Back to Journals » Journal of Inflammation Research » Volume 14
Urinary Trypsin Inhibitor Protects Tight Junctions of Septic Pulmonary Capillary Endothelial Cells by Regulating the Functions of Macrophages
Authors Wang R, Song W, Xie C, Zhong W ![]() , Xu H, Zhou Q, Deng Y
, Xu H, Zhou Q, Deng Y ![]() , Hong Y, Li X, Fang M
, Hong Y, Li X, Fang M
Received 28 January 2021
Accepted for publication 19 March 2021
Published 17 May 2021 Volume 2021:14 Pages 1973—1989
DOI https://doi.org/10.2147/JIR.S303577
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ning Quan
Ruijie Wang,1,2,* Wenliang Song,3,* Chengyuan Xie,2,4,* Wenhong Zhong,2 Hui Xu,2,5 Qiuping Zhou,1,2 Yiyu Deng,2 Yimei Hong,2 Xin Li,2 Ming Fang1,2
1Department of Intensive Care Unit, Guangdong Provincial People’s Hospital, Guangdong Academy of Medical Sciences, School of Medicine, South China University of Technology, Guangzhou, People’s Republic of China; 2Department of Emergency and Critical Care Medicine, Guangdong Provincial People’s Hospital, Guangdong Academy of Medical Sciences, Guangzhou, People’s Republic of China; 3Department of Critical Care Medicine, The First Affiliated Hospital of Sun Yat-Sen University, Guangzhou, 510080, People’s Republic of China; 4The Second School of Clinical Medicine, Southern Medical University, Guangzhou, People’s Republic of China; 5Shantou University Medical College, Shantou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Ming Fang
Department of Intensive Care Unit, Guangdong Provincial People’s Hospital, Guangdong Academy of Medical Sciences, School of Medicine, South China University of Technology, Guangzhou, 510080, People’s Republic of China
Tel +8613527774075
Email [email protected]
Background: Our previous study found that urinary trypsin inhibitor (ulinastatin, UTI) protected tight junctions (TJs) of lung endothelia via TNF-α inhibition, thereby alleviating pulmonary capillary permeability in septic rats. As the activated macrophage is the main source of TNF-α in sepsis, we speculate that UTI may exert the above effects by regulating the functions of macrophages.
Methods: Bone-marrow derived macrophages (BMDM) were divided into control, lipopolysaccharide (LPS), UTI+LPS and UTI groups. TNF-α, TGF-β, IL-10, CD86, CD206 and MCP-1 expression were assessed by Western blot. The phagocytosis and migration of BMDM were detected. Pulmonary microvascular endothelial cells (PMVECs) were cultured with the conditioned medium (CM) from each group of BMDM above. Sprague-Dawley rats were divided into sham, cecal ligation and puncture (CLP), and UTI+CLP groups. Western blot and immunofluorescence were used to detected zonula occludens-1 (ZO-1), occludin and claudin-5 expression in PMVECs, as well as TNF-α, TGF-β, iNOS, CD86 and CD206 expression in lungs. Pulmonary capillary permeability was assessed by extravasated Evans blue, lung injury score (LIS), wet-to-dry weight ratio and electron microscope.
Results: TNF-α and CD86 expression were increased in LPS-treated BMDM, but were reversed by UTI pretreatment. TGF-β, IL-10 and CD206 expression were the opposite. UTI markedly decreased phagocytosis and migration of LPS-treated BMDM. ZO-1, occludin and claudin-5 expression were markedly decreased in PMVECs of the CM-LPS group, but significantly increased in the CM-UTI+LPS group. TNF-α, iNOS and CD86 expression were increased in the lungs of CLP-rats but decreased with UTI pretreatment, while TGF-β and CD206 expression were the opposite. UTI markedly ameliorated the lung EB leakage, improved LIS, reduced the wet-to-dry ratio and revised the damaged TJs of PMVECs in CLP-rats.
Conclusion: UTI effectively inhibits the conversion of M1 macrophage but increases M2, reduces the phagocytosis and migration, which helps to protect endothelia TJs and reduce pulmonary capillary permeability during sepsis.
Keywords: ulinastatin, sepsis, macrophage, pulmonary capillary endothelial cells, tight junctions
Introduction
At present, sepsis continues to be one of the most frequent conditions encountered in intensive care units,1 and lung injury is one of the most frequent consequences of sepsis.2 Acute respiratory distress syndrome (ARDS) is the most severe form of lung injury caused by sepsis, characterized by lung epithelial and endothelial cell injury. Generally, pulmonary microvascular endothelial cells (PMVECs) line the luminal surface of the vasculature and are tightly held, providing a translucent membrane between alveolar tissue and circulating blood. Due to overwhelmed inflammatory response in sepsis, the disrupted endothelial barrier can lead to increased permeability of pulmonary tissue and result in neutrophil influx and alveolar flooding,3 which contributes to acute pulmonary edema and leads to high mortality.4
Tight junctions (TJs) between the endothelium play an important role in the acellular permeability of the endothelial barrier.5 The TJs in endothelial cells are composed of occludin, claudins and zonula occludens (ZO-1, ZO-2, and ZO-3),6 which interact to seal the intercellular space and maintain vascular homeostasis.7 A lot of evidence supports the fact that TJs of endothelial cells decrease in sepsis and result in increased paracellular permeability in the brain and umbilical veins.8
Urinary trypsin inhibitor (ulinastatin, UTI), which is isolated and purified from human urine,9 was proven to have a protective role against sepsis-induced multi-organ injuries, such as septic ARDS.10 Our previous study demonstrated that UTI could attenuate the permeability of pulmonary capillary endothelial cells in sepsis via protecting the TJs between the endothelium, which attributed to inhibition of the release of tumor necrosis factor-α (TNF-α).11
TNF-α initiated inflammatory cascade and destroyed the alveolar capillary barrier,12 leading to increased endothelial barrier permeability and pulmonary edema. As we know, the pro-inflammatory cytokines, such as TNF-α, are mainly derived from macrophage but not endothelial cells in septic lungs. Whether UTI exerts its effects by regulating the function of macrophages is worth considering. Macrophages are monocytes derived from innate immune cells, which exist in most human body tissues and display high plasticity and function as regulating tissue homeostasis and adaptive responses. Generally, there are two types of macrophage: proinflammatory (M1) and anti-inflammatory (M2).13 As reported, the ratio of M1 to M2 macrophage is highly regulated in normal tissues.14 The expression profile of M1 macrophages is that they characteristically release high levels of TNF-α, interleukin-6 (IL-6), monocyte chemoattractant protein-1 (MCP-1) and inducible nitric oxide synthase (iNOS). M2 macrophages express high levels of arginase and IL-10, which play an important role in tissue remodeling and repair.15,16
In this study, LPS-stimulated primary macrophages of mice, conditioned medium from bone-marrow derived macrophages (BMDM), PMVECs and cecal ligation and puncture (CLP)-induced sepsis model in rats were adopted to demonstrate that UTI ameliorates pulmonary capillary leakage in sepsis through regulating the transformation of M1 macrophage to M2, as well as the functions of macrophages.
Materials and Methods
Isolation of BMDM
The tibias and femurs of 6 C57Bl/6 mice were used to isolate BMDM, according to standard protocols,17,18 that is, using a syringe containing the medium and a 21G needle to flush bone marrow from bone cavity. The medium was composed of Roswell Park Memorial Institute (RPIM) 1640 liquid medium and contained 10% fatal bovine serum (FBS, Gibco, Life Technologies, United States, 10099–141). Primary macrophages were seeded onto 25 cm2 plate. They were cultured for 7 days with RPMI-1640 medium, which contained macrophage colony-stimulating factor (100 ng/mL, Abcam, United Kingdom, ab129146).
Cell Culture
Primary mouse BMDM were cultured in 25 cm2 culture flasks with an RPMI-1640 medium containing 10% fetal bovine serum (FBS, Gibco, Life Technologies, United States, 10099–141) at 37°C in humidified 5% CO2/95% air. The medium was partially changed every 48 h. Primary rat PMVECs (Bena Culture Collection, Suzhou, China, BNCC338210) were cultured in a 25 cm2 plate with the complete medium at 37°C in humidified 5% CO2/95% air. There were 3 components in the complete medium: Dulbecco’s Modified Eagle Medium (DMEM, Gibco, Thermo Fisher Scientific, United States, C11995500BT), 1% antibiotic (Gibco, Life Technologies, United States, 15140–122) and 10% fetal bovine serum (FBS). The medium was partially changed every 48 h.
Cell Viability Assay
The viability of BMDM was assessed by Cell Counting kit-8 (CCK-8, Beyotime, C0038). To determine the cytotoxic effect of UTI and LPS, BMDM were cultured in 96-well microplates (105 cells/well), treated with lipopolysaccharide (LPS, Sigma, United States, L-2880) (ranging from 0.01 to 2 µg/mL), UTI (Ulinastatin for Injection, Techpool, Guangdong, China) (ranging from 500 to 104 U/mL) for 0h, 3h, 6h and 9h in triplicates. After removing the medium and adding 90 µL of new basic medium and 10 µL CCK8 solution to each well, the plate was incubated for another 3 hours. The optical density at 450 nm was then read with a microplate reader. The experiment was performed 3 times.
