Back to Journals » Blood and Lymphatic Cancer: Targets and Therapy » Volume 7
Uptake of lymphoma-derived exosomes by peripheral blood leukocytes
Authors Ferguson Bennit HR, Gonda A, Oppegard LJ, Chi DP, Khan S ![]() , Wall NR
, Wall NR ![]()
Received 21 December 2016
Accepted for publication 27 January 2017
Published 28 February 2017 Volume 2017:7 Pages 9—23
DOI https://doi.org/10.2147/BLCTT.S130826
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor David Dingli
Heather R Ferguson Bennit,1,2 Amber Gonda,1,3 Laura J Oppegard,2 David P Chi,2 Salma Khan,1,2 Nathan R Wall1,2
1Center for Health Disparities & Molecular Medicine, 2Division of Biochemistry, Department of Basic Sciences, 3Department of Anatomy, Loma Linda University School of Medicine, Loma Linda, CA, USA
Abstract: Exosomes are nanosized lipid vesicles secreted into blood and other body fluids and serve as vehicles for intercellular communication. Despite being an important component of the tumor microenvironment (TME), exosomal targeting and uptake into recipient cells are still not fully understood. Few studies have looked at lymphoma exosomes and their interactions with circulating blood cells. In this study, we examine the exosomal uptake distribution among peripheral blood leukocytes (PBLs) using vesicles derived from a diffuse large B cell lymphoma cell line, WSU-DLCL2. Lymphoma cells survive, proliferate, and are protected from the cytotoxic effects of chemotherapeutic agents by soluble factors or by direct contact with inflammatory and stromal cells within the TME. In an attempt to close the gap in knowledge concerning lymphoma TME immunosuppression, we have treated normal human PBLs with PKH67-labeled lymphoma exosomes and monitored the uptake by measuring fluorescence at different time points using flow cytometry and fluorescent microscopy. Our results show that of the four populations examined, B cells and monocytes demonstrated uptake of PKH67-labeled exosomes, while T cells and NK cells displayed significantly less uptake.
Keywords: exosome, non-Hodgkin’s lymphoma, B cell
Introduction
Diffuse large B cell lymphoma (DLCL) is an intermediate grade and the most common form of non-Hodgkin’s lymphoma (NHL), affecting 40–50% of adult lymphoma patients in the US.1 Chemokines, cytokines, and growth factors are critical for the growth and survival of these malignant B cells.2 In addition, specific oncogenes such as c-Myc have been shown to have proliferation regulatory ability in malignant B cells both ex vivo and in vivo.3 The cross talk between the tumor microenvironment (TME) and the DLCL cells is mainly mediated by direct cell-to-cell interactions but has recently been shown to be facilitated through extracellular vesicle-trafficked soluble factors.4,5
Exosomes are small 30–150 nm sized extracellular vesicles important in the intercellular communication between cells.6–9 Communication can occur both by transfer of nucleic acids and proteins and by binding cell surface receptors and inducing cell signaling pathways. Both normal and tumor cells release exosomes, although tumor-derived exosomes (TEXs) have been the subject of a wide range of studies. TEXs have been shown to be involved in many aspects of the TME, including immune suppression,10,11 antigen presentation,12–16 a means of acquiring chemotherapeutic resistance,17–21 as biomarker reservoirs,22–28 inducers of angiogenesis,29–31 and vehicles of niche preparation for metastasis.32–36
However, the modes and mechanisms of uptake are not completely understood. Cells appear to internalize exosomes through several endocytic pathways, including clathrin- and caveolin-dependent endocytosis, phagocytosis, and lipid raft-mediated internalization. It is likely that cells utilize multiple routes to take up exosomes, depending on the proteins, glycoproteins, and lipids found on the surface of the vesicles and the target cell itself.37 Numerous studies show proficient uptake of TEXs by endothelial cells,38–40 epithelial cells,41 fibroblasts,42 myeloid precursors in bone marrow,35,40 mesenchymal stem cells,40 and other tumor cells.43
There have been few studies investigating uptake of exosomes by peripheral blood cell populations. Zech et al44 found that rat pancreatic adenocarcinoma exosomes could be taken up by all leukocyte subpopulations examined, with CD11b+ cells demonstrating higher internalization than T or B cells. At this time, there is only one other publication addressing peripheral blood uptake of lymphoma exosomes – a study by Hazan-Halevy et al45 looking at mantle cell lymphoma exosomes and their preferential uptake by B-lymphocytes. DLCL, an aggressive form of lymphoma representing >40% of adult lymphoma patients, has not been investigated. It is therefore important to investigate these interactions between the lymphoma cells and the TME in order to find and exploit new prognostic factors and to design new therapeutic approaches.
Methods
Cell culture
Human lymphoma cell lines WSU-DLCL2 and WSU-FSCCL were developed at Wayne State University and are Epstein–Barr virus-negative.46 Cell lines were grown in Roswell Park Memorial Institute (RPMI) 1640 media supplemented with 10% United States Department of Agriculture-sourced heat-inactivated fetal bovine serum (FBS; Mediatech, Manassas, VA, USA), 4 mM L-glutamine, 0.1 mg/mL streptomycin, and 100 units/mL penicillin and incubated at 37°C and 5% CO2. Trypan blue staining was used to measure cell density (confluent at 1×106/mL) and viability (>90%).
Peripheral blood from healthy apheresis blood donors were obtained from the Life Stream Blood Bank (San Bernardino, CA, USA) according to our approved Loma Linda University institutional review board (IRB) protocols. The red blood cells were lysed using an ACK lysis buffer containing 8.3 g/L NH4Cl, 1 g/L KHCO3, and 1.8 mL 5% EDTA and centrifuged for 5 minutes at 1,500 rpm at 4°C in a Beckman Coulter Allegra X-15R centrifuge, equipped with an SX4750 rotor to obtain a pellet of peripheral blood leukocytes (PBLs). These PBLs were allowed to rest overnight before exosome treatment in 5×106/mL complete RPMI, with or without 100 IU/mL IL-2. This study, in its entirety, was approved by Loma Linda University’s IRB.
