Back to Journals » International Journal of Nanomedicine » Volume 14
Variable Effects Of LDL Subclasses Of Cholesterol On Endothelial Nitric Oxide/Peroxynitrite Balance – The Risks And Clinical Implications For Cardiovascular Disease
Authors Hua J ![]() , Malinski T
, Malinski T ![]()
Received 13 August 2019
Accepted for publication 9 October 2019
Published 18 November 2019 Volume 2019:14 Pages 8973—8987
DOI https://doi.org/10.2147/IJN.S223524
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Jiangzhou Hua , Tadeusz Malinski
Nanomedical Research Laboratory, Ohio University, Athens, OH, USA
Correspondence: Tadeusz Malinski
Nanomedical Research Laboratory, Biochemistry Research Facility, Ohio University, 350 West State Street, Athens, OH 45701, USA
Tel/fax +1 740 597 1247
Email [email protected]
Background: Elevated levels of low density lipoprotein (LDL), “bad cholesterol”, is not an accurate indicator of coronary disease. About 75% of patients with heart attacks have cholesterol levels that do not indicate a high risk for a cardiovascular event. LDL is comprised of three subclasses, with particles of different size and density. We used nanomedical systems to elucidate the noxious effects of LDL subclasses on endothelium.
Experimental: Nanosensors were employed to measure the concentrations of nitric oxide (NO) and peroxynitrite (ONOO−) stimulated by LDL subclasses in HUVECs. N-LDL and ox-LDL (subclass A: 1.016–1.019 g/mL, subclass I: 1.024–1.029 g/mL, and subclass B: 1.034–1.053 g/mL) stimulated NO and ONOO− release. The concentrations ratio of (NO)/(ONOO−) was used to evaluate the noxious effects of the subclasses on endothelium.
Results: In HUVECs, the (NO)/(ONOO−) ratio for normal endothelium is about 5, but shifts to 2.7±0.4, 0.5±0.1, and 0.9±0.1 for subclasses A, B, and I, respectively. Ratios below 1.0 indicate an imbalance between NO and ONOO−, affecting endothelial function. LDL of 50% B and 50% I produced the most severe imbalance (0.45±0.04), whereas LDL of 60% A, 20% B, and 20% I had the most favorable balance of 5.66±0.69. Subclass B significantly elevated the adhesion of molecules and monocytes. The noxious effect was significantly higher for ox-LDL than n-LDL.
Conclusion: Subclass B of “bad cholesterol” is the most damaging to endothelial function and can contribute to the development of atherosclerosis. Contrary to the current national guidelines, this study suggests that it’s not the total LDL, rather it is the concentration of subclass B in relation to subclasses A and/or I, that should be used for diagnosis of atherosclerosis and the risk of heart attack. By utilizing specific pharmacological therapy to address the concentration of subclass B, there is a potential to significantly reduce the risk of heart attack and atherosclerosis.
Keywords: low density lipoprotein, nitric oxide, endothelium, peroxynitrite, cell adhesion
Introduction
Low density lipoprotein (LDL) transports fat molecules through the bloodstream. Both native-LDL (n-LDL) and oxidized-LDL (ox-LDL) have been considered as bad cholesterol because of an association with several cardiovascular diseases. Of the large number of patients hospitalized with coronary artery disease, about half are admitted with LDL levels below 100 mg/dL. In addition, 75% of all heart attack patients have LDL levels that give no indication of cardiovascular risk.1 Though they are not homogenous, it has recently been suggested that some of the subclasses of n-LDL and ox-LDL may differently increase a cardiovascular risk.2–4 Clinical studies show that a high concentration of small dense LDL particles correlated positively with cardiovascular events.5 There are three major subclasses of LDL with distinct densities: n-LDL subclass A contains more of the larger and less dense LDL particles (density of 1.025–1.034 g/mL); an intermediate group, n-LDL subclass I has density of 1.034–1.044 g/mL; and finally, n-LDL subclass B, which has more smaller and denser LDL particles (density of 1.044–1.060 g/mL).6–8 In clinical studies, Griffin et al9 found that the concentration of subclass B was high in coronary artery disease patients, and it was associated with a low concentration of high density lipoprotein (HDL) cholesterol, suggesting that it may be used as a risk marker for coronary artery disease. Although it’s possible that subclass B particles carry the same cholesterol content as subclass A particles, subclass B can be considered as a higher risk factor for coronary heart disease (CHD) than subclass A. This is not only because subclass B can accelerate the growth of atheroma and the progression of atherosclerosis, but it also causes much more severe cardiovascular damage.8 The small and dense particles of subclass B may penetrate the membrane of the endothelium much easier, where they are more susceptible to be oxidized than the larger less dense particles of subclass A.4 ox-LDL is known to further increase oxidative stress10 and up-regulates the expression of adhesion molecules as compared to n-LDL,11–13 and finally, accelerates the premature development of atherosclerosis.14,15 Generally, endothelial dysfunction is associated with increased levels of n-LDL and ox-LDL and may trigger many forms of cardiovascular disease, such as atherosclerosis,16,17 peripheral artery disease,18 hypertension,19 and coronary artery disease.14
The heterogeneity of LDL was first found by Lindgren et al20 and then confirmed by other groups.9,21,22 It has been shown that small and dense LDL is strongly associated with increased cardiovascular risk.7,23,24 However, the molecular effect of each of the different subclasses of LDL on endothelium and its dysfunction has not yet been investigated. Thus, the purpose of this study is to elucidate the fundamental molecular mechanism of interactions of different LDL fractions with the endothelium. We utilized a nanomedical approach, employing nanosensors with a diameter of <300 nm, to simultaneously measure, near-real time, the concentration of nitric oxide (NO) and peroxynitrite (ONOO−) released from a single endothelial cell exposed to each of the LDL subclasses (A, B, and I). The ratio of cytoprotective NO concentration to cytotoxic ONOO− concentration (NO)/(ONOO−) was used as a marker of oxidative stress and the dysfunction of endothelial nitric oxide synthase (eNOS). We revealed that all n-LDL and ox-LDL subclasses unfavorably shift the balance of the (NO)/(ONOO−) ratio, imposing noxious effects such as: elevated oxidative stress, a shortage of cytoprotective NO, and the up-regulation of adhesion molecules in the endothelium. However, one particular subclass (B) dramatically shifted (NO)/(ONOO−) balance to a very low level, causing significant damage to endothelial function. It seems that subclass B is an extremely bad component of LDL – “the bad cholesterol”. Therefore, the relatively high content/level of subclass B LDL in total cholesterol can be a major determinant of potential risk for the cardiovascular system. We suggest that, with further analysis, this relative content of subclasses could be used as the best marker in assessing the risk of LDL in the cardiovasculature.
Methods
Cell Culture
Human umbilical vein endothelial cells (HUVECs) and human monocytoid cells (THP-1) were purchased from American Type Culture Collection. HUVECs were cultured as a monolayer in MCDB-131 Complete Medium (VEC tech) at 37°C in a humidified atmosphere enriched with 5% CO2. The THP-1 cells were cultured in RPMI-1640 medium containing 10% FBS (ATCC), 100 U/mL penicillin, and 100 U/mL streptomycin at 37°C in a humidified atmosphere enriched with 5% CO2.
N-LDL Isolation, Oxidation, And Analysis
Normal human plasma (Innovative Research) was mixed with 12% of OptiPrep density gradient medium (Sigma) at the ratio (v:v) of 1 to 1. The mixture was loaded to the centrifuge tube and placed in a NVT65 rotor (Beckman Coulter), then centrifuged at 60,000 rpm (342,000 g) for 4 hours at 16°C in an Optima L-90K ultracentrifuge (Beckman Coulter) set at slow acceleration and slow deceleration. Samples were fractionated within 1 hour after centrifugation. Fractions were collected from each gradient by downward displacement using a syringe tip piercing the bottom of the tube and pumped out. The fractions were collected into Eppendorf tubes with 1.5 mL per fraction. The density and concentration of each fraction were measured by using a refractometer (ATAGO) and cholesterol assay kit (Invitrogen), respectively. Oxidized-LDL (ox-LDL) was prepared according to a previously reported method.25 CuSO4 was added to native LDL (n-LDL) with a final concentration of 10 µmol/L. Oxidation was carried out at room temperature over 24 hours until oxidation was complete. The ox-LDL was then placed in ultra-centrifuge tubes (Sigma-Aldrich, Ultra-4, MWCO 30 kDa) and centrifuged at 3,000 rpm for 20 minutes to remove CuSO4. All of the LDL samples were filtered and stored at 4°C.
