Back to Journals » Clinical Epidemiology » Volume 12
Validity of First-Time Diagnoses of Inherited Ichthyosis in the Danish National Patient Registry and the Danish Pathology Registry
Authors Kristensen MH ![]() , Schmidt SAJ
, Schmidt SAJ ![]() , Kibsgaard L, Hove H, Sommerlund M
, Kibsgaard L, Hove H, Sommerlund M ![]() , Koppelhus U
, Koppelhus U ![]()
Received 30 September 2019
Accepted for publication 12 May 2020
Published 19 June 2020 Volume 2020:12 Pages 651—657
DOI https://doi.org/10.2147/CLEP.S232956
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Henrik Toft Sørensen
Mattias Hedegaard Kristensen,1 Sigrun Alba Johannesdottir Schmidt,2 Line Kibsgaard,1 Hanne Hove,3 Mette Sommerlund,1 Uffe Koppelhus1
1Department of Dermatology, Aarhus University Hospital, Aarhus, Denmark; 2Department of Clinical Epidemiology, Aarhus University Hospital, Aarhus, Denmark; 3Centre for Rare Diseases, Copenhagen University Hospital, Copenhagen, Denmark
Correspondence: Uffe Koppelhus
Department of Dermatology, Aarhus University Hospital, F3, Plan 2,202,Palle Juul-Jensens Boulevard 99, DK 8200, Denmark
Tel +45 2494 6245
Fax +45 7846 1860
Email [email protected]
Purpose: Inherited ichthyosis is a monogenetic disease characterized by hyperkeratosis and scaling of the skin, with large interindividual variation in severity. It can affect quality of life for patients and their families. Population-based data on inherited ichthyosis are lacking, which hampers studies into its epidemiology.
Patients and Methods: Based on medical record review, we validated diagnoses of inherited ichthyosis in two nationwide population-based registries commonly used for epidemiological research: The Danish National Patient Registry and the Danish Pathology Registry. The study period was January 1, 1977, through December 31, 2015. Validation samples were taken from one regional hospital without a specialized dermatological department and two specialized dermatological departments. Positive predictive values (PPVs) were estimated overall and for each coding system (ICD-8, ICD-10 and SNOMED), including for specific ICD-10 codes.
Results: We identified 1772 first-time diagnoses of inherited ichthyosis; 363 patients were diagnosed at the departments selected for validation, and 307 of these patients (84.6%) had medical records enabling validation. We observed an overall PPV of 73.3% (95% CI: 68.1– 77.9). For ICD-8, ICD-10, and SNOMED diagnoses, the PPVs were 73.2% (95% CI: 58.1– 84.3), 74.7% (95% CI: 69.0– 79.7), and 46.2% (95% CI: 22.1– 71.7), respectively. In analyses for ICD-10 diagnoses, we observed much higher validity of diagnoses from the specialized departments (PPV 79.7%; 95% CI: 74.1– 84.3) than the regional hospital (PPV 5.9%; 95% CI: 0.6– 24.3). The PPVs for specific diagnoses were 80.1% for ichthyosis vulgaris and 96.6% for X-linked ichthyosis but below 45% for remaining, rarer, subtypes.
Conclusion: The PPV of first-time diagnosis of inherited ichthyosis made at specialized dermatological departments in the Danish National Patient Registry is approximately 80%. Diagnoses from the Danish Pathology Registry had low PPVs precluding their use for research.
Keywords: Denmark, diagnosis, ichthyosis, health administrative data, registration, validity
Introduction
Inherited ichthyosis is a group of rare genetic skin diseases (genodermatosis) with known monogenetic causes.1 Ichthyosis is defined as a disease with abnormal terminal keratinocyte differentiation, which primarily presents as hyperkeratosis and/or scaling of the skin.2 Associated morbidity varies between subtypes, with the most severe cases of ichthyosis requiring highly specialized levels of care.3,4 The clinical and genetic variability, as well as unclear genotype-phenotype correlations, make further investigations into the epidemiology, pathogenesis, comorbidity and prognostic factors of this disease warranted. Furthermore, validated cohorts of certain subtypes, such as X-linked ichthyosis, are needed for clinical studies.
