Back to Journals » Psoriasis: Targets and Therapy » Volume 12
Updated Perspectives on Keratinocytes and Psoriasis: Keratinocytes are More Than Innocent Bystanders
Authors Ortiz-Lopez LI, Choudhary V, Bollag WB ![]()
Received 26 January 2022
Accepted for publication 8 April 2022
Published 2 May 2022 Volume 2022:12 Pages 73—87
DOI https://doi.org/10.2147/PTT.S327310
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Uwe Wollina
Laura I Ortiz-Lopez,1,2 Vivek Choudhary,1,3 Wendy B Bollag1,3– 5
1Department of Physiology, Medical College of Georgia at Augusta University, Augusta, GA, 30912, USA; 2Department of Neuroscience and Regenerative Medicine, Medical College of Georgia at Augusta University, Augusta, GA, 30912, USA; 3Charlie Norwood VA Medical Center, Augusta, GA, 30904, USA; 4Department of Dermatology, Medical College of Georgia at Augusta University, Augusta, GA, 30912, USA; 5Department of Medicine, Medical College of Georgia at Augusta University, Augusta, GA, 30912, USA
Correspondence: Wendy B Bollag, Department of Physiology, Medical College of Georgia at Augusta University, 1120 15th Street, Augusta, GA, 30912, USA, Tel +1 706 721-0698, Fax +1 706 721-7299, Email [email protected]
Abstract: Psoriasis is a complex disease triggered by genetic, immunologic, and environmental stimuli. Many genes have been linked to psoriasis, like the psoriasis susceptibility genes, some of which are critical in keratinocyte biology and epidermal barrier function. Still, the exact pathogenesis of psoriasis is unknown. In the disease, the balance between the proliferative and differentiative processes of keratinocytes becomes altered. Multiple studies have highlighted the role of dysregulated immune cells in provoking the inflammatory responses seen in psoriasis. In addition to immune cells, accumulating evidence shows that keratinocytes are involved in psoriasis pathogenesis, as discussed in this review. Although certain immune cell-derived factors stimulate keratinocyte hyperproliferation, activated keratinocytes can also produce anti-microbial peptides, cytokines, and chemokines that can promote their proliferation, as well as recruit immune cells to help initiate and reinforce inflammatory feedback loops. Psoriatic keratinocytes also show intrinsic differences from normal keratinocytes even after removal from the in vivo inflammatory environment; thus, psoriatic keratinocytes have been found to exhibit abnormal calcium metabolism and possible epigenetic changes that contribute to psoriasis. The Koebner phenomenon, in which injury promotes the development of psoriatic lesions, also provides evidence for keratinocytes’ contributions to disease pathogenesis. Furthermore, transgenic mouse studies have confirmed the importance of keratinocytes in the etiology of psoriasis. Finally, in addition to immune cells and keratinocytes, data in the literature support roles for other cell types, tissues, and systems in psoriasis development. These other contributors are all potential targets for therapies, suggesting the importance of a holistic approach when treating psoriasis.
Keywords: anti-microbial peptides, inflammation, innate immunity, Koebner phenomenon, skin
Introduction
The skin serves as a crucial barrier between the interior of an organism and the external environment. This largest organ of the body mediates various functions ranging from protection against chemical, biological and physical insults, to regulation of water loss and the entrance of solutes. The skin’s unique architecture and cell arrangement greatly influence its selective permeability as well as the homeostasis of the tissue. Skin is subdivided into three principal layers: the epidermis, dermis and hypodermis (Figure 1). The epidermis contains primarily keratinocytes, but also melanocytes (pigment cells), Langerhans cells (immune cells) and Merkel cells (sensory cells), whereas the dermis and hypodermis are comprised primarily of connective tissue and adipose tissue, respectively. The epidermis is further subdivided into layers called “strata”. These strata are denominated, from superficial to deep, as follows: stratum corneum, stratum lucidum (present only in thick skin, such as on the palms and soles), stratum granulosum, stratum spinosum, and stratum basale, also known as the cornified, clear, granular, spinous and basal layers, respectively (Figure 1). As noted, each cell layer in the epidermis is composed mostly of keratinocytes. The basal layer, composed of cuboidal-shaped basal keratinocytes attached to a basement membrane, possesses progenitor cells that continually undergo mitosis. Through this mechanism, new strata form; specific gene/protein markers indicate the stratum that the cells occupy and their differentiation status.1 As they proliferate, keratinocytes ascend through the epidermis, expressing the various differentiation genes/proteins until they are eventually shed into the environment as corneocytes, dead cells that have undergone programmed cell death and lost their organelles. These corneocytes constitute the “bricks” of a brick-and-mortar structure that allows the epidermis to function as a mechanical and water permeability barrier. The “mortar” is composed of lipids contained in lamellar bodies that are released from keratinocytes in the granular layer; the lipids are then processed in the stratum corneum to form a layer that inhibits water loss through the skin (reviewed in2).
