Back to Journals » Journal of Inflammation Research » Volume 14
Upregulation of Neuronal Cylindromatosis Expression is Essential for Electroacupuncture-Mediated Alleviation of Neuroinflammatory Injury by Regulating Microglial Polarization in Rats Subjected to Focal Cerebral Ischemia/Reperfusion
Authors Lin X, Zhan J, Jiang J, Ren Y
Received 23 February 2021
Accepted for publication 17 April 2021
Published 20 May 2021 Volume 2021:14 Pages 2061—2078
DOI https://doi.org/10.2147/JIR.S307841
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Ning Quan
Xing Lin,1 Jian Zhan,2 Jin Jiang,3 Yikun Ren3
1Department of Biological Immunotherapy, Chongqing University Cancer Hospital, Chongqing Cancer Institute, Chongqing Cancer Hospital, Shapingba District, Chongqing, 400030, People’s Republic of China; 2Department of Neurology, The Second Affiliated Hospital of Zunyi Medical College, Zunyi, Guizhou Province, 563000, People’s Republic of China; 3Department of Neurology, The First Affiliated Hospital of Chongqing Medical University, Chongqing, 400016, People’s Republic of China
Correspondence: Jin Jiang; Yikun Ren
The First Affiliated Hospital of Chongqing Medical University, Yuzhong District, Chongqing, 400016, People’s Republic of China
Tel +86 177 4992 3714
; +86 151 9609 7005
Email [email protected]; [email protected]
Background: Activated microglia are polarized into the M1 or M2 phenotype. We previously reported that electroacupuncture (EA) effectively prevented nuclear factor-κB (NF-κB) nuclear translocation and improved neuronal C-X-C motif 3 chemokine ligand 1 (CX3CL1) expression, repressing microglial activation by upregulating neuronal cylindromatosis (CYLD) expression in the periischemic cortex. However, the potential mechanisms are unclear. Therefore, we explored whether EA improved CYLD protein expression to regulate microglial polarization-mediated neuroinflammation and the potential mechanisms in an ischemic stroke model.
Methods: A middle cerebral artery occlusion/reperfusion (MCAO/R) model was established in male Sprague-Dawley (SD) rats. The rats were treated with EA at the Baihui, Hegu and Taichong acupoints once daily beginning 2 h after focal cerebral ischemia. CYLD gene interference was used to investigate the role of CYLD in microglial polarization. We used neurobehavioral evaluations and TTC staining to examine the neuroprotective effect of EA via CYLD upregulation. Immunofluorescence and RT-qPCR were used to measure NLRP3 activation, M1/M2 microglial activation, pro-/anti-inflammatory gene mRNA expression and crosstalk (CX3CL1/CX3CR1 axis) between neurons and microglia. Western blotting was used to assess the underlying molecular mechanism.
Results: CYLD inhibited M1 microglial activation and improved M2 microglial activation after 72 h of reperfusion. CYLD overexpression decreased the NLRP3 mRNA level. CYLD suppressed microglial overactivation by inhibiting NLRP3 activation. CYLD gene silencing partially weakened EA improvement of neurological function deficits and reduction of infarct volumes after 72 h reperfusion. In addition, EA inhibited M1-like phenotypic microglial activation and promoted M2-like phenotypic microglia through upregulating CYLD expression. Finally, EA-mediated modulation of the CX3CL1/CX3CR1 axis and NLRP3 inflammasome was reversed by CYLD gene silencing in the periischemic cortex.
Conclusion: EA-induced upregulation of neuronal CYLD expression plays anti-inflammatory and neuroprotective roles and regulates the interaction between neurons and microglia, thereby suppressing M1 and improving M2 microglial activation in the periischemic cortex.
Keywords: microglial polarization, cerebral ischemia, neuroinflammation, CYLD, electroacupuncture, NLRP3 inflammasome, CX3CL1/CX3CR1 axis
Introduction
Under normal conditions, microglia exhibit a neural-specific phenotype and maintain an ostensibly quiescent surveillance phenotype to supervise the brain parenchyma.1,2 Microglia exhibit a resting state with a ramified morphology in the absence of stimuli.3,4 As resident cells of the central nervous system (CNS), microglia play an important role in regulatory processes in response to stimulation and repair.4 Microglia are thought to be the first cells that respond to various stimuli.5 To detect alterations in CNS homeostasis induced by inflammatory stimuli, microglia extend and retract cellular processes.6–8 Similar to other tissue-specific resident macrophages, microglia induce an immune response to pathogens, mediate exogenous injury and maintain homeostasis by eliminating agents, apoptotic cells, debris and denatured proteins.8,9 Interestingly, stimulated neurons activate various signaling pathways, including cytokines and chemokines, and the receptors of these cytokines and chemokines are exclusively expressed by microglia.
CX3CL1 is a neuroinflammatory chemokine exclusively expressed in neurons.10,11 CX3CR1, which is a unique CX3CL1 receptor, is exclusively expressed by microglia in the CNS.11,12 A recent study showed that CX3CL1 plays a neuroprotective role after ischemic stroke.13 Previous studies have suggested that the CX3CL1/CX3CR1 axis plays a crucial role in microglial-neuronal crosstalk.14 Neuronal CX3CL1 activation leads to microglial activation via crosstalk with CX3CR1, often occurring within minutes after ischemic stroke.15,16 Activated microglia polarize into classic (M1) and alternative (M2) phenotypes. The M1 phenotype often secretes proinflammatory cytokines, which are harmful to neurons. M2 microglia release anti-inflammatory cytokines to protect neurons from inflammatory injuries. Emerging evidence indicates that M1-like microglia are increased during the first 24 h in the ischemic core.17–19 However, during the first week, the M2 phenotype is predominant in the ischemic core, while the M1 phenotype is predominant in the periischemic areas.20 Thus, alleviating inflammatory injury in the border of ischemic cortex by inhibiting M1 microglial activation and promoting the M2 phenotype during the acute stage of the ischemic stroke is critical.
Cylindromatosis (CYLD) is a tumor suppressor involved in familial cylindromatosis.21 A recent study showed that gadolinium chloride inhibits nuclear factor-κB (NF-κB) activation to ameliorate lung injury in rats mainly by upregulating CYLD.22 The function of CYLD is related to the deubiquitination of K63-linked chains to break RIP1 and IKKγ binding, which negatively regulates IKK activation.22,23 Nikolaou et al suggest that CYLD gene overexpression alleviates inflammation during the acute or chronic stage after liver injury.24,25 Recently, CYLD was shown to negatively regulate oxygen-glucose deprivation (OGD)-induced necroptosis in primary cortical neurons.26 In our previous study, we demonstrated that neuronal CYLD could suppress microglial activation by negatively regulating the NF-κB signaling pathway after focal cerebral ischemia/reperfusion.27 However, silencing CYLD expression exacerbated ischemic stroke outcomes. Limited studies indicate that effects of the CYLD protein on inflammatory injury due to modulating microglial polarization during acute focal cerebral ischemia/reperfusion in rats, which may provide a potential strategy to regulate neuroinflammatory injury in the clinic.
Electroacupuncture (EA), as a traditional and nondrug therapy, has often been used in China. EA is often used as an adjuvant treatment to improve stroke prognosis and neuroinflammatory injuries in China.28–30 We reported that EA inhibited the expression of IKKα and IKKβ proteins to negatively regulate activation of the NF-κB signaling pathway, increase anti-inflammatory cytokines (IL-4 and IL-10) and ameliorate neuroinflammatory injuries during the acute stages of focal cerebral ischemia/reperfusion.31 Additionally, we previously demonstrated that EA could regulate the NF-κB signaling pathway to alleviate microglial activation-mediated neuroinflammatory injury in ischemic stroke model rats by upregulating zinc finger protein A20.32 Recently, our previous study indicated that upregulating neuronal CYLD expression was essential for EA-induced inhibition of microglial overactivation and decreasing detrimental proinflammatory cytokines.27 Thus, we hypothesize that EA improves CYLD protein expression to regulate microglial polarization.
Materials and Methods
Ethics Statement
All studies were performed in accordance with an experimental protocol approved by the Ethics Committee for Animal Experimentation of Chongqing Medical University and the guidelines of the National Institutes for Animal Research. The experimental process followed randomized and blinded guidelines.
Animal Model
Specific pathogen-free (SPF) male Sprague-Dawley (SD) rats (280–300 g) were purchased from the Experimental Animal Center of Chongqing Medical University. The rats were anesthetized with pentobarbital sodium (60 mg/kg i.p.). The right common and right external carotid arteries were exposed using a ventral midline neck incision. An ischemic stroke model was established by inserting a 2.0 monofilament nylon suture into the right external carotid artery (ECA) and extending it to the beginning of the middle cerebral artery (MCA). The rats were anesthetized with pentobarbital sodium to alleviate pain when the nylon monofilament was removed. Reperfusion was accomplished by withdrawing the suture after 2 h of ischemia. Then, the incision was sutured and sterilized. The animals were sacrificed after 72 h of reperfusion. During the surgical procedure, body temperatures were monitored rectally and maintained at approximately 37.5 °C with a thermostatic pad. A successful middle cerebral artery occlusion (MCAO) model was confirmed by a laser Doppler computerized main unit (PeriFlux 5000, Perimed AB, Sweden) when the regional cerebral blood flow (rCBF) decreased to 20% and recovered to >80% of the baseline (preischemic) level.
Lentivirus Production and Administration
Lentivirus administration was performed as previously described. Lentiviruses carrying exogenous CYLD (LV-CYLD, 2×108 transduction units, TU/mL) and CYLD shRNA (LV-shCYLD, 3×108 transduction units, TU/mL) to overexpress and silence CYLD, respectively, and the control (LV-control) were purchased from GeneChem (Shanghai, China) and stored at −80 °C until use. Lentiviruses were immediately centrifuged and placed on ice prior to injection. The viral dose was determined according to an approximate 80% infection rate in the brain. Two weeks before surgery, the rats received an intracerebral ventricular injection of 5 μL of LV-CYLD, LV-shCYLD, or LV-control (Figure 1A and B). The rats were anesthetized by intraperitoneal administration of pentobarbital sodium and placed onto a stereotaxic frame (Stoelting, USA). According to stereotactic parameters and the actual sizes of the rats, a cranial hole located 1.3 mm lateral and 1.5 mm posterior to the bregma was drilled in the right hemisphere, and the right lateral ventricle was slowly exposed to a depth of 3.8 mm beneath the dural surface. Then, the lentivirus (LV-CYLD or LV-shCYLD) or vehicle (LV-control) was injected into the right lateral ventricle at a rate of 0.5 μL/min. The effects of gene interference on CYLD overexpression or silencing were verified by real-time quantitative polymerase chain reaction (RT-qPCR) (Figure 1C).