Unfortunately, studies of rare diseases, such as inherited ichthyosis, are hampered by the low disease prevalence and the lack of validated databases. The Danish National Patient Registry (DNPR) and The Danish Pathology Registry (DPR) are two large nationwide databases which provide a potential framework; however, in a recent study of another genodermatosis, epidermolysis bullosa (EB), we found evidence of insufficient validity of diagnostic codes.5 For the DNPR, the positive predictive value (PPV) was 77% for diagnoses registered after the introduction of the 10th edition of International Classification of Disease (ICD-10) and only 31% for diagnoses registered in the preceding 8th edition (ICD-8). The PPV was only 0% for SNOMED codes used in the Danish Pathology Registry. Diagnosis and registration of inherited ichthyosis in nationwide registries may be associated with similar challenges.5 The first consensus report on nomenclature and classification of inherited ichthyosis was not published until 2010,2 illustrating the ambiguity of the clinical diagnostics that this rare genetic disease has been subject to over the years.
In the current study, we therefore investigated the validity of a first-time diagnosis of inherited ichthyosis in the DNPR and the DPR using review of medical records as reference.
Patients and Methods
Setting
Denmark has a universal healthcare system, guaranteeing residents unrestricted access to general practitioners and hospitals, including highly specialized treatments. Health services are routinely recorded in the various registries using the Civil Personal Registration (CPR) number, which is a unique personal identifier assigned to all Danish residents by the Civil Registration System.6,7 In this study, we identified all patients with a first-time diagnosis of inherited ichthyosis recorded in the DNPR and the DPR during the study period from January 1, 1977 to December 31, 2015. Subsequently, patient records were acquired and reviewed to validate the diagnoses registered.
Identification of Study Population
We searched the DNPR for first-time contacts with inherited ichthyosis recorded as either a main or secondary diagnosis. The DNPR includes data on all somatic hospital admissions since 1977 and all visits to emergency rooms and outpatient clinics since 1994.7 For each hospital contact, start and end dates for the contact, the hospital department, and relevant diagnoses (one main and optional secondary diagnoses) are registered. Diseases are registered by the physician in charge of discharge or outpatient contact using the Danish version of the International Classification of Diseases (ICD). The ICD-8 revision was used from 1977 and was replaced by the ICD-10 after 1993. We considered all inpatient, emergency room, and outpatient clinic contacts, including those that were ongoing at the end of the study period. We included ICD-8 code 757.20 labelled “Ichthyosis” and ICD-10 code Q80 for “Inherited ichthyosis”. Although the consensus statement suggests that some rare diagnoses of inherited ichthyosis are not included within this code list,2 in our experience, “other” or “unspecified” ichthyosis (ie, Q80.8 or Q80.9) are the most commonly used in clinical practice. Of note, only syndromic forms of ichthyosis were within the scope of this study.
We also searched the DPR for patients registered with ichthyosis. The DPR includes results from histopathological investigations completed at all Danish departments of pathology since 1997. The Registry was established to supplement other Danish registries (eg, DNPR) and contains information relating to patient diagnosis and treatment from both hospitals and the primary sector. Registration is performed by the investigating pathologist using the Danish version of the Systematized Nomenclature of Medicine (SNOMED).8 We included all patients registered with the SNOMED code M74410 labelled “Ichthyosis”. There are no other Danish SNOMED codes consistent with ichthyosis. As for the ICD-8 code, the Danish SNOMED code for ichthyosis is less specific for inherited ichthyosis than the ICD-10 code and may thus include other ichthyosis forms.