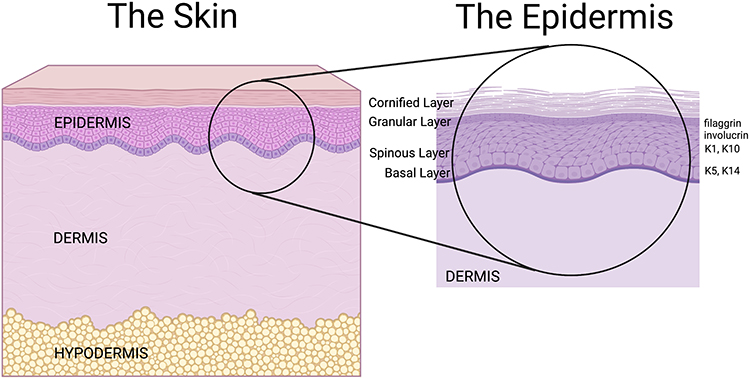 |
Figure 1 The structure of the skin and the epidermis. Shown on the left is a schematic of normal human skin illustrating the three skin layers: the upper epidermis, the middle dermis, and the lower hypodermis. The schematic on the right is an amplified schematic of the epidermis showing the various strata/layers of the epidermis and some of the protein markers expressed in each stratum. The uppermost epidermal layer is the cornified layer (stratum corneum) consisting of essentially dead cells that help to provide a barrier function, followed by the granular layer (stratum granulosum) expressing filaggrin. The next deeper layer is the spinous layer (stratum spinosum) expressing involucrin more superficially as well as keratin (K)-1 (K1) and K10 more deeply, followed by the lowermost basal layer (stratum basale) expressing K5 and K14. Note that this schematic does not show the stratum lucidum or clear layer, which lies between the stratum corneum and stratum granulosum only in the thick skin of the palms and feet. Created using Biorender.com. |
The homeostasis of these proliferative and differentiative processes can become altered in skin pathologies such as psoriasis, a common inflammatory skin disorder estimated to occur in about 2–3% of the population worldwide.3 In terms of its etiology, psoriasis is a complex disease triggered by multiple factors, such as genetic, immunologic and environmental parameters, and its exact cause is still an unresolved issue (reviewed in4–6). Psoriasis patients characteristically develop thickening of the epidermis (acanthosis), in large part due to the accelerated proliferation and poor differentiation of keratinocytes. Indeed, psoriatic keratinocytes can ascend through the epidermis in only 4 to 5 days,7 whereas in healthy individuals, a complete epidermal replacement requires approximately a month (37 days).8 The formation of the red plaques and silvery-white, dry and scaly psoriatic lesions are the result of the inflammatory response as well as the erratic differentiation and proliferation of keratinocytes. In addition, there is an abnormal shedding cycle of the epidermis, with consequent accumulation of skin cells, especially at the extensor surfaces, causing a burning and stinging sensation often accompanied by pruritus.9 In addition, improper maturation of the keratinocytes can lead to a dysfunctional epidermal barrier, which can trigger inflammation.10,11
As noted above, the pathogenesis of psoriasis is not yet completely understood. Until the 1970s, psoriasis was considered primarily a disease of keratinocyte dysfunction.12 However, significant evidence has accumulated throughout the years suggesting that the etiology of this disease may involve inflammatory responses brought about by dysregulated immune cells from skin-associated lymphoid tissue; that is, the innate and adaptive immune systems exhibit a pathological interaction with the dermis and epidermis. Thus, psoriasis is considered an immune-mediated condition13 and perhaps even an autoimmune disease (e.g.,14,15). Triggers may vary by patient, including stressful events, physical trauma, streptococcal infections and some medications, although there is also a significant genetic component (reviewed in16,17). Ultimately, these can provoke an atypical immune response in keratinocytes.18 Affected keratinocytes proceed to release LL37 peptide, the carboxy terminus of the