Exosome isolation
Lymphoma cells were cultured for 24 hours in media depleted of exosomes from FBS (Hyclone Laboratories, Inc., South Logan, Utah, USA) by overnight ultracentrifugation at 100,000× g. This conditioned medium was subjected to serial centrifugation, removing cells (300× g, 5 minutes) and removing noncellular debris (2,000× g for 10 minutes). The supernatant was then centrifuged at 10,000× g for 30 minutes. Exosomes were isolated using the commercially available ExoQuick-TC™ (System Biosciences, Mountain View, CA, USA) at a 1:5 ratio of reagent to conditioned medium and incubated overnight at 4°C. A low speed spin at 1,500× g for 30 minutes was sufficient to pellet the precipitated vesicles. Exosome pellets were resuspended in 40–70 μL PBS and protein quantified by bicinchoninic acid assay protein assay (#23225; Pierce/Thermo Scientific, Rockford, IL, USA). Size of the vesicles was examined using dynamic light scattering (DLS) with a Nicomp N3000 nanoparticle sizing instrument (Particle Sizing Systems, Port Richey, FL, USA).
Uptake of exosomes
The exosome pellet, obtained from ExoQuick isolation, was labeled with PKH67 Green Fluorescent Cell Linker Kit (Sigma Aldrich; Saint Louis, MO, USA) as per manufacturer’s protocol, with modifications. Briefly, exosomes in PBS corresponding to 200 µg were added to 250 μL of diluent C. As a control, the same volume of PBS was also added to 250 μL of diluent C (no exosome control) and processed in parallel. The exosome suspension was added to an equal volume of 2× PKH67 dye mixture and mixed well for 4 minutes. The dye reaction was stopped by addition of 9 mL of media depleted of bovine exosomes by ultracentrifugation and then spun for 90 minutes at 110,000× g using an SW41 rotor. The pellet was washed in PBS with a second ultracentrifugation. The PKH67-labeled vesicles were incubated either with PBLs for 1, 4, or 24 hours for dose–curve experiments or with NK cells for 30, 60, or 240 minutes. Cells were washed with PBS, stained with surface marker antibodies, and fixed in 2% paraformaldehyde before proceeding with further analysis by flow cytometry or microscopy.
Flow cytometry
Antibodies directed against the following markers and directly labeled with indicated fluorophore were used to stain PBLs for flow cytometry analysis: CD3-PE, CD1a-PE (Becton, Dickinson [BD] Biosciences, San Diego, CA, USA); CD14-antigen-presenting cells (APC, M5E2; BioLegend; San Diego, CA, USA); CD56-APC (MY31; Tonbo; San Diego, CA, USA); and CD19-APC (H1B19). Live cell exosome binding was distinguished from dead cells using fixable viability dye eFluor 780 (eBioscience; San Diego, CA, USA). Cells were run on MACSQuant Analyzer (Miltenyi Biotec; Bergisch Gladbach, Germany) and data analyzed using FlowJo software (Tree Star; Ashland, OR, USA). The percentage of cells positive for PKH67+ exosomes was determined for each cell population. The gating strategy used to assess uptake of PKH67-labeled exosomes is shown in Figure S2.
Microscopy
Samples were spun onto poly-l-lysine slides using StatSpin CytoFuge 2 (Beckman Coulter; Brea, CA USA) set at 800 rpm for 4 minutes. One drop of mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) stain to visualize nuclear structures (Vectashield, Vector Laboratories, Burlingame, CA, USA) was placed onto the glass slide before adding glass coverslip and sealing with nail polish. Slides were imaged using a fluorescence light microscope (BIOREVO BZ7000; Keyence; Osaka, Japan) and a Zeiss LSM 710 NLO confocal microscope.
Statistical analysis
All the quantitative data of this study were expressed as mean ± SD, and statistical analysis was conducted using GraphPad Prism software v.5.01 for Windows (San Diego, CA, USA). To test for statistical significance, nonparametric two-tailed Mann–Whitney analysis was performed. Comparisons between groups were performed using Student’s t-test with probability p<0.05 considered to indicate a statistically significant difference. Each experiment was repeated at least twice to assess the level of reproducibility.
Results
Cellular uptake characterization of DLCL2 exosomes
Exosome uptake by peripheral blood cells was measured using flow cytometry and fluorescent microscopy, with demonstrated uptake differing between cell lineages and in a dose- and time-dependent manner. Specifically, to study the uptake of lymphoma-derived exosomes by PBLs, vesicles were isolated from the conditioned media from the WSU-DLCL2 cell line. Vesicle size was evaluated by DLS and confirmed to be consistently in the reported range of exosomes (30–150 nm; Figure S1).
The exosomes were labeled with a lipophilic dye, PKH67, after which 25, 50, 100, 200, or 400 μg of these exosomes were incubated with PBLs for 1, 4, and 24 hours. Internalization by confocal microscopy was performed and analyzed. Exosomal internalization was observed as early as 1 hour postincubation with longer incubation times and higher concentrations resulting in higher accumulation of exosomes inside the cells (Figure 1).