BMDM Groups and Treatments
The BMDM (6.5×105 cells/well) were divided randomly into four groups: control, LPS, UTI+LPS and UTI. It was reported that 5000U/mL UTI was used on BMDM19 and 104U/mL on PMVECs.19 We used 104 U/mL UTI for the in vitro experiments in this study. In the control, BMDM were cultured with basic a medium (RPIM-1640) for 8 h, whereas the cells in the UTI group were treated with UTI (104 U/mL) for 2 h and then cultivated with a new basic medium for 6 h. In the UTI+LPS group, cells were pretreated with UTI (104 U/mL) for 2 h and then treated with LPS (1µg/mL) for 6 h, while BMDM in LPS were incubated to LPS (1µg/mL) for 6 h. The cells in the 6-well plates were then collected for immunofluorescence and protein extraction.
Preparation of BMDM Conditioned Medium
Three kinds of conditioned medium were prepared at 24 h after BMDM (6.5×105 cells/well) seeded in 6-well plates: (1) BMDM conditioned medium (CM): BMDM were incubated with 2 mL basic medium (DMEM medium) for 8 h; (2) BMDM conditioned medium + LPS (CM-LPS): BMDM were cultured with 2 mL basic medium containing 1µg/mL LPS for 6 h; (3) BMDM conditioned medium with UTI pretreatment + LPS (CM-UTI+LPS): BMDM were pretreated with 2 mL medium with UTI (104 U/mL) for 2 h, then the supernatant was discarded and it was cultured with 1µg/mL LPS for 6 h. Before use, the 3 conditioned mediums should be collected and filtered with a 0.22 mm syringe filter.
Treatment of PMVECs with BMDM Conditioned Medium
PMVECs were seeded on coverslips in 6-well plates (3.5×105 cells/well) with the complete medium (DMEM containing 10% FBS) for 10–14 days until they were confluent. Every 48 h, the medium was partially replaced. PMVECs were divided into 3 groups: CM, CM-LPS, CM-UTI+LPS. Then PMVECs were cultured in the corresponding conditioned medium of BMDM for 48 hours.
Ethical Use of Animals
The use and handling of rats for CLP or isolated for BMDM all followed the ethical guidelines stated in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Animal use and experimental protocols were approved by the approval authority at Jinan University (No. 20180225016). We made our best effort to reduce the number of rats and their suffering.
Animals and Treatments
Fifty adult male Sprague-Dawley rats (Table 1) weighing 220–280 g were obtained from the Animal Experiment Center of Jinan University. They were fed with water adlibitum and a standard laboratory diet. The rats were randomly divided into 3 groups: sham, CLP and UTI+CLP. We used the CLP method to construct a sepsis rat model according to our previous experiment: in brief, after intraperitoneal injection of 3% sodium pentobarbital (30 mg/kg) anesthesia, the abdominal cavity was opened to expose the cecum and then, using an 18G needle in the middle part with 3–0 silk thread ligation, it was perforated 5 times (exposing a small amount of feces). After the cecum was reset, the abdomen was sutured with 2–0 silk suture. The rats were then resuscitated using a subcutaneous injection of saline (5 mL/100 g; 37°C). The health and general condition (hair, mobility, weight, posture, alertness and startle reflex) of the rats were evaluated every 8 h after CLP surgery. In the sham operation group, the rats underwent a similar operation, but the cecum was neither ligated nor perforated. For the purpose of minimizing the differences among the experiments, CLP surgery was performed by the same researcher. Within 48 h after the CLP procedure, all 18 rats in the sham group survived; in the CLP group, 13 rats were successfully modeled and 7 rats died; and in the UTI+CLP group, 15 rats were successfully modeled and 3 rats died. The daily dose range of UTI was 105~106U/day for patients in the clinic.20 To find the corresponding dose range in rats, the body surface area of a human should be converted to that of a rat. The ratio of a rat’s (200g) body surface area to a human's (70kg) is 1/56,21 thus the calculation formula for the dose of a 200g rat is as follows: 105~106U×0.018/0.2kg=0.9~9×104 U/kg. In this study, we treated SD rats with 5×104 U/kg UTI for vivo experiment, as reported in other studies.22
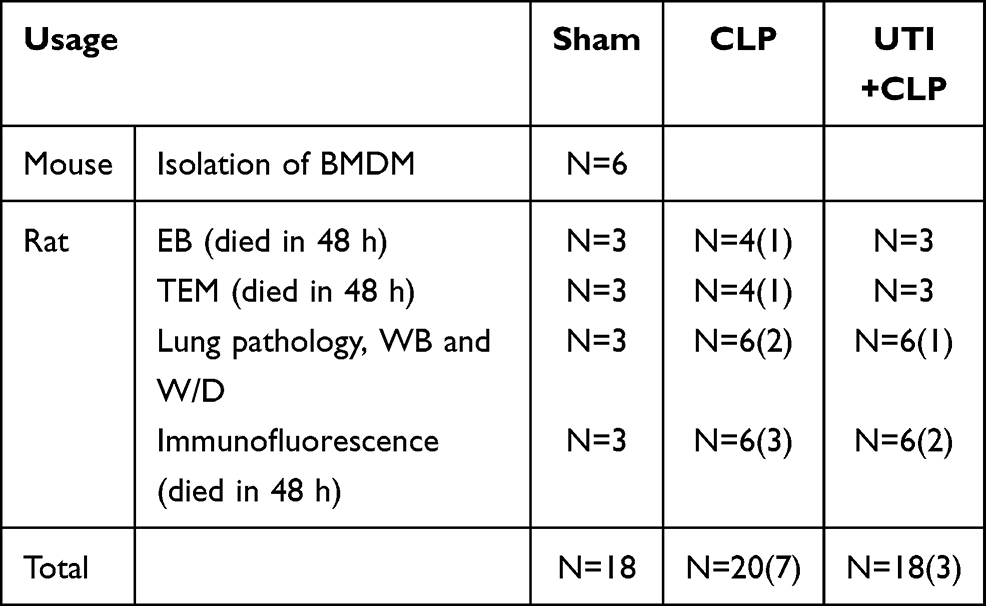 |
Table 1 Number of Animals Used in Various Treatments |
Rats in the UTI+CLP group were given UTI dissolved in 2 mL 0.9% saline (China Guangdong Dexing Biochemical Pharmaceutical Co, Ltd.) (5×104 U/kg) via an indwelling vein needle (24G, BD) every 24 h, at 72 h before the CLP procedure. In the CLP group, the rats were injected with saline, while the rats in the sham group received no treatment. All rats were euthanized at 48 h after CLP.
Western Blotting
For different groups of BMDM, after discarding the medium, the cells were washed with 1×PBS more than twice; a lysis buffer was used to lyse cells. Then a rubber scraper was used to scrap off the cells, and cell debris were centrifuged at 15,000 rpm for 15 min to collect the supernatant. For a different group of PMVECs, after being incubated with the conditioned medium for 48 h, the old medium was discarded and the same treatment with BMDM was performed. For different groups of rats, the left lung was stored in liquid nitrogen at −80°C. The total proteins were extracted with protein extraction reagents (Pierce; Thermo Fisher Scientific, Bloomingdale, Illinois, USA) that contain protease inhibitors.
The protein concentration of BMDM, PMVECs and tissues was assessed by the Pierce TM BCA protein assay kit (Thermo Fisher Scientific, United States, Product # 23227). After that, samples of 40 µg protein were heated to 100°C for 10 min. Then they were separated on 10% gels using sodium dodecyl sulfate-poly-acrylamide gel electrophoresis. The protein bands were electrically attracted to the polyvinylidene fluoride (PVDF) membrane. Subsequently, the membrane was blocked with 5% milk at room temperature for 1 h.
The membranes were incubated with TNF-α (Abcam, United Kingdom, ab66579), iNOS (Abcam, United Kingdom, ab49999), MCP-1 (Abcam, United Kingdom, ab25124), IL-10 (Abcam, United Kingdom, ab9969), β-Actin (13E5) rabbit mAb (Cell Signaling Technology, United States, #4970S), as well as rabbit anti-mannose receptor antibody (Abcam, United Kingdom, ab125028), TGF-β (Abcam, United Kingdom, ab31013), ZO-1 (Thermo Fisher Scientific, United States, #40-2200), claudin-5 (Thermo Fisher Scientific, United States, #35-2500), occludin (Thermo Fisher Scientific, United States, #71-1500), and CD86 antibody (Thermo Fisher Scientific, United States, #MA5-32078). Primary antibodies were diluted in 5% milk or 5% BSA overnight at 4°C. Then the membranes were incubated with secondary antibodies, such as anti-mouse IgG, HRP-linked antibody (Cell Signaling Technology, United States, 7076S) or anti-rabbit IgG, HRP-linked antibody (Cell Signaling Technology, United States, 7074S). A SuleLumia ECL kit (Abbkine, USA, K22020) was used to detect immunoblots. Image J software was used to quantify band intensity. Details of antibodies used are given in Table 2. The experiment was performed at least 3 times.