anti-microbial peptide cathelicidin. This peptide forms a complex with nucleic acids that can activate plasmacytoid dendritic cells (pDCs). These pDCs can then produce interferon (IFN)-α and IFN-β, activating myeloid dendritic cells (DC).19 Myeloid DC activation results in the production and release of critical cytokines like interleukin (IL)-12 and IL-23. These pro-inflammatory cytokines are found to be elevated in lesional psoriatic plaques. In addition, LL37 complexes can activate and up-regulate the innate immune system-stimulating toll-like receptor-9 (TLR9) in keratinocytes to further promote the production of pro-inflammatory mediators.15 Furthermore, IL-12 leads to the differentiation of naïve T cells to T helper type 1 (Th1) cells that produce IFN-γ. At the same time, IL-23 is important for the ongoing stimulation of Th17 cells that produce IL-17A and F.20 Finally, for some of these T cells, LL37 appears to serve as an autoantigen: Lande et al14 detected the presence of autoantibodies to this peptide in roughly two-thirds of psoriasis patients with moderate-to-severe diseases. These authors found that both IFN-γ- and IL-17-producing T cells recognize LL37.14
Although immune cells are clearly critically important for psoriasis pathogenesis and in some patients may instigate the disease, data in the literature also support the involvement of keratinocytes in disease initiation and maintenance, and in fact, there seems to be crosstalk between these cells and cells of the innate and adaptive immune system. Thus, activated keratinocytes produce cytokines and chemokines that recruit and activate immune cells, which in turn produce cytokines that activate keratinocytes and promote their hyperproliferation. The result of this crosstalk is that in the chronic form of the disease, mature dermal dendritic cells produce IL-12 and IL-23, activating Th17, Th1, and Th22 cells. These T cells stimulate the proliferation of keratinocytes via the production of TNF-α and IL-17, IFN-γ, and IL-22, respectively.7 In addition, the IFN-γ made by Th1 cells supports the recruitment of more Th1 cells and magnifies the stimulation of keratinocytes. IL-17 facilitates neutrophil migration into the skin and stimulates keratinocytes to produce anti-microbial peptides.21 Additionally, keratinocyte hyperproliferation, a hallmark of psoriasis, is promoted by the IL-22 secreted by Th22 cells. Also, the pro-inflammatory role of TNF-α involves its activation of macrophages and DCs in the skin, as well as recruitment of additional inflammatory cells into the tissue.22 Indeed, the importance of TNF-α in psoriasis is highlighted by the ability of anti-TNF-α drugs to successfully treat the disease.23 Lastly, keratinocytes contribute to the progression of psoriasis through the release of chemokines and anti-microbial peptides, which can activate the innate immune system.24 This series of events amplifies the immune feedback loop that increases the severity and complexity of psoriasis.7,25 In other words, the cytokines, chemokines and anti-microbial peptides produced by keratinocytes are indispensable for the inflammatory process in psoriasis since they reinforce the continuous attraction and activation of leukocytes. Thus, although psoriasis seems to be immune-mediated, keratinocytes likely play a critical role in the initiation and maintenance of psoriasis. Below we summarize the data that suggest the involvement of keratinocytes in the disease process.