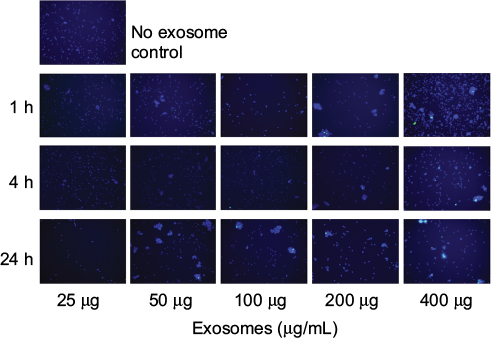 | Figure 1 Peripheral blood leukocytes take up DLCL2 exosomes. Notes: Peripheral blood leukocytes were treated with DLCL2 exosomes (25–400 μg/mL) and harvested at 1, 4, and 24 hours. Cells were cytospun onto poly-L-lysine-coated slides. Microscopy images acquired with BIOREVO BZ7000 fluorescent microscope (Keyence), 20× magnification. Nucleic acids are stained with DAPI (blue), and exosomes bind and internalization is visualized with PKH67 (green). Cells without the addition of exosomes were used as a negative control. Several fields were analyzed for each labeling condition, and representative results are presented. The data are representative of two independent experiments. Abbreviations: DLCL, diffuse large B cell lymphoma; DAPI, 4′,6-diamidino-2-phenylindole; h, hour. |
To study the kinetics of exosome accumulation, we performed quantitative flow cytometry. PKH67-labeled DLCL2 exosomes were incubated with PBLs, and the fluorescence intensity was detected. Uptake was most prominent by B cells and myeloid-derived cells and less so in T cells and NK cells (Figure 2). The uptake of exosomes from healthy B cells was rapid in the higher exosome concentrations, with 28% positive after 1 hour at 400 μg/mL (Figure 2A and B) and increasing to 69% at 24 hours at 400 μg/mL (Figure 2B). Cell specificity for uptake of DLCL2-derived exosomes was further studied in a single experiment using B cells (CD19+), T cells (CD3+), NK cells (CD56+), and monocytes (CD14+; Figure 2C and S3). Compared to B cells where 28% to 70% of cells showed exosome uptake in the 400 μg/mL exosomes over the 24-hour study, NK cells, monocytes, and T cells only maximized 6%, 8%, and 3% uptake, respectively (Figure 2C and S3). After a second independent experiment was concluded, CD19+ B cells and CD14+ monocytes maximized nearly 40% in the 400 μg/mL incubation while CD3+ T cells and CD 56+ NK cells only proved to be able to uptake nearly 10% (Figure 2D).
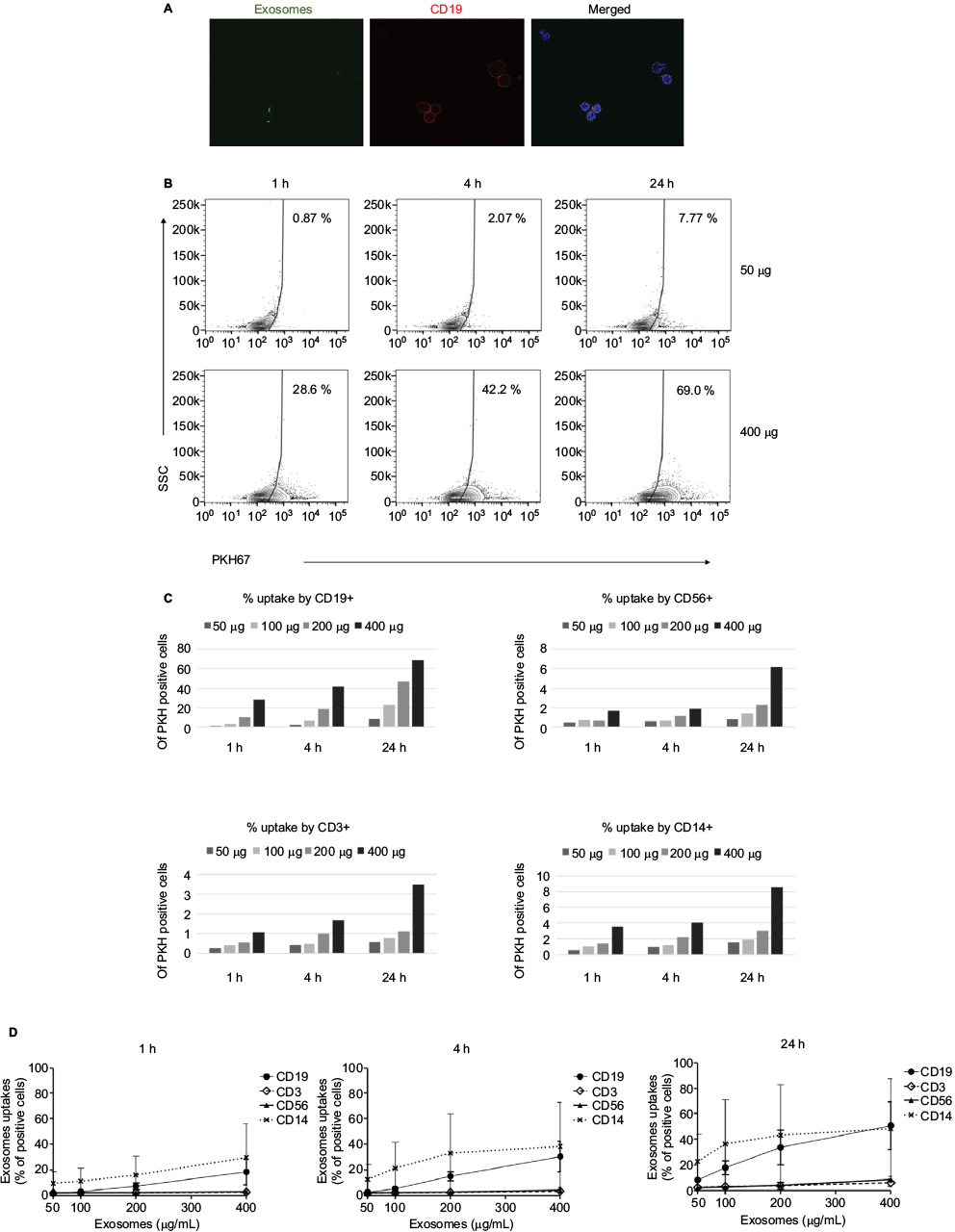 | Figure 2 Uptake of PKH67-labeled exosomes in a time- and dose-dependent manner. Notes: DLCL2 exosomes were labeled with PKH67 and then added to peripheral blood cells for various lengths of time (1, 4, and 24 hours) and treatment amounts (25, 50, 100, 200, and 400 μg/mL). As measured by flow cytometry, the uptake of labeled exosomes proceeded in a time- and dose-dependent manner. (A) Uptake of PKH67-labeled exosomes after 4 hours by CD19-APC cells (red). Microscopy images acquired with Zeiss LSM 710 NLO confocal microscope, 60× magnification. (B) Representative flow cytometry data of CD19+ cells. (C) Graphical representation of the percentage of PKH+ cells in each of the four lineages derived from one donor: B cells (CD19+), monocytes (CD14+), NK cells (CD56+), and T cells (CD3+). (D) Combined data from two separate experiments depicting the disparity in uptake between each cell population. Results are expressed as mean ± SD. Abbreviations: DLCL, diffuse large B cell lymphoma; APC, antigen-presenting cells; h, hours. |
The specificity of DLCL2 exosomes was further tested using exosomes derived from FSCCL and HeLa cells. Coculture of B cells and NK cells with 200 mg/mL PKH67-labeled DLCL2, FSCCL, and HeLa-derived exosomes for 1-hour and 4-hour time points showed very similar results to those previously recorded using DLCL2 exosome (Figure 3). In this study, B cells were able to rapidly and preferentially take up PKH67-stained DLCL2, FSCCL, and HeLa cell-derived exosomes in a time-dependent manner, while the NK cells were significantly less capable of this internalization. Four hours postincubation, 22%±8, 20%±10, and 12%±9 of B cells had taken up DLCL2, FSCCL, and HeLa exosomes, respectively (Figure 3).