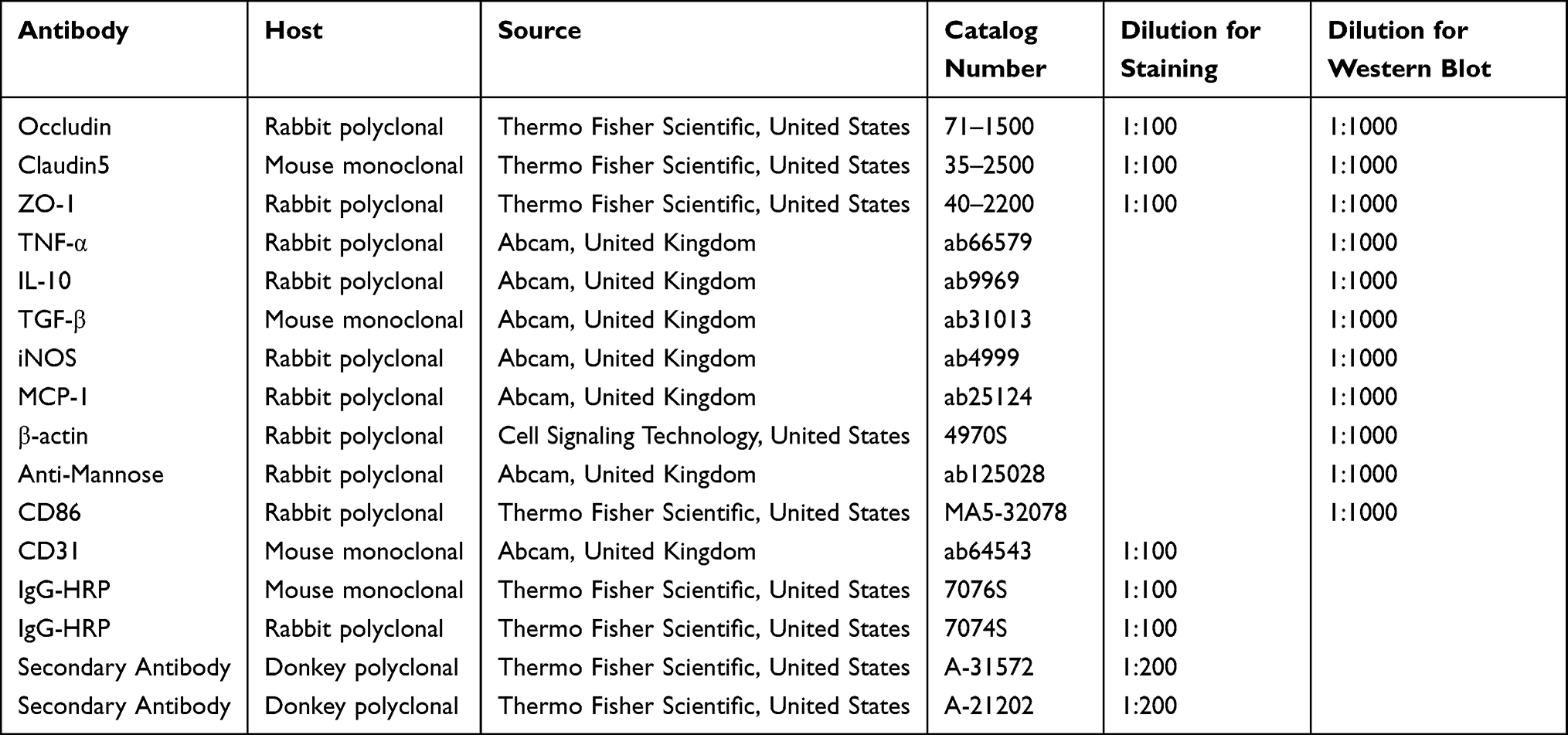 |
Table 2 Antibodies Used for Western Blotting and Immunofluorescence |
Phagocytosis Assay
BMDM were incubated in a 96-well plate and then treated with a phagocytosis assay kit (Abcam, United Kingdom, ab234053). Cells were treated as described previously. After 5µL of Zymosan slurry was added to all wells, the plate was incubated at 37°C and 5% CO2 for 2 h. After that, the plate was washed 3 times with an ice cold phagocytosis assay buffer. The plate was imaged using a fluorescence microscope. The experiment was performed at least 3 times.
Cell Migration Assay
BMDM was placed in a 24-well plate. Transwell™ permeable support polycarbonate membrane inserts (8.0 µm pore size; Costar, USA) were used to assess the effect of LPS and UTI on BMDM migration. First, 200μL of cell suspension (104 cells) was added to the inside of the insert. Second, the cells were processed as described previously. Third, after washing the cells with PBS 2 or 3 times, 500μL of the serum-free medium was added to the upper chamber and 500μL of the complete medium was added to the lower chamber. After incubating at 37°C and 5% CO2 for 24 h, the old medium was discarded and the remaining cells in the insert were carefully removed. Subsequently, the cells were fixed with 100% methanol and stained with 0.5% of crystal violet. The cells were washed with distilled water to remove the excess dye. The migrated cells were imaged using a microscope and ImageJ software was used to count the cells in the five fields of each insert. The experiment was performed at least three times.
Scratch Wound Assay
BMDM were incubated in 6-well plates with the complete medium. When they grew to confluence, a 200-μL pipette tip was used to form a scratch of the same width through the entire well. Subsequently, BMDM were washed carefully with PBS to remove cell debris. Then cells were cultured with a basic medium at 37°C in humidified 5% CO2/95% air. After 24 h, the migration of BMDM into the wound area was observed. The experiment was performed at least 3 times.
Double Immunofluorescence
After different treatments with the conditioned medium for 48 h, PMVECs were fixed in 4% paraformaldehyde and 0.1 M PBS for 15 min at room temperature. Then the cells were blocked with 10% donkey serum for 1 h, and each washed with 1×PBS 3 times for 5 min. Subsequently, the cells on the coverslips were incubated overnight with the corresponding primary antibodies at 4°C. Next, the cells were incubated with FITC/Cy3-conjugated secondary antibodies in the dark for 1 h at room temperature. The coverslips were each washed with 1×PBS 3 times for 5 min. DAPI was used to mount the coverslips. The images were captured at a magnification of 200×. The experiment was performed at least 3 times.
A total of 15 rats were used for double immunofluorescence labeling. They were anesthetized with 3% pentobarbital sodium (30 mg/kg) at 48 h after the CLP procedure. Then the rats were sacrificed using 2% paraformaldehyde. After gradient dehydration, the lung tissue was embedded with OCT and sectioned at 7µm thickness on a microtome (Model: 2165; Leica, Bensheim, Germany). Tissue sections were blocked with 5% normal goat serum for 1 h at room temperature and then incubated with primary antibodies at 4°C overnight. After 3 rinses with PBS, the sections were incubated with fluorescent secondary antibody at room temperature for 1 h. After washing in PBS, the sections were mounted with DAPI. The images were captured at a magnification of 400×. The experiment was performed at least 3 times.
Assessment of Pulmonary Capillary Leakage
Extravasated Evans blue (EB) was used to assess pulmonary capillary permeability. In brief, rats were anesthetized with 3% pentobarbital sodium (30 mg/kg) at 48 h after the CLP procedure. They were injected with 2.5% EB dissolved in 2 mL 0.9% saline (20 mg/kg) via the tail vein. After the EB injection, the rats were euthanized at 30 min. Next, the right lung tissue was weighed, homogenized and treated with formamide (4 mL/g [wet tissue]) at room temperature for 24 h. The samples were centrifuged for 30 min at 4000 g. A microplate reader (620 nm) was then used to detect dye absorbance values. The concentration of Evans blue was calculated from the absorbance according to a standard curve.
Transmission Electron Microscopy (TEM)
A total of 10 rats were subjected to TEM. The rat tissue was removed on ice, cut into 1 mm3 pieces and immersed in 2.5% glutaraldehyde. Then the tissue was subjected to the following procedures. First, 1% oxidizing fixative was used to fix it. Second, it was stained with 1% uranyl acetate. Finally, the tissue was dehydrated and immersed in a gradient acetone solution. The tissue was imaged using a transmission electron microscope. The experiments were repeated at least 3 times.
Histological Examination and Lung Injury Score (LIS)
A total of 15 rats rats were anesthetized with 3% pentobarbital sodium (30 mg/kg) at 48 h after the CLP procedure. The right tissue was isolated and fixed in 4% paraformaldehyde, embedded in paraffin and then cut into 4µm slices. The sections were routine-stained with hematoxylin-eosin (H&E). As shown in Table 3, the lung injury score (LIS) was quantified in a blinded manner.23
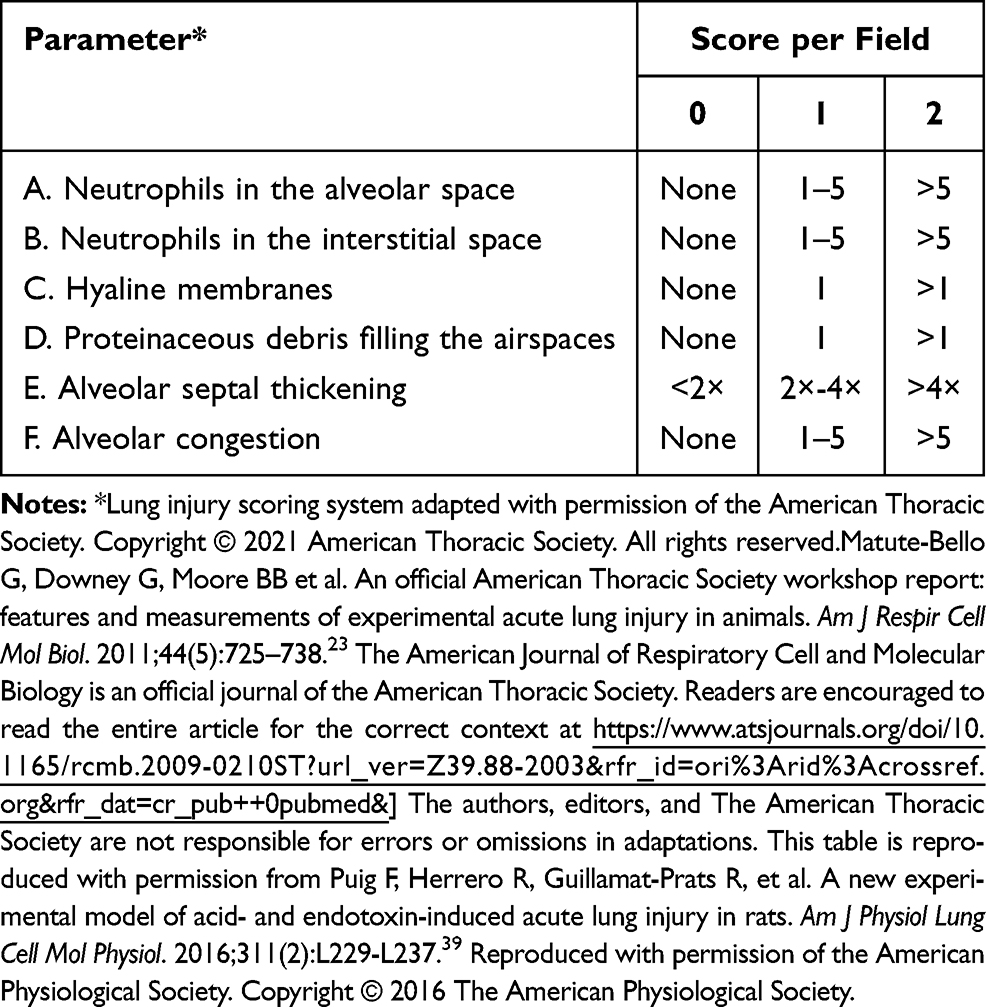 |
Table 3 Histological lung injury results |
Measurement of Lung Wet-to-Dry (W/D) Weight Ratio
The right lungs were weighed and then dried at 65°C for 72 h until their weight did not change. The ratio of dry-to-wet weight was calculated.