Psoriasis Susceptibility Genes and the Importance of the Epidermal Barrier
As noted above, there is a distinct genetic predisposition for psoriasis.16,17 Genome-wide association, linkage and other genetic studies in humans have identified several genes with mutations/variants that are associated with psoriasis (reviewed in16,26). The gene first identified to be linked to psoriasis is psoriasis susceptibility-1 (PSORS1), identified as the gene encoding the human leukocyte antigen (HLA)-C major histocompatibility complex in humans, for which the HLA-Cw6 variant is especially associated with the disease (reviewed in17,27). This association between HLA-C and psoriasis suggests the importance of the immune system in the disease, although it should be noted that keratinocytes can also express HLA and act as antigen-presenting cells (reviewed in28,29). Additional immune-related PSORS genes have since been identified including PSORS316 (with a likely candidate gene identified as IRF2, encoding interferon regulatory factor-2), whereas some of these genes, such as the PSORS2 locus thought to be CARD14, play significant roles in keratinocyte biology.17 Indeed, PSORS4 has been localized to the epidermal differentiation complex (EDC), which encodes a number of genes, such as the late cornified envelope (LCE) genes LCE3B and LCE3C, involved in keratinocyte differentiation and formation of the epidermal barrier.16,17 A genome-wide search for copy number variants identified deletion of the LCE3B and LCE3C genes in psoriatic plaques; these variants have been associated with an increased risk of psoriasis.30 Expression of these LCE3 genes was reportedly low in normal skin; however, disruption of the skin barrier using tape stripping resulted in marked upregulation of their expression.30 Disruption of the barrier function of the skin is thought to be involved in the development of inflammatory skin diseases (reviewed in31), including psoriasis.11 Thus, barrier disruption is known to induce inflammation/inflammatory mediator production,11 and psoriasis is characterized by an impaired barrier (reviewed in9). Furthermore, psychological stress has been found to impair the barrier, and psoriasis can be initiated or exacerbated by such stress.32 These results suggest a link between the epidermal permeability functions mediated by keratinocytes and the inflammatory processes characterizing psoriasis (and other inflammatory skin diseases; reviewed in33). Overall, more than 60 genes have been identified that are linked to psoriasis susceptibility (reviewed in16,17).
In addition to the involvement of cornified envelope proteins, such as the LCE family members mentioned above, in the formation of the water permeability barrier, lipids also play a critical role. For example, the importance of ceramides and acylceramides to the barrier is shown by the phenotype of mice that are genetically engineered to lack the gene encoding FATP4. FATP4 is an acyl-CoA synthetase that converts fatty acids to acyl-CoAs that can be used to synthesize, for instance, acylceramides; defects in this enzyme have been linked to ichthyosis prematurity syndrome. Yamamoto et al34 found that FATP4 knockout mice show reduced levels of several acylceramides and an impaired barrier that results in enhanced dye penetration and elevated trans-epidermal water loss (TEWL). In fact, these mice exhibit perinatal lethality from dehydration. Similar perinatal lethality has been observed in ELOVL4 knockout mice, also because of an impaired barrier and dehydration.35 ELOVL4 is a fatty acid elongase that synthesizes very-long chain fatty acids. Together, these results indicate the importance of lipids in the formation of the epidermal permeability barrier.
Additional evidence links ceramides to psoriasis as well (reviewed in36). For example, a recent study by Luczaj et al37 used lipidomics to examine lipid profiles in keratinocytes isolated from psoriatic skin lesions versus healthy skin (although changes in very long chain fatty acids were not investigated). These authors found by principal component analysis that the lipid profile could distinguish between the psoriatic and healthy keratinocytes. Differences between psoriatic and healthy keratinocytes were found in several ceramide species, with some ceramides increased and others decreased in keratinocytes from psoriatic lesions.37 These data suggest that changes in lipids may underlie the pathology of psoriasis as well. Another group examined differences in glycerophospholipid profiles between psoriatic and normal skin and demonstrated significant alterations in 17 glycerophospholipids, including phosphatidylcholine, phosphatidic acid, phosphatidylinositol and lysophosphatidylcholine.38 Thus, lipids derived from keratinocytes play an important role in barrier homeostasis and appear to be altered in psoriasis, suggesting another way in which keratinocytes may contribute to the disease.
