Back to Journals » Drug Design, Development and Therapy » Volume 17
Ursolic Acid Ameliorated Neuronal Damage by Restoring Microglia-Activated MMP/TIMP Imbalance in vitro
Authors Qiu L, Wang Y, Wang Y, Liu F, Deng S, Xue W, Wang Y
Received 7 March 2023
Accepted for publication 5 August 2023
Published 22 August 2023 Volume 2023:17 Pages 2481—2493
DOI https://doi.org/10.2147/DDDT.S411408
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Yan Zhu
Luying Qiu,1 Yaxuan Wang,2 Yuye Wang,1,3 Fang Liu,1 Shumin Deng,1 Weishuang Xue,1 Yanzhe Wang1
1Department of Neurology, Key Laboratory for Neurological Big Data of Liaoning Province, The First Affiliated Hospital of China Medical University, Shenyang, People’s Republic of China; 2Department of Anesthesiology, The First Affiliated Hospital of China Medical University, Shenyang, People’s Republic of China; 3Department of Neurology, China-Japan Friendship Hospital, Peking Union Medical College and Chinese Academy of Medical Sciences, Beijing, People’s Republic of China
Correspondence: Yanzhe Wang, Department of Neurology, Key Laboratory for Neurological Big Data of Liaoning Province, The First Affiliated Hospital of China Medical University, No. 155, Nanjing North Street, Heping District, Shenyang, Liaoning Province, 110001, People’s Republic of China, Tel +86-15040289417, Email [email protected]
Purpose: The oxygen and glucose deprivation-reoxygenation (OGDR) model is widely used to evaluate ischemic stroke and cerebral ischemia-reperfusion (I/R) injury in vitro. Excessively activated microglia produce pro-inflammatory mediators such as matrix metalloproteinases [MMPs] and their specific inhibitors, tissue inhibitors of metalloproteinases [TIMPs], causing neuronal damage. Ursolic acid (UA) acts as a neuroprotective agent in the rat middle cerebral artery occlusion/reperfusion (MCAO/R) model keeping the MMP/TIMP balance with underlying mechanisms unclear. Our study used OGDR model to determine whether and how UA reduces neuronal damage by reversing MMP/TIMP imbalance caused by microglia in I/R injury in vitro.
Methods: SH-SY5Y cells were first cultured with 95% N2 and 5% CO2 and then cultivated regularly for OGDR model. Cell viability was tested for a proper UA dose. We established a co-culture system with SH-SY5Y cells and microglia-conditioned medium (MCM) stimulated by lipopolysaccharide (LPS) and interferon-gamma (IFNγ). MMP9 and TIMP1 levels were measured with ELISA assay to confirm the UA effect. We added recombinant MMP9 (rMMP9) and TIMP1 neutralizing antibody (anti-TIMP1) for reconfirmation. Transmission electron microscopy was used to observe cell morphology, and flow cytometry and Annexin V-FITC and PI labeling for apoptotic conditions. We further measured the calcium fluorescence intensity in SH-SY5Y cells.
Results: The MCM significantly reduced cell viability of SH-SY5Y cells after OGDR (p< 0.01), which was restored by UA (0.25 μM) (p< 0.05), whereas lactate dehydrogenase activity, intraneuronal Ca2+ concentration, and apoptosis-related indexes were showed significant improvement after UA treatment (p< 0.01). UA corrected the MMP/TIMP imbalance by decreasing MMP9 expression and increasing TIMP1 expression in the co-culture system (p< 0.01) and the effects of UA on SH-SY5Y cells were mitigated by the administration of rMMP9 and anti-TIMP1 (p< 0.01).
Conclusion: We demonstrated that UA inhibited microglia-induced neuronal cell death in an OGDR model of ischemic reperfusion injury by stabilizing the MMP9/TIMP1 imbalance.
Keywords: glucose deprivation-reoxygenation, matrix metalloproteinase, microglia, neuroinflammation, oxygen, ursolic acid
Introduction
Ischemic stroke, which is defined by thromboembolic blockage of the cerebral artery, is a condition with a high rate of morbidity, disability, recurrence, and fatality.1 When the blood supply is restored, the condition typically worsens, leading to a post-ischemia brain damage (often referred to as an ischemia/reperfusion (I/R) injury).2 Numerous studies conducted yet have mostly examined methods that attempted to stop the harmful processes to avoid ischemia neuronal death.3–5 However, no clinical trials were successful up to now.6–8 It implies that single target therapy may not be sufficient to protect neurons. Even greater consideration should be given to finding innovative therapeutics that can successfully lower excessive neuroinflammation in cerebral I/R injury in acute ischemic stroke.
After an acute ischemic stroke, microglia, the primary resident immuno-modulatory cells in the central nervous system (CNS), are quickly activated and release various inflammatory mediators and cytotoxic substances (such as tumor necrosis factor [TNF]-α, interleukin [IL]-1, and matrix metalloproteinases [MMPs]).9,10 MMPs and their specific inhibitors, tissue inhibitors of metalloproteinases (TIMPs), act in a coordinated fashion to regulate several pathological processes. MMP9, one of the most widely investigated MMPs, is a type IV collagenase known to encourage neuroinflammation and hastens neuronal degeneration.11,12 TIMP-1 is functionally relevant as a crucial MMP9 regulator since it has been shown to compromise the MMP9/TIMP1 ratio, whose imbalance is a key factor in the etiology of ischemic stroke.13,14 Recent evidence has further shown that the infarct size was reduced in MMP9-deficient mice compared with control mice, and this finding suggests that excessive activation of MMP9 is deleterious to the brain and that MMP9 inhibitors may serve as therapeutic agents for ischemia and reperfusion injury.15,16
Ursolic acid (UA, 3b-hydroxy-urs-12-ene-28-oic acid) was found to play a neuroprotective role in many neurological disorders. Researchers proved that UA could alleviates blood–brain barrier disruption in traumatic brain injuries.17 In Parkinson’s disease, UA functioned on mitochondrial and reducing dopaminergic neurons loss.18 In our earlier research, we found that the naturally occurring pentacyclic triterpenoid UA showed an anti-inflammatory action that could keep the MMP/TIMP balance of the ischemic penumbra in the rat middle cerebral artery occlusion/reperfusion (MCAO/R) model.19 However, based on the evidences we found, the link between the neuroprotective effects of UA and the microglial-inflammation suppression has to be further examined in vitro because active microglia are the main cellular contributors to the MMP/TIMP imbalance.