Validation
To validate the ichthyosis diagnoses, we selected all patients with a first-time diagnosis from two dermatologic departments (at Aarhus and Bispebjerg University Hospitals) and one regional hospital (Herning Central Hospital). One author (MK) reviewed the medical records and collected relevant data on a predefined set of clinical descriptions, family histories, and results from histopathological examinations and molecular genetic tests. The clinical findings extracted included signs of scaling, color, shape and localization of scales; age at manifestation; syndromal signs; and other significant specific manifestations (see Supplementary Table 1).2 Family history was considered positive if there was one or more similar or confirmed cases in the family. Presence of one or more of the following histopathological characteristics was consistent with positive histology: orthohyperkeratosis, absent stratum granulosum, thickened stratum granulosum, retention hyperkeratosis, lymphohistiocytic infiltrates, or epidermolytic hyperkeratosis.2 Genetic verification was considered present if the medical record included information stating that there was a positive molecular genetic test consistent with inherited ichthyosis.
Based on the information extracted, we classified diagnoses as ‘not inherited ichthyosis’, ‘probable inherited ichthyosis’ and/or “confirmed inherited ichthyosis”. A diagnosis was considered to be “probable” inherited ichthyosis if the clinical information described scaling of the skin and an age of manifestation before adulthood. Patients who in addition to such manifestations had positive family history, histology and/or genetic verification (all considered of equal importance) were categorized as “confirmed”. Of note, confirmed cases formed a subgroup of probable cases (ie, groups were not exclusive). The remaining patients were classified as “not inherited ichthyosis”. In cases where available information was cause for doubt (eg, wording or other causes), MS or UK (both with broad expertise in diagnosis of patients with genodermatoses) were consulted. Thus, we based the first level of confirmation primarily on clinical description, while family history and paraclinical findings had higher diagnostic specificity. The exact clinical information, eg, the distribution and morphology of the scaling, other clinical features such as accentuated palmoplantar markings, presence of eczema or incidence of cryptorchidism, together with the description of the histology and the specific genetic variant, if known, were used to classify cases into ICD-10 subgroups of inherited ichthyosis according to the classification proposed by Oji et al.2
We collected and processed data from medical records through the REDCap (Research Electronic Data Capture)—an electronic tool hosted by Aarhus University to ensure secure data capture for research studies.9 The study involved no patient contact.
Statistical Analysis
As an estimate of the validity of diagnoses, we computed the positive predictive value (PPV) for first-time diagnoses of inherited ichthyosis in the DNPR and the DPR, respectively. The PPV was defined as the proportion of patients with a probable or confirmed diagnosis of inherited ichthyosis. We used the Wilson’s method to compute 95% confidence intervals for proportions based on data for at least 40 patients; otherwise, we used the Jeffrey’s method.10 We estimated PPVs for each registry overall, for the individual coding systems, and for ICD-10 codes, in subgroups defined by type of department (specialized or regional), sex, age at diagnosis (below 1 year, 1–5 years, 6–15 years, 16–64 years, and above 64 years), calendar year of diagnosis (before 2001, between 2001 and 2008, and from 2009 to 2015), type of diagnosis (primary or secondary), type of contact (admitted or outpatient clinic), and the most specific level of the diagnosis code.
Statistical analysis was performed by MK using Stata software (StataCorp. 2015. Stata Statistical Software: Release 14.2. College Station, TX: StataCorp LP).
The study was approved by the Danish Data Protection Agency (journal number 2013-58-0026, case number 1-16-02-668-15) and collection of the relevant data from the medical files was approved by the National Board of Health (case number 3-3013-1606/1/). Per Danish law, no written informed consent is needed in studies based on registry data. The study was carried out in accordance with the principles of the Declaration of Helsinki.