Statistical Analysis
All statistical analyses were performed using SPSS 16.0 statistical software. Data were presented as means ± SD. After the homogeneity test of variances, one-way analysis of variance (ANOVA) followed by multi-factor comparison of Tukey’s test were used to determine the statistical significance between different groups. All experiments were conducted at least 3 times with different groups. Differences were considered statistically significant when p < 0.05.
Results
Effects of UTI and LPS on Viability of BMDM
The MTS assay was used to obtain the cytotoxicity data for the effects of LPS and UTI on BMDM. When BMDM were treated with UTI at the concentration of 500–104 U/mL for 9 h (Figure 1B), and with LPS (10–2000 ng/mL) for 9 h (Figure 1A), cell viability remained relatively unchanged. Therefore, we used UTI (104 U/mL) for 2 h and LPS (1μg/mL) for 6 h for all subsequent in vitro analysis.
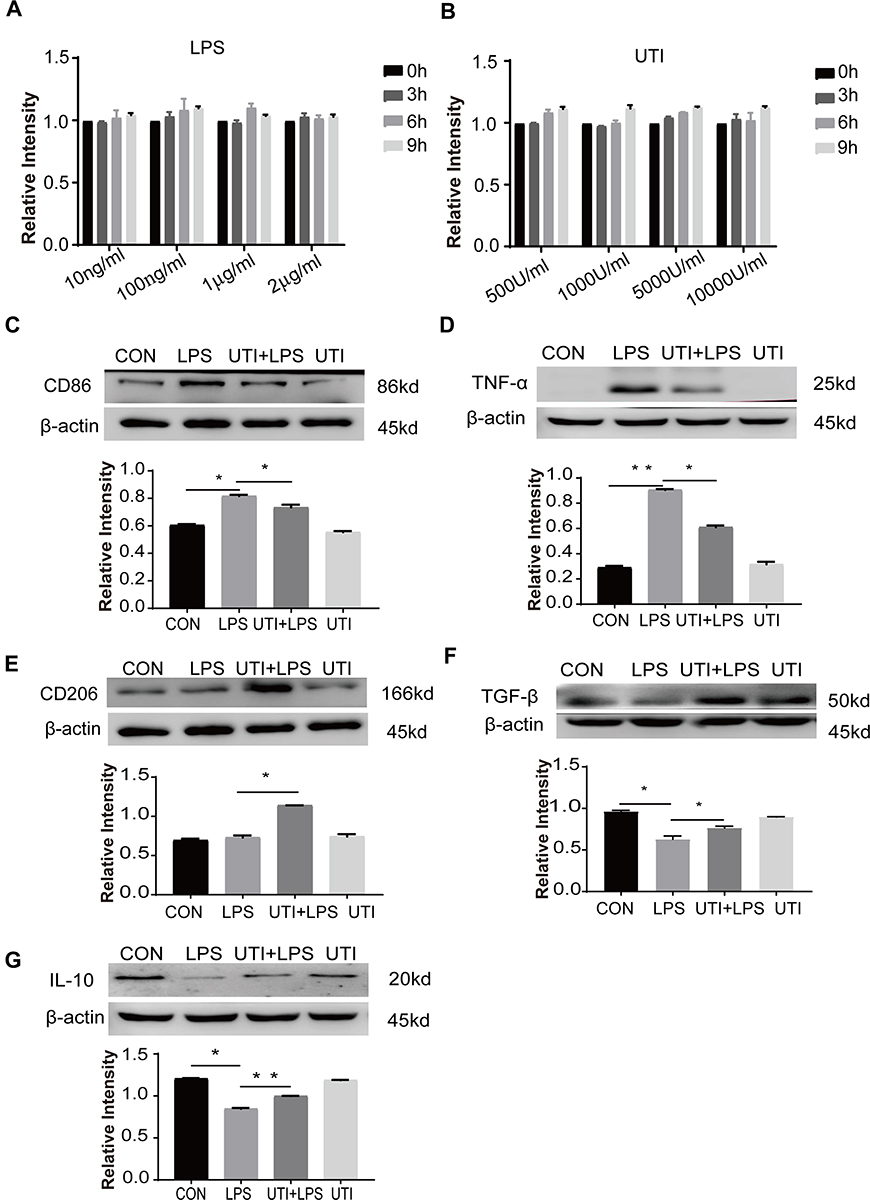 |
Figure 1 Effects of UTI and LPS on viability of BMDM. In the range of 10–2000 ng/mL, LPS (A) did not affect the cell viability at 0–9 h. UTI (B) at the concentration between 500 U/mL and 104 U/mL did not affect the viability of cells at 0–9 h. UTI regulated polarization of M1 to M2 macrophage when incubated with LPS. Western blot showing expression level of CD86 (C) and TNF-α (D) that was markedly increased in the LPS group when compared with the control group; it was significantly decreased when pretreated with UTI. CD206 (E), TGF-β (F) and IL-10 (G) expression was markedly decreased in the LPS group in comparison with the control group; it was augmented when pretreated with UTI. *p<0.05; **p<0.01. |
LPS Increased M1 Macrophages but Decreased by UTI
Western blot showed that markers of M1 macrophage CD86 (Figure 1C) and inflammatory factors TNF-α (Figure 1D) increased in the BMDM of the LPS group in comparison with the control group, while they decreased in response to UTI pretreatment. Markers of M2 macrophage CD206 (Figure 1E) and anti-inflammatory cytokines TGF-β and IL-10 (Figures 1F and 1G) decreased in the LPS group but increased in the UTI+LPS group.
LPS Increased the Phagocytosis of BMDM but UTI Reversed It
The phagocytosis of BMDM was significantly increased in the LPS group compared with the control group but decreased in the UTI+LPS group (Figure 2).
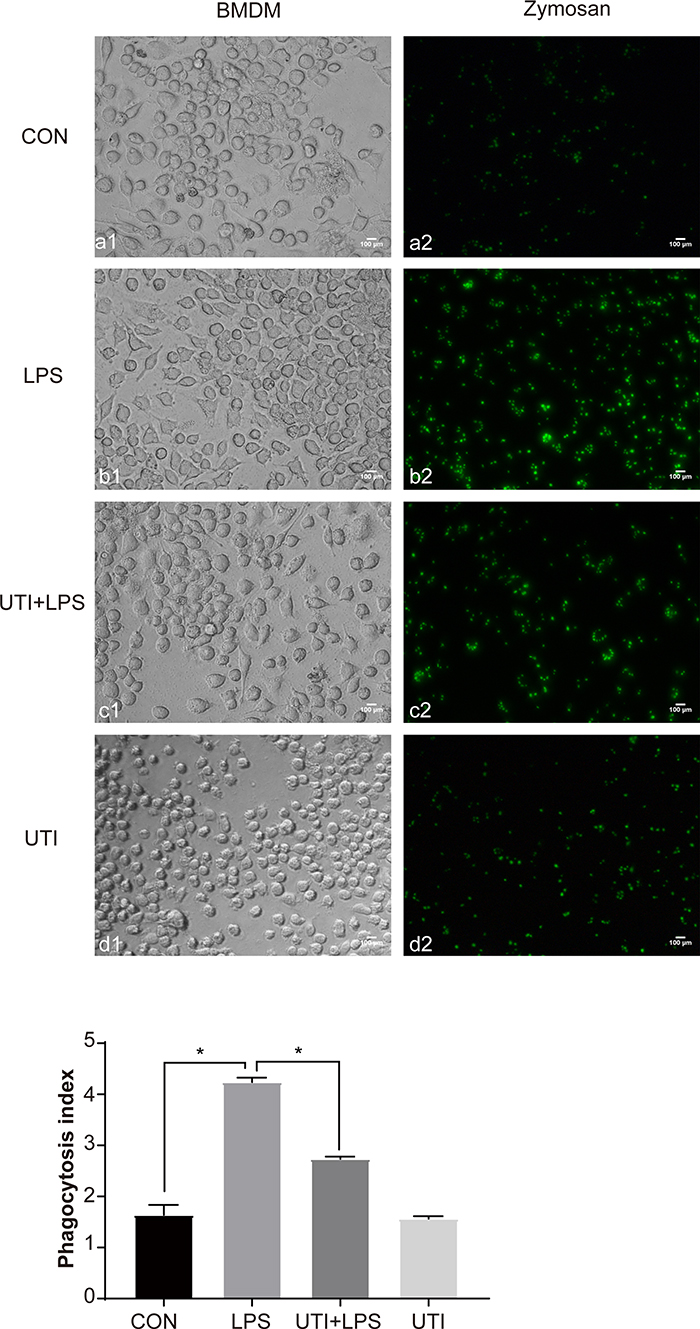 |
Figure 2 UTI markedly decreased the phagocytosis of BMDM caused by LPS. As the phagocytosis assay kitshowed, the phagocytosis of BMDM was significantly increased in LPS-treated BMDM when compared with the control and UTI groups; it was markedly decreased in the UTI+LPS group. *p<0.05. |
UTI Decreased Migration in BMDM
The scratch wound healing assay at time points 0, 24, 48 and 72 h also determined that LPS significantly promoted the migration of BMDM while UTI effectively inhibited migration (Figure 3A). Expression of the chemokine MCP-1 in BMDM significantly increased in the LPS group compared with the control group but sharply decreased in the UTI+LPS group (Figure 3B). Transwell migration assay also showed that migration of BMDM in the LPS group was markedly increased compared with the control group, but significantly decreased in the UTI+LPS group (Figure 3C).
