Back to Journals » Clinical Epidemiology » Volume 8
Validation of diagnostic codes for Charcot-Marie-Tooth disease in the Danish National Patient Registry
Authors Vaeth S ![]() , Jensen UB, Christensen R, Andersen H
, Jensen UB, Christensen R, Andersen H ![]()
Received 22 June 2016
Accepted for publication 17 August 2016
Published 21 November 2016 Volume 2016:8 Pages 783—787
DOI https://doi.org/10.2147/CLEP.S115565
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Henrik Toft Sørensen
Signe Vaeth,1 Uffe Birk Jensen,1 Rikke Christensen,1 Henning Andersen2
1Department of Clinical Genetics, Aarhus University Hospital, Aarhus N, 2Department of Neurology, Aarhus University Hospital, Aarhus C, Denmark
Purpose: To validate the diagnostic codes for Charcot–Marie–Tooth disease (CMT) in the Danish National Patient Registry (DNPR) using positive predictive value (PPV) as a measure of validity.
Patients and methods: We used the DNPR to identify all patients diagnosed with at least one primary CMT diagnosis at a specialized department in the Central Denmark Region during the period 1977–2012. From this population, we randomly selected 123 patients for the validation study. Medical files were reviewed and used as reference standard. We estimated the PPV of the CMT diagnoses and stratified the analysis according to age at diagnosis, gender, and calendar time.
Results: In the DNPR, 275 patients were identified. We were able to retrieve 96 medical files from the random sample of 123 patients, and 85 CMT diagnoses were confirmed. The average age at diagnosis was 42.5 years, and 34% were female. The PPV was 88.5% (95% confidence interval: 80.4–94.1).
Conclusion: The CMT diagnoses in the DNPR have high validity. The DNPR can be used as a data source for epidemiologic research on CMT.
Keywords: registries, positive predictive value, International Classification of Disease codes, epidemiology
Introduction
Charcot–Marie–Tooth disease (CMT) is a clinically and genetically heterogeneous group of neuropathies, and is also known as hereditary motor and sensory neuropathy. The classic CMT phenotype is characterized by slowly progressive distal muscle weakness and atrophy, mild-to-moderate sensory loss, foot deformities (pes cavus and hammer toes), and loss of deep tendon reflexes.1 However, symptoms vary greatly between patients and sometimes overlap with other neurologic disorders. The severity of CMT varies from mild to severe, and the age at onset ranges from early childhood to late adulthood. Pathogenic variants in more than 70 genes are associated with CMT, and all types of Mendelian inheritance patterns are seen.2 There is no golden standard for diagnosing CMT, and due to the heterogeneity of the disease, CMT can pose a great diagnostic challenge.
A recent review on epidemiologic studies of CMT worldwide underscored the lack of epidemiologic knowledge in CMT.3 The different epidemiologic studies vary in quality and methodology, and prevalence estimates vary greatly ranging from 9.74 to 82.25 per 100,000. Few studies were performed nationwide, and no study had used nationwide register-based data.3
For the purpose of conducting an epidemiologic study on CMT in Denmark, we aimed to identify patients diagnosed with CMT using the Danish National Patient Registry (DNPR). The DNPR is considered one of the finest national health registers worldwide,6 but the usability of secondary data in registries such as the DNPR is highly dependent on the validity of the data.7 This study aims to examine the quality of the CMT discharge diagnosis in the DNPR by estimating the positive predictive value (PPV), using medical record data as reference standard.
Materials and methods
Setting
Denmark has 5.6 million citizens and the country is divided into five regions. In 2012, there were 1,271,223 inhabitants in the Central Denmark Region (CDR), roughly 23% of the Danish population.8 The Danish National Health Service provides all inhabitants with tax-supported health care. Since 1968, all Danish citizens have been registered in the Civil Personal Registry and given a unique 10-digit civil registry number (CPR number). The CPR number contains information on birth date and gender, and enables unique identification and matching of data between databases.9
The study was approved by the Danish Data Protection Agency (record number 1-16-02-18-12) and the Regional Ethics Commitee for the Central Denmark Region (1-10-72-331-12). Study protocol can be accessed by contacting the authors.