Results
Figure 1 shows the study flowchart. During the study period, 1,772 patients had a first-time diagnosis of inherited ichthyosis in the DNPR and/or the DPR, of which 637 were identified at specialized dermatological departments and 1135 from non-dermatological departments. Of all diagnoses, 363 (20.5%) were recorded at one of the three departments selected for validation. We were unable to retrieve the medical records for 56 (14.4%) of these patients: 4/238 (1.7%) at the Department of Dermatology in Aarhus University Hospital, 45/96 (46.9%) at the Department of Dermatology at Bispebjerg University Hospital, 7/29 (24.1%) at Herning Central Hospital. Thus, the final sample validated included 307 persons. Compared with the total study population, this validated population had a younger median age at diagnosis, a lower proportion of females, and a greater proportion of diagnoses identified with the ICD-10 system (Table 1). The low proportion of records identified at Bispebjerg University Hospital was due to erasing of records from early calendar periods (Supplementary Table 2), as record-keeping was not legally required by the time of review (Supplementary Table 2). Comparing the validation samples at the specialized departments, there was a lower proportion of females and lower median diagnosis age for those identified at Aarhus University Hospital than at Bispebjerg University Hospital (Supplementary Tables 3 and 4).
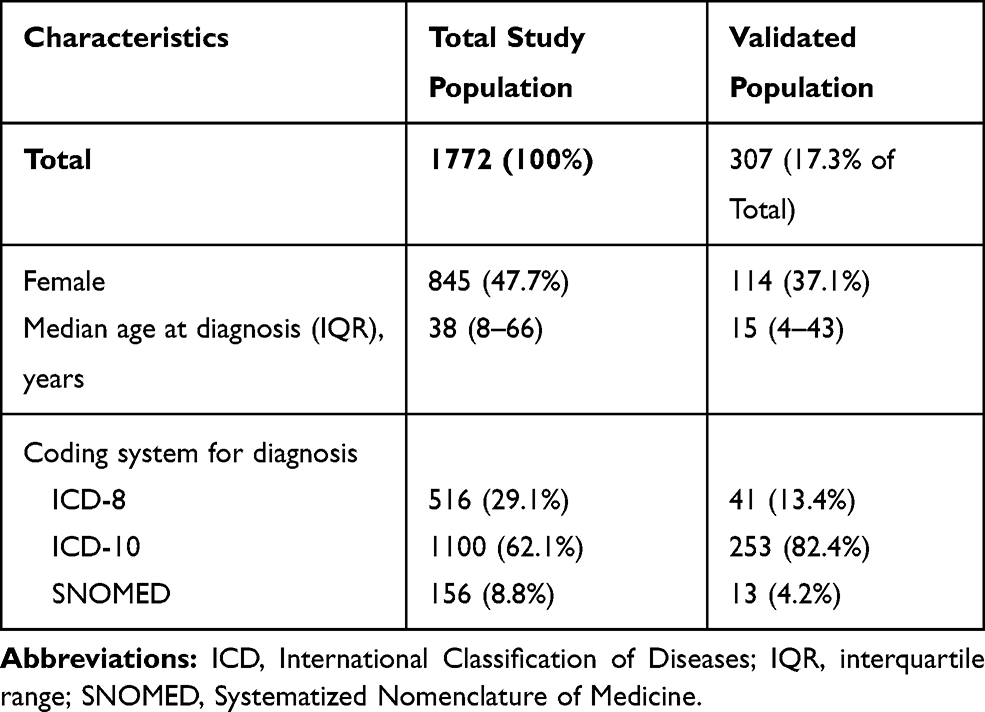 |
Table 1 Characteristics of Persons with First-Time Diagnosis of Inherited Ichthyosis in the Danish National Patient Registry and Pathology Registry, Total Study Population and Validated Population |