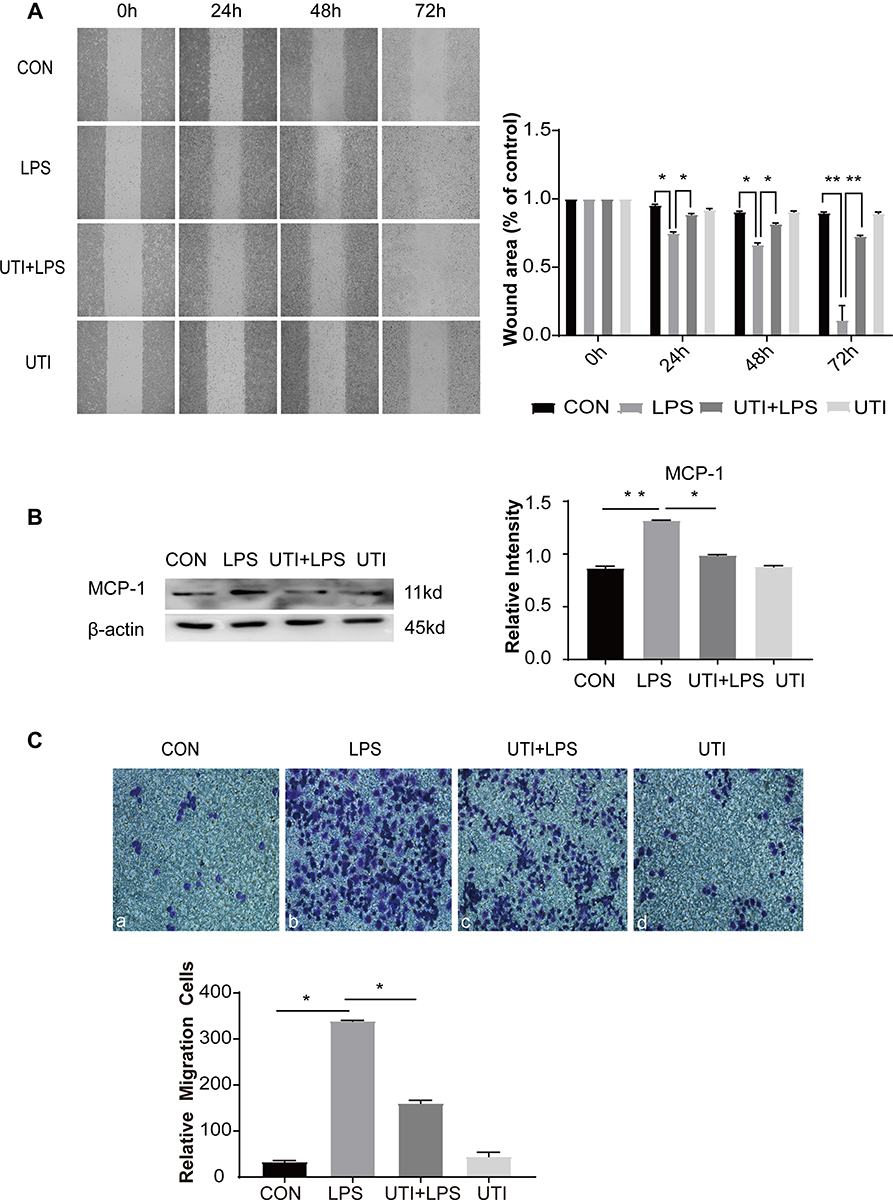 |
Figure 3 UTI markedly decreased the migration of BMDM caused by LPS. Cell migration ability evaluated by wound healing scratch (A) showed that the area of wound was noticeably reduced in the LPS group in comparison with the control group at 24 h, 48 h and 72 h; it was reversed in the UTI+LPS group at each time point. The result was analyzed using ImageJ software. Protein expression of MCP-1 (B) increased in LPS-treated BMDM and decreased in the UTI+LPS group. As revealed by the Transwell test, the number of migration cells significantly increased in LPS-treated BMDM in comparison with the control group, but markedly decreased in the UTI+LPS group (C). *p<0.05; **p<0.01. |
UTI Pretreated Conditioned Medium Derived from LPS-Treated BMDM Increased Tight Junctional Protein Expression in PMVECs
Expression of junctional proteins (claudin-5, ZO-1 and occludin) was assessed in PMVECs incubated with differently conditioned medium (Figure 4). Western blot showed that ZO-1 (Figure 4D) occludin (Figure 4E), claudin-5 (Figure 4F) expression was obviously decreased in the CM-LPS group in comparison with the CM group; it was significantly increased in the CM-UTI+LPS group. The immunofluorescence intensity of ZO-1 (Figure 4A), occludin (Figure 4B) and claudin-5 (Figure 4C) in the CM-LPS group was significantly decreased compared with the CM group; it was obviously augmented in the CM-UTI+LPS group.
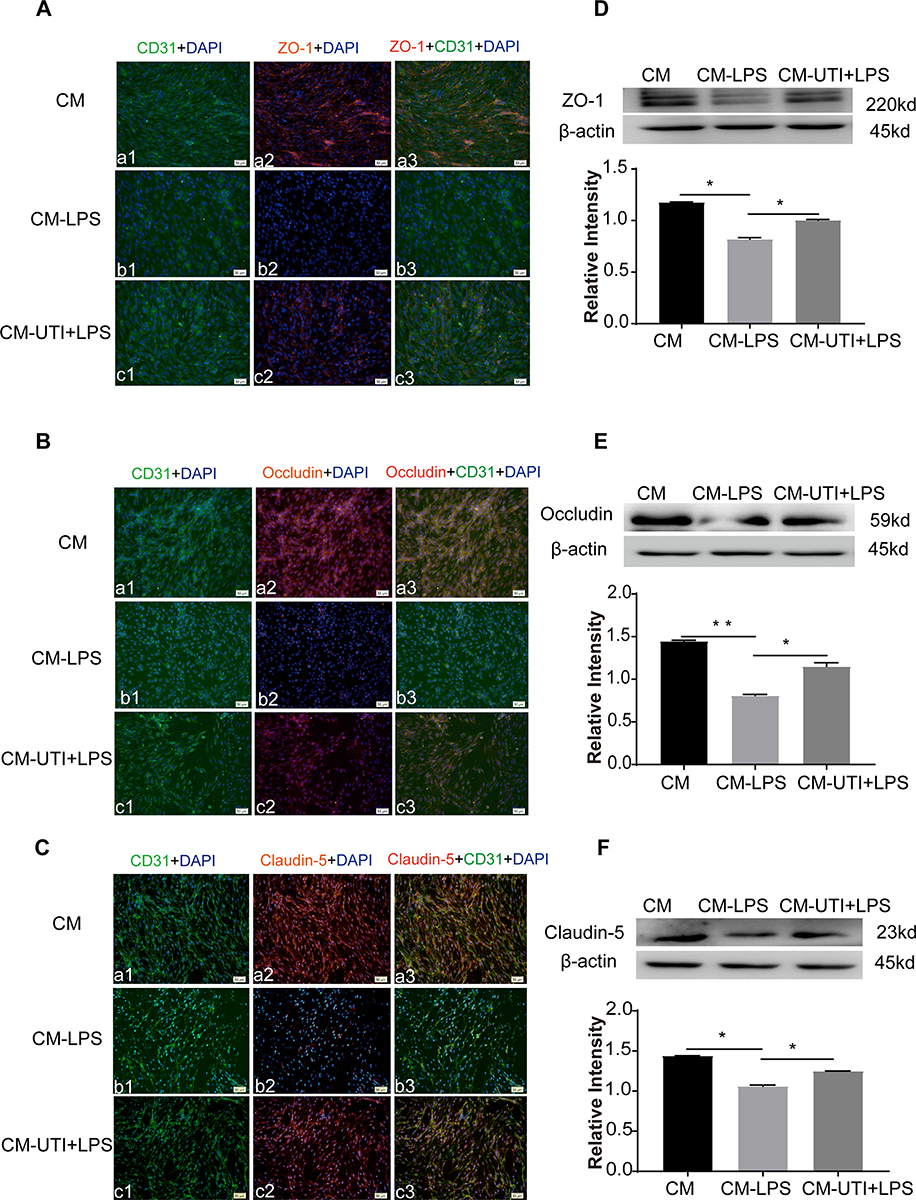 |
Figure 4 UTI pretreated conditioned medium derived from LPS-treated BMDM reversed tight junctional protein expression of PMVECs. ZO-1 (Aa1–3), occludin (Ba1–3), and claudin-5 (Ca1–3) expression (red) was intensely labeled in PMVECs (green) of the CM (conditioned medium) group. It was hardly detected in the CM-LPS group (Ab1–3, Bb1–3, Cb1–3). The expression increased sharply in the CM-UTI+LPS group (Ac1–3, Bc1–3, Cc1–3). Correspondingly, Western blot showing expression levels of ZO-1 (D), occludin (E), and claudin-5 (F) was markedly decreased in the CM-LPS group in comparison with the control group; it was significantly increased in the CM-UTI+LPS group. CM group: PMVECs incubated with BMDM conditioned medium of basic medium for 8 h; CM-LPS group: PMVECs incubated with BMDM conditioned medium of 1µg/mL LPS for 6 h; CM-UTI+LPS: PMVECs incubated with BMDM conditioned medium of UTI pretreatment (104 U/mL) for 2 h followed by 1µg/mL LPS for 6 h. Scale bars: 50 µm, DAPI-blue. *p<0.05; **p<0.01. |
UTI Increased M2 Macrophages in Lung Tissue of CLP-Induced Rats
Expression of inflammatory factors was detected in CLP-induced rat lungs. Immunofluorescence intensity of CD86 (Figure 5A), TNF-α (Figure 5B) and iNOS (Figure 5C) were increased in the CLP group when compared with the sham group; it was obviously decreased in the UTI+CLP group. Immunofluorescence intensity of CD206 (Figure 6A) and TGF-β (Figure 6B) were markedly decreased in the CLP group compared with the sham group; it was augmented in the UTI+CLP group. Western blot showed that expression of the M1 macrophage marker CD86 (Figure 5D), pro-inflammation cytokines TNF-α (Figure 5E) and iNOS (Figure 5F) were markedly increased in the CLP group when compared with the sham group; these were significantly decreased in the UTI+CLP group. The expression level of CD206 (Figure 6C) and TGF-β (Figure 6D) decreased in the CLP group,but reversed following UTI pretreatment.