Nanosensors For Measurement Of NO And ONOO−
Concurrent measurements of NO and ONOO− were performed with electrochemical nanosensors (diameter: 200–300 nm). The designs of nanosensors are based on previously well-developed chemically modified carbon-fiber technology.26–30 Each of those sensors was made by depositing a sensing material on the tip of the carbon fiber. A conductive film of polymeric nickel (II) tetrakis (3-methoxy-4hydroxy-phenyl) porphyrinic was used for the NO sensor and a polymeric film of Mn (III)–paracyclophanyl-porphyrin was used for the ONOO− sensor. NO and ONOO− release from its basal level were measured by using amperometry with time (detection limit of 1 nmol/L and resolution time <50 ms). Each sensor was calibrated by using linear calibration curves from 50 nmol/L to 1,000 nmol/L and/or standard addition methods before and after measurements with aliquots of NO or ONOO− standard solutions, respectively.
Determination Of N-LDL/ox-LDL Stimulated NO And ONOO− Production In Endothelial Cells
Endothelial cells were seeded to 24 well plates and cultured in complete medium until a confluent monolayer formed. Then the study was carried out as follows: Endothelial cells were stimulated with direct injection of n-LDL with different densities (subclass A: 1.016–1.019 g/mL, subclass I: 1.024–1.029 g/mL, and subclass B: 1.034–1.053 g/mL) and different concentration (50, 100, 250, 500, 750, 1000 µg/mL), and the release of NO/ONOO− was measured by placing NO/ONOO− nanosensors at a close proximity (5±2µm) from the surface of endothelial cells and measuring the electrical current generated by these NO/ONOO− nanosensors. Endothelial cells were also stimulated with direct injection of n-LDL with different combinations of subclasses A, B, and I LDL (800 µg/mL) as follows: (1) 60% A, 20% B, and 20% I; (2) 20% A, 60% B, and 20% I; (3) 20% A, 20% B, and 60% I; (4) 50% A and 50% B; (5) 50% A and 50% I; (6) 50% B and 50% I; (7) 33% A, 38% B, and 29% I (simulation of original constituent from general human plasma), and the release of NO/ONOO− was also measured with nanosensors. Endothelial cells were pre-treated with superoxide dismutase covalently linked to polyethylene glycol (PEG-SOD, 400 U/mL, Sigma), L-arginine (300 µmol/L, Sigma), a precursor of endothelial nitric oxide synthase (eNOS) cofactor tetrahydrobiopterin (sepiapterin, 200 µmol/L, Sigma), L-NG-arginine methyl ester (L-NAME, 100 µmol/L, Sigma) as an inhibitor of eNOS, and a selective nicotinamide adenine dinucleotide phosphate (NADPH) oxidase inhibitor (VAS2870, 10 µmol/L, Sigma) in endothelial basal medium (EBM) at 37ºC for 30 minutes. A control group was incubated in EBM only. After incubation, endothelial cells were stimulated with direct injection of subclasses A, B, and I (800 µg/mL), and the release of NO/ONOO− was measured with nanosensors. Also, endothelial cells were stimulated with direct injection of n-LDL/ox-LDL (800 µg/mL) and the release of NO/ONOO− was measured in the same way as described above. In separate experiments, the maximal NO and ONOO− concentrations which could be produced by HUVECs was measured after stimulation with 1.0 µmol/L calcium ionophore (A23187, Sigma).
Measurement Of Monocyte Adhesion To HUVECs
Endothelial cells were seeded in 96 well plates with complete medium until a confluent monolayer formed. THP-1 cells were cultured in RPMI medium 1640 containing 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37°C in a humidified atmosphere of 5% CO2. THP-1 cells were pre-labeled with 2ʹ,7ʹ-bis-(2-carboxyethyl)-5-(and-6)-carboxyl-fluorescein acetoxymethyl ester (BCECF-AM) (Molecular Probes, Life Technology) for quantitative adhesion assay. Fluorescence labeling of THP-1 cells was done by incubating cells (5×106 cells/mL) with 5 μmol/L BCECF-AM in RPMI-1640 medium for 30 minutes at 37°C and 5% CO2. After incubation, cells were washed three times with PBS to remove excess dye. Cells were then re-suspended in EBM at a density of 106 cells/mL. Then the study was carried out as follows: confluent HUVECs were incubated with constant concentration (400 μg/mL) of n-LDL at 37°C for 5 hours. Then cells were washed with PBS twice to remove LDL. Fluorescently labeled THP-1 cells were added to the surface of confluent endothelial monolayer as 105/well and co-incubated at different time intervals (from 10 to 60 minutes); and then the co-cultured cells were washed twice with PBS in order to eliminate the non-adherent cells. The fluorescence intensity of each well was measured by using a fluorescence multi-well plate reader set at excitation and emission wavelengths of 485 and 528 nmol/L, respectively. In addition, confluent endothelial cells were incubated with LDL at a final concentration of 50, 100, 200, or 400 μg/mL at 37°C for 5 hours. Then cells were washed with PBS twice to remove LDL. Fluorescently labeled THP-1 cells were added to the surface of confluent endothelial monolayer as 105/well and co-incubated at 37°C for 1 hour, then the co-cultured cells were washed twice with PBS in order to eliminate the non-adherent cells. The fluorescence intensity of each well was measured in the same way as described above. In a set of separate experiments, confluent endothelial cells were incubated with n-LDL or ox-LDL (400 μg/mL) at 37°C for 5 hours. After that, cells were washed with PBS twice to remove LDL and fluorescently labeled THP-1 cells were added to the confluent endothelial monolayer (105/well) and co-incubated at 37°C for 1 hour. The co-cultured cells were then washed twice with PBS in order to eliminate the non-adherent cells. The fluorescence intensity of each well was measured in the same way as described above.
Measurements Of Adhesion Molecules
Cell ELISA was used to measure the expression of adhesion molecules. Endothelial cells were seeded in 96 well plates with complete medium until a confluent monolayer formed. Then cells were incubated with n-LDL/ox-LDL (400 μg/mL) at 37°C for 5 hours. Control is EBM with 3% iodixanol. After stimulation with LDL, endothelial cells were washed with phosphate buffered saline (PBS) twice and fixed with 4% formaldehyde solution for 20 minutes at room temperature. After fixation, HUVECs were washed twice with phosphate buffered saline with tween 20 (PBST) and incubated with blocking buffer (4% BSA in PBST) for 1 hour at room temperature. The plate was washed three times with PBST and primary monoclonal antibody against ICAM-1 and VCAM-1 (Santa Cruz) diluted in PBST (0.5 µg/mL for ICAM-1, and 2 µg/mL for VCAM-1) were added to the cells at 4°C overnight. The plate was washed three times with PBST and incubated with horseradish peroxidase-conjugated goat anti-mouse IgG (Santa Cruz) diluted at 1:1,000 in PBST for 1 hour at room temperature. The cells were washed again three times, and 4,4ʹ-Bi-2,6-xylidine;4,4ʹ-Diamino-3,3ʹ,5,5ʹ-tetramethylbiphenyl (TMB) solution was added to each well and incubated at room temperature. After then, 2M citric acid solution was added to each well. The absorbance was measured at 450 nm wavelength in a microplate reader. Each experiment was performed in six duplicates and repeated at least three times.