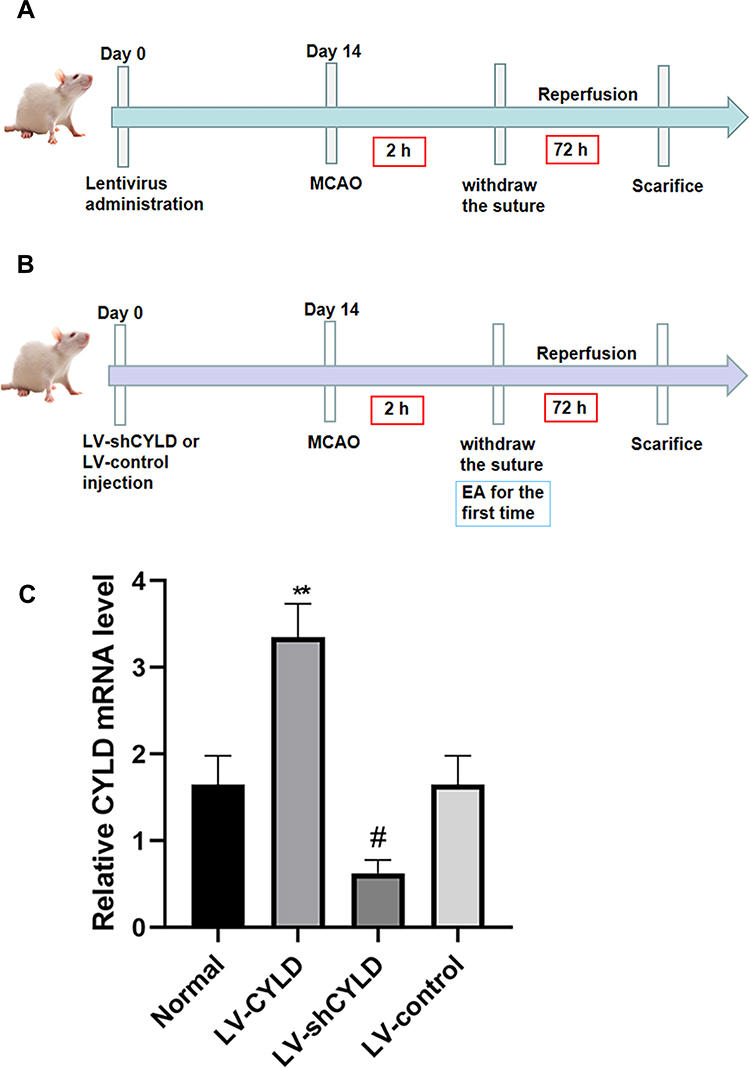 |
Figure 1 The timeline of experiment. The nylon monofilament was removed after 2 h of ischemia. (A) Schematic representation of part 1 experiment about the effect of CYLD gene interference on microglial polarization after 72h reperfusion. (B) Schematic representation of part 2 experiment. The rats were initially treated with EA when the nylon monofilament was withdrawn and then once daily until sacrifice. The effect of EA on neuroinflammatory injury was explored at 72 h reperfusion. (C) The expression of CYLD mRNA before inducing MCAO/R model was clearly enhanced/decreased via lentivirus administration. The rats were randomly divided into four groups: Normal, LV-CYLD, LV-shCYLD and LV-control group. **p<0.01 and #p<0.05 vs Normal group. |
Electroacupuncture Treatment
The Baihui (GV 20) acupoint (intersection of the sagittal midline and the line between the ears), the Hegu (LI 4) acupoint (the radial side of the left second metacarpal midpoint) and the Taichong (LR 3) acupoint (the dent between the first and second left metatarsal) were selected according to Experimental Animals Meridians Mapping (Figure 2B). The rats were stimulated at an intensity of 1 mA and a frequency of 20 Hz for 5 min, followed by 2 Hz for 30 min, using a G6805-2 EA instrument. The rats were initially treated with EA when the nylon monofilament was removed and then once daily until sacrifice (Figure 1B). Rats in the MCAO/R+EA, MCAO/R+LV-CYLD+EA, MCAO/R+LV-shCYLD+EA, and MCAO/R+LV-control+EA groups but not the MCAO/R group were treated with EA.
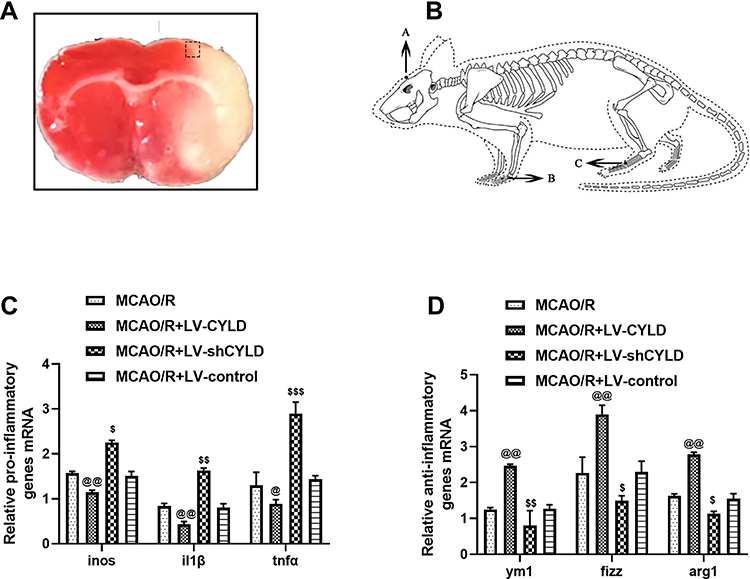 |
Figure 2 CYLD regulates microglial polarization after 72 h of reperfusion in ischemic stroke rats. (A) The coronal brain section stained with TTC is labeled with a box to show the periischemic cortex analyzed. (B) Schematic representation of the Baihui (GV 20) acupoint ((A) intersection of the sagittal midline and the line between the ears), the Hegu (LI 4) acupoint ((B) the radial side of the left second metacarpal midpoint) and the Taichong (LR 3) acupoint ((C) the dent between the first and second left metatarsal) were selected according to Experimental Animals Meridians Mapping. The mRNA level of M1 microglia related pro-inflammatory genes ((C), n=5) and M2 microglia related anti-inflammatory genes ((D), n=5) were measured with RT-qPCR at 72 h reperfusion in the border region of the ischemic cortex. The graph shows the relative mRNA levels after normalization to the housekeeping gene β-actin. All values are presented as the means ± SEMs. @@p<0.01, @p<0.05, $p<0.05, $$p<0.01 and $$$p<0.001 vs the MCAO/R group. |
Neurobehavioral Evaluation
The rats underwent neurological assessment after 72 h of reperfusion by a researcher who was blinded to the animal treatments. The Longa score was used to detect motor functions (Table 1).33 The higher the Longa score, the more severe is the damage. Animals that scored 2 and 3 were included in this study, and the MCAO rats that scored 0, 1, or 4 were excluded. The Bederson score was applied to evaluate global neurological function (Table 2).34 The higher the Bederson score, the more severe is the damage. The Garcia score was used to evaluate the sensorimotor function (Table 3).35 The lower the Garcia score, the more severe is the damage.
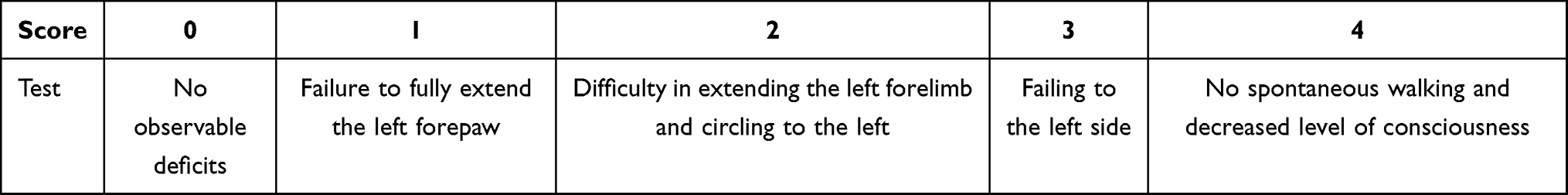 |
Table 1 The Longa Score for Neurological Scores |
 |
Table 2 The Bederson Score for Neurological Scores |
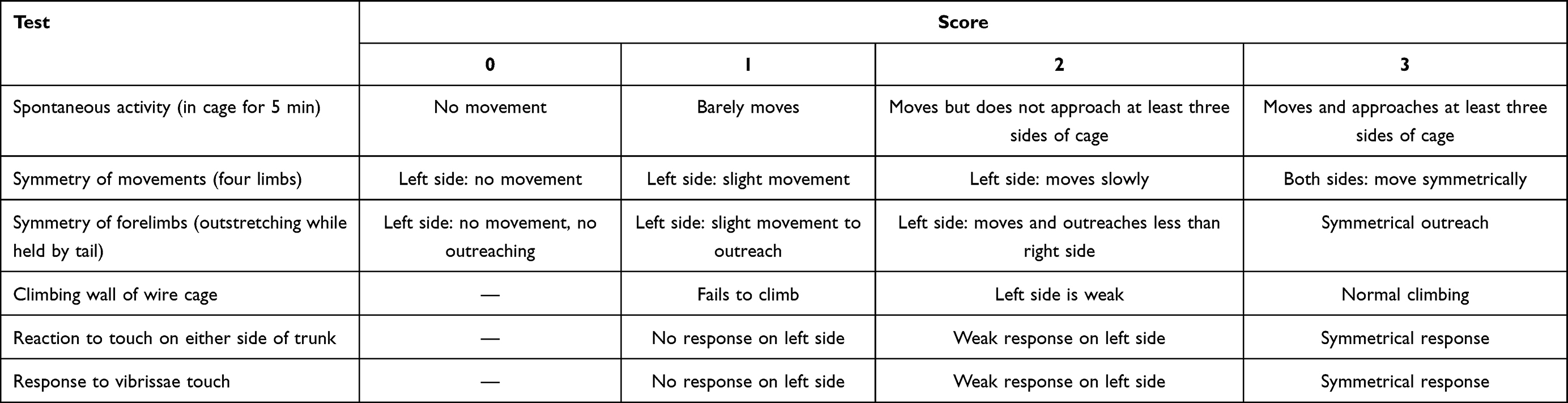 |
Table 3 The Garcia Score for Neurological Scores |
Infarct Volume Assessment
The rats were anesthetized as described and rapidly beheaded after 72 h of reperfusion. The intact brain was immediately frozen at −20 °C for 20 min and then sliced into 2 mm-thick coronal sections, which were stained with 2% 2,3,5-triphenyltetrazolium chloride for 15 min at 37 °C. Images of the brain slices were collected using a digital camera and uploaded for analysis. Pale brain areas were regarded as infarcts, and the corresponding volumes were measured using imaging analysis software by a researcher who was blinded to the animal treatment groups. The infarction volume is shown as the percentage of intact hemispheres.