The DNPR
The DNPR was established in 1977, and contains data on all somatic hospital admissions, including CPR number, admission and discharge dates, hospital department, primary discharge diagnosis code, and up to 19 supplementary diagnoses. Data on outpatient contacts were added in 1995. Data are recorded prospectively, independent of specific research questions. Medical diagnoses were registered in the DNPR using the International Classification of Disease, version 8 (ICD-8) until 1993 and the International Classification of Disease, version 10 (ICD-10) thereafter.6
Identification of study population
We chose to perform our validation study on cases diagnosed with CMT in the CDR. Using the DNPR, we identified all discharge diagnoses between 1977 and 2012 consistent with CMT: ICD-10 DG600 (hereditary motor and sensory neuropathy) and ICD-8 33009 (atrophia mm. neuropathica, Charcot–Marie–Tooth). In order to improve the validity of our study population, we included only cases that fulfilled the following criteria: 1) one or more primary CMT diagnoses, as they are considered more accurate than secondary diagnoses; and 2) only cases diagnosed at departments of neurology, neurophysiology, clinical genetics, or pediatrics in the CDR, as we consider these departments to be the ones most familiar with CMT diagnostics.10 From this study population, we produced a random sample of 20 patients for every 5 calendar years during the entire study period. However, for some of the earlier periods, less than 20 cases were diagnosed in the DNPR, and the final study population therefore included 123 patients. The validation process is described in Figure 1.
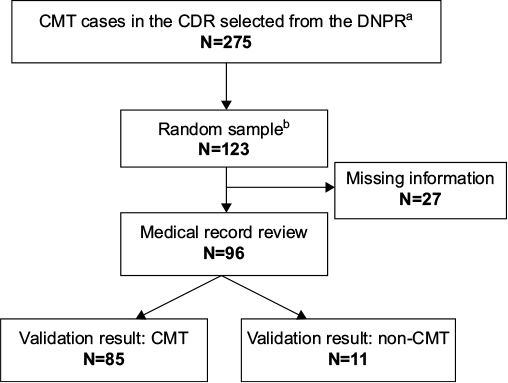 | Figure 1 Flowchart of the validation process Notes: aSelection in the DNPR: at least one primary diagnosis given at a department of neurology, neurophysiology, clinical genetics, or pediatrics during 1977–2012; brandom sample: 20 cases for every 5 years from 1977 to 2012. Abbreviations: CMT, Charcot-Marie-Tooth disease; CDR, Central Denmark Region; DNPR, Danish National Patient Registry; N, number of cases. |
Validation
Validation was performed on the random sample, using medical record data as reference standard. Medical records were identified using CPR number and manually reviewed. Access to medical records without written informed consent from the patients was approved by the Regional Ethics Commitee for the Central Denmark Region and The Danish Data Protection Agency. All records were reviewed by the same physician (SV), and uncertain cases were reviewed together with another physician (HA). Cases were categorized as CMT or non-CMT according to a list of seven criteria, based on the European CMT Consortium criteria11,12 and the classic CMT phenotype. The seven criteria are listed in Table 1. To validate a diagnosis, we determined that at least four of the seven criteria had to be fulfilled, and at least one of the fulfilled criteria had to be criteria number 1, 2, or 3 (Table 1). Genetically confirmed cases were all categorized as CMT regardless of the number of fulfilled criteria.
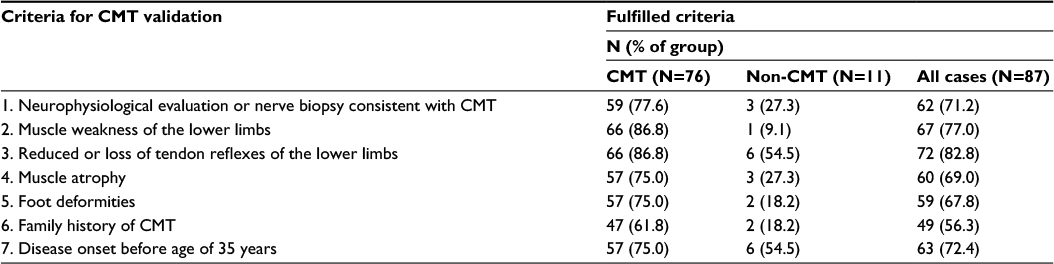 | Table 1 Criteria for validationa and distribution of fulfilled criteria among the 87 cases without genetically confirmed diagnosis Note: aIn order to validate a case as CMT, at least four out of the seven criteria had to be fulfilled, and at least one of the fulfilled criteria had to be criteria number 1, 2, or 3. Abbreviations: CMT, Charcot-Marie-Tooth disease; N, number of cases. |
Statistical analysis
PPV was used as a measure of diagnostic validity. The PPV was estimated as the proportion of CMT cases identified in the DNPR sample, which fulfilled our criteria for CMT according to the medical files. Exact 95% confidence intervals (CIs) based on the binomial distribution were also computed. The analysis was stratified by age at diagnosis, gender, and calendar time, to evaluate differences in PPV. A 5% level of significance was used for hypothesis testing. Data were analyzed using STATA version 13.1.