Statistical Analysis
All data are expressed as means±SD. Unpaired Student’s t-test was used to measure statistical differences. A P-value less than 0.01 was considered statistically significant. Data analysis was performed using Excel version 2013 (Microsoft, Seattle, WA). Asterisks in the figure are represented as follows: *P<0.01.
Results
N-LDL Subclasses Stimulated NO And ONOO− Release In Endothelial Cells
To determine the distinct effect of different subclasses of n-LDL on NO and ONOO− release from HUVECs, we measured the real-time production of NO and ONOO− from endothelial cells with nanosensors. A rapid release of NO/ONOO− was detected within 0.1 seconds after injection of n-LDL, and the maximal concentrations of NO and ONOO− were reached within 1.0 seconds (Figures 1A and B). The maximal concentrations of NO and ONOO− released from endothelial cells varied significantly among LDL subclasses A, B, and I. Subclass A contains particles with larger size and is less dense than subclass B; and produced the highest concentration of NO. Subclass B consists mainly of n-LDL particles with smaller size and higher density and stimulated the lowest concentration of NO. NO release stimulated by the injection of subclass I is between subclasses A and B. In contrast to NO production, subclass B stimulated the highest level of ONOO−, while subclass A produced the lowest level of ONOO− (Figure 2A). The highest (NO)/(ONOO−) ratio was observed for A, while the lowest was observed for subclass B. The ratio of NO concentration, (NO) to the concentration of peroxynitrite, (ONOO−) was used to reflect the balance/imbalance between cytoprotective NO and cytotoxic ONOO−. A high (NO)/(ONOO−) ratio indicates a high level of bioavailable, diffusible NO and/or a low level of cytotoxic ONOO− (Figure 2B). The highest (NO)/(ONOO−) ratio was observed for A, while the lowest was observed for subclass B. Maximal (NO) and (ONOO−) is dose-dependent (Figures 3A and B). The ratio of (NO)/(ONOO−) maintained a low and narrow range of about 0.29–0.52 for subclass B (Figure 3C). For subclasses I and A, ratios increased from 0.50 to 0.93 (plateau) for subclass I and from about 1.37 to 2.66 for subclass A. Apparently at very low LDL concentrations (about 50 μg/mL) the balance of (NO)/(ONOO−) is highly unfavorable for the B and I subclasses. A plateau for (NO)/(ONOO−) is established at about 100–150 μg/mL. However, the level for this plateau is favorable (higher than one) only for subclass A. For both subclasses I and B, the (NO)/(ONOO−) plateau level is below one. Also, at very low concentrations (around 50 µg/mL and lower) the (NO)/(ONOO−) ratio is particularly low (below 0.50) and is an indicator of severe endothelial function. Meaning at both high and low LDL, B, and I are damaging and contribute to the dysfunction of endothelium.
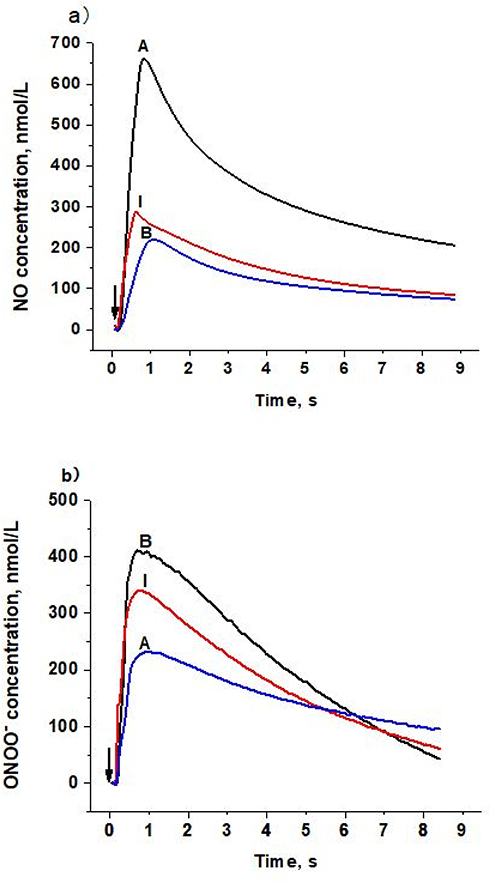 |
Figure 1 Amperograms (current calibrated as concentration vs time) of NO and ONOO− release stimulated by LDL with different patterns on the surface of endothelial cells. a) NO release from endothelial cells stimulated by LDL (Patterns A, B, and I, 1,000 µg/mL). b) ONOO− release from endothelial cells stimulated by LDL (Patterns A, B, and I, 1,000 µg/mL). Arrows indicate LDL injection. |
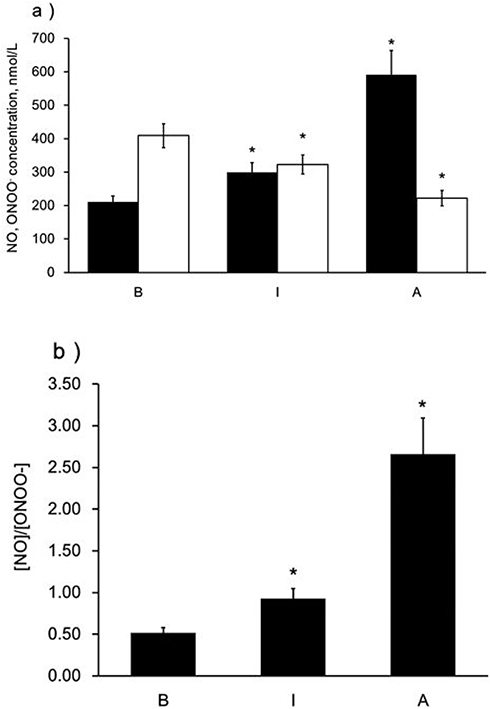 |
Figure 2 Maximal NO and ONOO− release from the surface of endothelial cells stimulated by LDL with different patterns. a) Maximal NO and ONOO− release from endothelial cells stimulated by LDL (Patterns A, B, and I, 1,000 µg/mL), solid bar indicates NO and open bar indicates ONOO−. b) A ratio of maximal NO to ONOO−. Data are expressed as mean±SD. Significance was determined using Student’s t-test. *P<0.01 vs B. |
 |
Figure 3 Dose-dependent NO and ONOO− release from the surface of endothelial cells stimulated by LDL. a) Production of NO stimulated by LDL with different patterns (A, B, and I) and different concentrations (from 50 µg/mL to 1,000 µg/mL). b) Production of ONOO− stimulated by LDL with different patterns (A, B, and I) and different concentrations (from 50 µg/mL to 1,000 µg/mL). c) The ratio of NO to ONOO−. Black triangle, white circle, and black dot indicate LDL injection of pattern A, B, and I, respectively. |
Effects Of The Combinations Of Different N-LDL Subclasses On NO And ONOO− Release
The experiment was carried out by stimulating cells with a different combination of n-LDL subclass, seven combinations were studied: (1) 60% A, 20% B, and 20% I; (2) 20% A, 60% B, and 20% I; (3) 20% A, 20% B, and 60% I; (4) 50% A and 50% B; (5) 50% A and 50% I; (6) 50% B and 50% I; and (7) 33% A, 38% B, and 29%. Our data showed that group 1(60% A, 20% B, and 20% I) produced the lowest concentrations of ONOO− (77±8 nmol/L) and highest concentration of NO (436±28 nmol/L), while group 6 (50% B and 50% I) generated the highest level of ONOO− (369±25 nmol/L) and lowest level of NO (166±10 nmol/L). The ratio of (NO) to (ONOO−) concentration was about 5.5 for (1), and about 0.45 for (6) (Figure 4).
 |
Figure 4 NO and ONOO− release stimulated by LDL mixture with different combinations. a) NO and ONOO− production stimulated by LDL mixture (800 µg/mL). Solid bar indicates NO and open bar indicates ONOO−. b) Ratio of NO to ONOO−. Data are expressed as mean±SD. |
Effect Of Modulation In eNOS Pathway On N-LDL Stimulates NO And ONOO− Release
In order to elucidate kinetics and dynamics of LDL stimulated of NO and ONOO− production, we used different modulators of eNOS. All reagents except L-NAME (eNOS inhibitor) increased NO production after injection of subclasses A, B, or I (Figure 5A). ONOO− production diminished in the presence of PEG-SOD, L-arginine, sepiapterin, L-NAME, and VAS2870 in all standard subclasses (Figure 5B). With subclass A, the (NO)/(ONOO−) ratio remained above one for all treatments. The ratio for subclass I was greater than one for treatments with L-arginine, sepiapterin, and VAS2870, but lower than one for PEG-SOD and L-NAME treatment groups. Subclass B revealed a ratio that was below one for all of treatments except for L-arginine (Figure 5C).