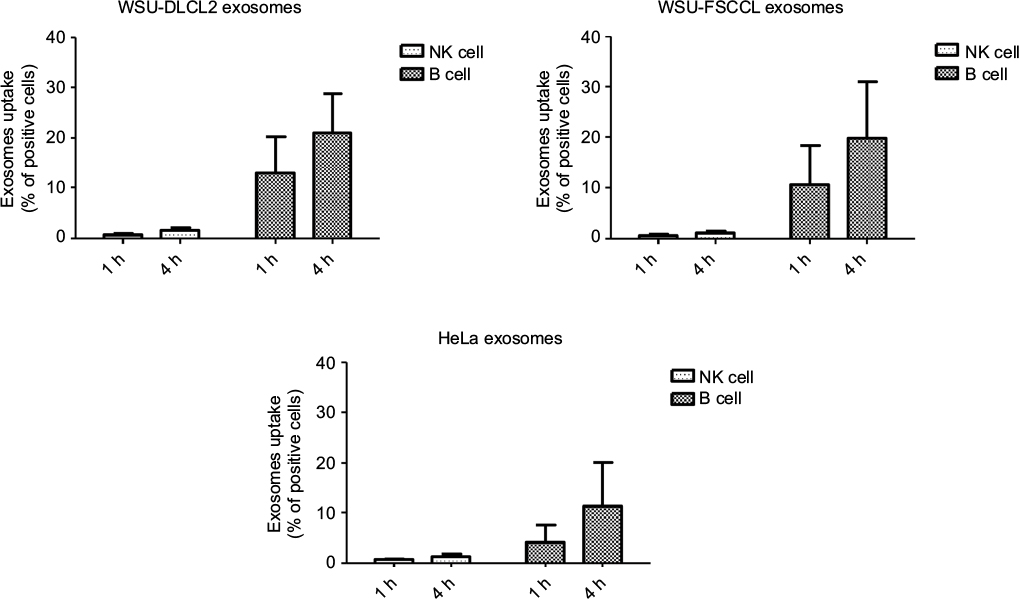 | Figure 3 Differences in uptake by NK cells and B cells are not unique to DLCL2 exosomes. Notes: DLCL2, FSCCL, and HeLa cell-derived exosomes were labeled with PKH67 and then added to peripheral blood for 1 and 4 hours at 200 μg/mL. As measured by flow cytometry, the uptake of labeled exosomes proceeded in a time- and dose-dependent manner as before in lymphocyte lineage B cells but not the NK cells. Abbreviations: DLCL, diffuse large B cell lymphoma; h, hours. |
Discussion
Although the spleen, liver, and lymph nodes take up the majority of exosomes produced by organs and hematopoietic cells, plasma and other body fluids still contain large quantities of exosomes.47 Cancer cells in particular secrete large quantities of TEXs, which can be found in peripherally circulating blood.48 Consequently, blood cells are exposed to many exosomes from both normal and malignant cells,49 which play key roles in modulating the immune system.8
While previous studies have investigated exosomal interactions with leukocytes in lymphoid organs, such as APCs in the spleen50 and follicular dendritic cells (DCs) in the lymph nodes,51,52 there is less work done regarding uptake of exosomes by peripheral blood cell populations.
In this study, we tested the hypothesis that some populations of white blood cells will be more receptive to interact with B cell lymphoma exosomes and, therefore, will be more vulnerable to TME modulating effects of these vesicles. In an attempt to close the gap in knowledge concerning lymphoma TME immunosuppression, we have treated normal human PBLs with PKH67-labeled lymphoma exosomes and monitored uptake by measuring fluorescence at different time points using flow cytometry and fluorescent microscopy. We expected to observe a disparity in exosome uptake between blood cells of lymphoid and myeloid lineages that we hypothesized was perhaps due to myeloid-derived cells such as monocytes and macrophages, being better equipped for exosome uptake than lymphocytes such as B cells, T cells, and NK cells. However, this was not what we observed with B cells being the most effective and efficient at TEX uptake.
