenital hypothyroidism in the Korean population. Ann Lab Med. 2016;36(2):145–153.
47. Park E, Jung J, Araki O, et al. Concurrent TSHR mutations and DIO2 T92A polymorphism result in abnormal thyroid hormone metabolism. Sci Rep. 2018;8(1):10090.
48. Long W, Lu G, Zhou W, et al. Targeted next-generation sequencing of thirteen causative genes in Chinese patients with congenital hypothyroidism. Endocr J. 2018;65(10):1019–1028.
49. Chang WC, Liao CY, Chen WC, et al. R450H TSH receptor mutation in congenital hypothyroidism in Taiwanese children. Clin Chim Acta. 2012;413(11–12):1004–1007.
50. Li L, Zhang W, Zang Y, Yan S, Kong B, Liu S. Identification of a novel TSHR mutation from a Chinese baby with congenital hypothyroidism due to ectopy. Int J Clin Exp Pathol. 2016;9(1):153–158.
51. Cangul H, Aycan Z, Saglam H, et al. TSHR is the main causative locus in autosomal recessively inherited thyroid dysgenesis. J Pediatr Endocrinol Metab. 2012;25(5–6):419–426.
52. Persani L, Calebiro D, Cordella D, et al. Genetics and phenomics of hypothyroidism due to TSH resistance. Mol Cell Endocrinol. 2010;322(1):72–82.
53. Kostopoulou E, Miliordos K, Spiliotis B. Genetics of primary congenital hypothyroidism-a review. Hormones (Athens). 2021;5:1–2.
54. Abe K, Narumi S, Suwanai AS, et al. Association between monoallelic TSHR mutations and congenital hypothyroidism: a statistical approach. Eur J Endocrinol. 2018;178(2):137–144.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.