Double-Immunofluorescent Labeling
The rats were deeply anesthetized with pentobarbital sodium after 72 h of reperfusion. The bodies were then perfused with saline, followed by 4% paraformaldehyde (PFA, 4 °C). Intact brains were rapidly harvested, postfixed in 4% PFA for 48 h, dehydrated in 30% sucrose, and cut into 10 μm-thick coronal slices. The sections were permeabilized with 0.3% Triton X-100 for 20 min at 37 °C and blocked with 5% donkey serum for 1 h at 37 °C. Then, the sections were incubated at 4 °C overnight with the following primary antibodies: rabbit anti-CX3CL1 (ab25088, Abcam, United Kingdom, 1:100), mouse anti-CX3CR1 (sc-377227, Santa Cruz, United States, 1:50), rabbit anti-NLRP3 (NBP2-12446, Novus, United States, 1:50), goat anti-Iba1 (to label microglia, NB100-1028SS, Novus, United States, 1:50), rabbit anti-iNOS (18985-1-AP, Proteintech, China, 1:50) and rabbit anti-CD206 (18985-1-AP, Proteintech, China, 1:50). After the sections were rewarmed at 37 °C for 1 h, each section was incubated with the following secondary antibodies at 37 °C for 1 h in the dark: Alexa Fluor 488-conjugated goat anti-mouse IgG (H+L) (SA00006-1, Proteintech, China, 1:200), Alexa Fluor 594-conjugated donkey anti-rabbit IgG (H+L) (SA00006-8, Proteintech, China, 1:200), and FITC-conjugated AffiniPure donkey anti-goat IgG (H+L) (SA00003-3, Proteintech, China, 1:200). DAPI was used to stain the nuclei (C1005, Beyotime Biotechnology, China) for 10 min at room temperature. All images were obtained using an immunofluorescence microscope (LSM-800, Carl Zeiss Micro-Image Co., Germany). Cell counts are expressed as numbers/mm2 in images at 200× magnification. Five randomly acquired fields of the periischemic cortex were analyzed in each group (Figure 2A). ImageJ was used by a researcher who was blinded to the treatments to count the positive cells.
RT-qPCR
Total RNA was extracted from the periischemic cortex of brain tissue (Figure 2A) with TRIzol reagent (TaKaRa, Japan) after 72 h of reperfusion. The RNA was reverse transcribed into cDNA with a PrimeScript RT reagent kit with gDNA Eraser. RT-qPCR was performed using an iQ5 Gradient Real-Time PCR detection system (Bio-Rad Co., city and state required) and SYBR Green (SYBR Premix Ex TaqII, TaKaRa, Japan) to analyze NLRP3, proinflammatory genes (inos, il1β, and tnfα) and anti-inflammatory genes (ym1, arg1, and fizz1) (Table 4). The following cycling conditions were used: 1) 30 s at 95 °C; 2) 5 s at 95 °C; 3) 30 s at 60 °C; 4) 30 s at 72 °C, followed by a plate read; 5) repeat step 2) for 39 cycles; 6) 10 s at 95 °C; and 7) melt curve at 60 °C to 95 °C in increments of 0.5 °C. Then, the plates were read. The melting curve of each sample was analyzed to determine the primer-target specificity. The data were normalized to the mean of β-actin. Relative mRNA expression was calculated using the 2−ΔΔCt method.
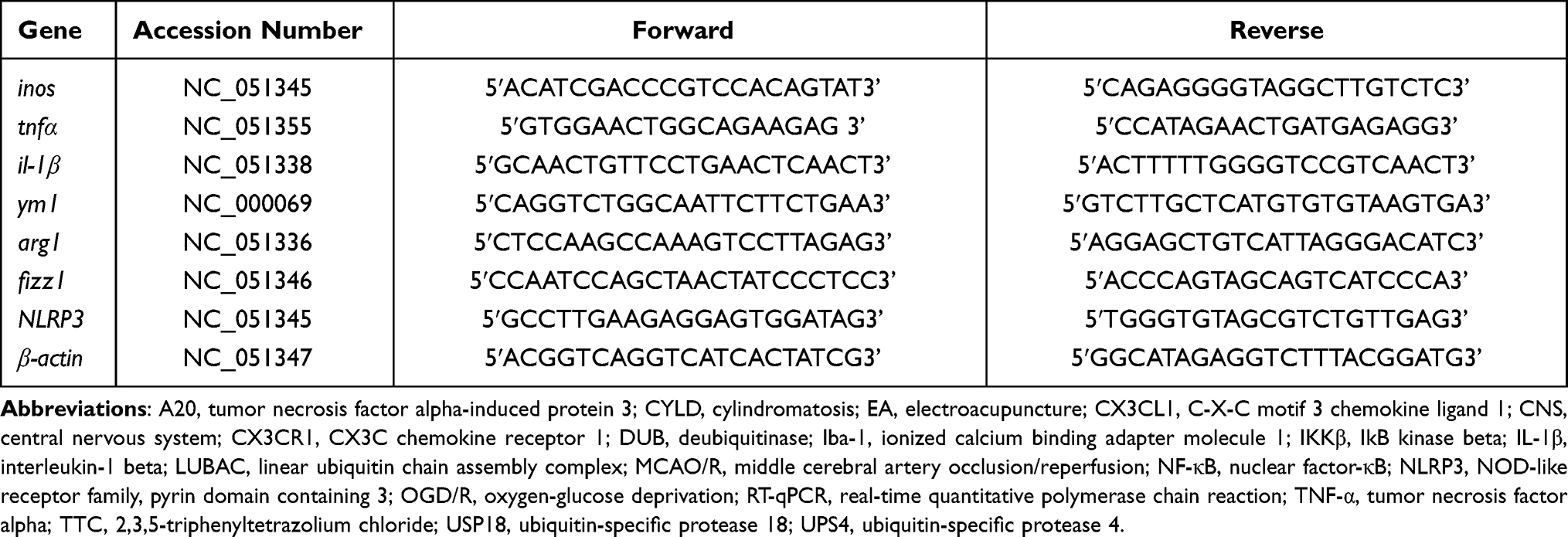 |
Table 4 List of Primers Used for RT-qPCR |
Western Blot Analysis
Rats were treated as described and perfused with saline. Brain tissues were extracted from the periischemic cortex (Figure 2A). Tissues were homogenized in radioimmunoprecipitation assay (RIPA) lysis buffer (no. P0013B, Beyotime, China) containing phenylmethane sulfonyl fluoride (PMSF) (Beyotime, Shanghai, China). Total protein was extracted from the supernatants on ice after centrifugation at 12,000 rpm for 15 min. Western blotting was performed as previously described (Bio-Rad Co., California, USA). The total protein (50–100 μg) was separated by 8–12% SDS-PAGE (Beyotime, Shanghai, China) and then transferred to 0.46 μm polyvinylidene fluoride (PVDF) membranes (IPVH00010, Millipore Co., Massachusetts, USA). After blocking nonspecific epitopes, the membranes were incubated at 4 °C overnight with the following primary antibodies: mouse anti-β-actin (66009-1-Ig, 1:5000, Proteintech), mouse anti-GAPDH (66009-1-Ig, 1:5000, Proteintech), rabbit anti-CX3CL1 (ab25088, Abcam, UK, 1:1000), mouse anti-CX3CR1 (sc-377227, Santa Cruz, United State,1:500), rabbit anti-NLRP3 (NBP2-12446, Novus, USA,1:1000) and rabbit anti-IL-1β (bs-0812, Bioss, China). The membranes were then incubated with the corresponding secondary antibodies, which were horseradish peroxidase-conjugated goat anti-rabbit (Abclonal, 1:3000) or goat anti-mouse (Abclonal, United State, 1:3000), for 2 h at 37 °C. A gel imaging apparatus (Vilber Lourmat fusion FX 7 Spectra, France) was used to scan the immunoblots. Analysis software (FUSION-CAPT, France) was used to analyze each band.
Statistical Analysis
GraphPad Prism Version 8.0 was used for all statistical analyses. The Longa score, Bederson score, and Garcia score were analyzed using Kruskal–Wallis tests followed by post hoc Dunn’s multiple comparison tests. All other data are expressed as the means ± SEM. Additional data were analyzed by one-way ANOVA followed by LSD multiple-range test. A value of p<0.05 was considered statistically significant.
Results
CYLD Regulates Microglial Polarization After Focal Ischemia/Reperfusion
We previously demonstrated that the protein CYLD could attenuate microglial overactivation after 72 h of reperfusion. To examine the effect of CYLD expression on microglial polarization in the border region of the ischemic cortex in rats after ischemic stroke, we used RT-qPCR to analyze anti-inflammatory and proinflammatory genes after 72 h of reperfusion. The animals were randomly divided into four groups: MCAO/R, MCAO/R+LV-shCYLD, MCAO/R+LV-CYLD, and MCAO/R+LV-control. After 72 h of reperfusion, the mRNA expression of M1 microglia-related proinflammatory genes (inos, il1β, and tnfα) was reduced (p<0.01, p<0.01 and p<0.05, Figure 2C) and that of M2 microglia-related anti-inflammatory genes (ym1, arg1, and fizz1) was increased (p<0.01, p<0.01 and p<0.01, Figure 2D) by CYLD overexpression compared with that of the MCAO/R group. However, compared with the MCAO/R group, the MCAO/R+LV-shCYLD group showed increased mRNA expression of proinflammatory genes and decreased mRNA expression of anti-inflammatory genes (Figure 2C and D). There was no significant difference in inflammatory gene expression between the MCAO/R and MCAO/R+LV-control groups (p>0.05, Figure 2C and D). These results indicated that CYLD could change phenotypic patterns by inhibiting the M1-like phenotype and potentiating M2-like microglia. Thus, we hypothesized that the neuroprotective effect of CYLD was due to its ability to regulate microglial polarization after focal cerebral ischemia/reperfusion.