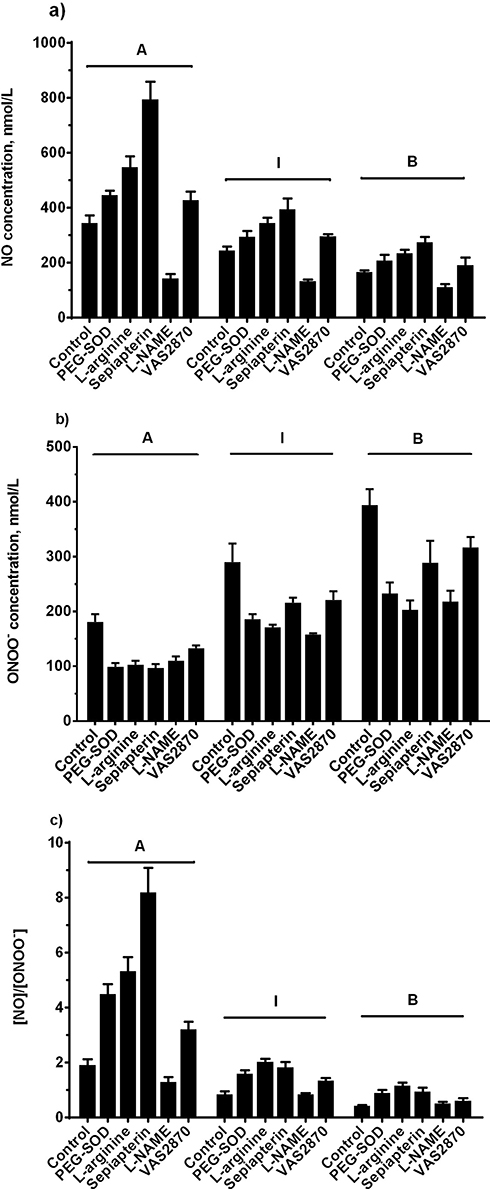 |
Figure 5 NO and ONOO− release from endothelial cells stimulated by LDL after incubation with different treatments. Endothelial cells were incubated with control EBM, PEG-SOD (400 U/mL), L-arginine (300 µM), sepiapterin (200 µM), L-NAME (100 µM), and VAS2870 (10 µM) at 37ºC for 30 minutes. a) NO production stimulated by LDL (Patterns A, B, and I, 800 µg/mL). b) ONOO− production stimulated by LDL (Patterns A, B, and I, 800 µg/mL). c) Ratio of NO to ONOO−. Data are expressed as mean±SD. |
Differences Between N-LDL And Ox-LDL Stimulated NO And ONOO− Release In Endothelial Cells
We also investigated and compared the effects of different subclasses of n-LDL with those of oxidized LDL (ox-LDL). Ox-LDL stimulated NO release at a much lower level than n-LDL, 267±11 vs 418±16 nmol/L for subclass A, 95±7 vs 152±10 nmol/L for subclass I, and 65±3 vs 85±3 nmol/L for subclass B (Figure 6A). However, ox-LDL stimulated much higher levels of ONOO− production than n-LDL, 145±6 vs 86±5 nmol/L for subclass A, 284±18 vs 208±13 nmol/L for subclass I, and 432±18 vs 347±20 nmol/L for subclass B (Figure 6B). Therefore, the ratio of (NO) to (ONOO−) is 1.84 vs 4.86, 0.33 vs 0.73, and 0.15 vs 0.24 (ox-LDL vs n-LDL) for subclasses A, B, and I, respectively (Figure 6C). Consequently, the deleterious effects of ox-LDL on endothelial dysfunction is much more substantial than that observed for n-LDL, especially in subclasses I and B.
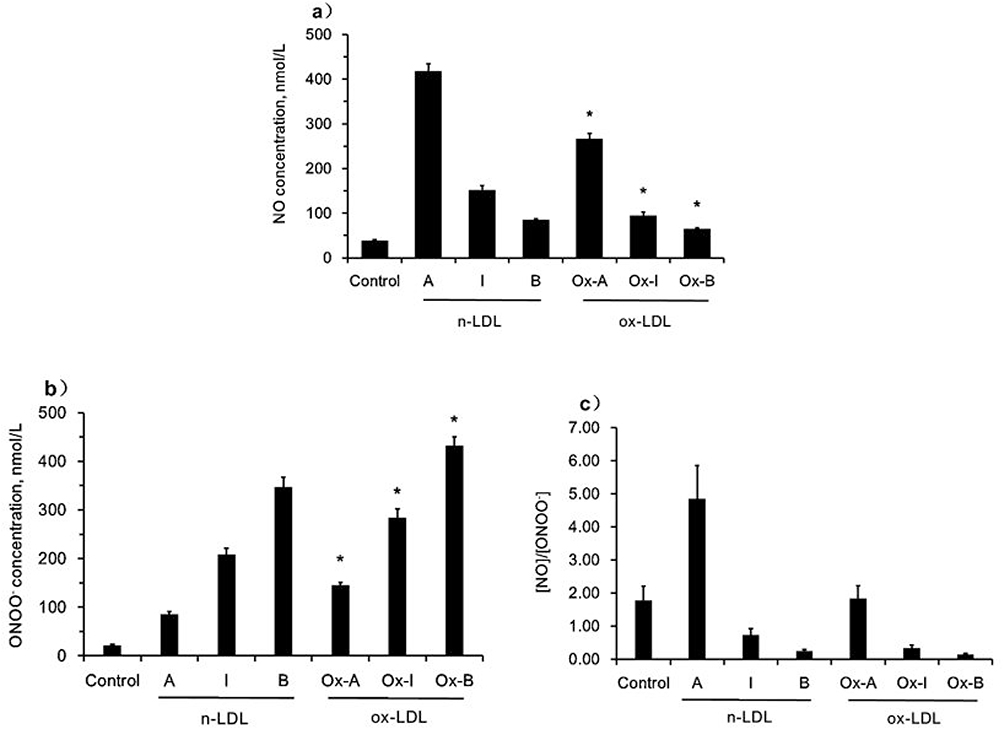 |
Figure 6 NO and ONOO− release stimulated by ox-LDL/n-LDL. a) NO production stimulated by ox-LDL/n-LDL (800 µg/mL). b) ONOO− production stimulated by ox-LDL/n-LDL (800 µg/mL). c) Ratio of NO to ONOO−. Data are expressed as mean±SD. Significance was determined using Student’s t-test. *P<0.01 vs n-LDL. |
N-LDL-Stimulated Cell Adhesion In Endothelial Cells
To investigate the effect of different subclasses of n-LDL and the expression of ICAM-1 and VCAM-1 on monocytes adhesion to endothelial cells, fluorescently pre-labeled THP-1 cells were used. Data showed that the adhesion of monocytes to endothelial cells increased significantly. For subclasses I and A monocytes adhesion was similar, but less extensive than that observed for subclass B (Figure 7). This adhesion increased with time, and, after 60 minutes, the differences of THP-1 cells adhesion among treatments with all subclasses (A, B, and I) was most significant. The result suggests that THP-1 cells adhesion is dose-dependent, 400 µg/mL LDL treatment stimulated the maximal monocytes adhesion, while 50 µg/mL LDL treatment stimulated the minimal adhesion. At the same concentration level, n-LDL of different subclasses stimulated monocytes adhesion differently, subclass B stimulated cell adhesion was the highest, while subclass A was the lowest MFI (Figure 7). Ox-LDL subclasses A, I, and B stimulated more monocytes adhesion than the n-LDL subclasses A, I, and B, by 21%, 63%, and 73%, respectively. Among different subclasses, ox-LDL showed similar results with n-LDL. Subclass B triggered the highest level of cell adhesion (4-fold increase from control), while subclass A showed the lowest level of cell adhesion (2-fold increase from control), and in between, subclass I stimulated cell adhesion about 3-fold from control (Figure 8). These patterns of change were similar for n-LDL and ox-LDL.