Our data are not consistent with those of previous studies, which show a disparity in uptake between myeloid-derived cells and lymphocytes.44,53 We demonstrated that NK and T cells have lower levels of internalization than monocytes and B cells. In our study model, B cells showed a high level of uptake, possibly due to the B cell origin of the lymphoma exosomes, supporting the idea of uptake specificity given the exosomes were collected primarily from cells originating from B cell lymphoma disease. These findings are in line with those of Hazan-Halevy et al45 and Gutzeit et al54 concerning B cells and Riches et al55 in their work with breast tissue. In this study, we observed that DLCL2 exosomes were taken up rapidly and preferentially into CD19+ B lymphocytes and CD14+ monocytes. Only a small percentage of T cells and NK cells showed internalization or binding to exosomes, even after 24-hour incubations. We have further investigated to see if the origin of the exosome would influence the preference or rate of uptake on the peripheral cell (Figure 3). Exosomes were taken from two lymphoma cell lines (DLCL2 and FSCCL) and from the cervical cancer cell line HeLa. In our hands there was little difference recorded in exosomal uptake. This may indicate that the uptake is controlled by something specific to the exosome rather than the cell of origin.
The mechanisms of uptake into B lymphocytes remain to be elucidated, whether it be caveolin, clathrin, cholesterol, lipid-raft, or receptor-mediated endocytosis, or something completely novel and not proposed. In this work, we have demonstrated a natural preference of TEXs to B cells, further supporting the concept of targeting therapy to this lymphocyte population. However, to fully appreciate and dissect the mechanism, many more cell line-derived as well as patient-procured exosomes, from varying pathologies, will need to be investigated, a process that has only just begun in our laboratory. Understanding the structure and marker/receptor profiles on the exosome, the cell of origin as well as the recipient cell’s membranes and the protein, RNA, and DNA contents from within the exosome will further the ability to regulate the role of TEXs in the pathobiology of hematologic malignancies and to identify novel therapeutic approaches.
In addition to the indirect effects of exosomes through interactions with APCs, and the limited ability of NK cells and T cells for exosome uptake, these cell populations seem to have a wide variety of responses to direct exposure to exosomes. Whether these responses are the result of surface interactions rather than uptake or due to secondary effects from other cells which more readily internalize exosomes is not always apparent. There has been evidence for both possibilities, and it is likely that exosomes interacting with lymphocytes deliver signals by direct surface contact more frequently than internalization.42,56 The surface interactions between exosomes and recipient cells can occur via membrane-bound activating or inhibitory proteins that directly signal through relevant receptors and initiating downstream pathways. TEXs are enriched in proteins specialized for surface interactions, such as integrins, MHC class I and II molecules, co-stimulatory molecules (CD40, CD86), various growth factor receptors, such as epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER-2), death receptor ligands such as Fas ligand (FasL), tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), and programmed cell death ligand 1 (PDL-1) and inhibitory factors such as prostaglandin E2 (PGE2). Therefore, uptake is not necessary for a cell to be altered by exosomes, and may likely be the case for changes induced by exosomes in T cells and NK cells. In fact, much of the immune suppression mediated by TEXs occurs through surface molecules such as FasL, TGF-β1, and IL-10.57–59
Perhaps an equally important aspect could be that exosome uptake has downstream direct and indirect effects on PBLs. Through producing cytokines like TNFα, presenting antigen on MHC I and II, and providing costimulatory signals via CD80/CD86 and CD40,53,60 many of the exosomal effects observed in T and NK cells may be an indirect result from primary changes induced in macrophages, monocytes, B cells, and DCs, which actually internalize the vesicles. Uptake of exosomes induces monocytes to produce TNFα,53 macrophages and DCs capture and present antigen to T cells,60 and B cells can be activated by antigen-carrying exosomes with subsequent Th1 cell stimulation.61 Antigen-bearing TEXs seem to require uptake and processing by DCs before they can efficiently stimulate a specific cytotoxic T lymphocyte response.12,62 The exosomes from DCs carry MHC I, MHC II, CD80, and CD86 and are therefore equipped to elicit T cell activation through surface interactions.63
To summarize, in the present study, we characterized DLCL2 cell line-derived exosomes on different PBL populations showing that there is preference of targeted uptake. However, it is still not clear whether exosomes are still inducing cellular signaling pathways in the T cell and NK cells through direct cell to cell contact. From these findings, we hope a better understanding of tumor cell/TME communication may result, further leading to increased knowledge of how the tumor cells communicate with and manipulate the TME. By better understanding these signaling pathways, we may better prepare therapeutic modalities to enhance immune cell surveillance and killing of these tumors, which up to now seem immune.
Acknowledgments
The authors would like to thank the Center for Health Disparities & Molecular Medicine for supporting this project and the graduate students that it involved. They would also like to thank members of the Wall Laboratory for the careful review of our manuscript, Johnny Figueroa for the use of Keyence microscope, and Kimberley Payne for the use of the Flow Cytometry Core. Confocal imaging was performed in the LLUSM Advanced Imaging and Microscopy Core with support of NSF Grant MRI-DBI 0923559 and the Loma Linda University School of Medicine. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The research reported in this publication was supported by NIH award P20MD006988 (NRW).
Disclosure
The authors report no conflicts of interest in this work.