Differences between psoriatic and normal keratinocytes
It is clear that keratinocytes in patients with psoriasis exhibit hyperproliferation and abnormal differentiation, but whether these effects result entirely from the production of keratinocyte-activating signals from the immune system or whether there are intrinsic differences between psoriatic and normal keratinocytes has been unclear. However, accumulating evidence suggests that keratinocytes derived from psoriatic epidermis demonstrate differences that persist for some time in the absence of the inflammatory milieu in the patient. For example, keratinocytes differentiated from induced pluripotent stem cells (iPSCs) show different genetic profiles when derived from normal individuals versus those with psoriasis.39 Thus, iPSCs were generated from the peripheral blood mononuclear cells of controls versus psoriatic patients over more than 3 weeks. These iPSCs were then differentiated into keratinocytes over the course of another 2 to 4 weeks, and their gene expression profiles compared. iPSC-derived keratinocytes from patients with psoriasis demonstrated a large number of differentially expressed genes in comparison to those from controls, and many of the observed changes were similar to those found in freshly obtained psoriasis skin lesions.39 In addition, keratinocytes derived from psoriatic iPSCs showed enhanced proliferation, even almost 2 months removed from the inflammatory psoriasis environment.39 Although there are concerns about the small number of donors used to create the iPSCs, as well as the imperfect matching of the individuals with psoriasis and the control subjects, this study suggests the possibility that there are genetic differences or perhaps epigenetic changes that persist even once the keratinocyte is removed from the in vivo inflammatory milieu. Indeed, there is emerging interest in the role of epigenetics in various physiological and pathophysiological processes. Epigenetics refers to heritable mechanisms that can alter gene expression without modification of the genetic code (i.e., the DNA sequence) itself. Such mechanisms include DNA methylation, histone modifications (methylation and acetylation) and non-coding RNAs, and there is increasing evidence of an important role of these processes in normal skin biology and pathologies such as psoriasis (reviewed in40,41), as discussed below.
Calcium metabolism has also been found to be abnormal in psoriatic keratinocytes and in psoriatic skin. The extracellular calcium concentration is known to regulate keratinocyte growth and differentiation, with low calcium concentrations supporting continued keratinocyte proliferation and elevated levels promoting differentiation in vitro (reviewed in42). This effect of calcium is thought to be physiologically relevant, as a calcium gradient has been reported in the epidermis, also with lower levels in the proliferating basal layer that increase in the differentiating spinous and granular layers.43 This calcium gradient has been found to be dysregulated in psoriasis.44 The effect of calcium appears to be mediated through the calcium-sensing receptor, a G protein-coupled receptor for which calcium acts as a ligand to induce phosphoinositide turnover, resulting in keratinocyte differentiation (45,46 and reviewed in 47,48) Consistent with a role for altered keratinocyte calcium metabolism in psoriasis, the calcium-sensing receptor has been found to be a psoriasis susceptibility gene in some patient populations.16 In addition, cultured psoriatic keratinocytes exhibit abnormal calcium homeostasis despite being removed from their inflammatory environment for at least 5 passages: thus, these cells show decreased capacitative calcium influx and an increased cytosolic calcium response to ATP.49 A more recent study has shown downregulation of all transient receptor potential canonical (TRPC) channels (and reduced calcium influx) in psoriatic keratinocytes compared to normal keratinocytes ex vivo, as well as decreased immunoreactivity in psoriasis skin samples.50
Another way in which keratinocytes may initiate the development of psoriasis is through their production of anti-microbial peptides. Several anti-microbial peptides are known to be danger/damage-associated molecular patterns (DAMPs) capable of activating pattern recognition receptors, in particular toll-like receptors (TLRs), of the innate immune system (reviewed in51). TLR2 activation by the anti-microbial peptide, S100A9, has been shown to result in the induction of inflammatory mediator expression in both keratinocytes and a macrophage cell line.52 The β-defensins are also reported to be DAMPs (52 and reviewed in 51) providing a potential mechanism whereby excessive anti-microbial peptide production by keratinocytes could result in increased pro-inflammatory molecule release to recruit immune cells into the skin. Further, these anti-microbial peptides are elevated in patients with psoriasis (reviewed in53): for instance, S100A proteins are consistently upregulated in psoriatic skin and improve (decrease) with effective treatment;54–56 in addition, several cytokines that are elevated in psoriasis also induce the expression of S100 proteins.56–58 In various animal models of psoriasis, the anti-microbial peptides are also consistently elevated.54–57,59,60 β-Defensin-2 has also been reported to serve as a serum biomarker for psoriatic disease activity,61 and indeed, the levels of several anti-microbial peptides decrease with successful treatment.55,61,62 Finally, as discussed previously, LL37/cathelicidin can bind the DNA released by damaged skin cells to upregulate TLR9,15 and LL37 is recognized by autoantibodies in some patients with psoriasis.14 Therefore, it seems likely that the activation of keratinocyte TLRs by DAMPs, such as the S100 proteins and cathelicidin/LL37, may increase the production of cytokines/chemokines, which then recruit and activate the immune system to establish a cycle of continued inflammation, immune activation and keratinocyte stimulation.