 |
Figure 7 Monocyte adhesion and cell adhesion molecule expression stimulated by n-LDL. a) Monocyte adhesion stimulated by subclass A, B, and I of n-LDL (400 µg/mL) measured at different incubation time (from 10 to 60 minutes). b) Dose-dependent monocyte adhesion stimulated by patterns A, B, and I of n-LDL (50, 100, 200, 400 µg/mL). Data are expressed as mean±SD. MFI indicates mean fluorescence intensity. c) Effect of LDL with different patterns on the expression of ICAM-1 and VCAM-1. Endothelial cells were incubated with LDL of patterns A, B, and I (400 µg/mL) at 37ºC for 5 hours. After incubation, the cells were washed with DPBS and fixed with 4% formaldehyde solution. ICAM-1 and VCAM-1 expression were determined by cell ELISA. Data are expressed as mean±SD. Solid bar indicates ICAM-1, open bar indicates VCAM-1. OD indicates optical density. Significance was determined using Student’s t-test. *P<0.01 vs control. |
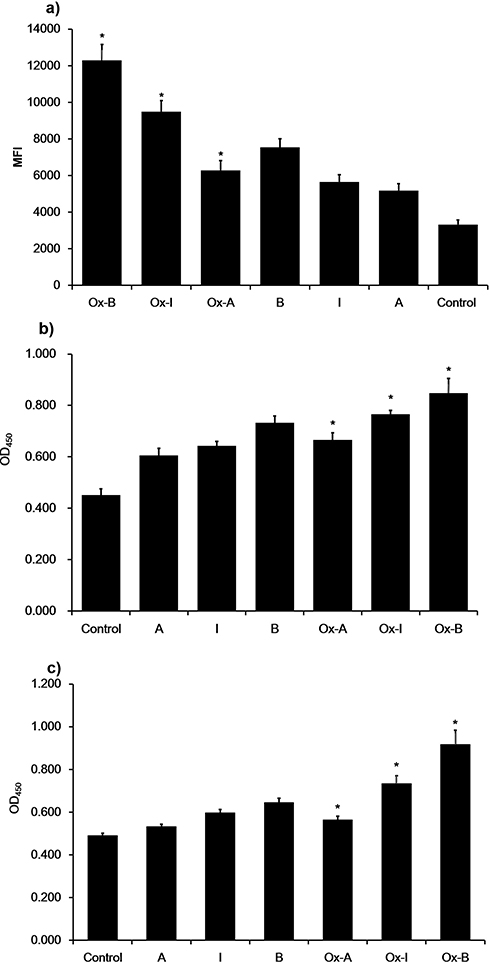 |
Figure 8 Monocyte adhesion and cell adhesion molecular expression stimulated by n-LDL or ox-LDL. a) Monocyte adhesion stimulated by n-LDL or ox-LDL of patterns A, B, and I (400 µg/mL). Data are expressed as mean±SD. MFI indicates mean fluorescence intensity. b) Effect of n-LDL/ox-LDL with different patterns on the expression of ICAM-1. Endothelial cells were incubated with n-LDL/ox-LDL of patterns A, B, and I (400 µg/mL) at 37ºC for 5 hours. After incubation, the cells were washed with DPBS and fixed with 4% formaldehyde solution. ICAM-1 and VCAM-1 expression were determined by cell ELISA. c) Effect of n-LDL or ox-LDL with different subclasses on the expression of VCAM-1. Data are expressed as mean±SD. OD450 indicates optical density. Significance was determined using Student’s t-test. *P<0.01 vs n-LDL. |
To determine the effect of LDL with different subclasses on ICAM-1 and VCAM-1 expression, endothelial cells were incubated with basal medium containing 400 µg/mL n-LDL or ox-LDL (subclasses A, B, and I) for 5 hours and the expression of ICAM-1 and VCAM-1 was measured by cell ELISA. Compared with control, ICAM-1 expression increased by 55±11%, 74±14%, and 90±7% versus control for LDL of subclasses A, I, and B, respectively. Ox-LDL subclasses increased ICAM-1 expression nearly 20% more than that observed in n-LDL (Figure 8). VCAM-1 expression stimulated by ox-LDL increased by about 20%, 50%, and 90% of control for subclasses A, I, and B. Also, VCAM-1 expression was 6%, 23%, and 42% higher than that observed n-LDL subclasses, respectively.
Discussion
This study has shown, for the first time, a distinct difference between three major subclasses of n-LDL and ox-LDL in the process of their interactions with the endothelium. The nanomedical approach employed here shows, in situ, that after colliding with the membrane of endothelial cells, subclasses A, B, and I of LDL can trigger calcium flux, NO production and the subsequent production of ONOO− by uncoupled eNOS in the membrane of endothelial cells. . The maximal concentrations of protective NO and cytotoxic ONOO− released differs significantly between each of the subclasses and the relative content of each subclass. The deleterious effect on endothelium is about 20% higher for subclasses of ox-LDL than n-LDL.
We have successfully used the ratio of (NO)/(ONOO−) for the precise measurement of eNOS uncoupling, endothelial dysfunction, and nitroxidative stress levels (ONOO− vs protective NO).31 The nanoanalytical system that was applied here allows the simultaneous measurements, in nmol/L, of both NO and ONOO− at near real time (several microseconds) in the femtoliter volume (about 10−15L) at a constant distance of 5±2 µm from the surface of endothelial cells. The simultaneous measurement of NO and ONOO− allowed us to use the ratio of the (NO)/(ONOO−) as the marker of a balance/imbalance between those two molecules, dysfunction of endothelium, and level of high oxidative stress. The production of NO by eNOS is always accompanied by the generation of ONOO−, which is the product of the reaction between superoxide (O2−) and NO.31 This rapid, diffusion controlled reaction between NO and O2− in the biological system prevents the overproduction of NO and/or O2−. In normal, functional endothelium, the maximal concentration of ONOO− is about 4–6 times lower than the maximal concentration of NO. The half-life of ONOO− in the biological milieu is less that 1 second, much shorter than the t1/2 of NO (about 3–4 seconds). At low concentrations, ONOO− molecules cannot diffuse any significant distance and are rapidly converted to nontoxic NO3−. At a high (NO)/(ONOO−) ratio in a normal endothelium, NO signaling, as well as anti-adhesion properties are efficient, and the potential for cellular damage by ONOO− (nitroxidative stress) is negligible. However, at high concentrations, the oxidative effect of ONOO− can be severe, especially at low levels of cytoprotective NO. At these high concentrations, ONOO− can be protonated and can diffuse, collide with biological molecules, and isomerize to initiate a cascade of highly oxidative species – causing oxidative damage to cells, enzymes, and DNA leading to endothelial dysfunction, as well as hindered NO signaling and diminished anti-adhesive properties.32,33
It has been well established by our studies and others31,34,35 that at extensive NO production, the dimeric form of eNOS became uncoupled and can produce concomitantly NO and O2−, becoming an efficient generator of ONOO−. With an increase in eNOS uncoupling and endothelial dysfunction, the efficiency of NO signaling decreases exponentially, while the nitroxidative damage to the endothelium increases significantly. We found that with a (NO)/(ONOO−) ratio below 1, the peroxynitrite starts to control the redox environment and the protective anti-adhesion role and signaling of NO are greatly diminished.