CYLD Inhibits the NLRP3 Inflammasome and Microglial Activation
We previously indicated that neuronal CYLD could be a negative regulator of the NF-κB signaling pathway.27 Previous studies have shown that NLRP3 transcription was markedly upregulated via NF-κB nuclear translocation.36 To examine the effect of CYLD on NLRP3 transcription in the border region of ischemic areas in rats, we used RT-qPCR to measure the mRNA expression of NLRP3 after 72 h of reperfusion. The animals were also randomly divided into four groups: MCAO/R, MCAO/R+LV-shCYLD, MCAO/R+LV-CYLD, and MCAO/R+LV-control. Figure 3A shows that NLRP3 mRNA expression was lower in the MCAO/R+LV-CYLD group than in the MCAO/R group (p<0.01, Figure 3A). Interestingly, we found that CYLD silencing improved the mRNA expression of NLRP3 compared with that of the MCAO/R group (p<0.01, Figure 3A). However, there was no obvious difference in NLRP3 mRNA expression between the MCAO/R and MCAO/R+LV-control groups (Figure 3A).
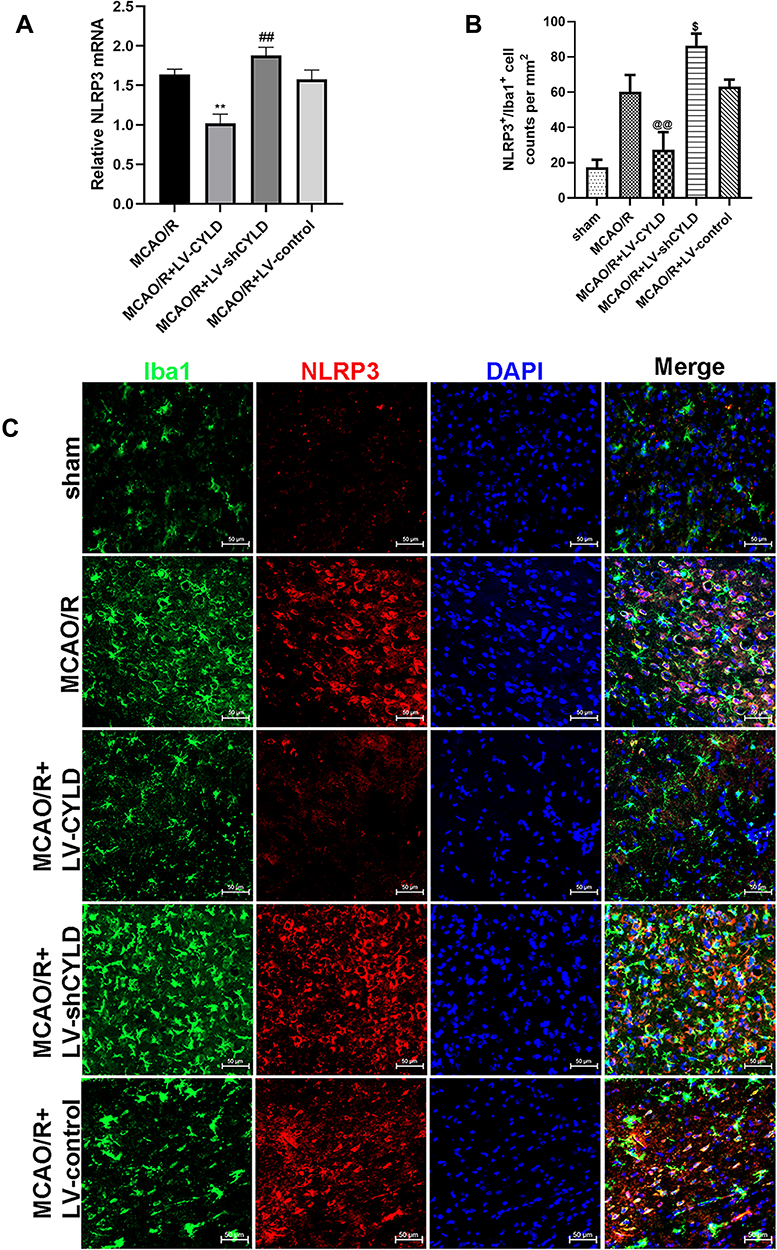 |
Figure 3 CYLD inhibits the NLRP3 inflammasome and microglial activation after 72 h reperfusion in rats. (A) NLRP3 mRNA expression were measured with RT-qPCR at 72 h reperfusion in the periischemic cortex (n=5). The graph shows the relative mRNA levels after normalization to the housekeeping gene β-actin. (B) NLRP3+/Iba+ cell counts were expressed as number/mm2. (C) Immunofluorescence staining showed co-expression of NLRP3 (red) and microglia (green, Iba 1) in the periischemic cortex (White arrows indicate co-expression between NLRP3 and microglia, scale bar = 50 µm, n=5). All values are presented as the means ± SEMs. **p<0.01, ##p<0.01,$p<0.05 and @@p<0.01 vs the MCAO/R group. |
We further investigated the effect of CYLD on the inhibition of microglial activation by suppressing NLRP3 expression. The rats were randomly divided into five groups: sham, MCAO/R, MCAO/R+LV-shCYLD, MCAO/R+LV-CYLD, and MCAO/R+LV-control. The expression of NLRP3 in microglia was investigated by using double-immunofluorescence labeling after 72 h of reperfusion. In the present study, there were fewer NLRP3 and Iba1 double-labeled cells in the MCAO/R+LV-CYLD group, while there were more activated microglia with larger cell bodies and retracted, thickened protuberances in the ischemic border region and an increased number of double-labeled cells in the MCAO/R group (p<0.01, Figure 3B and C). Moreover, an increase in double-labeled cells was observed in the MCAO/R+LV-shCYLD group compared with the MCAO/R group (p<0.05, Figure 3B and C). There was no obvious difference in the number of NLRP3 and Iba1 double-labeled cells between the MCAO/R and MCAO/R+LV-control groups (Figure 3B and C). Therefore, these data suggested that the CYLD protein could suppress microglial overactivation by inhibiting NLRP3 activation in the periischemic cortex.
CYLD Silencing Partially Weakens the Neuroprotective Effects of EA After Focal Cerebral Ischemia/Reperfusion
In a previous study, we demonstrated that CYLD could play a neuroprotective role in ischemic stroke.27 To explore whether CYLD is required for the neuroprotective effect of EA, neurobehavioral evaluations and infarct volumes were measured in the five groups (sham, MCAO/R, MCAO/R+LV-shCYLD+EA, MCAO/R+LV-control+EA and MCAO/R+EA) after 72 h of reperfusion. Neurological function improvements were observed in the MCAO/R+EA group compared to the MCAO/R group in terms of the Longa score (p<0.01, Figure 4C), Bederson score (p<0.001, Figure 4D) and Garcia score (p<0.001, Figure 4E). Moreover, rats in the MCAO/R+EA group displayed significantly smaller infarct volumes compared to those in the MCAO/R group (p<0.01, Figure 4A and B). In contrast, poor neurological function and larger infarct volumes were observed in the MCAO/R+LV-shCYLD+EA group compared to the MCAO/R+EA group (Figure 4A–E). There was no significant difference in neurobehavioral evaluations or infarct volumes between the MCAO/R+LV-control+EA and MCAO/R+EA groups (Figure 4A–E). These data indicated that upregulation of CYLD expression is essential for EA-mediated improvement of neurological function after ischemic stroke in rats.
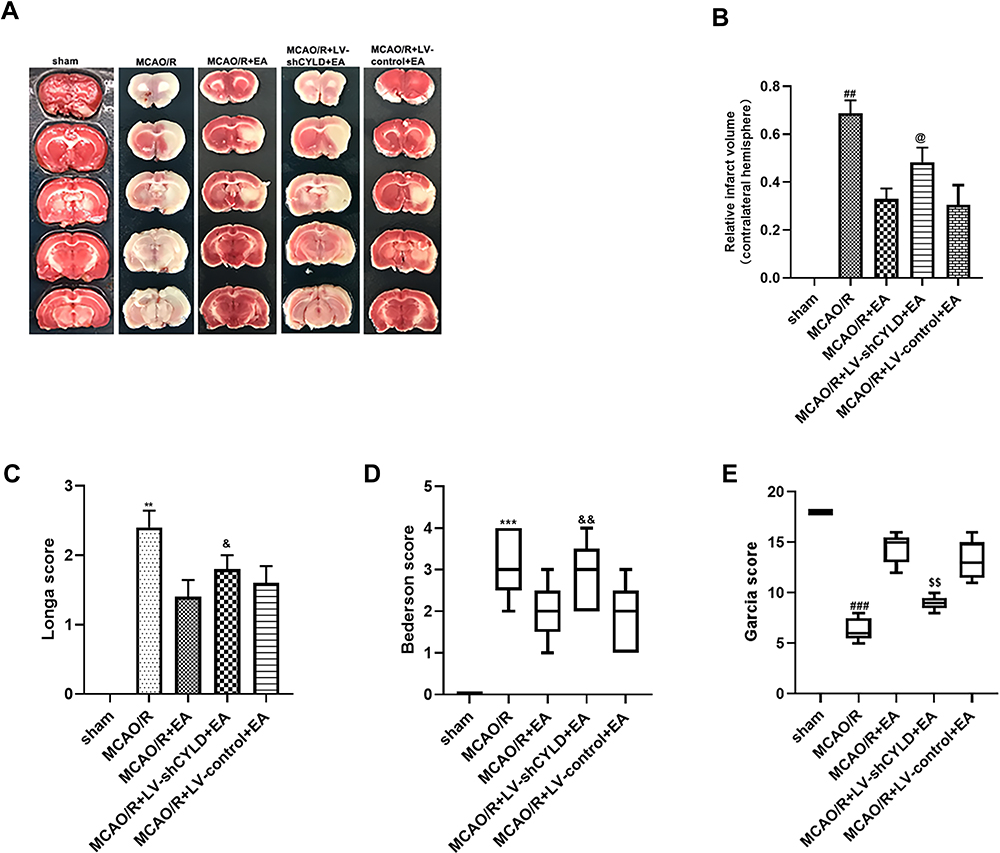 |
Figure 4 CYLD silencing partially weakens the neuroprotective effects of EA after 72 h reperfusion in ischemic stroke rats. (A) Images of cerebral infarction stained with 2% TTC (white, infarct tissue; red, non-infarct tissue) at 72 h reperfusion. (B) Infarct volumes are percent of intact hemispheres at 72 h reperfusion (n=3). (C) Longa score for neurological evaluation at 72 h reperfusion (n=5). (D) Bederson score for neurological evaluation at 72 h reperfusion (n=5). (E) Garcia score for neurological evaluation at 72 h reperfusion (n=5). All values are presented as the means ± SEMs. ##p<0.01, @p<0.05,**p<0.01, &p<0.05, ***p<0.001, &&p<0.01, ###p<0.001 and $$p<0.01 vs the MCAO/R+EA group. |
CYLD Silencing Partially Weakens EA-Mediated Modulation of Microglial Phenotypes
To investigate whether CYLD was essential for EA-mediated modulation of microglial phenotypes, the levels of inflammatory genes were measured by RT-qPCR, and microglial phenotypes were investigated by double-immunofluorescence labeling after 72 h of reperfusion. Rats were randomly divided into four groups: MCAO/R, MCAO/R+EA, MCAO/R+LV-shCYLD+EA, and MCAO/R+LV-control+EA. RT-qPCR showed increased expression of anti-inflammatory genes (ym1, fizz1 and arg1) (p<0.01, p<0.01 and p<0.001, Figure 5B) and decreased expression of proinflammatory genes (inos, tnfα and il1β) (p<0.001, p<0.01 and p<0.001, Figure 6B) in the MCAO/R+EA group compared to the MCAO/R group. The expression of proinflammatory genes (p<0.05, p<0.05 and p<0.05, Figure 6B) was significantly increased in the MCAO/R+LV-shCYLD+EA group compared with the MCAO/R+EA group. However, reduced expression of anti-inflammatory genes was clearly observed in the MCAO/R+LV-shCYLD+EA group compared with the MCAO/R+EA group (p<0.01, p<0.01 and p<0.01, Figure 5B). There was no significant difference between the MCAO/R+ LV-control+EA and MCAO/R+EA groups (Figures 5–6B).