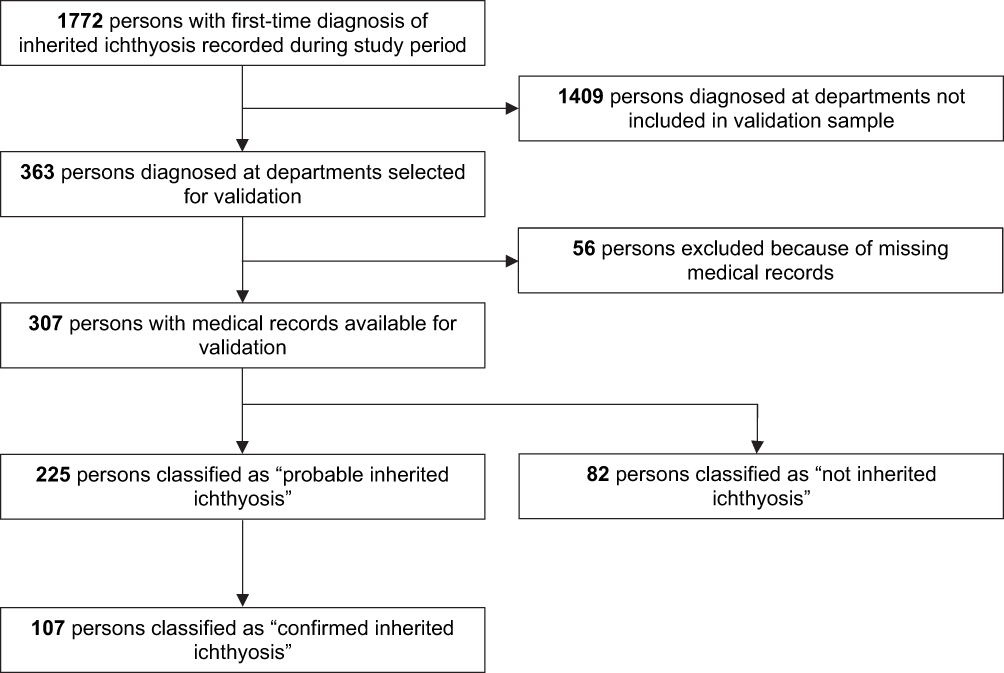 |
Figure 1 Flowchart illustrating the validation process of the inherited ichthyosis diagnoses. |
Of the 307 first-time diagnoses validated, we classified 225 as probable and 82 as not inherited ichthyosis. Of the 225 classified as probable, 107 were further classified as confirmed, yielding PPVs of 73.3% (95% CI: 68.1–77.9) and 34.9% (95% CI: 29.7–40.3), respectively (Table 2). Among confirmed cases, 75 had histological verification, 25 had positive family history, and 56 had genetic verification. Of the 82 records classified as “not inherited ichthyosis”, 46 (56%) represented other diagnoses, 18 (22%) had insufficient evidence to satisfy validation, and 18 (22%) had too sparse material for validation. The misclassified other diagnoses included acquired ichthyosis (n=14), cerebral palsy (n=14; ICD-10 code: G80.9); unspecified contact dermatitis (n=2); psoriasis (n=2); paraneoplastic acanthosis (n=1); Sjögrens syndrome (n=6); infant harlequin phenomenon (n=1); CHILD syndrome (n=2); unspecified congenital linear nevi (n=2); and localized palmoplantar keratodermas (n=2). Of 18 records with insufficient data to satisfy validation, medical records suggested that 2 were syndromic ichthyoses, 3 were X-linked ichthyosis, 4 were autosomal recessive ichthyoses, 2 were other ichthyoses, and 7 were ichthyosis vulgaris.
 |
Table 2 PPVs (95% Confidence Interval) for the Coding of Inherited Ichthyosis in the DNPR and the DPR, Overall and by Coding System |
The overall PPV was driven by the results for the ICD-8 (PPV 73.2%; 95% CI: 58.1–84.3) and ICD-10 (PPV 74.7%; 95% CI: 69.0–79.7), whereas the PPV for SNOMED codes was low (PPV 46.2%; 95% CI: 22.1–71.7).