Therefore, as depicted in the schematic diagram (Figure 1), the goal of our investigation was to determine whether and how UA reduces neuronal damage by reversing the imbalance in MMP/TIMP caused by microglia in brain I/R injury. We activated BV2 cells with lipopolysaccharide (LPS)/interferon gamma (IFNγ) like the activation of microglia in brain I/R injury and collected the microglia-conditioned medium (MCM) containing the released cytokines to establish an SH-SY5Y co-culture system. As a well-known cerebral I/R in vitro model, we used oxygen and glucose deprivation reoxygenation (OGDR) treated SH-SY5Y. UA was applied for observation. The study flowchart is shown in Figure 2. We investigated that how UA affected activated microglia that released MMP9 and TIMP1 as well as the effects of recombinant MMP9 treatment or TIMP1 neutralization used in co-culture systems to explore the underlying mechanism.
 |
Figure 1 Schematic diagram of this study. |
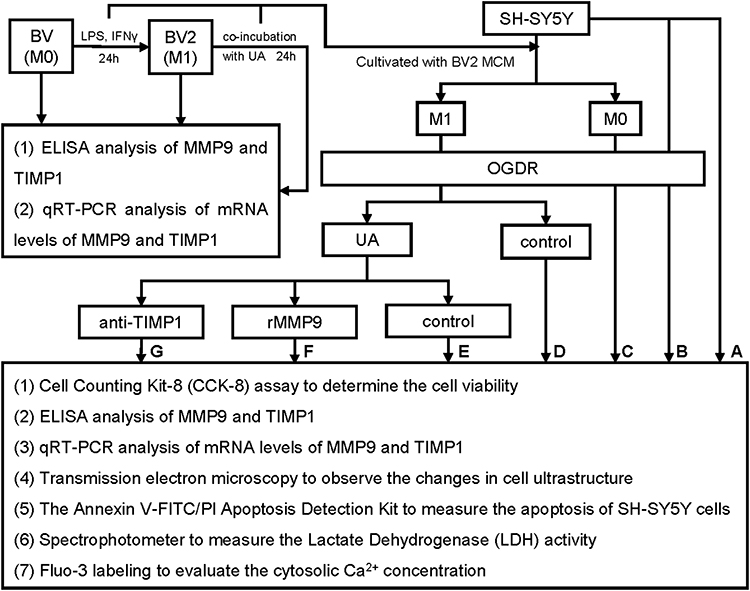 |
Figure 2 Flowchart of this study. Abbreviation: MCM, microglia-conditioned medium. Notes: (A) Control, (B): Model, (C) M0, (D) M1, (E) M1+UA, (F) M1+UA+rMMP9, (G) M1+UA+TIMP1. |
Materials and Methods
Cell Cultures
BV2 cells (murine microglial cells, CBP60922) were purchased from CoBioer Biosciences Co., Ltd. (Nanjing, China). The BV2 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, Grand Island, NY, United States) supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 IU/mL penicillin G (Invitrogen, Carlsbad, CA, United States), and 100 g/mL penicillin-streptomycin (PS, Invitrogen, Carlsbad, CA, United States). The human neuroblastoma cell line SH-SY5Y was obtained from the American Type Culture Collection (ATCC, United States). Cells were cultivated in Nutrient Mixture Ham’s F-12 (DMEM/F12, 1:1) medium containing 10% (v/v) FBS and 100 g/mL PS in a humidified environment with 5% CO2 and 95% air at 37°C.
Grouping
SH-SY5Y cells were cultivated into seven groups from A-G with different treatments (Figure 2): (A) Control: Non-treated SH-SY5Y cells, (B) Model: OGDR-exposed cells, (C) M0: OGDR-exposed cells in M0-MCM, (D) M1: OGDR-exposed cells in M1-MCM, (E) M1+UA OGDR-exposed cells in M1-MCM pre-treated with UA, (F) M1+UA+rMMP9: OGDR-exposed cells in M1-MCM plus rMMP9 pre-treated with UA, (G) M1+UA+TIMP1: OGDR-exposed cells in M1-MCM plus anti-TIMP1 pre-treated with UA.
Microglial Stimulation
LPS (100 ng/mL, Sigma Aldrich, St. Louis, MO, United States) and IFNγ (50 ng/mL, Sigma Aldrich, St. Louis, MO, United States) were administered to BV2 cells in order to promote the M1 phenotype.20 The BV2 cells were stimulated for 24 hours to stimulate from M0 to M1 phenotypes, then UA (quality 99.8%, Selleck Chemicals, Shanghai, China) diluted in dimethyl sulfoxide (DMSO, Sigma Aldrich, St. Louis, MO, United States) was added to the culture medium for additional 24 hours. Subsequently, microglia-conditioned medium (MCM) was collected and stored at −80°C for upcoming experiments. MCM and SH-SY5Y cells were used to create the co-culture system. The cells were cultured at 37°C for additional 24 hours before to OGDR.21,22
The OGDR Model
The OGDR model was constructed in SH-SY5Y cells as previously described.23,24 SH-SY5Y cells were first cultured in DMEM without sugar or serum at a density of 2×106 cells/60 mm dish under a specialized chamber (MCO-170MUVHL-PC) with 95% N2 and 5% CO2 for 8 hours. Then, cells were regularly cultivated (95% air, 5% CO2) for another 24 hours.
Recombinant MMP9 Protein and TIMP1 Neutralizing Antibody
The mouse recombinant MMP9 protein (2 g/mL) was used to treat the cells in the co-culture system as previously described.25 The cells were further treated with a TIMP1 neutralizing antibody (3 g/mL) (R&D System, Minneapolis, MN, United States).26
Cell Viability Assay
The SH-SY5Y cell viability after OGDR-induced injury was determined by using the Cell Counting Kit-8 (CCK-8) assay. The SH-SY5Y cells were seeded in a 96-well plate at a density of 1×105 cells/well. Briefly, the CCK-8 buffer (10 μL, Beyotime Institute Biotech., Beijing, China) was added into each well. The cells were incubated at 37°C for 2 hours. The T cells were washed in DMEM and 10 µL of CCK-8 solution was added to each well for incubation. Subsequently, the optical density (OD) of each well was measured at a wavelength of 450 nm using a spectrophotometer (BioTek Instruments, Inc., Winooski, VT, United States). The cell viability was calculated according to the following formula: cell viability (%) = [(OD sample − OD blank) − (OD control − OD blank)] / (OD control − OD blank) × 100%.27
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Around 24 hours after the last treatment, the total RNA was extracted from SH-SY5Y cells using the Trizol reagent (Sangon Biotech, China). The reverse transcription was performed using the Prime Script RT reagent kit (Takara Bio). The qRT-PCR was performed using the TB Green Premix Ex Taq kit (Takara Bio). The transcript abundance was normalized to the transcription of the endogenous control ACTB gene. The 2−ΔΔCt method was used to determine relative RNA abundance. The qRT-PCR experiments were run in triplicate to ensure accurate results. The amplification primers were as follows:
Mouse MMP9 Forward: TGGGACCATCATAACATCAC
Reverse: ATGACAATGTCCGCTTCG
Mouse TIMP1 Forward: TGGGAAATGCCGCAGATA
Reverse: GCCAGGGAACCAAGAAGC
Mouse ACTB Forward: CTGTGCCCATCTACGAGGGCTAT
Reverse: TTTGATGTCACGCACGATTTCC
Enzyme-Linked Immunosorbent Assay (ELISA)
The MMP9 and TIMP1 concentrations in cell culture supernatants were analyzed by using commercially available ELISA kits (Abcam, Cambridge, UK). After processing the raw data, results are provided as the total protein concentration (ng/mL).