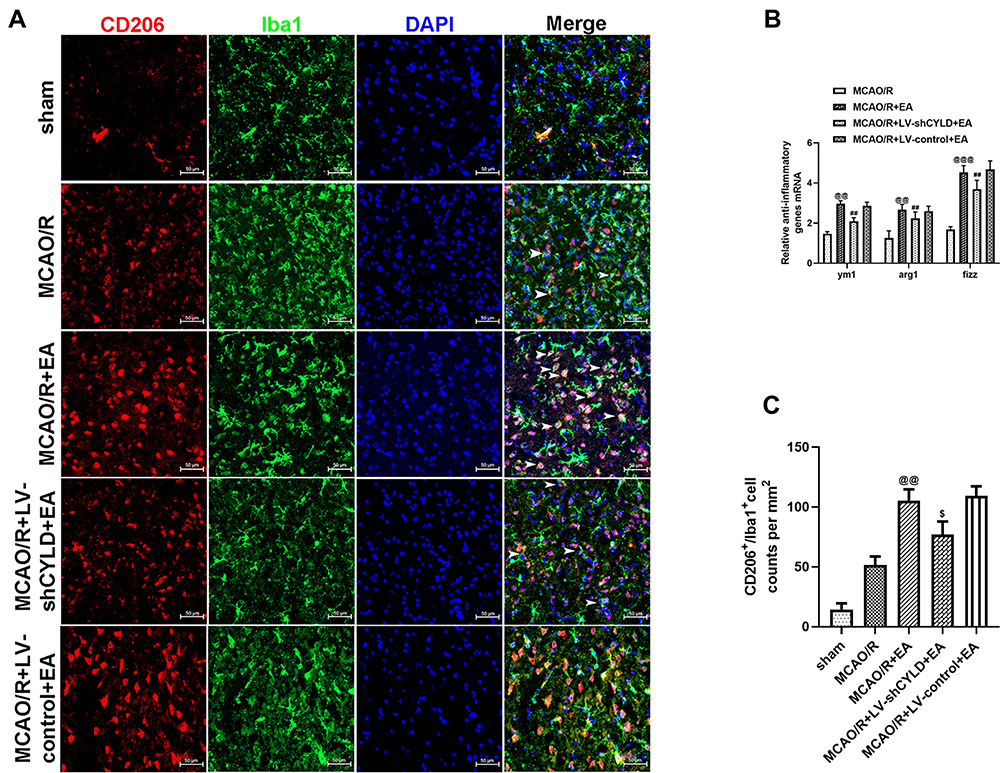 |
Figure 5 CYLD silencing partially weakens EA-mediated improvement of M2 microglial phenotype in the periischemic cortex. (A) Immunofluorescence staining showed co-expression of CD206 (red) and microglia (green, Iba 1) in the periischemic cortex (n=5, white arrows indicate M2 (➤). Scale bar = 50 µm). (B) Anti-inflammatory genes ((B), n=6) mRNA expression were measured with RT-qPCR at 72 h reperfusion in the periischemic cortex. The graph shows the relative mRNA levels after normalization to the housekeeping gene β-actin. (C) CD206+/Iba+ cell counts were expressed as number/mm2. All values are presented as the means ± SEMs. @@p<0.01 and @@@p<0.001 vs the MCAO/R group. ##p<0.01 and $p<0.05 vs the MCAO/R +EA group. |
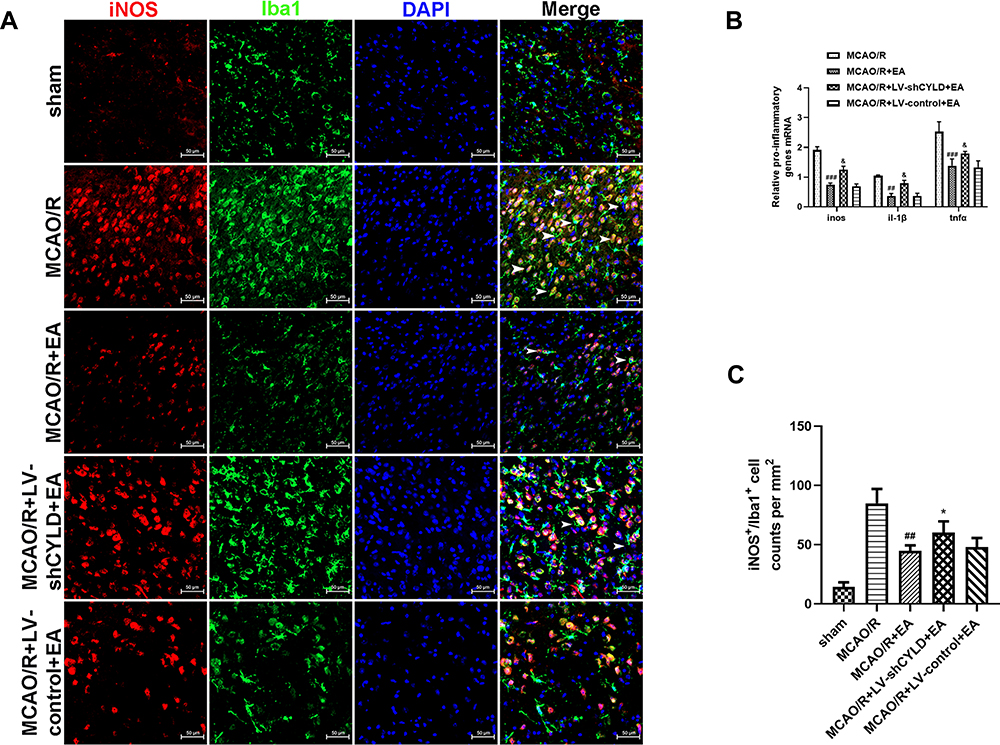 |
Figure 6 CYLD silencing partially weakens EA-mediated inhibition of M1 microglial phenotype in the periischemic areas. (A) Immunofluorescence staining showed co-expression of iNOS (red) and microglia (green, Iba 1) in the periischemic cortex (n=5, white arrows indicate M1 (➤) microglia. Scale bar = 50 µm). (B) Pro-inflammatory genes mRNA expression were measured with RT-qPCR at 72 h reperfusion in the periischemic cortex. The graph shows the relative mRNA levels after normalization to the housekeeping gene β-actin. (C) iNOS+/Iba+cell counts were expressed as number/mm2. All values are presented as the means ± SEMs. ##p<0.01 and ###p<0.001 vs the MCAO/R group. &p<0.05 and *p<0.05 vs the MCAO/R +EA group. |
We further investigated the effect of EA on the regulation of microglial phenotypes. After 72 h of reperfusion, Iba1/iNOS-immunopositive cells (M1) and Iba1/CD206-immunopositive cells (M2) in the periischemic cortex were measured by double-immunofluorescence labeling. Rats were randomly divided into five groups: sham, MCAO/R, MCAO/R+EA, MCAO/R+LV-shCYLD+EA, and MCAO/R+LV-control+EA. Iba1+/CD206+ cells (p<0.01, Figure 5A and C) were increased in the MCAO/R+EA group, whereas Iba1+/iNOS+ cells (p<0.01, Figure 6A and C) were decreased compared to those in the MCAO/R group. Moreover, CYLD silencing+EA resulted in a significant decrease in Iba1+/CD206+ cells (M2) (p<0.05, Figure 5A and C), whereas the number of Iba1/iNOS-immunopositive cells (M1) (p<0.05, Figure 6A and C) significantly increased compared with that of the MCAO/R+EA group. There was no significant difference between the MCAO/R+ LV-control+EA and MCAO/R+EA groups. These results indicated that EA may induce M2-like microglial activation and inhibit M1-like microglia by upregulating CYLD protein expression after 72 h of reperfusion in the periischemic cortex.
CYLD Silencing Partially Weakens EA-Mediated Modulation of the CX3CL1/CX3CR1 Axis and NLRP3 Activation After Focal Cerebral Ischemia/Reperfusion
The CX3CL1/CX3CR1 axis is significantly upregulated by activation of the NF-κB signaling pathway, which is the key step in the neuronal-microglial crosstalk after focal cerebral ischemia/reperfusion.14 We confirmed whether neuronal CYLD was essential for EA-mediated modulation of neuronal-microglial crosstalk via the CX3CL1/CX3CR1 axis. Rats were randomly divided into four groups: MCAO/R, MCAO/R+LV-shCYLD+EA, MCAO/R+EA and MCAO/R+LV-control+EA. After 72 h of reperfusion, Western blot analysis showed that CX3CR1 protein expression was obviously reduced and CX3CL1 protein expression was markedly increased in the MCAO/R+EA group than in the MCAO/R group (p<0.001, p<0.01, respectively; Figure 7B and C). In contrast, CX3CL1 protein expression decreased and CX3CR1 expression increased in the MCAO/R+LV-shCYLD+EA group compared to the MCAO/R+EA group (p<0.01, p<0.05, respectively; Figure 7B and C). We also found decreased NLRP3 protein expression in the MCAO/R+EA group compared with the MCAO/R group (p<0.01, Figure 7B and C). However, increased NLRP3 protein expression was observed in the MCAO/R+LV-shCYLD+EA group compared with the MCAO/R+EA group (p<0.01, Figure 7B and C). Rats were randomly divided into five groups: sham MCAO/R, MCAO/R+LV-shCYLD+EA, MCAO/R+EA and MCAO/R+LV-control+EA. After 72 h of reperfusion, immunofluorescence showed that CX3CL1-positive CX3CR1 cells were markedly increased in the MCAO/R group than in the MCAO/R+EA group (p<0.001, Figures 8 and 7A). Upon CYLD gene silencing, the decrease in CX3CL1-positive CX3CR1 cells were abrogated in the MCAO/R+LV-shCYLD+EA group compared with MCAO/R+EA group (p<0.01, Figures 8 and 7A). Thus, neuronal CYLD was essential for EA-induced modulation of neuronal-microglial crosstalk (CX3CL1/CX3CR1 axis) and inhibited NLRP3 activation in the border of the ischemic cortex after ischemic stroke.