Using this particular criterion, we find a very significant distinction between the molecular effects of subclasses A, B, and I of LDL with their interaction with the endothelium. Subclass A produces a very mild effect in its interaction with the endothelium, efficiently stimulating small concentrations of ONOO−. This indicates that the coupling status of eNOS and the efficiency in generating superoxide is minimal. The (NO)/(ONOO−) balance is shifted slightly to 2.66±0.43 for subclass A. Therefore, the toxic effect of subclass A of both n-LDL and ox-LDL is negligible. There is a further small decrease in the (NO)/(ONOO−) ratio, to 0.93±0.12 for subclass I. But we still have a relatively well-functioning endothelium, because (NO)/(ONOO−) is about one.
Contrary to subclasses A and I, subclass B has a very significant effect on the (NO)/(ONOO−) ratio and imbalance between these two molecules. First, subclass B decreases the (NO)/(ONOO−) ratio well below 1.0. Under these conditions, the ONOO− becomes a dominating factor in controlling a cytotoxic redox environment in and around endothelial cells. Also, low production of bioavailable NO hinders the rate of diffusion, decreasing the distance and speed of NO signaling. The diminished role of NO in the dysfunction of eNOS is accompanied by an exponential increase in nitroxidative stress imposed by ONOO−.
The net result of action of LDL subclass B on the endothelium is the decrease of NO stimulated vasorelaxation as well as an increase in the adhesion of LDL, platelets, and leukocytes to the endothelium – all promoted by ONOO−. We have demonstrated contrasting levels of the (NO)/(ONOO−) ratio between subclasses A, B, and I of LDL cholesterol. Among these three major subclasses, we found that B imposes the most severe effect on eNOS and endothelial function. The net effect of a mixture of LDL subclasses A, B, and I has on the endothelium is additive, and depends on the content of each of the subclasses. The present study also shows the subclass-specific differences in both n-LDL and ox-LDL. Subclass B of ox-LDL produced the lowest ratio of (NO)/(ONOO−) – about 20% lower than that observed with n-LDL and the lowest among all of the subclasses studied here. As expected, in comparison to n-LDL, the effect of decreasing the (NO)/(ONOO−) was observed for all ox-LDL subclasses.
Clinical studies suggest that elevated levels of LDL do not correlate well with increases in cardiovascular risk, however, as seen in the data presented here, evidence has accumulated that a small and dense subclass of LDL may be the key factor that is in strong association with the development of atherosclerosis and other CVD events.2–5,36–38 Understanding the role of distinct subclasses of LDL in triggering endothelial dysfunction as well as the progress of atherosclerosis may facilitate improving accuracy of diagnosis for the evaluation of CVD risk rate. Our main purpose was to investigate the role of LDL with different subclasses in induction of NO and ONOO− imbalance in endothelial cells. In our study, the densities of subclasses A, B, and I were a little lower than those in a previous report.9 This is due to the ios-osmotic iodixanol gradients we used for separation of LDL subclasses. Protein molecules of LDL will keep water inside to maintain their native hydrate status, rather than loss of water in highly hyper-osmotic salt gradients, which results in increasing density. The present study shows subclass-specific differences in both n-LDL and ox-LDL stimulated NO and ONOO− release from endothelial cells. Our data suggest that subclass B can stimulate endothelial cells to produce the highest level of ONOO− and the lowest level of NO, resulting in an imbalance of the (NO)/(ONOO−) ratio, which can lead to severe endothelial dysfunction and aggravate the oxidative stress in endothelial cells. On the contrary, subclass A stimulated the lowest level of ONOO− and the highest level of NO, keeping the ratio of (NO)/(ONOO−) in balance, thus maintaining the functionality of endothelium.
To investigate the effect of LDL with different constituents on stimulating NO and ONOO− release from endothelial cells, we tried different mixture combinations of LDL with subclasses A, B, and I. The most severe combination of LDL consisted of 50% B and 50% I. We may draw the conclusion that the constituents of LDL mixture containing all of three subclasses is related with the release of NO and ONOO−. A high percentage of subclass B stimulated a high level of ONOO− and a low level of NO; while a high percentage of subclass A stimulated more NO production than ONOO−. Therefore, analyzing the constituents of LDL with different subclasses may provide a parameter-based model for an early medical diagnosis of estimating the risk of cardiovascular disease.
Reagents which can modulate the L-arginine/NO pathway, such as PEG-SOD, L-arginine, and sepiapterin, were used in experiments to boost the level of bioavailable NO and simultaneously limit the concentration of ONOO−, thus favorably increasing the ratio of (NO)/(ONOO−).
NO is biosynthesized from L-arginine by eNOS, and thereby as the substrate for NO production, increasing the supplementation of L-arginine can partially restore the normal status of eNOS and balance of the (NO)/(ONOO−) ratio by enhancing NO production. Our previous studies have already showed that L-arginine treatment with endothelial cells before LDL incubation can increase NO production and decrease ONOO− generation.25 These results suggest that sufficient supplementation of L-arginine coupled with eNOS can partially restore normal activity of eNOS and bioavailability of NO, therefore the synthesis pathway of ONOO− is turned down at the presence of L-arginine, leading to the reduction of ONOO− production.
Sepiapterin is a precursor of cNOS cofactor tetrahydrobiopterin (BH4), which can convert to BH4 via salvage pathway by sepiapterin reductase and dihydrofolate reductase,39 thereby it can help endothelial NOS maintain functional status with catalytic activity and normal balance between NO and ONOO− by increasing NO biosynthesis from L-arginine. Our data suggest that uncoupling of NOS stimulated by LDL subclasses can be inhibited or reversed by supplementation of sepiapterin. By restoring the catalytic function of NOS, endothelial cells in the sepiapterin treatment group released a higher level of NO and a lower level of ONOO− than the control group after direct injection of LDL with subclasses A, I, and B. However, among all of LDL subclasses’ injections, subclass A still stimulated the highest level of NO and the lowest level of ONOO−, and on the contrary, subclass B stimulated the lowest level of NO and the highest level of ONOO−, suggesting that subclass B is more severe than subclass A in the induction of NOS dysfunction and imbalance of the (NO)/(ONOO−) ratio.
As an L-arginine analog and nonspecific inhibitor of cNOS, L-NAME can bind to the active site of cNOS to block its catalytic activity, resulting in reducing the production of both NO and ONOO−.40 However, this substrate analog-mediated inhibition of NOS activity is reversible with sufficient supplementation of L-arginine.41 It is very interesting that some studies reported increased NOS activity in low dose treatment of L-NAME, which may upregulate NO production via feedback regulatory mechanisms, as well as increase the expression level of NOS.42–44 However, it does not mean that higher bioavailability of NO is necessarily in association with increased NOS expression and/or NOS activity. Due to the fact that NO biosynthesis is determined by many factors, for instance, the lack of cofactors needed for NOS activation, oxidation and/or inactivation of BH4 and the presence of highly reactive ROS can reduce NO production.45,46
As the major product of NADPH oxidases and reactive oxygen species (ROS), O2− can oxidize NO to form ONOO−, which contributes to bring oxidative stress to endothelium and lead to endothelial dysfunction.46 VAS2870 not only permeates cell membrane and inhibits NADPH oxidase activity in a rapid and reversible way, but also repeals agonist-stimulated ROS production and thereby provides protection against oxidative stress generated by ROS.47–49 In this present study, NO concentration was increased by 15–24% of the control group, suggesting that this portion of NO produced by endothelial cells is consumed by O2− generated by NADPH oxidase to form ONOO−. Meanwhile, ONOO− concentration was decreased by 20–27% of the control group, which was consistent with the increase of NO production.