Furthermore, there is evidence that the expression of certain of these anti-microbial peptides may be dysregulated in psoriasis, often at the epigenetic level. For instance, the S100A9 gene was found to be hypomethylated in psoriasis (63,64 and reviewed in 6); this resulted in S100A9 expression correlating inversely with gene methylation,64 since DNA methylation results in repression. Other S100A proteins have also been reported to exhibit differential expression and epigenetic modification in psoriasis (e.g.,63,65 and reviewed in 41). Other genes have been found to be down-regulated epigenetically, with genes such as CARD14 showing hypomethylation.64 Furthermore, in the case of the β-defensins, higher genomic copy number has been associated with a greater risk of psoriasis.16,66 These results suggest that epigenetic changes leading to up-regulation of anti-microbial peptides, and perhaps other gene products, could contribute to the inflammatory processes observed in psoriasis. Thus, the capacity for excessive production of anti-microbial peptides may be another way in which psoriatic keratinocytes initiate and/or maintain the disease process. Intrinsic differences between psoriatic and normal keratinocytes, such as reduced TRPC channel function and capacitative calcium influx, increased cytosolic calcium responses to ATP and expression of S100A proteins due to gene hypomethylation, are summarized in Figure 2.
 |
Figure 2 The structure of psoriatic skin with intrinsic differences between psoriatic and normal keratinocytes highlighted. On top is shown a schematic of psoriatic skin illustrating the scaly stratum corneum and the overall thickened epidermis, including elongated papillae and rete ridges with dilated capillaries in the dermis, as well as the immune cell infiltration and inflammation. In the inset, a keratinocyte from the basal layer of the epidermis is enlarged to illustrate intrinsic differences observed in psoriatic versus normal keratinocytes. The changes in psoriatic keratinocytes are indicated by the red arrows and include: abnormal calcium metabolism via decreased capacitative calcium influx [for example, decreased transient receptor potential canonical (TRPC) channel levels] and an increased cytosolic calcium response to ATP, as well as enhanced secretion of anti-microbial peptides, apparently upregulated at least in part as a result of epigenetic changes (e.g., hypomethylation of the S100A9 gene). All of these alterations in psoriatic keratinocytes amplify the inflammatory immune feedback loop that initiates and sustains chronic inflammation in psoriasis. Created using Biorender.com. |
Keratinocytes and the Koebner Phenomenon
The well-known Koebner phenomenon also suggests a likely role of keratinocytes in psoriasis. The Koebner phenomenon refers to the fact that injury of the skin in individuals predisposed to psoriasis often results in the development of a psoriatic plaque at the site of damage.67 This phenomenon may also explain the propensity for plaques to develop on the knees and elbows, which are sites of mechanical stress,68 in patients with psoriasis. Injured or stressed keratinocytes can release a number of factors that may help to promote inflammation and initiate the development of psoriasis. For example, it was recently shown that mechanical stress can induce the release of ATP from keratinocytes,69 and in fact, purinergic signaling has been suggested to play a role in psoriasis pathogenesis (reviewed in70). Injury can also trigger keratinocytes to release innate immune-system activating cytokines and chemokines, including CCL-20, IL-8, TNF-α and others (71 and reviewed in 29). This phenomenon may also relate to a recently discovered ability of epidermal stem cells to “remember” previous inflammatory insults.72 The mechanism was shown to involve a persistent association of particular transcription factors with certain genomic loci even after inflammation resolution, thereby maintaining these loci in an easily activated state, such that subsequent stimuli rapidly induce the expression of these, often pro-inflammatory, genes.72