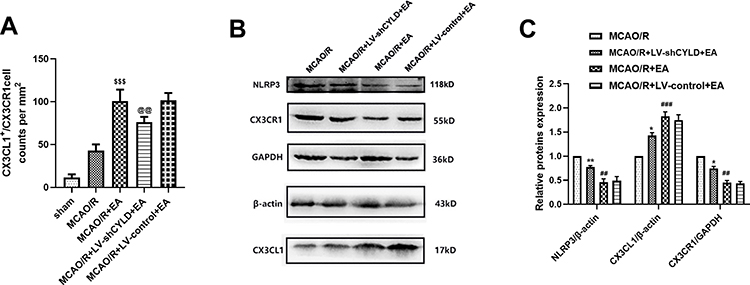 |
Figure 7 CYLD silencing partially weakens EA-mediated modulation of the CX3CL1/CX3CR1 axis and NLRP3 activation after 72 h reperfusion. (A) CX3CL1+/CX3CR1+ cell counts were expressed as number/mm2 (n=3). (B) Western blot of NLRP3, CX3CL1 and CX3CR1 protein expression. (C) NLRP3 and CX3CL1 proteins normalized with β-actin. CX3CR1 protein normalized with GAPDH (n=5). All values are presented as the means ± SEMs. @@p<0.01, **p<0.01 and *p<0.05 vs the MCAO/R+EA group. $$$p<0.001, ###p<0.001 and ##p<0.01 vs the MCAO/R group. |
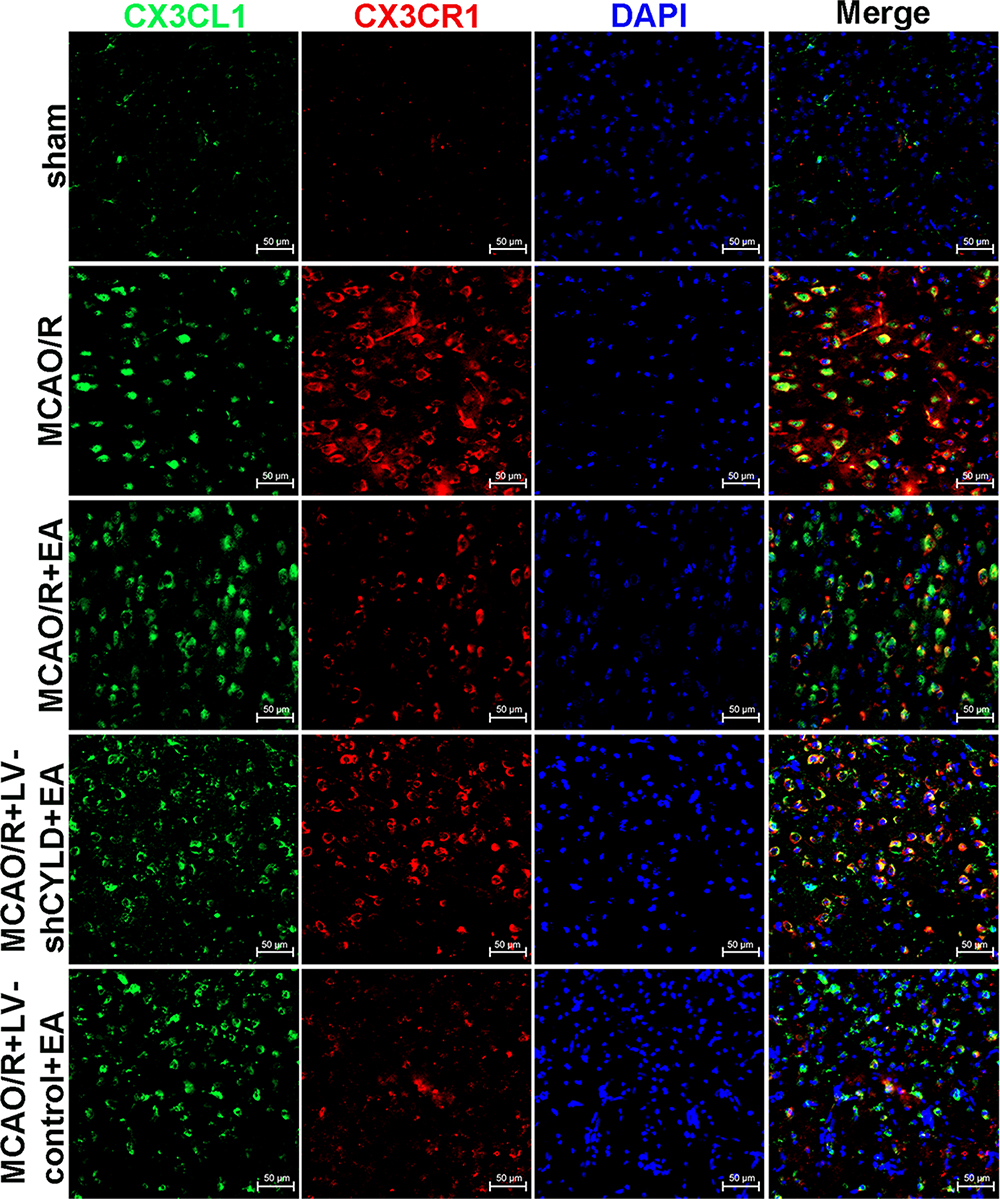 |
Figure 8 Co-expression of CX3CL1 (red) and CX3CR1 (green) in the periischemic cortex (n=5. Scale bar = 50 µm). |
Discussion
CYLD is a member of the deubiquitinase (DUB) family that negatively regulates inflammatory injury.21,37–39 In our previous study, we demonstrated that CYLD was mainly expressed in the cytoplasm of cortical neurons in periischemic areas during the acute stage of focal cerebral ischemia/reperfusion.27 Moreover, we found that CYLD overexpression inhibited microglial activation and proinflammatory cytokines in periischemic areas.27 Here, we further report that CYLD not only regulates microglial polarization but is also essential for EA-mediated neuroprotection and anti-inflammatory effects via the NLRP3 inflammasome and CX3CL1/CX3CR1 axis after focal cerebral ischemia/reperfusion in rats.
Neuroinflammation exerts beneficial or detrimental impacts on brain tissues after ischemic stroke.40–43 Microglia play a critical role as a double-edged sword in neurological recovery after ischemic stroke.2,3,43–45 M1 microglia secrete the proinflammatory cytokines TNF-α and IL-1β, which accelerate NF-κB-induced NLRP3 inflammasome activation.46 The M2 phenotype releases IL-4 and IL-10 to reduce inflammation. M1-type microglia, which are detrimental to brain tissues, are predominant in the periischemic areas during the first week. Therefore, it is essential to reduce inflammatory injury in the border of ischemic areas during the acute stage. Zinc inhibits LPS-induced inflammatory responses by upregulating expression of A20, which also belongs to the DUB family, in microglial BV2 cells.47 Ubiquitin-specific protease 18 (USP18) overexpression protects against focal cerebral ischemia injury in mice by suppressing microglial activation.48 A recent study demonstrated that downregulation of ubiquitin-specific protease 4 (USP4) expression could promote microglial activation and subsequent neuronal inflammation in rat spinal cord injury.49 A series of studies was conducted to examine the anti-inflammatory effects of DUBs against microglial activation after ischemic damage. However, there have been few reports about the effect of CYLD on microglial polarization in the brain after ischemic stroke. We found that CYLD overexpression may hinder microglial activation. In addition, our results showed that CYLD overexpression not only promoted microglial M2 polarization but also definitively inhibited the expression of proinflammatory (M1) genes after 72 h of reperfusion. Therefore, we suggest that neuronal CYLD may be a promising potential therapeutic target for the regulation of microglial polarization in the acute stage of ischemic brain injury. However, the underlying mechanism is not clear. Emerging evidence has suggested that NLRP3 inflammasome activation, which is involved in microglial polarization, requires NF-κB-mediated NLRP3 activation.50–53 In our previous study, we demonstrated that CYLD could negatively regulate the NF-κB signaling pathway.27 A recent report showed that CYLD deficiency in macrophages profoundly promoted IL-1β secretion, active caspase-1 generation, apoptosis-associated speck-like protein containing speck (ASC) formation, and pyroptotic cell death in response to canonical activation of the NLRP3 inflammasome in a peritonitis mouse model.54 Consistent with these studies, our results showed that CYLD overexpression inhibited NLRP3 inflammasome-induced microglia in an ischemic stroke rat model. CYLD protein exerts its downregulating effects through interaction with SPATA2.55 SPATA2 recruits CYLD to the linear ubiquitin chain assembly complex (LUBAC) to deconjugate K63-linked polyubiquitin chains from RIP1 and IKKγ, thereby preventing phosphorylation of IκB and subsequent NF-κB activation.56,57 However, it is likely that CYLD overexpression inhibits neuroinflammation if excessively regulated.39 Thus, we hypothesize that moderate regulation must occur, depending on the accurate recruitment of SPATA2. We first reported the lowest expression at 24 h of reperfusion after ischemic stroke in a previous study. It is not sufficient to depend on endogenous CYLD protein during the acute stage of ischemic stroke. Interestingly, we demonstrated that EA could increase neuronal CYLD protein expression in the ischemic cortical border region from 24 h to 72 h of reperfusion.