Among different subclasses of LDL, subclass B is the most susceptible to be oxidized.3 Incubation with ox-LDL/n-LDL can stimulate ONOO− release and inhibit NO production from endothelial cells.25 However, the real-time effect of ox-LDL with different subclasses during direct injection to endothelial cells remains unclear. In this study, our data show for the first time that injection with ox-LDL stimulated less NO production and more ONOO− release than n-LDL, suggesting that ox-LDL is more cytotoxic than n-LDL in induction of endothelial dysfunction and imbalance of the (NO)/(ONOO−) ratio, which may play an important role in the pathogenesis of atherosclerosis.
Previous studies have shown that LDL can increase monocyte adhesion to endothelial cells by enhancing the expression level of ICAM-1 and/or VCAM-1.49–53 In this work, we elucidated the effect of LDL with different subclasses on inducing ICAM-1/VCAM-1 expression and monocyte adhesion to endothelial cells. We revealed that LDL significantly up-regulated the expression level of ICAM-1 and VCAM-1, leading to enhancement of monocyte adhesion to endothelial cells. Our data also showed that monocyte adhesion was positive correlated with the concentration of LDL. Subclass B stimulated the highest level of monocyte adhesion, while subclass A stimulated the lowest level of adhesion at the same concentration of LDL incubation. Compared with the n-LDL treatment group, ox-LDL can stimulate a higher level of ICAM-1 and VCAM-1 expression, suggesting that ox-LDL is more likely to cause monocyte adhesion on the surface of endothelial cells. Our data from monocyte adhesion is consistent with the result of ICAM-1 and VCAM-1 expression.
This study reveals that n-LDL and ox-LDL with different density can differently alter NO and ONOO− production, and the effect is dose-dependent in a narrow range of their concentrations. The decrease in cytoprotective NO and the increase of cytotoxic ONOO− suggests that subclass B uncouples eNOS bioactivity more significantly than subclasses I and A, causing severe dysfunction in endothelial cells. In addition, subclass B not only stimulated higher expression of ICAM-1 and VCAM-1 than subclasses I and A, but also stimulated maximal monocyte adhesion. Based on data from this research, subclass B can cause more serious damage to endothelial cells than subclasses A and I, and the distribution of those three LDL subclasses in human blood may play a crucial role in the pathology of cardiovascular diseases. It appears that elevated levels of subclass B is the leading contributor/component of bad cholesterol.
Conclusion
Based on the studies presented here, one can conclude that the concentration of subclass B, in relation to the concentrations of subclasses I and A, may be a very valuable tool in the early diagnosis of atherosclerosis and the potential risk of heart attack. Our studies are coherent with published clinical findings and can explain why a correlation of total “bad” cholesterol with a risk of heart attack is poor and dangerously misleading (which is wrong in about 75% of cases). National guidelines may seriously underestimate the noxious effect of LDL cholesterol, especially in cases where the content of subclass B in total LDL is high (50% or higher).
Acknowledgment
We would like to thank Dr. J. Jose Corbalan and Hazem Dawoud for technical assistance, as well as Collin Arocho for his assistance in the preparation of this manuscript.
Disclosure
Dr Tadeusz Malinski reports a pending patent 60609-US-PSP/OU-19018. The authors report no other conflicts of interest in this work.
References
1. Sachdeva A, Cannon CP, Deedwania PC, et al. Lipid levels in patients hospitalized with coronary artery disease: an analysis of 136,905 hospitalizations in get with the guidelines. Am Heart J. 2009;157(1):111–117.
2. Nishikura T, Koba S, Yokota Y, et al. Elevated small dense low-density lipoprotein cholesterol as a predictor for future cardiovascular events in patients with stable coronary artery disease. J Atheroscler Thromb. 2014;21:755–767. doi:10.5551/jat.23465
3. Hoogeveen RC, Gaubatz JW, Sun W, et al. Small dense low-density lipoprotein-cholesterol concentrations predict risk for coronary heart disease: the atherosclerosis risk in communities (aric) study. Arterioscler Thromb Vasc Biol. 2014;34:1069–1077. doi:10.1161/ATVBAHA.114.303284
4. Austin MA. Small, dense low-density-lipoprotein as a risk factor for coronary heart-disease. Int J Clin Lab Res. 1994;24:187–192. doi:10.1007/BF02592460
5. Blake GJ. Low-density lipoprotein particle concentration and size as determined by nuclear magnetic resonance spectroscopy as predictors of cardiovascular disease in women. Circulation. 2002;106:1930–1937. doi:10.1161/01.cir.0000033222.75187.b9
6. Yee MS, Pavitt DV, Tan T, et al. Lipoprotein separation in a novel iodixanol density gradient, for composition, density, and phenotype analysis. J Lipid Res. 2008;49:1364–1371. doi:10.1194/jlr.D700044-JLR200
7. Campos H, Genest JJ, Blijlevens E, et al. Low density lipoprotein particle size and coronary artery disease. Arterioscler Thromb Vasc Biol. 1992;12:187–195. doi:10.1161/01.ATV.12.2.187
8. Austin MA, Breslow JL, Hennekens CH, Buring JE, Willett WC, Krauss RM. Low-density lipoprotein subclass patterns and risk of myocardial infarction. JAMA. 1988;260:1917–1921.
9. Griffin BA, Caslake MJ, Yip B, Tait GW, Packard CJ, Shepherd J. Rapid isolation of low density lipoprotein (ldl) subfractions from plasma by density gradient ultracentrifugation. Atherosclerosis. 1990;83:59–67. doi:10.1016/0021-9150(90)90131-2
10. Pritchard KA, Groszek L, Smalley DM, et al. Native low-density lipoprotein increases endothelial cell nitric oxide synthase generation of superoxide anion. Circ Res. 1995;77:510–518. doi:10.1161/01.res.77.3.510
11. Apostolov EO, Shah SV, Ok E, Basnakian AG. Carbamylated low-density lipoprotein induces monocyte adhesion to endothelial cells through intercellular adhesion molecule-1 and vascular cell adhesion molecule-1. Arterioscler Thromb Vasc Biol. 2007;27:826–832. doi:10.1161/01.ATV.0000258795.75121.8a
12. O’Byrne D, Devaraj S, Islam KN, et al. Low-density lipoprotein (ldl)-induced monocyte-endothelial cell adhesion, soluble cell adhesion molecules, and autoantibodies to oxidized-ldl in chronic renal failure patients on dialysis therapy. Metabolism. 2001;50:207–215. doi:10.1053/meta.2001.19486
13. Haller H, Schaper D, Ziegler W, et al. Low-density lipoprotein induces vascular adhesion molecule expression on human endothelial cells. Hypertension. 1995;25:511–516. doi:10.1161/01.hyp.25.4.511
14. Al-Benna S, Hamilton CA, McClure JD, et al. Low-density lipoprotein cholesterol determines oxidative stress and endothelial dysfunction in saphenous veins from patients with coronary artery disease. Arterioscler Thromb Vasc Biol. 2006;26:218–223. doi:10.1161/01.ATV.0000193626.22269.45
15. Holvoet P. Endothelial dysfunction, oxidation of low-density lipoprotein, and cardiovascular disease. Ther Apher. 1999;3:287–293. doi:10.1046/j.1526-0968.1999.00169.x
16. Davignon J, Ganz P. Role of endothelial dysfunction in atherosclerosis. Circulation. 2004;109:III27–III32. doi:10.1161/01.CIR.0000131515.03336.f8
17. Witztum JL, Steinberg D. Role of oxidized low density lipoprotein in atherogenesis. J Clin Invest. 1991;88:1785–1792. doi:10.1172/JCI115499
18. Thanyasiri P, Celermajer DS, Adams MR. Endothelial dysfunction occurs in peripheral circulation patients with acute and stable coronary artery disease. Am J Physiol. 2005;289:H513–H517. doi:10.1152/ajpheart.01086.2004
19. McIntyre M, Bohr DF, Dominiczak AF. Endothelial function in hypertension: the role of superoxide anion. Hypertension. 1999;34:539–545. doi:10.1161/01.hyp.34.4.539
20. Lindgren FT, Elliott HA, Gofman JW. The ultracentrifugal characterization and isolation of human blood lipids and lipoproteins, with applications to the study of atherosclerosis. J Phys Colloid Chem. 1951;55:80–93.