References
Solimando AG, Ribatti D, Vacca A, Einsele H. Targeting B-cell non Hodgkin lymphoma: new and old tricks. Leuk Res. 2016;42:93–104. | ||
Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–899. | ||
Miller DM, Thomas SD, Islam A, Muench D, Sedoris K. c-Myc and cancer metabolism. Clin Cancer Res. 2012;18(20):5546–5553. | ||
Valenzuela MM, Ferguson Bennit HR, Gonda A, et al. Exosomes secreted from human cancer cell lines contain inhibitors of apoptosis (IAP). Cancer Microenviron. 2015;8(2):65–73. | ||
Khan S, Bennit HF, Wall NR. The emerging role of exosomes in survivin secretion. Histol Histopathol. 2015;30(1):43–50. | ||
Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2(8):569–579. | ||
Ratajczak J, Miekus K, Kucia M, et al. Embryonic stem cell-derived microvesicles reprogram hematopoietic progenitors: evidence for horizontal transfer of mRNA and protein delivery. Leukemia. 2006;20(5):847–856. | ||
Iero M, Valenti R, Huber V, et al. Tumour-released exosomes and their implications in cancer immunity. Cell Death Differ. 2008;15(1):80–88. | ||
Bobrie A, Krumeich S, Reyal F, et al. Rab27a supports exosome-dependent and -independent mechanisms that modify the tumor microenvironment and can promote tumor progression. Cancer Res. 2012;72(19):4920–4930. | ||
Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9(8):581–593. | ||
Zhang HG, Grizzle WE. Exosomes and cancer: a newly described pathway of immune suppression. Clin Cancer Res. 2011;17(5):959–964. | ||
Wolfers J, Lozier A, Raposo G, et al. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat Med. 2001;7(3):297–303. | ||
Mallegol J, van Niel G, Heyman M. Phenotypic and functional characterization of intestinal epithelial exosomes. Blood Cells Mol Dis. 2005;35(1):11–16. | ||
Hao S, Bai O, Li F, Yuan J, Laferte S, Xiang J. Mature dendritic cells pulsed with exosomes stimulate efficient cytotoxic T-lymphocyte responses and antitumour immunity. Immunology. 2007;120(1):90–102. | ||
Zeelenberg IS, Ostrowski M, Krumeich S, et al. Targeting tumor antigens to secreted membrane vesicles in vivo induces efficient antitumor immune responses. Cancer Res. 2008;68(4):1228–1235. | ||
Zeelenberg IS, van Maren WWC, Boissonnas A, et al. Antigen localization controls T cell-mediated tumor immunity. J Immunol. 2011;187(3):1281–1288. | ||
Safaei R, Larson BJ, Cheng TC, et al. Abnormal lysosomal trafficking and enhanced exosomal export of cisplatin in drug-resistant human ovarian carcinoma cells. Mol Cancer Ther. 2005;4(10):1595–1604. | ||
Shedden K, Xie XT, Chandaroy P, Chang YT, Rosania GR. Expulsion of small molecules in vesicles shed by cancer cells: association with gene expression and chemosensitivity profiles. Cancer Res. 2003;63(15):4331–4337. | ||
Khan S, Aspe JR, Asumen MG, et al. Extracellular, cell-permeable survivin inhibits apoptosis while promoting proliferative and metastatic potential. Br J Cancer. 2009;100(7):1073–1086. | ||
Wang J, Hendrix A, Hernot S, et al. Bone marrow stromal cell–derived exosomes as communicators in drug resistance in multiple myeloma cells. Blood. 2014;124(4):555–566. | ||
Yu D-D, Wu Y, Zhang X-H, et al. Exosomes from adriamycin-resistant breast cancer cells transmit drug resistance partly by delivering miR-222. Tumor Biol. 2016;37(3):3227–3235. | ||
Mitchell P, Welton J, Staffurth J, et al. Can urinary exosomes act as treatment response markers in prostate cancer? J Transl Med. 2009;7(1):4. | ||
Taylor DD, Gercel-Taylor C. MicroRNA signatures of tumor-derived exosomes as diagnostic biomarkers of ovarian cancer. Gynecol Oncol. 2008;110(1):13–21. | ||
Skog J, Wurdinger T, van Rijn S, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10(12):1470–1476. | ||
Welton JL, Khanna S, Giles PJ, et al. Proteomics analysis of bladder cancer exosomes. Mol Cell Proteomics. 2010;9(6):1324–1338. | ||
Keller S, Ridinger J, Rupp A-K, Janssen J, Altevogt P. Body fluid derived exosomes as a novel template for clinical diagnostics. J Transl Med. 2011;9(1):86. | ||