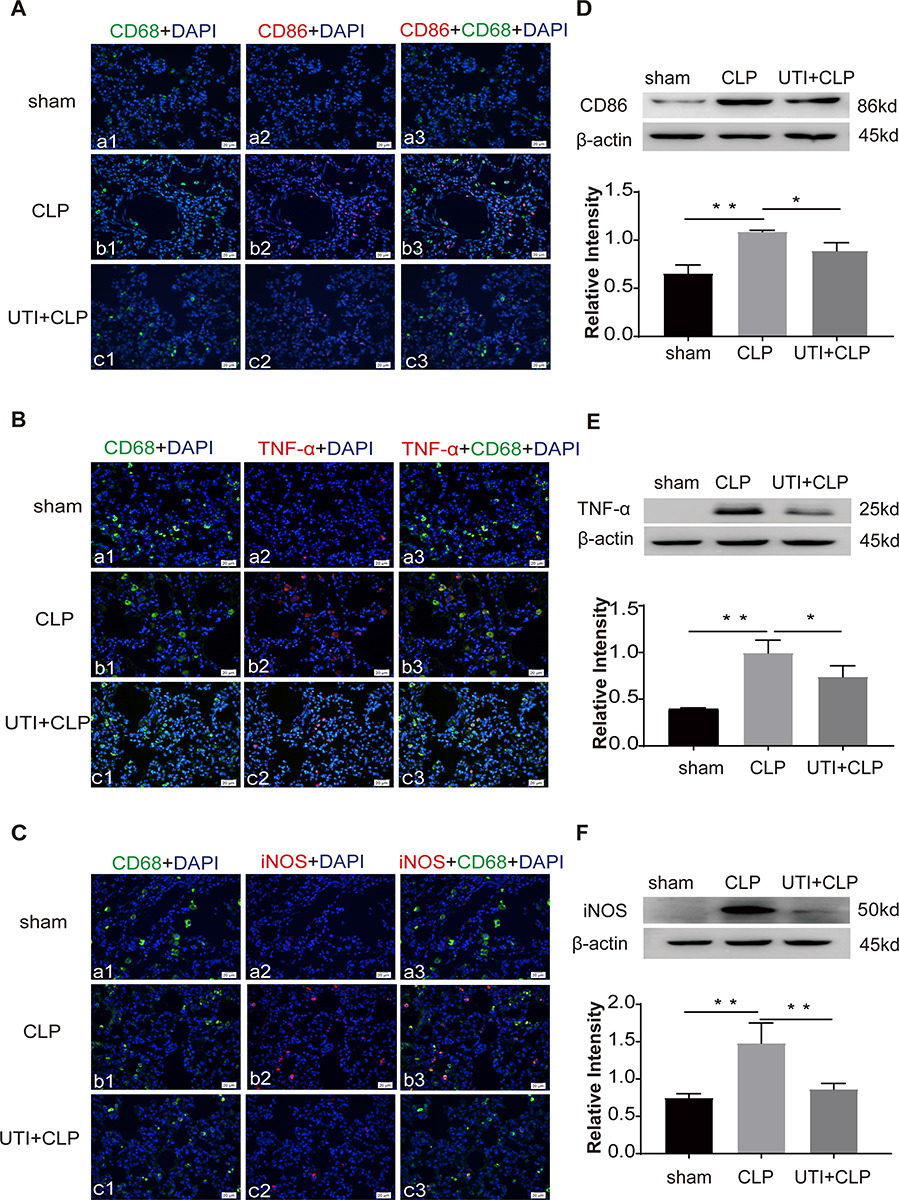 |
Figure 5 UTI decreased M1 macrophages in lung tissue in CLP-induced rat lungs. CD86 (Ab1–3), TNF-α (Bb1–3), iNOS (Cb1–3) expression (red) was intensely labeled in lung tissue of the CLP group. The expression decreased in the UTI+CLP group (Ac1–3, Bc1–3, Cc1–3). It was hardly detected in the sham group ((Aa1–3, Ba1–3, Ca1–3). Western blot showing the expression of CD86 (D), TNF-α (E) and iNOS (F) sharply increased in the CLP group in comparison with the sham group; it significantly decreased in the UTI+CLP group. *p<0.05; **p<0.01. |
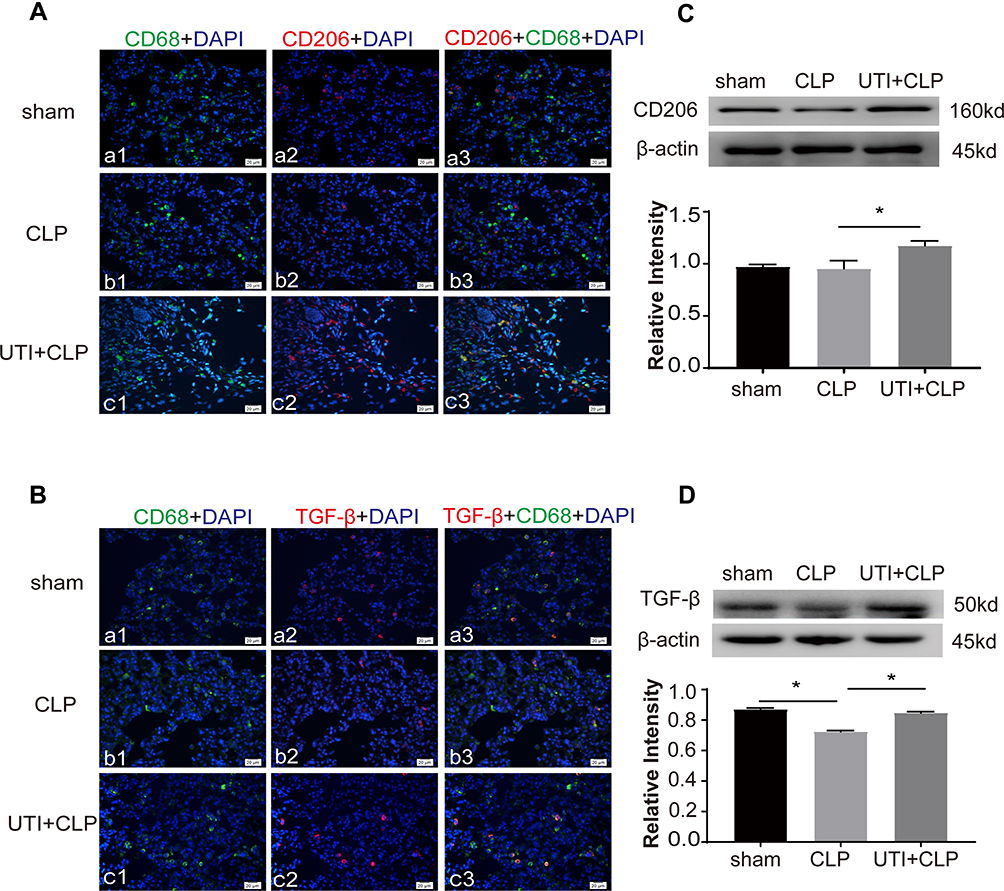 |
Figure 6 UTI increased M2 macrophages in lung tissue in CLP-induced rat lungs. CD206, TGF-β expression was labeled (red) in lung tissue. The expression of CD206 (Ab1–3), TGF-β (Bb1–3) decreased in the CLP group compared with the sham group (Aa1–3, Ba1–3). It significantly increased in the UTI+CLP group (Ac1–3, Bc1–3). Western blot showing expression of CD206 (C) and TGF-β (D) markedly decreased in the CLP group and reversed in the UTI+CLP group. *p<0.05. |
UTI Repaired the Endothelial Barrier Structure and Relieved Pulmonary Capillary Endothelial Leakage in CLP-Induced Rat Lungs
The extravasated Evans blue in the CLP group was significantly higher than in the other groups, while it was reduced after UTI treatment (Figure 7A). To detect the pulmonary capillary endothelial, we analyzed it using TEM (Figure 7B). In the sham group, it showed tight connections with a rivet-like structure, while the barrier structure of CLP-induced rats was severely damaged in the CLP group, the gap between endothelial cells had increased. UTI treatment nicely repaired the damage caused by sepsis as evidenced by the gaps between endothelial cells being narrower and the barrier structure reversed in the UTI+CLP group.