When stratifying results for ICD-10 codes, PPVs for probable diagnoses were remarkably higher for specialized departments and increased with decreasing age at diagnosis (Table 3). We observed no substantial variation by sex, calendar period of diagnosis, or the type or setting of diagnoses. Much lower estimates where found when restricting to confirmed diagnoses. Comparing the specialized departments, estimates were higher for Aarhus University Hospital (PPV 84.0%; 95% CI: 78.9–88.5) than for Bispebjerg University Hospital (PPV 63.3; 95% CI: 49.3–75.3) (Supplementary Table 5).
 |
Table 3 PPVs (95% Confidence Intervals) for Probable and Confirmed ICD-10 Diagnoses of Inherited Ichthyosis, Stratified by Characteristics |
Considering probable diagnoses, PPVs for the complete (subtype-specific) ICD-10 code was 80.1% (95% CI: 72.7–86.0) for ichthyosis vulgaris; 96.6% (95% CI: 85.0–99.6) for X-linked ichthyosis; 42.9% (95% CI: 20.3–68.1) for lamellar ichthyosis; and 40.0% (95% CI: 9.4–79.1) for congenital ichthyosiform erythroderma (Supplementary Table 6). For the remaining ICD diagnoses, PPVs were 0%, as no cases were classified as probable during this validation.
Discussion
In this validation study of inherited ichthyosis, we found a PPV of 73–75% for first-time ICD diagnoses recorded in the DNPR when considering diagnoses classified as at least probable based on medical record review. The results were driven by findings at dermatological departments. The validity of SNOMED diagnoses in the DPR was only 46%.
No previous study has examined the validity of ichthyosis diagnoses in the Danish registries. Our findings are, however, similar to our recently published study concerning validity of EB,5 except for better validity of ichthyosis diagnosis codes from the ICD-8 system in the current study.
We investigated validity using two reference standards. The PPV for probable diagnoses likely represents a more full clinical spectrum, including milder forms of inherited ichthyosis where diagnosis is based solely on clinical presentation. Meanwhile, we expect that the confirmed group consists mainly of cases with a severe clinical presentation or a need for confirmation with respect to the choice of treatment or to implement prenatal molecular diagnosis for patients’ offspring. Thus, the probable group may be the most relevant measure of the precision of the diagnostic process as it takes place in the clinical practice in Denmark.
The PPVs of inherited ichthyosis were higher for the ICD diagnoses compared to the SNOMED diagnoses. Histopathological findings are generally considered nondiagnostic for inherited ichthyosis.2 The low PPV of SNOMED codes reflects that most cases identified by SNOMED codes did not have the sufficient characteristic clinical features recorded in their medical records.
For the ICD-10 codes, the PPV was highest for the specialized dermatological departments, which is expected both because of experience with the condition and because of a greater disease prevalence. Notably, many of the cases identified at the regional department were cerebral palsy (14 of 17 cases), which is probably due to the alphanumerical similarity of the ICD-10 codes (G80.9 for cerebral palsy and Q80.9 for unspecified ichthyosis), thus representing miscoding rather than misdiagnosis. The pattern with better PPV for the lowest diagnosis age is expected for congenital conditions where many cases manifest with characteristic symptoms at birth or in early childhood and where especially the patients with the most severe phenotypes are likely to be referred to dermatological departments. The PPVs of the disease-specific ICD-10 diagnoses showed that the diagnoses of ichthyosis vulgaris and X-linked ichthyosis were above 80%. PPVs for other subtypes were below 43% and precision was poor based on a very low number of registered cases.
There are some potential sources of bias in our study. Some records included insufficient information to judge the correct diagnosis, which may have caused us to underestimate the validity. On the other hand, the criteria for fulfilling a “probable” diagnosis were quite inclusive in order to accommodate the clinical variability in ichthyosis; this may have caused overestimation. Furthermore, records were evaluated by only one person; it is difficult to predict in which direction this procedure may have biased the results. Precision of our estimates is also a concern, as illustrated by the low number of cases and wide confidence intervals for especially subgroup analyses and specific diagnoses.