Results
At the departments described earlier in the CDR, a total of 275 cases had received a primary CMT diagnosis. The random sample consisted of 123 cases, whereof 96 medical files could be located. All but 3 of the 27 missing records were from the period before 1995 (data not shown).
The average age at diagnosis was 42.5 years, and 34.4% were female. In 85 cases (88.5%), the diagnosis had been made at a neurologic department, 9 cases (9.4%) were diagnosed at a neurophysiologic department, and 2 cases (2.1%) were diagnosed at a pediatric department. The diagnosis codes ICD-8 33009 and ICD-10 DG600 were observed in 21 and 75 cases, respectively. In 53 cases (55.2%), the diagnosis was made after the year 2000.
In 85 cases, the diagnosis was found to be CMT. Nine cases were genetically confirmed at the time of first diagnosis. Of the remaining 76 cases, 57 had a neurophysiologic evaluation consistent with CMT and in 2 cases, findings at nerve biopsy were consistent with CMT. Among the 17 cases without any neurophysiological evaluation or nerve biopsy supporting the diagnosis, all but one had both muscle weakness and reduced tendon reflexes of the lower limbs. The distribution of validation criteria in the cases without genetically confirmed diagnosis is shown in Table 1.
The PPV of the entire sample was 88.5% (95% CI: 80.4%–94.1%). When stratifying the data according to age at diagnosis, gender, and calendar time, we observed trends for a higher PPV (nonsignificant) in the following conditions: cases diagnosed between ages 30 and 49 years (PPV 96.9%), cases diagnosed after the year 2000 (PPV 90.6%), and among females (PPV 92.1%) (Table 2).
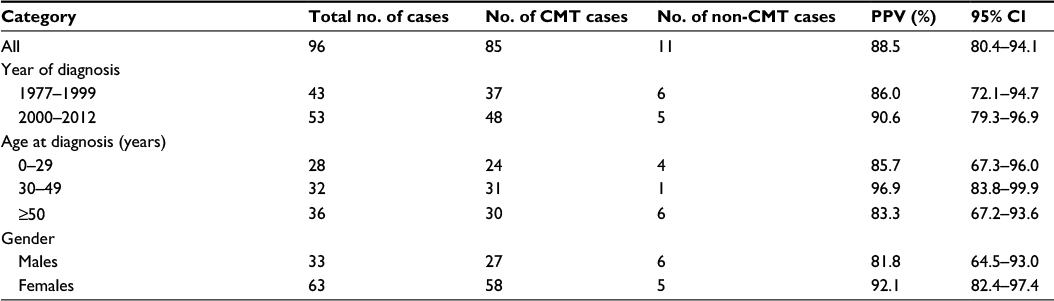 | Table 2 Validity of the Charcot-Marie-Tooth diagnosis in the DNPR Abbreviations: DNPR, Danish National Patient Registry; CMT, Charcot-Marie-Tooth disease; PPV, positive predictive value; CI, confidence interval. |
Discussion
In this validation study, a great majority of the CMT diagnoses could be confirmed by review of medical records, yielding a high PPV. Our data suggest a slightly higher PPV in the age group of 30–49 years, in cases diagnosed after the year 2000, and among females, although our study population is too small to reveal statistically significant differences. To our knowledge, this is the first validation study of the CMT diagnosis. The use of national registry-based data minimized self-selection bias, and allowed us to sample patients at date of first diagnosis, so length-time bias and other problems related to cross-sectional sampling were avoided.
Limitations
Our study has a number of limitations to be considered. First, our study was performed using data from only one of the five regions in Denmark. Regional differences in diagnostic tradition may affect the PPV, but due to the uniform nature of the Danish health care system and the structure of record keeping, we consider the results generalizable to the whole country. Second, our results do not apply to other registries. Third, the medical records were not perfect or uniform and some records could not be located. The missing cases were primarily from the earlier period before 1995; hence, it is most likely that they have been lost or destroyed. However, it is possible that the missing records were less thoroughly studied, and could have introduced bias. Fourth, there is no golden standard for the CMT diagnosis. Although a Charcot–Marie–Tooth neuropathy score has been developed to study the natural history and response to therapy in CMT,13 the data available in our study does not allow calculation of a score for each patient. Validation in our study was performed according to a list of criteria based on the classical CMT phenotype.11,12 This may lead to an underestimation of the PPV, as atypical and mild cases may be classified as non-CMT. Finally, we did not include data on undiagnosed patients in this study, and therefore were unable to estimate the negative predictive value (NPV), sensitivity, and specificity of the CMT diagnosis. Due to the heterogeneous nature of CMT, it is reasonable to assume that many patients are undiagnosed or misdiagnosed in cases with mild symptoms or atypical clinical presentation. The proportion of false-negatives might thus be high, and would yield a low sensitivity. Due to the low prevalence of CMT, the a priori risk of having CMT is low, and the number of false-positives and false-negatives will therefore be low relative to the large population of undiagnosed people without CMT, resulting in a high specificity and a high NPV.10,14
Conclusion
This study shows that registration of CMT in the DNPR has high validity, and supports the use of the DNPR as a data source for epidemiological research on CMT.