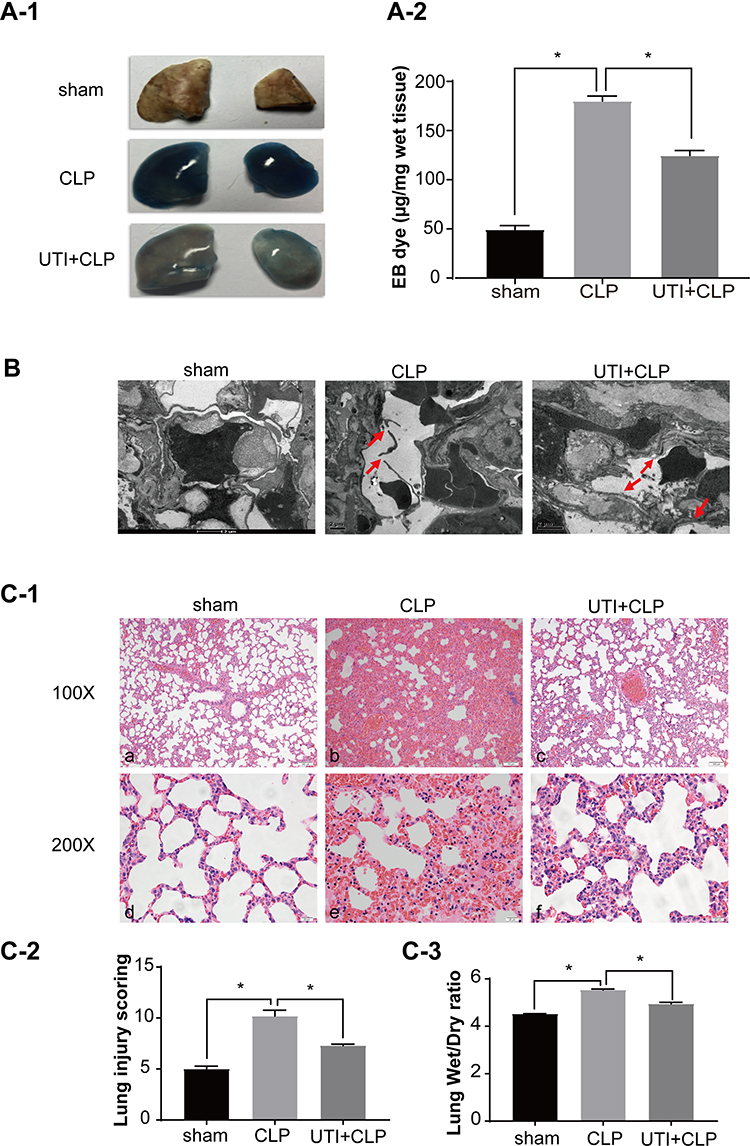 |
Figure 7 UTI repaired the barrier structure and relieved pulmonary capillary endothelial leakage in CLP-induced rat lungs. In the CLP group, EB inundated the lung tissue, while it was noticeably reduced in theUTI+CLP group (A). Electron microscope (B) showed that the barrier structure of CLP-induced rats was severely damaged (red arrow) when compared with the sham group; the gaps between endothelial cells were evidently narrowed and the barrier structure was reversed in the UTI+CLP group. Histopathological morphology was assessed by performing hematoxylin and eosin staining (C-1). The lung W/D ratio varied in different groups of rats (C-3). In the CLP group, severe pulmonary edema, interstitial hemorrhage, alveolar collapse, a large number of inflammatory cell infiltration (C-1b,e), higher lung injury score (C-2) and W/D ratio (C-3) occurred. Compared with the CLP group, the UTI+CLP group showed less destruction of lung structure (C-1c,f), lower lung injury score (C-2) and lower W/D ratio (C-3). *p<0.05. Abbreviation: W/D, wet to dry. |
UTI Alleviated Histopathological Alterations, Lung Injury Score and Reduced W/D Weight Ratio
Lung sections stained with hematoxylin and eosin showed drastic changes in histopathology in the CLP group. The main manifestations were obvious thickening of the alveolar septa, infiltration of a large number of lymphocytes and neutrophils in the interstitium, interstitial hemorrhage, obvious alveolar collapse and high LIS. But following UTI treatment, the inflammation and destruction in lung tissue were reduced (Figures 7C-1 and C-2). The W/D ratio was significantly increased in the CLP group compared with the sham group. After UTI pretreatment, the W/D ratio decreased (Figure 7C-3).
Discussion
Our previous research demonstrated that UTI can protect tight junction proteins of endothelial cells and reduce TNF-α expression in the lungs of septic rats.11 When exploring the mechanism in PMVECs, we noticed that blocking the TNF-α receptor has a similar protective effect as UTI, which decreases the damage of tight junction proteins. The results suggested that TNF-α may be one of the core factors contributing to damage of tight junction proteins in endothelial cells. It is known that macrophages are the major source of TNF-α in sepsis, thus we put forward a reasonable hypothesis that UTI participates in the type transformation of macrophages, regulate the function of macrophages and influences endothelial cells.
As CD86, TNF-α and CD206, TGF-β are cell surface markers which can differentiate M1 and M2 polarization,24 in the present study LPS stimulation increased the expression of cell surface markers of M1 macrophages, such as CD86 and TNF-α. Interestingly, UTI significantly reduced CD86 and TNF-α expression, but increased the expression of M2 markers, such as CD206 and TGF-β, and anti-inflammatory cytokines IL-10. These results indicated that UTI has a regulatory effect on the polarization of macrophages, which inhibits the excessive activation of macrophages to M1 under sepsis conditions, and increases the proportion of M2. Correspondingly, it reduced pro-inflammatory cytokine expression induced by LPS and increased anti-inflammatory cytokine expression. Recent studies reported that UTI can decrease the gene levels of pro-inflammatory factors (TNF-α) and M1 macrophage markers (IL-6, CD68, and ROS) whilst also increasing the gene levels of anti-inflammatory factors (IL-10) and M2 macrophage markers (Arg-1, MgI-1, and CD209) in bronchoalveolar lavage fluid.25 Thus, UTI played an important role in reducing the inflammatory response in sepsis.
The regulation of UTI on macrophage functions needs to be further explored. Migration is one of the important functions of macrophages, and it is necessary for activated macrophages to invade injured or inflamed sites through various substrates26,27 and serve a vital role in wound healing and immune responses. In severe sepsis, however, macrophages and inflammatory cells are activated and recruited in tissues and expand the inflammatory response.28,29 It is recognized that an excessive migration of macrophages and the following inflammatory response (“cytokine storm”) contributes to tissue damage, tumor spread and so on.30 A recent study showed that suppressed macrophage migration induced by miR-25 could decrease the secretion of pro-inflammatory cytokines in sepsis.31 In our study, the results of Transwell and scratch wound assay demonstrated that UTI inhibited migration and invasion of LPS-treated macrophages. In addition, the present study showed, in comparison with the LPS group, that UTI pretreatment markedly decreased the phagocytosis rate of LPS-incubated macrophages, indicating that UTI could inhibit the phagocytosis of macrophages in sepsis. The phagocytosis of macrophages is an important component of innate immunity. Increased phagocytosis is a key feature of activated macrophages.32 However, excessive phagocytosis may lead to increased tissue damage and impaired repair.33 It has been reported that anti-inflammatory agent (Dp44mT) could significantly reduce the phagocytosis of LPS-treated macrophages.34 Our study found similar experimental results demonstrating that UTI can reduce phagocytosis of LPS-activated macrophages. The above results showed that UTI can not only regulate macrophage polarization but also its functions, suggesting the potential therapeutic prospects for UTI in sepsis.
Does the regulation of macrophage function by UTI directly affect the tight junction of endothelial cells in sepsis? To answer this question, we used the conditioned medium of macrophages under different conditions to cultivate PMVECs. The results showed that ZO-1, claudin-5 and occludin expression sharply decreased when incubated with CM-LPS, but reversed when pretreated with UTI in UTI-LPS CM. Evidence showed that tight junction proteins played a crucial role in the integrity of the alveolar–endothelia barrier and the reduction of TJ molecules caused by sepsis usually leads to enhanced pulmonary vascular permeability.35,36 Our findings provide direct evidence of UTI protecting tight junction proteins in pulmonary capillary endothelial cells by regulating the function of macrophages. It is reported that a conditioned medium of M2 type macrophages can upregulate neonatal cardiomyocyte proliferation37 and a conditioned medium of peritoneal macrophage from CO-treated rats stimulated endothelial cell proliferation.38 Such evidence supports the view that macrophages can interfere with endothelial cells.