Observation of the Changes in Cell Ultrastructure
After the cells were subjected to the test conditions, they were fixed in glutaraldehyde (at a concentration of 2.5%). To observe changes in cell ultrastructure using a Libra200 microscope, samples were dehydrated, soaked, and embedded before being cut and stained for transmission electron microscopy (TEM) (Zeiss GmbH, Jena, Germany).
Apoptosis Assay
The apoptosis of SH-SY5Y cells was measured by using the Annexin V-FITC/PI Apoptosis Detection Kit (BD Biosciences, CA). A total of 1×106 cells were seeded into 6-well plates for 12 hours. Then, cells were pre-treated with α-cyperone for 2 hours and treated with H2O2 for the following 24 hours. After two gentle washes with ice-cold PBS, cells were collected and centrifuged at 1500 rpm for 5 minutes at 4°C. Subsequently, the apoptotic cell percentage was determined by using the flow cytometry.
Lactate Dehydrogenase (LDH) Assay
Around 24 hours after the last treatment, the release of lactate dehydrogenase (LDH) into the medium was measured using a colorimetric assay (Pierce LDH assay kit, Thermo Scientific, Waltham, United States). The procedure was performed according to the manufacturer’s instructions. The LDH activity was measured at 490 nm using a spectrophotometer (FLUOstar Omega, BMG Labtech, Ortenberg, Germany). The experiment was run in triplicate.
Cytosolic Calcium Concentration
Fluo-3 acetomethoxyester (Fluo-3 AM, Molecular Probes) was used to label Ca2+ (Eugene, OR, United States). Hydrolysis of Fluo-3 AM produced Fluo-3, which was used as an indicator for cytosolic Ca2+. Cells were washed twice in PBS and then incubated with Fluo-3 AM (4 M) in FBS-free media at 37°C for 40 minutes in the dark. Then, cells were washed twice with PBS. Flow cytometry was used to determine how brightly Fluo-3 fluoresced.
Statistical Analysis
The Prism 9.0 software was used for statistical analysis (Graph Pad Software, Inc., United States). When comparing three or more groups, we used the one-way analysis of variance (ANOVA). When comparing two groups, we utilized the unpaired Student’s t-test. The data were presented as means and standard deviations. The p value < 0.05 was used to denote statistical significance.
Results
UA Ameliorated the OGDR-Induced Injury in Microglia-Conditioned SH-SY5Y Cells
To find a proper concentration of UA for treatment, SH-SY5Y cells were treated with UA at various doses (0.5, 1, 5, 10, 20, 40 and 60 µM) for 24 hours. The CCK-8 assay was used to determine the cytotoxicity of UA by measuring the concentration at which 50% of SH-SY5Y cells were killed (LC50 = 60.87 µM) (Figure 3A). Then, we evaluated the effects of UA on SH-SY5Y survival in the OGDR model. Around 24 hours after OGDR, the SH-SY5Y cell viability dropped to 66.6 ± 8.3%. The SH-SY5Y cell viability was not influenced by UA treatment at the dose under 2.5µM (Figure 3B). We created a microglia-conditioned co-culture system to further evaluate the neuroprotective effects of UA on SH-SY5Ycells after OGDR. The experimental flowchart is displayed in Figure 4A and B. The treatment of SH-SY5Y cells with MCM isolated from M1 microglia (M1-MCM) significantly reduced cell viability to 41.9 ± 5.5% (## p < 0.01 vs M0 group) (Figure 4C). The cell viability of SH-SY5Y cells treated with UA (0.25, 0.5, 1, and 2.5 µM) was higher than the cell viability of SH-SY5Y cells treated with M1-MCM (M1 group). By treating the cells with UA (0.25 µM), the cell viability was restored to 55.6 ± 4.6% (** p < 0.05 vs M1 group) (Figure 4C). UA protected SH-SY5Y cells from the OGDR-induced injury in a microglia-conditioned co-culture system. We chose to utilize 0.25 µM of UA in the following experiments.