EA has been reported to regulate M1/M2 microglial polarization and decrease Aβ plaques to improve learning and memory at the Shenting and Baihui acupoints in mild Alzheimer’s disease (AD) mice.58 In addition, EA improved learning and memory abilities and protected neurons by upregulating Triggering receptor expressed on myeloid cells 2 (TREM2) expression in the hippocampus, which was essential for the anti-inflammatory effects in an AD animal model.59 EA administration at Baihui (GV 20) and Dazhui (GV 14) attenuated neuroinflammation after ischemic stroke by inhibiting NF-κB-mediated activation of M1 microglia.60 We observed not only improved mRNA expression of anti-inflammatory genes and numbers of CD206-positive microglia but also decreased mRNA expression of proinflammatory genes and numbers of iNOS-positive microglia after EA treatment at 72 h of reperfusion. However, CYLD silencing partially weakened the impact of EA on improving the M2 phenotype and inhibiting pro-inflammatory genes (M1-like phenotype). In this study, we suggest that CYLD is essential for EA-mediated suppression of M1 microglia and a shift in M1 microglia towards the M2 anti-inflammatory phenotype. Additionally, our data suggested that EA could inhibit NLRP3 inflammasome activation by upregulating CYLD after 72 h of reperfusion.
Interaction between neurons and microglia is essential for neuroinflammation. The CX3CL1/CX3CR1 axis is a well-known bridge in neuron-microglia crosstalk in the CNS.61 Recently, a number of studies have demonstrated that CX3CL1 has dual impacts on neurons, producing beneficial and detrimental effects.13 Liu et al established a bilateral common carotid artery stenosis (BCAS) model in mice and showed that exogenous CX3CL1 increased expression of TNF-α and IL-1β, which play detrimental roles in the ischemic brain via the p38 MAPK/PKC signaling pathway.62 More interestingly, we demonstrated that EA increased the number of CX3CL1-positive neuronal cells and CX3CL1 protein expression by upregulating CYLD in periischemic cortices after 72 h reperfusion. CYLD silencing partially weakened the effect of EA on improving neuronal CX3CL1 protein expression. Joanna et al showed that CX3CL1 administration exhibited anti-inflammatory effects, mainly in the frontal cortex of adult prenatally stressed rats, which was related to inhibition of the NLRP3 inflammasome.63 He et al found that ischemia-induced neuronal autophagy facilitated microglial inflammatory injury after ischemic stroke, and the efficacy of this process may be associated with downregulated CX3CL1 expression on autophagic neurons.64 Thus, we hypothesize that EA exerts anti-inflammatory effects on ischemic stroke through the CYLD/CX3CL1/NLRP3 inflammasome circuit. Emerging evidence suggests that CX3CR1 plays a harmful role in the ischemic brain.11,15 Recent studies have shown that CX3CR1-deficient mice show reductions in infarct size and neuronal death in the MCAO model.65 CX3CR1-deficient rats exhibit protection of neurological function after spinal cord injury.66 We report that EA inhibited CX3CR1 protein expression by increasing CYLD protein expression. Thus, we suggest that EA may regulate the interaction between neurons and microglia by modulating the CX3CL1/CX3CR1 axis. To date, the effect of the CX3CL1/CX3CR1 signaling pathway has been elusive.
The neuroprotective or deleterious effect of CX3CL1 depends on the activation state of microglia in the different stages of acute and chronic ischemic stroke. Increasing evidence demonstrates that metabolic reprogramming plays an important role in the regulation of the innate inflammatory response after the ischemic stroke.13 A recent study indicated polarization toward a proinflammatory phenotype microglia to change from oxidative phosphorylation to anaerobic glycolysis, leading to increased adenosine triphosphate (ATP) production.67 Indeed, CX3CL1 induces a metabolic switch toward oxidative metabolism in microglia, which accommodates the beneficial role of anti-inflammatory microglia.13 We speculate that the effect of EA on microglia polarization might be based on modification of metabolic functions via the CYLD/CX3CL1/CX3CR1 axis, some of which could be addressed in future studies.
How CYLD overexpression reduced microglia-mediated inflammatory injury at the ubiquitination level was not explored in the current study. K63-linked ubiquitylation is a crucial posttranslational modification in inflammation. The CYLD protein exerts its neuroprotective effects via interaction with SPATA2. SPATA2 recruits CYLD to LUBAC to deconjugate K63-linked polyubiquitin chains. The LUBAC complex is composed of longer isoforms of heme-oxidized iron-regulatory protein 2 ubiquitin ligase-1 (HOIL-1), HOIL-1 interacting protein (HOIP), and SHANK-associated RH domain-interacting protein (SHARPIN) and is critical in the assembly of ubiquitin chains.56,57 We hypothesize that moderate and accurate regulation must occur based on the SPATA2/LUBAC/CYLD circuit. Thus, some issues remain to be elaborated and could be addressed via future investigations.
Conclusion
Thus, neuronal CYLD plays neuroprotective and anti-inflammatory roles in periischemic areas during the acute stage of focal cerebral ischemia/reperfusion in rats. In addition, upregulating neuronal CYLD expression by EA may increase neuronal CX3CL1 expression and decrease microglial CX3CR1 expression and the NLRP3 inflammasome, thereby inhibiting M1 microglial activation and shifting microglia towards the M2 phenotype in periischemic areas.
Ethics Approval
Our manuscript data were collected from animals and the study was approved by the Ethics Committee for Animal Experimentation of Chongqing Medical University (number SCXK (Yu) 2018-0003).
Author Contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in drafting the article or revising it critically for important intellectual content; agreed to submit to the current journal; gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
The study was supported by the Natural Science Foundation of Chongqing, Grand NO. cstc2019jcyj-msxmX0630, to JJ; the Natural Science Foundation of Chongqing, Grand NO. cstc2020jcyj-msxmX0325, to XL; the Chinese Medicine Science and Technology project of Chongqing Municipal Health committee, Grand NO. 2021ZY3794, to XL.
Disclosure
Jin Jiang is the correspondence author, and Yikun Ren is the co-correspondence author in this study. The authors report no conflicts of interest in this work.
References
1. Chen Z, Zhong D, Li G. The role of microglia in viral encephalitis: a review. J Neuroinflammation. 2019;16(1):76. doi:10.1186/s12974-019-1443-2
2. Zhao SC, Ma LS, Chu ZH, et al. Regulation of microglial activation in stroke. Acta Pharmacol Sin. 2017;38:445–458. doi:10.1038/aps.2016.162
3. Harry GJ. Microglia during development and aging. Pharmacol Ther. 2013;139(3):313–326. doi:10.1016/j.pharmthera.2013.04.013
4. Kabba JA, Xu Y, Christian H, et al. Microglia: housekeeper of the Central Nervous System. Cell Mol Neurobiol. 2018;38(1):53–71. doi:10.1007/s10571-017-0504-2
5. Amani H, Shahbazi MA, D’Amico C, et al. Microneedles for painless transdermal immunotherapeutic applications. J of Con Rel. 2021;330.
6. Valles SL, Iradi A, Aldasoro M, et al. Function of Glia in Aging and the Brain Diseases. Int J Med Sci. 2019;16(11):1473–1479. doi:10.7150/ijms.37769
7. Liu X, Liu J, Zhao S, et al. Interleukin-4 Is Essential for Microglia/Macrophage M2 Polarization and Long-Term Recovery After Cerebral Ischemia. Stroke. 2016;47(2):498–504. doi:10.1161/STROKEAHA.115.012079
8. Ganbold T, Bao Q, Zandan J, et al. Modulation of Microglia Polarization through Silencing of NF-kappaB p65 by Functionalized Curdlan Nanoparticle-Mediated RNAi. ACS Appl Mater Interfaces. 2020;12:11363–11374. doi:10.1021/acsami.9b23004
9. Hopp SC. Targeting microglia L‐type voltage‐dependent calcium channels for the treatment of central nervous system disorders. J Neurosci Res. 2020;99(1):141–162. doi:10.1002/jnr.24585
10. Liu W, Jiang L, Bian C, et al. Role of CX3CL1 in Diseases. Arch Immunol Ther Exp (Warsz). 2016;64(5):371–383. doi:10.1007/s00005-016-0395-9
11. Fumagalli S, Perego C, Ortolano F, et al. CX3CR1 deficiency induces an early protective inflammatory environment in ischemic mice. Glia. 2013;61(6):827–842. doi:10.1002/glia.22474
12. Camargos QM, Silva BC, Silva DG, et al. Minocycline treatment prevents depression and anxiety-like behaviors and promotes neuroprotection after experimental ischemic stroke. Brain Res Bull. 2020;155:1–10. doi:10.1016/j.brainresbull.2019.11.009
13. Lauro C, Chece G, Monaco L, et al. Fractalkine Modulates Microglia Metabolism in Brain Ischemia. Front Cell Neurosci. 2019;13:414. doi:10.3389/fncel.2019.00414
14. Luo P, Chu SF, Zhang Z, et al. Fractalkine/CX3CR1 is involved in the cross-talk between neuron and glia in neurological diseases. Brain Res Bull. 2019;146:12–21. doi:10.1016/j.brainresbull.2018.11.017
15. Ahn J, Kim D, Park J, et al. Expression changes of CX3CL1 and CX3CR1 proteins in the hippocampal CA1 field of the gerbil following transient global cerebral ischemia. Int J Mol Med. 2019. doi:10.3892/ijmm.2019.4273
16. Sheridan GK, Murphy KJ. Neuron-glia crosstalk in health and disease: fractalkine and CX3CR1 take centre stage. Open Biol. 2013;3:130181. doi:10.1098/rsob.130181
17. Morrison HW, Filosa JA. A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J Neuroinflammation. 2013;10(1):4. doi:10.1186/1742-2094-10-4
18. Taylor RA, Sansing LH. Microglial responses after ischemic stroke and intracerebral hemorrhage. Clin Dev Immunol. 2013;2013:746068. doi:10.1155/2013/746068
19. Ma DC, Zhang NN, Zhang YN, et al. Kv1.3 channel blockade alleviates cerebral ischemia/reperfusion injury by reshaping M1/M2 phenotypes and compromising the activation of NLRP3 inflammasome in microglia. Exp Neurol. 2020;332:113399. doi:10.1016/j.expneurol.2020.113399