In addition, we used septic rat models to verify the phenotypic transformation of macrophages in vivo and effects on lung tight junctions. The results showed that UTI intervention increased the conversion of macrophages in the lungs to M2 type, ameliorated the destruction of rat lung endothelial cells, and relieved pulmonary capillary leakage. The evidence included: in CLP-induced rats, UTI treatment decreased the Evans blue exudation and W/D weight ratio of the lung, alleviated histopathological alterations and reduced the lung injury score. When observed through electron microscope, the barrier structure of pulmonary capillary endothelia in the CLP group was not continuous or even severely damaged but lost its normal form, while the normal structure could still be maintained when pretreated with UTI. The experiments in vivo confirmed the regulatory effect of UTI on macrophages, accompanied by structural changes in endothelial cells, which also corroborated the results of our cell experiments.
Conclusion
Taken together, the present study provided firm biochemical, morphological and experimental evidence that UTI protects TJs of pulmonary capillary endothelia through promoting the transformation of M2 type macrophages, inhibiting macrophages of pro-inflammatory cytokine expression, increasing anti-inflammatory cytokine expression, and reducing the phagocytosis and inhibiting the migration of macrophages, thereby attenuating the septic permeability of pulmonary capillaries.
Acknowledgment
Special thanks to Emeritus Professor Eng-Ang Ling, Department of Anatomy, Yong Loo Lin School of Medicine, National University of Singapore, for his contribution to this project and editing of the manuscript.
Funding
This work was funded by the Special Project on Emergency Response to Control of COVID-19 Infection of Guangdong Province (No. 2020B1111330006).
Disclosure
The authors report no other conflicts of interest in this work.
References
1. Mayr F, Yende S, Angus D. Epidemiology of severe sepsis. Virulence. 2014;5(1):4–11. doi:10.4161/viru.27372
2. Hudson L, Steinberg K. Epidemiology of acute lung injury and ARDS. Chest. 1999;116:74S–82S. doi:10.1378/chest.116.suppl_1.74S-a
3. Matthay M, Ware L, Zimmerman G. The acute respiratory distress syndrome. J Clin Invest. 2012;122(8):2731–2740. doi:10.1172/JCI60331
4. Matthay M, Zemans R, Zimmerman G, et al. Acute respiratory distress syndrome. Nat Rev Dis Primers. 2019;5(1):18.
5. Cong X, Kong W. Endothelial tight junctions and their regulatory signaling pathways in vascular homeostasis and disease. Cell Signal. 2020;66:109485.
6. Komarova Y, Malik A. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu Rev Physiol. 2010;72(1):463–493. doi:10.1146/annurev-physiol-021909-135833
7. Rochfort K, Collins L, McLoughlin A, Cummins P. Tumour necrosis factor-α-mediated disruption of cerebrovascular endothelial barrier integrity in vitro involves the production of proinflammatory interleukin-6. J Neurochem. 2016;136(3):564–572. doi:10.1111/jnc.13408
8. Karnad D, Bhadade R, Verma P, et al. Intravenous administration of ulinastatin (human urinary trypsin inhibitor) in severe sepsis: a multicenter randomized controlled study. Intensive Care Med. 2014;40(6):830–838. doi:10.1007/s00134-014-3278-8
9. Yoo J, Ryu J, Lee S, et al. Preventive effects of ulinastatin on post-endoscopic retrograde cholangiopancreatography pancreatitis in high-risk patients: a prospective, randomized, placebo-controlled trial. Pancreas. 2008;37(4):366–370. doi:10.1097/MPA.0b013e31817f528f
10. Zhou L-W, Wang Y-L, Yan X-T, He X-H. Urinary trypsin inhibitor treatment ameliorates acute lung and liver injury resulting from sepsis in a rat model. Saudi Med J. 2008;29(3):368–373.
11. Fang M, Zhong W, Song W, et al. Ulinastatin ameliorates pulmonary capillary endothelial permeability induced by sepsis through protection of tight junctions via inhibition of TNF-α and related pathways. Front Pharmacol. 2018;9:823. doi:10.3389/fphar.2018.00823
12. Turnage R, Nwariaku F, Murphy J, Schulman C, Wright K, Yin H. Mechanisms of pulmonary microvascular dysfunction during severe burn injury. World J Surg. 2002;26(7):848–853. doi:10.1007/s00268-002-4063-3
13. Sica A, Erreni M, Allavena P, Porta C. Macrophage polarization in pathology. Cell Mol Life Sci. 2015;72(21):4111–4126. doi:10.1007/s00018-015-1995-y
14. Fujisaka S, Usui I, Bukhari A, et al. Regulatory mechanisms for adipose tissue M1 and M2 macrophages in diet-induced obese mice. Diabetes. 2009;58(11):2574–2582. doi:10.2337/db08-1475
15. Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23(11):549–555. doi:10.1016/S1471-4906(02)02302-5
16. Martinez F, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. doi:10.12703/P6-13
17. Weischenfeldt J, Porse B. Bone marrow-derived macrophages (BMM): isolation and applications. CSH Protoc. 2008;2008:
18. Zhang X, Goncalves R, Mosser D. The isolation and characterization of murine macrophages. Curr Protoc Immunol. 2008;
19. Cao C, Yin C, Shou S, et al. Ulinastatin protects against LPS-induced acute lung injury by attenuating TLR4/NF-κB pathway activation and reducing inflammatory mediators. Shock. 2018;50(5):595–605. doi:10.1097/SHK.0000000000001104
20. Lü Q, Wang D, Xie D. [Effect of ulinastatin on perioperative glycocalyx and lung function in patients undergoing mitral valve replacement surgery]. Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2018;43(6):646–650. Chinese. doi:10.11817/j.issn.1672-7347.2018.06.011
21. Wei A, Wu X, Li Y. [Experimental Methodology of Pharmacology]. Fourth Edition. People's Health Publishing House; 2010. Chinese.
22. Li G, Zhou H, He Y, Sun S, Wu X, Yuan H. Ulinastatin inhibits the formation and progression of experimental abdominal aortic aneurysms. J Vasc Res. 2020;57(2):58–64. doi:10.1159/000504848
23. Matute-Bello G, Downey G, Moore BB, et al. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol. 2011;44(5):725–738. doi:10.1165/rcmb.2009-0210ST
24. Kigerl K, Gensel J, Ankeny D, Alexander J, Donnelly D, Popovich P. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29(43):13435–13444. doi:10.1523/JNEUROSCI.3257-09.2009
25. Liu W, Pang G, Wang S, Sun A. Protective effect of ulinastatin on severe pulmonary infection under immunosuppression and its molecular mechanism. Exp Ther Med. 2017;14(4):3583–3588. doi:10.3892/etm.2017.4993
26. Kharraz Y, Guerra J, Mann C, Serrano A, Muñoz-Cánoves P. Macrophage plasticity and the role of inflammation in skeletal muscle repair. Mediators Inflamm. 2013;2013:491497. doi:10.1155/2013/491497
27. Murray M, Birkland T, Howe J, et al. Macrophage migration and invasion is regulated by MMP10 expression. PLoS One. 2013;8(5):e63555. doi:10.1371/journal.pone.0063555
28. Bao M, Dai W, Li Y, Hu C. Rutaecarpine prevents hypoxia-reoxygenation-induced myocardial cell apoptosis via inhibition of NADPH oxidases. Can J Physiol Pharmacol. 2011;89(3):177–186. doi:10.1139/Y11-006
29. Delano M, Ward P. Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J Clin Invest. 2016;126(1):23–31. doi:10.1172/JCI82224
30. Condeelis J, Pollard J. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124(2):263–266. doi:10.1016/j.cell.2006.01.007
31. Zhu C, Chen T, Liu B. Inhibitory effects of miR-25 targeting HMGB1 on macrophage secretion of inflammatory cytokines in sepsis. Oncol Lett. 2018;16(4):5027–5033. doi:10.3892/ol.2018.9308
32. Henneke P, Golenbock DT. Phagocytosis, innate immunity, and host-pathogen specificity. J Exp Med. 2004;199(1):1–4. doi:10.1084/jem.20031256
33. Wang K, Li J, Zhang Y, et al. Central nervous system diseases related to pathological microglial phagocytosis. CNS Neurosci Ther. 2021. doi:10.1111/cns.13619
34. Lim JH, Kim HY, Lee JS, Kim HM, Jeong HJ. Dp44mT regulates the levels of inflammatory mediators through blocking NF-kappaB nuclear translocation in LPS-stimulated RAW 264.7 macrophages. In Vitro Cell Dev Biol Anim. 2021. doi:10.1007/s11626-021-00552-y
35. Dudek S, Garcia J. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol. 2001;91(4):1487–1500. doi:10.1152/jappl.2001.91.4.1487
36. Peter A, Fatykhova D, Kershaw O, et al. Localization and pneumococcal alteration of junction proteins in the human alveolar-capillary compartment. Histochem Cell Biol. 2017;147(6):707–719. doi:10.1007/s00418-017-1551-y
37. Zogbi C, Oliveira N, Levy D, et al. Beneficial effects of IL-4 and IL-6 on rat neonatal target cardiac cells. Sci Rep. 2020;10(1):12350. doi:10.1038/s41598-020-69413-0
38. Leake A, Salem K, Madigan M, et al. Systemic vasoprotection by inhaled carbon monoxide is mediated through prolonged alterations in monocyte/macrophage function. Nitric Oxide. 2020;94:36–47. doi:10.1016/j.niox.2019.10.003
39. Puig F, Herrero R, Guillamat-Prats R, et al. A new experimental model of acid- and endotoxin-induced acute lung injury in rats. Am J Physiol Lung Cell Mol Physiol. 2016;311(2):l229–l237.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.