21. MM KR S, Lindgren FT, Forte TM. Heterogeneity of serum low density lipoproteins in normal human. J Lipid Res. 1981;22:236–244.
22. Davies IG. Rapid separation of ldl subclasses by iodixanol gradient ultracentrifugation. Clin Chem. 2003;49:1865–1872.
23. Prado KB, Shugg S, Backstrand JR. Low-density lipoprotein particle number predicts coronary artery calcification in asymptomatic adults at intermediate risk of cardiovascular disease. J Clin Lipidol. 2011;5:408–413. doi:10.1016/j.jacl.2011.07.001
24. Sacks FM. Low-density lipoprotein size and cardiovascular disease: a reappraisal. J Clin Endocrinol Metab. 2003;88:4525–4532. doi:10.1210/jc.2003-030636
25. Vergnani L, Hatrik S, Ricci F, et al. Effect of native and oxidized low-density lipoprotein on endothelial nitric oxide and superoxide production: key role of l-arginine availability. Circulation. 2000;101:1261–1266. doi:10.1161/01.cir.101.11.1261
26. Brovkovych V, Patton S, Brovkovych S, Kiechle F, Huk I, Malinski T. In situ measurement of nitric oxide, superoxide and peroxynitrite during endotoxemia. J Physiol Pharmacol. 1997;48:633–644.
27. Kalinowski L, Dobrucki LW, Szczepanska-Konkel M, et al. Third-generation beta-blockers stimulate nitric oxide release from endothelial cells through ATP efflux: a novel mechanism for antihypertensive action. Circulation. 2003;107:2747–2752. doi:10.1161/01.CIR.0000066912.58385.DE
28. Kalinowski L, Malinski T. Endothelial NADH/-NADPH-dependent enzymatic sources of superoxide production: relationship to endothelial dysfunction. Acta Biochim Pol. 2004;51:459–469. doi:035001459
29. Malinski T, Taha Z, Moncada S. Direct electrochemical measurement of nitric oxide released from human platelets. Biochem Biophys Res Commun. 1993;194:960–965. doi:10.1006/bbrc.1993.1914
30. Malinski T, Taha Z. Nitric oxide release from a single cell measured in situ by a porphyrinic-based microsensor. Nature. 1992;358:676–678. doi:10.1038/358676a0
31. Corbalan JJ, Medina C, Jacoby A, Malinski T, Radomski MW. Amorphous silica nanoparticles trigger nitric oxide/peroxynitrite imbalance in human endothelial cells: inflammatory and cytotoxic effects. Int J Nanomedicine. 2011;6:2821–2835. doi:10.2147/IJN.S25071
32. Balbatun A, Louka FR, Malinski T. Dynamics of nitric oxide release in the cardiovascular system. Acta Biochim Pol. 2003;50:61–68. doi:035001061
33. Boisrame-Helms J, Kremer H, Schini-Kerth V, Meziani F. Endothelial dysfunction in sepsis. Curr Vasc Pharmacol. 2013;11:150–160.
34. Forstermann U, Munzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113:1708–1714. doi:10.1161/CIRCULATIONAHA.105.602532
35. Osto E, Matter CM, Kouroedov A, et al. C-jun n-terminal kinase 2 deficiency protects against hypercholesterolemia-induced endothelial dysfunction and oxidative stress. Circulation. 2008;118:2073–2080. doi:10.1161/CIRCULATIONAHA.108.765032
36. Yildirim E, Bugan B, Celik M, Yuksel UC, Yalcinkaya E. Small dense low-density lipoprotein could be used as a therapeutic marker for treatment in patients with acute coronary syndrome. Angiology. 2013;64:644. doi:10.1177/0003319713485806
37. Okumura K, Takahashi R, Taguchi N, et al. Small low-density lipoprotein cholesterol concentration is a determinant of endothelial dysfunction by peripheral artery tonometry in men. J Atheroscler Thromb. 2012;19:897–903. doi:10.5551/jat.13243
38. Lamarche B, Tchernof A, Moorjani S, et al. Small, dense low-density lipoprotein particles as a predictor of the risk of ischemic heart disease in men - prospective results from the quebec cardiovascular study. Circulation. 1997;95:69–75. doi:10.1161/01.cir.95.1.69
39. Thony B, Auerbach G, Blau N. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem J. 2000;347 Pt 1:1–16.
40. Pagano PJ, Tornheim K, Cohen RA. Superoxide anion production by rabbit thoracic aorta - effect of endothelium-derived nitric-oxide. Am J Physiol. 1993;265:H707–H712. doi:10.1152/ajpheart.1993.265.2.H707
41. Talarek S, Listos J, Fidecka S. Effect of nitric oxide synthase inhibitors on benzodiazepine withdrawal in mice and rats. Pharmacol Rep. 2011;63:680–689.
42. Kopincova J, Puzserova A, Bernatova I. L-name in the cardiovascular system - nitric oxide synthase activator? Pharmacol Rep. 2012;64:511–520.
43. Bernatova I, Kopincova J, Puzserova A, Janega P, Babal P. Chronic low-dose l-name treatment increases nitric oxide production and vasorelaxation in normotensive rats. Physiol Res. 2007;56 Suppl 2:S17–S24.
44. Pechanova O, Kojsova S, Jendekova L. Ambivalent effect of chronic l-name treatment in the heart and brain: the role of nuclear factor-kappa b. J Hypertens. 2008;26:S85–S85.
45. Milstien S, Katusic Z. Oxidation of tetrahydrobiopterin by peroxynitrite: implications for vascular endothelial function. Biochem Biophys Res Commun. 1999;263:681–684. doi:10.1006/bbrc.1999.1422
46. Oliveira MW, Minotto JB, de Oliveira MR, et al. Scavenging and antioxidant potential of physiological taurine concentrations against different reactive oxygen/nitrogen species. Pharmacol Rep. 2010;62:185–193.
47. Guzik TJ, West NE, Pillai R, Taggart DP, Channon KM. Nitric oxide modulates superoxide release and peroxynitrite formation in human blood vessels. Hypertension. 2002;39:1088–1094. doi:10.1161/01.hyp.0000018041.48432.b5
48. Schramm A, Matusik P, Osmenda G, Guzik TJ. Targeting nadph oxidases in vascular pharmacology. Vascul Pharmacol. 2012;56:216–231. doi:10.1016/j.vph.2012.02.012
49. Smalley DM, Lin JH, Curtis ML, Kobari Y, Stemerman MB, Pritchard KA
50. Erl W, Weber PC, Weber C. Monocytic cell adhesion to endothelial cells stimulated by oxidized low density lipoprotein is mediated by distinct endothelial ligands. Atherosclerosis. 1998;136:297–303. doi:10.1016/s0021-9150(97)00223-2
51. Weber C, Erl W, Weber PC. Enhancement of monocyte adhesion to endothelial cells by oxidatively modified low-density lipoprotein is mediated by activation of cd11b. Biochem Biophys Res Commun. 1995;206:621–628. doi:10.1006/bbrc.1995.1088
52. Takei A, Huang Y, Lopes-Virella MF. Expression of adhesion molecules by human endothelial cells exposed to oxidized low density lipoprotein. Influences of degree of oxidation and location of oxidized ldl. Atherosclerosis. 2001;154:79–86. doi:10.1016/s0021-9150(00)00465-2
53. BV PS K, Alexander RW, Medford RM. Modified low density lipoprotein and its constituents augment cytokine-activated vascular cell adhesion molecule-1 gene expression in human vascular endothelial cells. J Clin Invest. 1995;95:1262–1270. doi:10.1172/JCI117776
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.