20. Denes A, Vidyasagar R, Feng J, et al. Proliferating resident microglia after focal cerebral ischaemia in mice. J Cereb Blood Flow Metab. 2007;27(12):1941–1953. doi:10.1038/sj.jcbfm.9600495
21. Heideker J, Wertz IE. DUBs, the regulation of cell identity and disease. Biochem J. 2015;467(1):191. doi:10.1042/bj4670191
22. Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi:10.1016/j.cell.2008.01.020
23. Mukherjee S, Kumar R, Tsakem Lenou E, et al. Deubiquitination of NLRP6 inflammasome by Cyld critically regulates intestinal inflammation. Nat Immunol. 2020;21(6):626–635. doi:10.1038/s41590-020-0681-x
24. Hellerbrand C, Massoumi R. Cylindromatosis–A Protective Molecule against Liver Diseases. Med Res Rev. 2016;36:342–359. doi:10.1002/med.21381
25. Nikolaou K, Tsagaratou A, Eftychi C, et al. Inactivation of the deubiquitinase CYLD in hepatocytes causes apoptosis, inflammation, fibrosis, and cancer. Cancer Cell. 2012;21(6):738–750. doi:10.1016/j.ccr.2012.04.026
26. Ganjam GK, Terpolilli NA, Diemert S, et al. Cylindromatosis mediates neuronal cell death in vitro and in vivo. Cell Death Differ. 2018;25(8):1394–1407. doi:10.1038/s41418-017-0046-7
27. Jiang J, Luo Y, Qin W, et al. Electroacupuncture Suppresses the NF-κB Signaling Pathway by Upregulating Cylindromatosis to Alleviate Inflammatory Injury in Cerebral Ischemia/Reperfusion Rats. Front Mol Neurosci. 2017;10:10. doi:10.3389/fnmol.2017.00363
28. Chi L, Du K, Liu D, et al. Electroacupuncture brain protection during ischemic stroke: a role for the parasympathetic nervous system. J Cereb Blood Flow Metab. 2018;38(3):479–491. doi:10.1177/0271678X17697988
29. Zhu Y, Deng L, Tang H, et al. Electroacupuncture improves neurobehavioral function and brain injury in rat model of intracerebral hemorrhage. Brain Res Bull. 2017;131:123–132. doi:10.1016/j.brainresbull.2017.04.003
30. Zhu XL, Chen X, Wang W, et al. Electroacupuncture pretreatment attenuates spinal cord ischemia-reperfusion injury via inhibition of high-mobility group box 1 production in a LXA4 receptor-dependent manner. Brain Res. 2017;1659:113–120. doi:10.1016/j.brainres.2017.01.008
31. Qin WY, Luo Y, Chen L, et al. Electroacupuncture Could Regulate the NF-kappaB Signaling Pathway to Ameliorate the Inflammatory Injury in Focal Cerebral Ischemia/Reperfusion Model Rats. Evid Based Complement Alternat Med. 2013;2013:924541. doi:10.1155/2013/924541
32. Zhan J, Qin W, Zhang Y, et al. Upregulation of neuronal zinc finger protein A20 expression is required for electroacupuncture to attenuate the cerebral inflammatory injury mediated by the nuclear factor-kB signaling pathway in cerebral ischemia/reperfusion rats. J Neuroinflammation. 2016;13:258. doi:10.1186/s12974-016-0731-3
33. Longa EZ, Weinstein PR, Carlson S, et al. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi:10.1161/01.STR.20.1.84
34. Bederson JB, Pitts LH, Tsuji M, et al. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–476. doi:10.1161/01.STR.17.3.472
35. Garcia JH, Wagner S, Liu KF, et al. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke. 1995;26:627–634. doi:10.1161/01.STR.26.4.627
36. Ren J, Meng S, Yan B, et al. Protectin D1 reduces concanavalin A-induced liver injury by inhibiting NF-kappaB-mediated CX3CL1/CX3CR1 axis and NLR family, pyrin domain containing 3 inflammasome activation. Mol Med Rep. 2016;13:3627–3638. doi:10.3892/mmr.2016.4980
37. Massoumi R. CYLD: a deubiquitination enzyme with multiple roles in cancer. Future Oncol. 2011;7(2):285–297. doi:10.2217/fon.10.187
38. Courtois G. Tumor suppressor CYLD: negative regulation of NF-kappaB signaling and more. Cell Mol Life Sci. 2008;65:1123–1132. doi:10.1007/s00018-007-7465-4
39. Sun SC. CYLD: a tumor suppressor deubiquitinase regulating NF-kappaB activation and diverse biological processes. Cell Death Differ. 2010;17:25–34. doi:10.1038/cdd.2009.43
40. Jayaraj RL, Azimullah S, Beiram R, et al. Neuroinflammation: friend and foe for ischemic stroke. J Neuroinflammation. 2019;16:142.
41. Okada T, Suzuki H. Mechanisms of neuroinflammation and inflammatory mediators involved in brain injury following subarachnoid hemorrhage. Histol Histopathol. 2020;18208.
42. Le Thuc O, Blondeau N, Nahon J-L, et al. The complex contribution of chemokines to neuroinflammation: switching from beneficial to detrimental effects. Ann N Y Acad Sci. 2015;1351(1):127–140. doi:10.1111/nyas.12855
43. Mo Y, Sun YY, Liu KY. Autophagy and inflammation in ischemic stroke. Neural Regen Res. 2020;15:1388–1396. doi:10.4103/1673-5374.274331
44. Zhang W, Tian T, Gong SX, et al. Microglia-associated neuroinflammation is a potential therapeutic target for ischemic stroke. Neural Regen Res. 2021;16:6–11. doi:10.4103/1673-5374.286954
45. Collmann FM, Pijnenburg R, Hamzei-Taj S, et al. Individual in vivo profiles of microglia polarization after stroke, represented by the genes iNOS and Ym1. Front Immunol. 2019;10:1236. doi:10.3389/fimmu.2019.01236
46. Ye SY, Apple JE, Ren X, et al. Microglial VPS35 deficiency regulates microglial polarization and decreases ischemic stroke-induced damage in the cortex. J Neuroinflammation. 2019;16:235. doi:10.1186/s12974-019-1633-y
47. Hongxia L, Yuxiao T, Zhilei S, et al. Zinc inhibited LPS-induced inflammatory responses by upregulating A20 expression in microglia BV2 cells. J Affect Disord. 2019;249:136–142. doi:10.1016/j.jad.2019.02.041
48. Xiang J, Zhang X, Fu J, et al. USP18 overexpression protects against focal cerebral ischemia injury in mice by suppressing microglial activation. Neuroscience. 2019;419:121–128. doi:10.1016/j.neuroscience.2019.09.001
49. Jiang X, Yu M, Ou Y, et al. Downregulation of USP4 promotes activation of microglia and subsequent neuronal inflammation in rat spinal cord after injury. Neurochem Res. 2017;42(11):3245–3253. doi:10.1007/s11064-017-2361-2
50. Slusarczyk J, Trojan E, Wydra K, et al. Beneficial impact of intracerebroventricular fractalkine administration on behavioral and biochemical changes induced by prenatal stress in adult rats: possible role of NLRP3 inflammasome pathway. Biochem Pharmacol. 2016;113:45–56. doi:10.1016/j.bcp.2016.05.008
51. Cunha C, Gomes C, Vaz AR, et al. Exploring New Inflammatory Biomarkers and Pathways during LPS-Induced M1 Polarization. Mediators Inflamm. 2016;2016:1–17. doi:10.1155/2016/6986175
52. Qin Y, Qiu J, Wang P, et al. Impaired autophagy in microglia aggravates dopaminergic neurodegeneration by regulating NLRP3 inflammasome activation in experimental models of Parkinson’s disease. Brain Behav Immun. 2021;91:324–338. doi:10.1016/j.bbi.2020.10.010
53. Du X, Xu Y, Chen S, et al. Inhibited CSF1R Alleviates Ischemia Injury via Inhibition of Microglia M1 Polarization and NLRP3 Pathway. Neural Plast. 2020;2020:8825954. doi:10.1155/2020/8825954
54. Yang XD, Li W, Zhang S, et al. PLK4 deubiquitination by Spata2-CYLD suppresses NEK7-mediated NLRP3 inflammasome activation at the centrosome. EMBO J. 2020;39:e102201. doi:10.15252/embj.2019102201
55. Schlicher L, Brauns-Schubert P, Schubert F, et al. SPATA2: more than a missing link. Cell Death Differ. 2017;24(7):1142–1147. doi:10.1038/cdd.2017.26
56. Schlicher L, Wissler M, Preiss F, et al. SPATA2 promotes CYLD activity and regulates TNF-induced NF-kappaB signaling and cell death. EMBO Rep. 2016;17:1485–1497. doi:10.15252/embr.201642592
57. Kupka S, De Miguel D, Draber P, et al. SPATA2-Mediated Binding of CYLD to HOIP Enables CYLD Recruitment to Signaling Complexes. Cell Rep. 2016;16(9):2271–2280. doi:10.1016/j.celrep.2016.07.086
58. Li L, Li L, Zhang J, et al. Disease Stage-Associated Alterations in Learning and Memory through the Electroacupuncture Modulation of the Cortical Microglial M1/M2 Polarization in Mice with Alzheimer’s Disease. Neural Plast. 2020;2020:8836173. doi:10.1155/2020/8836173
59. Li Y, Jiang J, Tang Q, et al. Microglia TREM2: a Potential Role in the Mechanism of Action of Electroacupuncture in an Alzheimer’s Disease Animal Model. Neural Plast. 2020;2020:8867547. doi:10.1155/2020/8867547
60. Liu R, Xu NG, Yi W, et al. Electroacupuncture Attenuates Inflammation after Ischemic Stroke by Inhibiting NF-kappaB-Mediated Activation of Microglia. Evid Based Complement Alternat Med. 2020;2020:8163052.
61. He HY, Ren L, Guo T, et al. Neuronal autophagy aggravates microglial inflammatory injury by downregulating CX3CL1/fractalkine after ischemic stroke. Neural Regen Res. 2019;14:280–288. doi:10.4103/1673-5374.244793
62. Liu Y, Wu XM, Luo QQ, et al. CX3CL1/CX3CR1-mediated microglia activation plays a detrimental role in ischemic mice brain via p38MAPK/PKC pathway. J Cereb Blood Flow Metab. 2015;35:1623–1631. doi:10.1038/jcbfm.2015.97
63. Joanna S, Ewa T, Karolina W, et al. Beneficial impact of intracerebroventricular fractalkine administration on behavioral and biochemical changes induced by prenatal stress in adult rats: possible role of NLRP3 inflammasome pathway. Biochem Pharmacol. 2016;113:45–56.
64. He HY, Ren L, Guo T, et al. Neuronal autophagy aggravates microglial inflammatory injury by downregulating CX3CL1/fractalkine after ischemic stroke. Neural Regen Res. 2019;14(2):280–288.
65. Wang J, Gan Y, Han P, et al. Ischemia-induced neuronal cell death is mediated by chemokine receptor CX3CR1. Sci Rep. 2018;8(1):556. doi:10.1038/s41598-017-18774-0
66. Freria CM, Hall JC, Wei P, et al. Deletion of the Fractalkine Receptor, CX3CR1, Improves Endogenous Repair, Axon Sprouting, and Synaptogenesis after Spinal Cord Injury in Mice. J Neurosci. 2017;37:3568–3587. doi:10.1523/JNEUROSCI.2841-16.2017
67. Beth K, Luke AJ. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015;25(7):771–784. doi:10.1038/cr.2015.68
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.