 |
Figure 3 Effects of UA on the viability of SH-SY5Y cells. Abbreviations: UA, ursolic acid; LC50, half-maximal lethal concentration; OGDR, oxygen and glucose deprivation reoxygenation. Notes: (A) Toxicity of UA in SH-SY5Y cells. (B) 24 hours after OGDR, the survival rates of SH-SY5Y cell lines treated with different UA concentrations (0.5, 1.5, 10, 20, 40 or 60 µM) for 24 hours were evaluated. The cell viability is normalized to controls. Data are expressed as mean ± standard deviation. |
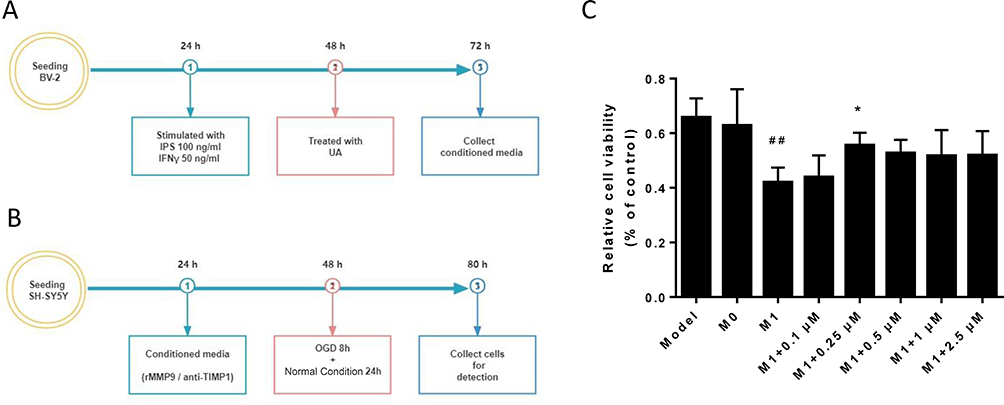 |
Figure 4 UA effects of the conditioned medium from LPS/IFNγ-stimulated BV2 cells in SH-SY5Y cells. Abbreviations: UA, ursolic acid; LPS, lipopolysaccharide; IFNγ, interferon gamma; rMMP9, recombinant metalloproteinase 9 protein; Anti-TIMP1, anti-tissue metallopeptidase inhibitor 1; M1-MCM, M1 microglia-conditioned medium; OGDR, oxygen and glucose deprivation reoxygenation. Notes: (A) Experimental flowchart of BV2 cells. (B) Experimental flowchart of SH-SY5Y cells. (C) 24 hours after OGDR, the survival rates of SH-SY5Y cells in M1-MCM with different concentrations of UA (0.1, 0.25, 0.5, 1 and 2.5 μM) are displayed. The cell viability is normalized to controls. Data are expressed as mean ± standard deviation. ##p < 0.01 vs the model group, *p < 0.05 vs the M1 group. |
UA Mitigated the Microglia-Induced MMP/TIMP Imbalance
The microglia-induced MMP/TIMP imbalance was investigated by measuring the release of MMP9 from activated BV2 cells. After 24 hours of co-incubation with UA (0.25 μM), BV2 cells were treated with LPS (100 ng/mL) and IFNγ (50 ng/mL). The ELISA assay was used to measure the MMP9 levels. Stimulated BV2 cells showed a more than 4-fold increase in the expression levels of MMP9 (M1 group, ## p < 0.01), whereas UA decreased the MMP9 expression by 43% (M1 + UA group, ** p < 0.01) (Figure 5A). The mRNA levels of MMP9 increased 4.76-fold in activated BV2 cells (## p < 0.01). UA administration reversed the increased mRNA levels of MMP9 (** p < 0.01, Figure 5B). After UA treatment, the mRNA levels of TIMP1 increased 1.93-fold (** p < 0.01) and the expression level of TIMP1 increased 2.28-fold (** p < 0.01) (Figure 5C and D). After microglia activation, the MMP9/TIMP1 ratio increased more than 13-fold (## p < 0.01), which was reversed by UA administration (** p < 0.01) (Figure 5E). UA corrected the MMP/TIMP imbalance by inhibiting MMP9 and upregulating TIMP1. We used a recombinant MMP9 protein (rMMP9) and a TIMP1 neutralizing antibody (anti-TIMP1) to further investigate the MMP/TIMP imbalance in microglia-conditioned SH-SY5Y cells. The M1-MCM augmented the levels of MMP9 and decreased the levels of TIMP1 (M1 group vs control group, ## p < 0.01) (Figure 6A and B). Levels of MMP9 and TIMP1 were significantly altered by UA (0.25 μM) (M1+UA group vs M1 group ** p < 0.01, Figure 6A and B). The ELISA results showed that the MMP9/TIMP1 ratio increased 5.09-fold in the rMMP9 group (▲▲ p < 0.01, vs the UA group) and 4.59-fold in the anti-TIMP1 group (▲ p < 0.05, vs the UA group) (Figure 6C), demonstrating that the MMP/TIMP imbalance was aggravated by administering rMMP9 anti-TIMP1.
 |
Figure 5 Effects of UA on the MMP9/TIMP1 ratio in BV2 cells. Abbreviations: UA, ursolic acid; MMP9, metalloproteinase 9; TIMP1, tissue metallopeptidase inhibitor 1. Notes: (A and B) ELISA analysis of MMP9 (A) and TIMP1 (B) in conditioned medium from M1 microglial cells treated with UA (0.25 μM). (C and D) qRT-PCR analysis of mRNA levels of MMP9 (C) and TIMP1 (D). (E) The mRNA level ratio of MMP9 to TIMP1. Data represent the mean ± standard error of the mean of 3 independent experiments. ##p < 0.01 vs the M0 group, **p < 0.01 vs the M1 group. |
 |
Figure 6 Effects of UA on the MMP9/TIMP1 ratio in a microglia-conditioned SH-SY5Y co-culture system. Abbreviations: UA, ursolic acid; MMP9, metalloproteinase 9; TIMP1, tissue metallopeptidase inhibitor 1; rMMP9, recombinant MMP9 protein; MCM, microglia-conditioned medium. Notes: (A and B) ELISA analysis of MMP9 (A) and TIMP1 (B) in MCM. (C) The levels of MMP9/TIMP1 ratio. Data represent the mean ± standard error of the mean of 3 independent experiments. ##p < 0.01 vs the M0 group, **p < 0.01 vs the M1 group, ▲p < 0.05 vs the M1 + UA group, ▲▲p < 0.01 vs the M1 + UA group. |
UA Attenuated the OGDR-Induced Injury by Restoring the MMP/TIMP Imbalance
Transmission electron microscopy showed nuclear convolution, chromatin margination and condensation along the nuclear membrane in model and M0 groups. M1-treated cells displayed morphological aberrations such as nuclear condensation, nuclear fragmentation and membrane blebbing. The UA treatment partially restored the morphology of M1-treated cells, while the administration of rMMP9 and anti-TIMP1 prevented these effects (Figure 7). According to the CCK-8 assay, SH-SY5Y cells treated with M1-MCM (M1 group) reported a significantly reduced cell viability (42.5 ± 7.7%, ## p < 0.01 vs the model group) (Figure 8A). The UA group showed significantly higher cell viability than the M1 group (** p < 0.01) (Figure 8A). The administration of rMMP9 and anti-TIMP1 reduced the effects of UA (▲▲ p < 0.01) (Figure 8A). The LDH expression increased 5-fold after OGDR in SH-SY5Y cells. The cells treated with M1-MCM showed a further increase in LDH expression (## p < 0.01 vs the model group) (Figure 8B). In accordance with the results of the cell viability assay, the UA group showed a decreased LDH expression (** p < 0.01) (Figure 8B). The administration of rMMP9 or anti-TIMP1 prevented the aforementioned UA effects (▲▲ p < 0.01 vs the UA group) (Figure 8B).