Hong C-S, Miller L, Whiteside TL, Boyiadzis M. Plasma exosomes as markers of therapeutic response in patients with acute myeloid leukemia. Front Immunol. 2014;5:160. | ||
Turay D, Khan S, Osterman CJD, et al. Proteomic profiling of serum-derived exosomes from ethnically diverse prostate cancer patients. Cancer Invest. 2016;34(1):1–11. | ||
Deregibus MC, Cantaluppi V, Calogero R, et al. Endothelial progenitor cell–derived microvesicles activate an angiogenic program in endothelial cells by a horizontal transfer of mRNA. Blood. 2007;110(7):2440–2448. | ||
Kawamoto T, Ohga N, Akiyama K, et al. Tumor-derived microvesicles induce proangiogenic phenotype in endothelial cells via endocytosis. PLoS One. 2012;7(3):e34045. | ||
Kosaka N, Iguchi H, Hagiwara K, Yoshioka Y, Takeshita F, Ochiya T. Neutral sphingomyelinase 2 (nSMase2)-dependent exosomal transfer of angiogenic microRNAs regulate cancer cell metastasis. J Biol Chem. 2013;288(15):10849–10859. | ||
Graves LE, Ariztia EV, Navari JR, Matzel HJ, Stack MS, Fishman DA. Proinvasive properties of ovarian cancer ascites-derived membrane vesicles. Cancer Res. 2004;64(19):7045–7049. | ||
Hood JL, San RS, Wickline SA. Exosomes released by melanoma cells prepare sentinel lymph nodes for tumor metastasis. Cancer Res. 2011;71(11):3792–3801. | ||
Luga V, Zhang L, Viloria-Petit Alicia M, et al. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell. 2012;151(7):1542–1556. | ||
Peinado H, Aleckovic M, Lavotshkin S, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18(6):883–891. | ||
Suetsugu A, Honma K, Saji S, Moriwaki H, Ochiya T, Hoffman RM. Imaging exosome transfer from breast cancer cells to stroma at metastatic sites in orthotopic nude-mouse models. Adv Drug Deliv Rev. 2013;65(3):383–390. | ||
Mulcahy LA, Pink RC, Carter DRF. Routes and mechanisms of extracellular vesicle uptake. J Extracell Vesicles. 2014;3. | ||
Nazarenko I, Rana S, Baumann A, et al. Cell surface tetraspanin Tspan8 contributes to molecular pathways of exosome-induced endothelial cell activation. Cancer Res. 2010;70(4):1668–1678. | ||
Rana S, Yue S, Stadel D, Zöller M. Toward tailored exosomes: the exosomal tetraspanin web contributes to target cell selection. Int J Biochem Cell Biol. 2012;44(9):1574–1584. | ||
Paggetti J, Haderk F, Seiffert M, et al. Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood. 2015;126(9):1106–1117. | ||
Obregon C, Rothen-Rutishauser B, Gerber P, Gehr P, Nicod LP. Active uptake of dendritic cell-derived exovesicles by epithelial cells induces the release of inflammatory mediators through a TNF-α-mediated pathway. Am J Pathol. 2009;175(2):696–705. | ||
Clayton A, Turkes A, Dewitt S, Steadman R, Mason MD, Hallett MB. Adhesion and signaling by B cell-derived exosomes: the role of integrins. FASEB J. 2004;18(9):977–979. | ||
Franzen CA, Simms PE, Van Huis AF, Foreman KE, Kuo PC, Gupta GN. Characterization of uptake and internalization of exosomes by bladder cancer cells. Biomed Res Int. 2014;2014:619829. | ||
Zech D, Rana S, Buchler MW, Zoller M. Tumor-exosomes and leukocyte activation: an ambivalent crosstalk. Cell Commun Signal. 2012;10(1):37. | ||
Hazan-Halevy I, Rosenblum D, Weinstein S, Bairey O, Raanani P, Peer D. Cell-specific uptake of mantle cell lymphoma-derived exosomes by malignant and non-malignant B-lymphocytes. Cancer Lett. 2015;364(1):59–69. | ||
Al-Katib AM, Sun Y, Goustin AS, et al. SMI of Bcl-2 TW-37 is active across a spectrum of B-cell tumors irrespective of their proliferative and differentiation status. J Hematol Oncol. 2009;2:8. | ||
Srinivasan S, Vannberg FO, Dixon JB. Lymphatic transport of exosomes as a rapid route of information dissemination to the lymph node. Sci Rep. 2016;6:24436. | ||
Sun Y, Liu J. Potential of cancer cell-derived exosomes in clinical application: a review of recent research advances. Clin Ther. 2014;36(6):863–872. | ||
De Toro J, Herschlik L, Waldner C, Mongini C. Emerging roles of exosomes in normal and pathological conditions: new insights for diagnosis and therapeutic applications. Front Immunol. 2015;6:203. | ||