A strength of our study is that we were able to retrieve a large proportion of records sought for validation. An exception is records from Bispebjerg in the early calendar period, but as this seem to have been related to archiving issues and therefore not the diagnosis itself, we find it unlikely to have biased our PPVs. This issue is likely to explain the higher proportion of ICD-10 diagnoses in the validated population compared with the total study population. The validated population also had a lower proportion of females and a lower median age at diagnosis. The lower proportion of females may be explained by the fact that the most prevalent single form of inherited ichthyosis is X-linked and therefore only manifest in males.2 The lower median age at diagnosis may be explained by overrepresentation of cases from dermatological departments where more severe cases are seen. Inference based on the results for the regional hospital is limited based on the low number of cases validated and our overall results should therefore not be generalized to non-specialized departments.
Finally, we were unable to examine completeness of diagnoses, which is another important measure of quality of registration. Incompleteness will exist and is expected to be related to severity of disease. Thus, cases with milder disease manifestations, that are inherited dominantly, may either go completely undiagnosed if the family has sufficient experience with handling the condition themselves or it may be managed by a general practitioner or a private practicing specialist.
Conclusion
In conclusion, we found that the validity of first-time inherited ichthyosis diagnoses recorded at specialized dermatological departments in the DNPR was at a moderate level of approximately 80%. However, SNOMED diagnoses in the DPR have low validity, precluding for identifying inherited ichthyosis for research. The only moderate validity of the first-time diagnoses even at the specialized departments, as well as lack of clinical and paraclinical data, indicates a need for specialized registers for these diseases. A new database, Danish National Database for Genodermatoses, has just been launched to address this need.
Ethical Statement
The study was carried out in accordance with the principles of the Declaration of Helsinki.
Acknowledgments
The authors would like to thank Mette Mogensen for her help. Also, the funding from Aage Bang and DDS and the cooperation with RareDis is much appreciated.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Leech SN, Moss C. A current and online genodermatosis database. Br J Dermatol. 2007;156(6):1115–1148. doi:10.1111/j.1365-2133.2007.07834.x
2. Oji V, Tadini G, Akiyama M, et al. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Soreze 2009. J Am Acad Dermatol. 2010;63(4):607–641. doi:10.1016/j.jaad.2009.11.020
3. Bodemer C, Bourrat E, Mazereeuw-Hautier J, et al. Short- and medium-term efficacy of specific hydrotherapy in inherited ichthyosis. Br J Dermatol. 2011;165(5):1087–1094. doi:10.1111/j.1365-2133.2011.10510.x
4. Lai-Cheong JE, Elias PM, Paller AS. Pathogenesis-based therapies in ichthyoses. Dermatol Ther. 2013;26(1):46–54. doi:10.1111/j.1529-8019.2012.01528.x
5. Kristensen MH, Schmidt SAJ, Kibsgaard L, Mogensen M, Sommerlund M, Koppelhus U. Validity of first-time diagnoses of congenital epidermolysis bullosa in the Danish National Patient Registry and the Danish Pathology Registry. Clin Epidemiol. 2019;11:115–124. doi:10.2147/CLEP.S184742
6. Schmidt M, Schmidt SAJ, Adelborg K, et al. The Danish health care system and epidemiological research: from health care contacts to database records. Clin Epidemiol. 2019;11:563–591. doi:10.2147/CLEP.S179083
7. Schmidt M, Pedersen L, Sørensen HT. The Danish civil registration system as a tool in epidemiology. Eur J Epidemiol. 2014;29(8):541–549. doi:10.1007/s10654-014-9930-3
8. Bjerregaard B, Larsen OB. The Danish pathology register. Scand J Public Health. 2011;39(7 Suppl):72–74. doi:10.1177/1403494810393563
9. Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)–a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377–381. doi:10.1016/j.jbi.2008.08.010
10. Brown LD, Cai TT, DasGupta A. Interval estimation for a binomial proportion. Stat Sci. 2001;16(2):101–117. doi:10.1214/ss/1009213286
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.