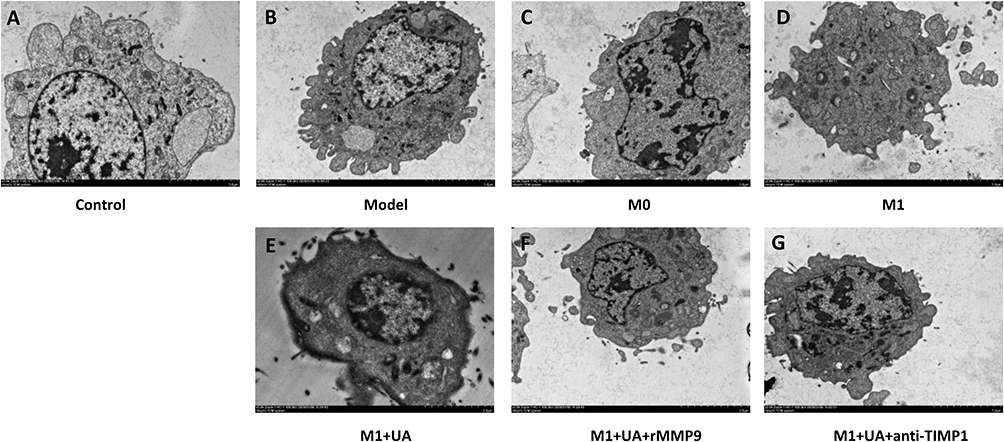 |
Figure 7 Transmission electron microscopy images of SH-SY5Y cells. Abbreviations: UA, ursolic acid; rMMP9, recombinant metalloproteinase 9 protein; Anti-TIMP1, Anti-tissue metallopeptidase inhibitor 1; MCM, microglia-conditioned medium; OGDR, oxygen and glucose deprivation reoxygenation. Notes: (A) Non-treated SH-SY5Y cells. (B) OGDR-exposed cells. (C) OGDR-exposed cells in M0-MCM. (D) OGDR-exposed cells in M1-MCM. (E) OGDR-exposed cells in M1-MCM pre-treated with UA (0.25 μM, 24 hours). (F) OGDR-exposed cells in M1-MCM plus rMMP9 pre-treated with UA (0.25 μM, 24 hours). (G) OGDR-exposed cells in M1-MCM plus anti-TIMP1 pre-treated with UA (0.25 μM, 24 hours). Magnification ×3000. |
 |
Figure 8 UA augmented the viability of SH-SY5Y cells by restoring the MMP/TIMP imbalance in a microglia-conditioned co-culture system. Abbreviations: UA, ursolic acid; rMMP9, recombinant metalloproteinase 9 protein; Anti-TIMP1, anti-tissue metallopeptidase inhibitor 1; MCM, microglia-conditioned medium; LDH, lactate dehydrogenase. Notes: (A) The survival rate of SH-SY5Y cells treated with MCM. (B) LDH release of SH-SY5Y cells treated with MCM. Data represent the mean ± standard error of the mean of 3 independent experiments. ##p < 0.01 vs the M0 group, **p < 0.01 vs the M1 group, ▲▲p < 0.01 vs the M1 + UA group. |
We determined the apoptotic rate in each group by using flow cytometry and Annexin V-FITC and PI labeling (Figure 9). Cells were divided into four groups: early apoptotic (Annexin positive/PI negative), necrotic (Annexin negative/PI positive), late apoptotic (Annexin positive/PI positive) and viable (Annexin negative/PI negative) groups. Compared to the control group, the percentage of SH-SY5Y cells at the early stage of apoptosis increased from 2.4% to 22.1% in the OGDR group, while the percentage of those cells at the late stage of apoptosis increased from 1.6% to 13.3% (Figure 9A and B). The apoptotic rates in SH-SY5Y cells drastically increased to 24.7% (early apoptotic) and 25.0% (late apoptotic) after treatment with M1-MCM (Figure 9D). Conversely, the apoptotic rate of cells at the late stage of apoptosis diminished from 22.0% to 13.6% after UA treatment (Figure 9E). The apoptotic rates of cells at the early stage of apoptosis increased to 37.6% and 29.3%, respectively, when the rMMP9 or anti-TIMP1 were used. Consistently, cells at the late stage of apoptosis showed decreased apoptotic rates (19.6% and 24.2%) (Figure 9F and G). The flow cytometry showed that UA reduced apoptosis of SH-SY5Y cells in a microglia-conditioned co-culture system by restoring the MMP/TIMP imbalance.
 |
Figure 9 Effects of UA on SH-SY5Y cell apoptosis detected by flow cytometry. Abbreviations: UA, ursolic acid; rMMP9, recombinant metalloproteinase 9 protein; anti-TIMP1, anti-tissue metallopeptidase inhibitor 1. Notes: (A–G) Representative plots of apoptosis detected by flow cytometry in each group. (H) Histograms and statistical results. Data represent the mean ± standard error of the mean of 3 independent experiments. ##p < 0.01 vs the M0 group, **p < 0.01 vs the M1 group, ▲▲p < 0.01 vs the M1 + UA group. |
We further measured the calcium fluorescence intensity in SHSY5Y cells to evaluate the cytosolic Ca2+ concentration and assess its contribution to UA-mediated effects (Figure 10). After M1-MCM treatment, the cytosolic Ca2+ was increased in SHSY5Y cells compared to the control group. This increase was greatly attenuated by UA treatment (Figure 10A and E). We also found that the administration of rMMP9 or anti-TIMP1 reversed the UA-induced changes (Figure 9F and G). It is possible that the anti-apoptotic effects of UA were achieved through decreasing the cytosolic Ca2+.