Morelli AE, Larregina AT, Shufesky WJ, et al. Endocytosis, intracellular sorting, and processing of exosomes by dendritic cells. Blood. 2004;104(10):3257–3266. | ||
Wubbolts R, Leckie RS, Veenhuizen PT, et al. Proteomic and biochemical analyses of human B cell-derived exosomes. Potential implications for their function and multivesicular body formation. J Biol Chem. 2003;278(13):10963–10972. | ||
Denzer K, van Eijk M, Kleijmeer MJ, Jakobson E, de Groot C, Geuze HJ. Follicular dendritic cells carry MHC class II-expressing microvesicles at their surface. J Immunol. 2000;165(3):1259–1265. | ||
Danesh A, Inglis HC, Jackman RP, et al. Exosomes from red blood cell units bind to monocytes and induce proinflammatory cytokines, boosting T-cell responses in vitro. Blood. 2014;123(5):687–696. | ||
Gutzeit C, Nagy N, Gentile M, et al. Exosomes derived from Burkitt’s lymphoma cell lines induce proliferation, differentiation, and class-switch recombination in B cells. J Immunol. 2014;192(12):5852–5862. | ||
Riches A, Campbell E, Borger E, Powis S. Regulation of exosome release from mammary epithelial and breast cancer cells – a new regulatory pathway. Eur J Cancer. 2014;50(5):1025–1034. | ||
Hwang I, Ki D. Receptor-mediated T cell absorption of antigen presenting cell-derived molecules. Front Biosci (Landmark Ed). 2011;16:411–421. | ||
Szajnik M, Czystowska M, Szczepanski MJ, Mandapathil M, Whiteside TL. Tumor-derived microvesicles induce, expand and up-regulate biological activities of human regulatory T cells (Treg). PLoS One. 2010;5(7):e11469. | ||
Szczepanski MJ, Szajnik M, Welsh A, Whiteside TL, Boyiadzis M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-beta1. Haematologica. 2011;96(9):1302–1309. | ||
Whiteside TL. Immune modulation of T-cell and NK (natural killer) cell activities by TEXs (tumour-derived exosomes). Biochem Soc Trans. 2013;41(1):245–251. | ||
Skokos D, Botros HG, Demeure C, et al. Mast cell-derived exosomes induce phenotypic and functional maturation of dendritic cells and elicit specific immune responses in vivo. J Immunol. 2003;170(6):3037–3045. | ||
Qazi KR, Gehrmann U, Domange Jordo E, Karlsson MC, Gabrielsson S. Antigen-loaded exosomes alone induce Th1-type memory through a B-cell-dependent mechanism. Blood. 2009;113(12):2673–2683. | ||
Dai S, Wan T, Wang B, et al. More efficient induction of HLA-A*0201-restricted and carcinoembryonic antigen (CEA)-specific CTL response by immunization with exosomes prepared from heat-stressed CEA-positive tumor cells. Clin Cancer Res. 2005;11(20):7554–7563. | ||
Viaud S, Thery C, Ploix S, et al. Dendritic cell-derived exosomes for cancer immunotherapy: what’s next? Cancer Res. 2010;70(4):1281–1285. |
Supplementary materials
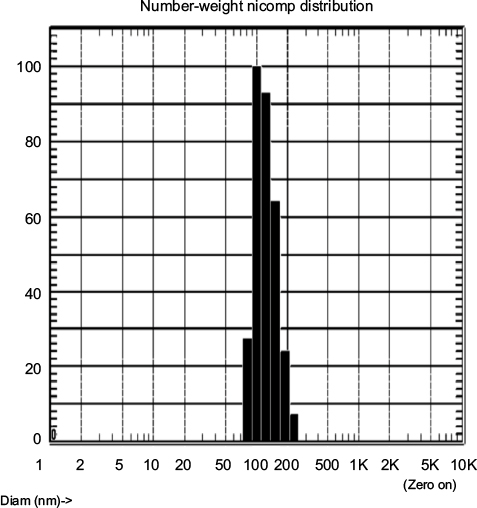 | Figure S1 Size determination of WSU-DLCL2 exosomes. Notes: Number–weight distribution of EV size using dynamic light scattering analysis with Nicomp BZ3000 instrument. Sample was read every minute for 30 minutes, with a calculated average diameter of 117 nm. One representative diameter histogram is shown. Abbreviations: DLCL, diffuse large B cell lymphoma; Diam, Diameter; EV, extracellular vesicle. |
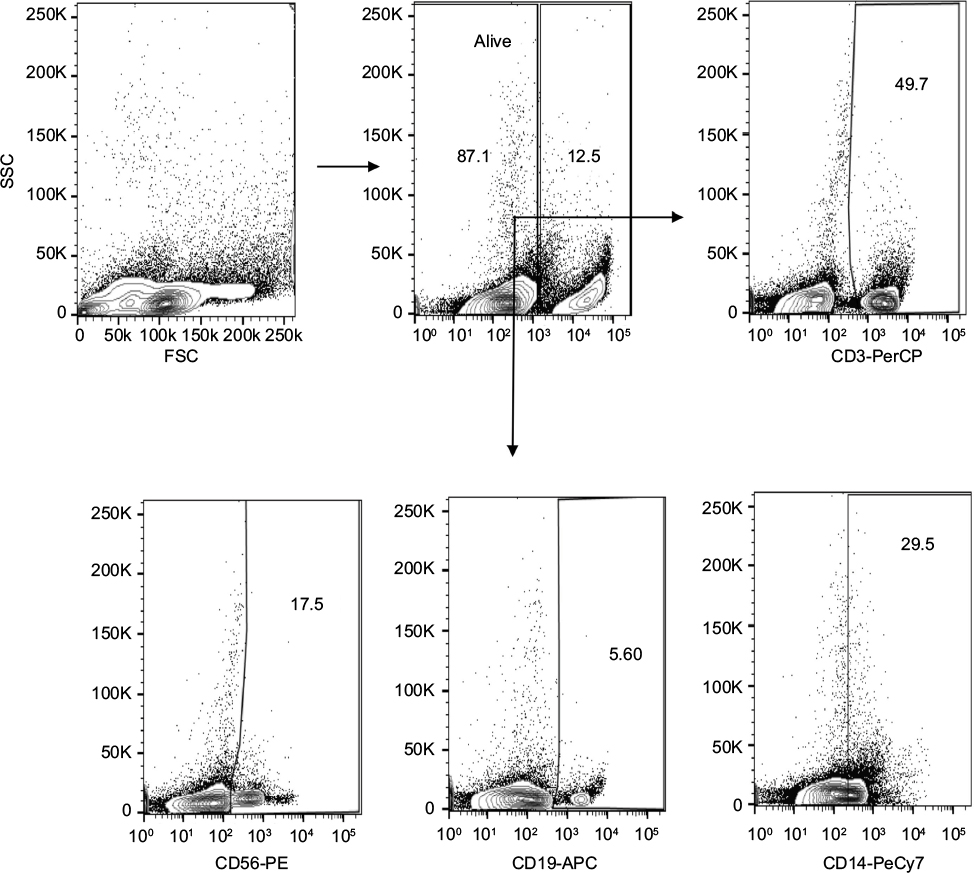 | Figure S2 Gating strategy used to assess binding of PKH67-labeled exosomes to human T cells (CD3+), NK cells (CD56+), B cells (CD19+), and monocytes (CD14+) within the peripheral blood leukocytes. Note: One representative dot plot is shown from two independent experiments. |
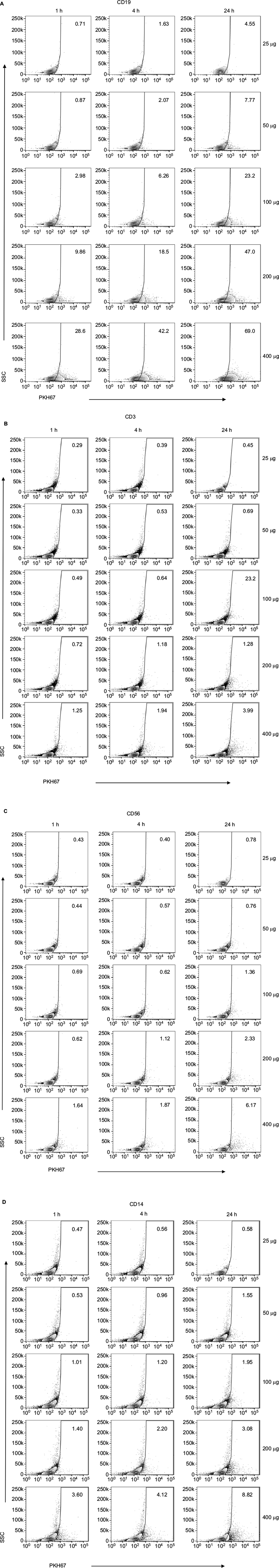 | Figure S3 Internalization of PKH67-labeled exosomes by healthy PBLs. Notes: Leukocytes were assayed by flow cytometry using specific antibodies to (A) B-lymphocytes (APC-anti-CD19), (B) T-lymphocytes (PerCP-anti-CD3), (C) NK cells (PE-anti-CD56), and (D) monocytes (PeCy7-anti-CD14). Representative dot plots are shown for each cell population. Abbreviation: PBLs, peripheral blood leukocytes; APC, antigen-presenting cell; h, hours. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.