Acknowledgments
The authors are very grateful to Michael Vaeth from The Department of Public Health, Section for Biostatistics at Aarhus University, for providing technical assistance related to the management of register data, and to Lars Pedersen from The Department of Clinical Epidemiology at Aarhus University Hospital for providing helpful advice regarding epidemiological issues. The authors would like to thank the following for assistance in the retrieval of medical records: Anita Højholt from The Department of Neurology at Aarhus University Hospital, Birgitte Forsom Sandal from The Department of Neurology at Holstebro Regional Hospital West Jutland, Friedrich Emanuel Schlesinger from The Department of Neurology at Viborg Regional Hospital Central Jutland, Hatice Tankisi from The Department of Neurophysiology at Aarhus University Hospital, John Rosendahl Østergaard from The Department of Pediatrics at Aarhus University Hospital, Thomas Helweg Thelle from The Department of Pediatrics at Viborg Regional Hospital Central Jutland, and Lene Vels Pedersen from The Department of Pediatrics at Herning Regional Hospital West Jutland.
Disclosure
The authors report no conflicts of interest in this work.
References
Harding AE, Thomas PK. The clinical features of hereditary motor and sensory neuropathy types I and II. Brain. 1980;103(2):259–280. | ||
Timmerman V, Strickland AV, Zuchner S. Genetics of Charcot-Marie-Tooth (CMT) disease within the frame of the human genome project success. Genes (Basel). 2014;5(1):13–32. | ||
Barreto LC, Oliveira FS, Nunes PS, et al. Epidemiologic study of Charcot-Marie-Tooth disease: a systematic review. Neuroepidemiology. 2016;46(3):157–165. | ||
Mladenovic J, Milic Rasic V, Keckarevic Markovic M, et al. Epidemiology of Charcot-Marie-Tooth disease in the population of Belgrade, Serbia. Neuroepidemiology. 2011;36(3):177–182. | ||
Braathen GJ, Sand JC, Lobato A, Hoyer H, Russell MB. Genetic epidemiology of Charcot-Marie-Tooth in the general population. Eur J Neurol. 2011;18(1):39–48. | ||
Lynge E, Sandegaard JL, Rebolj M. The Danish National Patient Register. Scand J Public Health. 2011;39(Suppl 7):30–33. | ||
Sorensen HT, Sabroe S, Olsen J. A framework for evaluation of secondary data sources for epidemiological research. Int J Epidemiol. 1996;25(2):435–442. | ||
Statistics Denmark. Available from: http://www.statistikbanken.dk/. Accessed June 1, 2016. | ||
Pedersen CB. The Danish Civil Registration System. Scand J Public Health. 2011;39(Suppl 7):22–25. | ||
Nielsen EH, Lindholm J, Laurberg P. Use of combined search criteria improved validity of rare disease (craniopharyngioma) diagnosis in a national registry. J Clin Epidemiol. 2011;64(10):1118–1126. | ||
De Jonghe P, Timmerman V, Van Broeckhoven C. 2nd Workshop of the European CMT Consortium: 53rd ENMC International Workshop on Classification and Diagnostic Guidelines for Charcot-Marie-Tooth Type 2 (CMT2-HMSN II) and Distal Hereditary Motor Neuropathy (distal HMN-Spinal CMT) 26-28 September 1997, Naarden, The Netherlands. Neuromuscul Disord. 1998;8(6):426–431. | ||
Haites NE, Nelis E, Van Broeckhoven C. 3rd workshop of the European CMT consortium: 54th ENMC International Workshop on genotype/phenotype correlations in Charcot-Marie-Tooth type 1 and hereditary neuropathy with liability to pressure palsies 28-30 November 1997, Naarden, The Netherlands. Neuromuscul Disord. 1998;8(8):591–603. | ||
Murphy SM, Herrmann DN, McDermott MP, et al. Reliability of the CMT neuropathy score (second version) in Charcot-Marie-Tooth disease. J Peripher Nerv Syst. 2011;16(3):191–198. | ||
Loong TW. Understanding sensitivity and specificity with the right side of the brain. BMJ. 2003;327(7417):716–719. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.