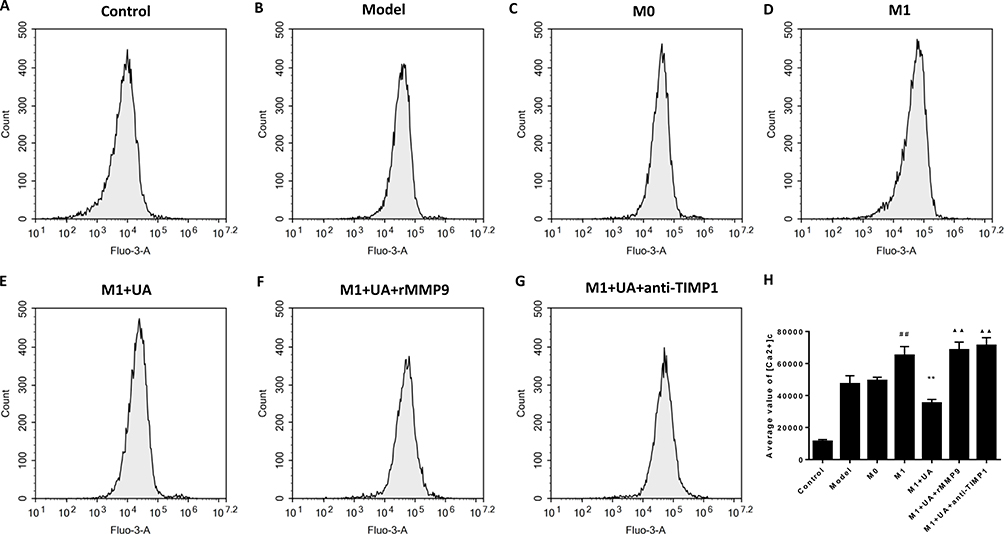 |
Figure 10 Effects of UA on SH-SY5Y cytosolic Ca2+ detected by flow cytometry. Abbreviations: UA, ursolic acid; rMMP9, recombinant metalloproteinase 9 protein; Anti-TIMP1, anti-tissue metallopeptidase inhibitor 1. Notes: (A–G) Intracellular calcium in each group. (H) Representative histograms of cytosolic Ca2+ measured by flow cytometry in each group. Data represent the mean ± standard error of the mean of 3 independent experiments. ##p < 0.01 vs the M0 group, **p < 0.01 vs the M1 group, ▲▲p < 0.01 vs the M1 + UA group. |
Discussion
In this study, we showed that UA attenuated the OGDR-induced injury, inhibited microglial activation, and reduced subsequent neuronal death in SHSY5Y cells and MCM co-culture system. The neuroprotective effects of UA were abolished in the presence of an MMP/TIMP imbalance. These findings suggested that UA suppressed neuroinflammation by restoring the MMP/TIMP imbalance and protecting neurons from the deleterious effects of inflammation. Targeting the microglial-induced MMP/TIMP imbalance might be a new therapeutic strategy against ischemic stroke, and these anti-inflammatory effects of UA may help with the microenvironment recovery.
Many different plants contain UA, a natural pentacyclic triterpenoid with demonstrated neuroprotective benefits.28 After reperfusion of ischemic tissues, we previously demonstrated that an intraperitoneal administration of UA improved neurological functions and reduced the infarct volume.19,29 Consistently with previous studies, we found that UA ameliorated the OGDR-induced injury in human SH-SY5Y cells. UA protected against cell death and apoptosis by decreasing LDH and intraneuronal Ca2+ concentrations. Although the mechanisms are still unclear, UA might be therapeutic against ischemia/reperfusion injury.
Meanwhile, we discovered that UA had no direct protective effects on SH-SY5Y survival. Based on these observations, we postulated that UA might have a positive effect on immunological microenvironment optimization. Because of their central role in the immune system, activated microglia are responsible for controlling the neuronal immunological milieu following an ischemic stroke by causing the production of a plethora of pro-inflammatory markers.30 Following an ischemic stroke, activated microglia are responsible for the production of several pro-inflammatory molecules. To determine the effects of UA on activated microglia, we created a co-culture system with SH-SY5Y neurons and MCM from BV2 cells pre-treated with LPS and IFN-γ, two powerful microglial activators.31 We showed that UA prevented the loss of SH-SY5Y neurons by the conditioned media from activated BV2 cells, suggesting that the neuroprotective potential of UA prevents excessive microglial activation.
We also showed that UA reduced MMP9 levels and increased TIMP1 levels in BV2 cells. Previous studies suggested that UA could block the vicious cycle of inflammation, leading to a continuous release of pro-inflammatory mediators.19,32,33 The hypothesized mechanisms involve many different signaling pathways, including the NF-κB, MAPK and ERK1/2 pathways.19,34,35 The proteolytic enzyme MMP9 is involved in the aforementioned pathways and has been studied as a prognostic factor in ischemic stroke.36 Of note, the stroke infarct size was reduced in MMP9-deficient mice.16,37,38 In this study, we found that UA reduced apoptosis of SH-SY5Y cells by restoring the MMP/TIMP imbalance, whereas the concomitant administration of rMMP9 increased the apoptotic rate. Overall, our findings demonstrated that UA inhibited microglia activation by restoring the MMP/TIMP imbalance, thereby protecting neurons against OGDR-induced injury, which is a possible reason why UA aids in treating strokes. For the first time, we demonstrate that UA can modulate the immunological milieu by controlling the activation of microglia and suppressing metalloproteinases.
However, this study is affected by some limitations. We employed a BV2 cell model to investigate the microglia activation in vitro. N9 and BV2 cell lines have been used to study microglia in many experimental settings.39,40 Previous in vitro and in vivo studies showed that BV2 cell lines and primary microglia share a number of phenotypic traits.41,42 However, further studies utilizing primary microglia are required to confirm our findings as BV2 cell lines are rather dissimilar to primary microglia in culture or the CNS. Secondly, the precise mechanisms underlying the correction of the MMP/TIMP imbalance remain unknown. The molecular docking approach might be used to predict the interactions between the involved molecular structures. However, it is possible that other proteins play a role in the adjustment of the MMP/TIMP imbalance. In the future, a probe with a peculiar chemical structure might be used to minimize the likelihood of off-target effects and find more about the components of this signaling pathway that contribute to SH-SY5Y cell protection. Thirdly, microglia and astrocytes play essential roles in the central nervous system contributing to many functions, including homeostasis, immune response, blood–brain barrier maintenance and synaptic support.43,44 More studies are needed to determine the comprehensive effects of UA on microglia and astrocytes.
Conclusion
In conclusion, this study demonstrated that UA inhibited microglia-induced neuronal cell death by stabilizing the MMP/TIMP imbalance in an OGDR model of ischemia/reperfusion injury. These effects of UA appeared to restore physiological neuroimmune interactions, indicating that UA might be a novel therapeutic target for neuroinflammatory disorders beyond ischemic stroke.
Abbreviations
OGDR, Oxygen and glucose deprivation-reoxygenation; I/R, ischemia-reperfusion; MMPs, matrix metalloproteinases; TIMPs, tissue inhibitors of metalloproteinases; MCAO/R, middle cerebral artery occlusion/reperfusion; UA, ursolic acid; MCM, microglia-conditioned medium; CNS, central nervous system; TNF, tumor necrosis factor; IL, interleukin; LPS, lipopolysaccharide; IFNγ, interferon gamma; FBS, fetal bovine serum; DMEM, Dulbecco’s modified Eagle’s medium; PS, penicillin-streptomycin; ATCC, American Type Culture Collection; DMSO, dimethyl sulfoxide; CCK-8, Cell Counting Kit-8; OD, optical density; TEM, transmission electron microscopy; LDH, Lactate Dehydrogenase; ANOVA, analysis of variance.
Acknowledgments
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. We acknowledge TopEdit LLC for the linguistic editing and proofreading during the preparation of this manuscript.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This study was supported by the National Natural Science Foundation of China, No. 81901189 (belongs to YZW) and the National Natural Science Foundation of China, No. 82201480 (belongs to WSX). The funding sources had no role in study conception and design, data analysis or interpretation, paper writing or deciding to submit this paper for publication.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Shah ASV, Lee KK, Perez JAR, et al. Clinical burden, risk factor impact and outcomes following myocardial infarction and stroke: a 25-year individual patient level linkage study. Lancet Reg Health Eur. 2021;7:100141. doi:10.1016/j.lanepe.2021.100141
2. Campbell BCV, Khatri P. Stroke. Lancet. 2020;396(10244):129–142. doi:10.1016/S0140-6736(20)31179-X
3. Chovsepian A, Berchtold D, Winek K, et al. A primeval mechanism of tolerance to desiccation based on glycolic acid saves neurons in mammals from ischemia by reducing intracellular calcium-mediated excitotoxicity. Adv Sci. 2022;9(4):e2103265. doi:10.1002/advs.202103265
4. Ni XC, Wang HF, Cai YY, et al. Ginsenoside Rb1 inhibits astrocyte activation and promotes transfer of astrocytic mitochondria to neurons against ischemic stroke. Redox Biol. 2022;54:102363. doi:10.1016/j.redox.2022.102363
5. Xu J, Zhao J, Wang R, et al. Shh and Olig2 sequentially regulate oligodendrocyte differentiation from hiPSCs for the treatment of ischemic stroke. Theranostics. 2022;12(7):3131–3149. doi:10.7150/thno.69217
6. Ahmed N, Nasman P, Wahlgren NG. Effect of intravenous nimodipine on blood pressure and outcome after acute stroke. Stroke. 2000;31(6):1250–1255. doi:10.1161/01.str.31.6.1250
7. Investigators RBT, Qiu Z, Li F, et al. Effect of intravenous tirofiban vs placebo before endovascular thrombectomy on functional outcomes in large vessel occlusion stroke: the RESCUE BT randomized clinical trial. JAMA. 2022;328(6):543–553. doi:10.1001/jama.2022.12584
8. Toul M, Mican J, Slonkova V, et al. Hidden potential of highly efficient and widely accessible thrombolytic staphylokinase. Stroke. 2022:101161STROKEAHA122040219. doi:10.1161/STROKEAHA.122.040219
9. Wang Y, Leak RK, Cao G. Microglia-mediated neuroinflammation and neuroplasticity after stroke. Front Cell Neurosci. 2022;16:980722. doi:10.3389/fncel.2022.980722
10. Wicks EE, Ran KR, Kim JE, Xu R, Lee RP, Jackson CM. The translational potential of microglia and monocyte-derived macrophages in ischemic stroke. Front Immunol. 2022;13:897022. doi:10.3389/fimmu.2022.897022
11. Leonardo CC, Hall AA, Collier LA, Gottschall PE, Pennypacker KR. Inhibition of gelatinase activity reduces neural injury in an ex vivo model of hypoxia-ischemia. Neuroscience. 2009;160(4):755–766. doi:10.1016/j.neuroscience.2009.02.080
12. Liou CJ, Yang CM, Lee TH, Liu PS, Hsieh HL. Neuroprotective effects of dehydroepiandrosterone sulfate through inhibiting expression of matrix metalloproteinase-9 from bradykinin-challenged astroglia. Mol Neurobiol. 2019;56(1):736–747. doi:10.1007/s12035-018-1125-6
13. Mashaqi S, Mansour HM, Alameddin H, et al. Matrix metalloproteinase-9 as a messenger in the cross talk between obstructive sleep apnea and comorbid systemic hypertension, cardiac remodeling, and ischemic stroke: a literature review. J Clin Sleep Med. 2021;17(3):567–591. doi:10.5664/jcsm.8928
14. Liu S, Liu J, Wang Y, et al. Differentially expressed genes induced by beta-caryophyllene in a rat model of cerebral ischemia-reperfusion injury. Life Sci. 2021;273:119293. doi:10.1016/j.lfs.2021.119293
15. Sanchez-Blazquez P, Pozo-Rodrigalvarez A, Merlos M, Garzon J. The sigma-1 receptor antagonist, S1RA, reduces stroke damage, ameliorates post-stroke neurological deficits and suppresses the overexpression of MMP-9. Mol Neurobiol. 2018;55(6):4940–4951. doi:10.1007/s12035-017-0697-x
16. Svedin P, Hagberg H, Sävman K, Zhu C, Mallard C. Matrix metalloproteinase-9 gene knock-out protects the immature brain after cerebral hypoxia-ischemia. J Neurosci. 2007;27(7):1511–1518. doi:10.1523/jneurosci.4391-06.2007
17. Gong QY, Cai L, Jing Y, et al. Urolithin A alleviates blood-brain barrier disruption and attenuates neuronal apoptosis following traumatic brain injury in mice. Neural Regenerat Res. 2022;17(9):2007–2013. doi:10.4103/1673-5374.335163
18. Qiu J, Chen Y, Zhuo J, et al. Urolithin A promotes mitophagy and suppresses NLRP3 inflammasome activation in lipopolysaccharide-induced BV2 microglial cells and MPTP-induced Parkinson’s disease model. Neuropharmacology. 2022;207:108963. doi:10.1016/j.neuropharm.2022.108963
19. Wang Y, He Z, Deng S. Ursolic acid reduces the metalloprotease/anti-metalloprotease imbalance in cerebral ischemia and reperfusion injury. Drug Des Devel Ther. 2016;10:1663–1674. doi:10.2147/dddt.S103829
20. Natunen TA, Gynther M, Rostalski H, Jaako K, Jalkanen AJ. Extracellular prolyl oligopeptidase derived from activated microglia is a potential neuroprotection target. Basic Clin Pharmacol Toxicol. 2019;124(1):40–49. doi:10.1111/bcpt.13094
21. Zhou X, Spittau B. Lipopolysaccharide-induced microglia activation promotes the survival of midbrain dopaminergic neurons in vitro. Neurotox Res. 2018;33(4):856–867. doi:10.1007/s12640-017-9842-6
22. Yin X, Feng L, Ma D, et al. Roles of astrocytic connexin-43, hemichannels, and gap junctions in oxygen-glucose deprivation/reperfusion injury induced neuroinflammation and the possible regulatory mechanisms of salvianolic acid B and carbenoxolone. J Neuroinflammation. 2018;15(1):97. doi:10.1186/s12974-018-1127-3
23. Wang R, Bao H, Zhang S, Li R, Chen L, Zhu Y. miR-186-5p promotes apoptosis by targeting IGF-1 in SH-SY5Y OGD/R model. Int J Biol Sci. 2018;14(13):1791–1799. doi:10.7150/ijbs.25352
24. Liang J, Yu Y, Wang B, et al. Ginsenoside Rb1 attenuates oxygen-glucose deprivation-induced apoptosis in SH-SY5Y cells via protection of mitochondria and inhibition of AIF and cytochrome c release. Molecules. 2013;18(10):12777–12792. doi:10.3390/molecules181012777
25. Ventayol M, Vinas JL, Sola A, et al. miRNA let-7e targeting MMP9 is involved in adipose-derived stem cell differentiation toward epithelia. Cell Death Dis. 2014;5:e1048. doi:10.1038/cddis.2014.2
26. Souvenir R, Fathali N, Ostrowski RP, Lekic T, Zhang JH, Tang J. Tissue inhibitor of matrix metalloproteinase-1 mediates erythropoietin-induced neuroprotection in hypoxia ischemia. Neurobiol Dis. 2011;44(1):28–37. doi:10.1016/j.nbd.2011.05.020
27. Lin L, Que Y, Lu P, et al. Chidamide inhibits acute myeloid leukemia cell proliferation by lncRNA VPS9D1-AS1 downregulation via MEK/ERK signaling pathway. Front Pharmacol. 2020;11:569651. doi:10.3389/fphar.2020.569651
28. Huang P, Wan H, Shao C, Li C, Zhang L, He Y. Recent advances in Chinese herbal medicine for cerebral ischemic reperfusion injury. Front Pharmacol. 2021;12:688596. doi:10.3389/fphar.2021.688596
29. Wang Y, Li L, Deng S, Liu F, He Z. Ursolic acid ameliorates inflammation in cerebral ischemia and reperfusion injury possibly via high mobility group Box 1/Toll-Like Receptor 4/NFkappaB pathway. Front Neurol. 2018;9:253. doi:10.3389/fneur.2018.00253
30. Rauchmann BS, Brendel M, Franzmeier N, et al. Microglial activation and connectivity in Alzheimer disease and aging. Ann Neurol. 2022;92(5):768–781. doi:10.1002/ana.26465
31. Nguyen HM, Grossinger EM, Horiuchi M, et al. Differential Kv1.3, KCa3.1, and Kir2.1 expression in “classically” and “alternatively” activated microglia. Glia. 2017;65(1):106–121. doi:10.1002/glia.23078
32. Slate JR, Chriswell BO, Briggs RE, McGill JL. The effects of ursolic acid treatment on immunopathogenesis following mannheimia haemolytica infections. Front Vet Sci. 2021;8:782872. doi:10.3389/fvets.2021.782872
33. Meng RY, Jin H, Nguyen TV, Chai OH, Park BH, Kim SM. Ursolic acid accelerates paclitaxel-induced cell death in esophageal cancer cells by suppressing Akt/FOXM1 signaling cascade. Int J Mol Sci. 2021;22(21):11486. doi:10.3390/ijms222111486
34. Njau F, Haller H. Calcium dobesilate modulates PKCdelta-NADPH Oxidase- MAPK-NF-kappaB signaling pathway to reduce CD14, TLR4, and MMP9 expression during monocyte-to-macrophage differentiation: potential therapeutic implications for atherosclerosis. Antioxidants. 2021;10:11.
35. Kim H, Roh Y, Yong Park S, et al. In vitro and in vivo anti-tumor efficacy of krill oil against bladder cancer: involvement of tumor-associated angiogenic vasculature. Food Res Int. 2022;156:111144. doi:10.1016/j.foodres.2022.111144
36. Zhang M, Meng X, Pan Y, et al. Predictive values of baseline matrix metalloproteinase 9 levels in peripheral blood on 3-month outcomes of high-risk patients with minor stroke or transient ischemic attack. Eur J Neurol. 2022;29:2976–2986. doi:10.1111/ene.15342
37. Morancho A, Rosell A, García-Bonilla L, Montaner J. Metalloproteinase and stroke infarct size: role for anti-inflammatory treatment? Ann N Y Acad Sci. 2010;1207:123–133. doi:10.1111/j.1749-6632.2010.05734.x
38. Fujimoto M, Takagi Y, Aoki T, et al. Tissue inhibitor of metalloproteinases protect blood-brain barrier disruption in focal cerebral ischemia. J Cereb Blood Flow Metab. 2008;28(10):1674–1685. doi:10.1038/jcbfm.2008.59
39. Lee J, Kim YS, Choi DH, et al. Transglutaminase 2 induces nuclear factor-kappaB activation via a novel pathway in BV-2 microglia. J Biol Chem. 2004;279(51):53725–53735. doi:10.1074/jbc.M407627200
40. Bi XL, Yang JY, Dong YX, et al. Resveratrol inhibits nitric oxide and TNF-alpha production by lipopolysaccharide-activated microglia. Int Immunopharmacol. 2005;5(1):185–193. doi:10.1016/j.intimp.2004.08.008
41. Henn A, Lund S, Hedtjarn M, Schrattenholz A, Porzgen P, Leist M. The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. ALTEX. 2009;26(2):83–94. doi:10.14573/altex.2009.2.83
42. Crotti A, Benner C, Kerman BE, et al. Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat Neurosci. 2014;17(4):513–521. doi:10.1038/nn.3668
43. Yew WP, Djukic ND, Jayaseelan JSP, Woodman RJ, Muyderman H, Sims NR. Differential effects of the cell cycle inhibitor, olomoucine, on functional recovery and on responses of peri-infarct microglia and astrocytes following photothrombotic stroke in rats. J Neuroinflammation. 2021;18(1):168. doi:10.1186/s12974-021-02208-w
44. Spurgat MS, Tang SJ. Single-Cell RNA-sequencing: astrocyte and microglial heterogeneity in health and disease. Cells. 2022;11(13):2021. doi:10.3390/cells11132021
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.