Transgenic mouse models suggesting keratinocyte involvement in psoriasiform skin lesion development
Technological advancements allowing genetic manipulation of animal models, and in particular the Cre-loxP technology, allowing tissue-specific deletion or over-expression of certain genes in mice, have resulted in the ability to generate novel mouse models of many diseases, including psoriasis. There are numerous such psoriasis models with features that both mimic the human psoriatic phenotype and those that do not, as has been elegantly and extensively reviewed previously.73–76 Several of these psoriasis models overexpress a particular gene in keratinocytes, usually under the control of keratinocyte-specific gene promoters, such as those for keratins 5 and 14 (expressed in the basal layer), keratins 1 and 10 (expressed in the spinous layer) or involucrin (expressed in the upper spinous/granular layer). For example, keratinocyte-specific overexpression of amphiregulin, a ligand of the epidermal growth factor receptor that is critically important in skin development (reviewed in40), has been shown to result in hyperproliferation and thickening of the epidermis, immune cell infiltration and even joint changes reminiscent of psoriatic arthritis.77 The overexpression of latent transforming growth factor-β1 targeted to keratinocytes with the keratin 5 promoter also promotes a skin phenotype resembling psoriasis.73 Nevertheless, there are limitations to models such as these. First, as the etiology of psoriasis in humans is not completely understood, the applicability of such mouse models to human psoriasis is sometimes questioned, especially since many of the models do not entirely recapitulate the phenotype observed in psoriasis;73 for this reason, the skin lesions in these models are referred to as psoriasiform, i.e., psoriasis-like. In addition, for the overexpression or knockout of a gene for which the product is secreted, like amphiregulin, the phenotype cannot necessarily be ascribed to a particular cell type, since the secreted protein may diffuse locally or even possibly enter the systemic circulation, thereby potentially affecting multiple cell types.
Transgenic mice with autonomous keratinocyte-specific modification of gene expression
On the other hand, genetically manipulated mouse models in which, for instance, a transcription factor or signaling enzyme/effector is overexpressed or deleted only in keratinocytes can provide strong evidence for the role of these proteins expressed in keratinocytes in the resultant psoriasiform phenotype (see Table 1). As an example, a transgenic mouse model was generated in which the AP-1 transcription factors c-jun and junB were deleted specifically in epidermal keratinocytes in a tamoxifen-inducible manner, using Cre recombinase driven by the keratin 5 promoter coupled to a mutant ttamoxifen-activatable estrogen receptor.78 These mice exhibit psoriasiform lesions as well as a psoriatic arthritis-like phenotype, and the skin lesions precede T cell recruitment.79 Interestingly, crossing these mice with Rag2 knockout mice so that they lack functional T and B cells resolves the arthritis, but a milder psoriasiform disease persists.78 These results argue for an important role of keratinocytes in the disease process. Similar results were observed with keratinocyte-specific overexpression of constitutively active signal transducer and activator of transcription-3 (Stat-3), which results in the development of psoriasiform skin lesions, either spontaneously or in response to wounding.80 Transgenic keratinocyte-specific expression of Tie-2, a receptor for pro-angiogenic angiopoietins, also promotes the development of skin lesions resembling psoriasis.81 The tissue specificity of the expression of these genes in transgenic mice suggests an important role for keratinocytes in the resultant psoriasis-like phenotype.
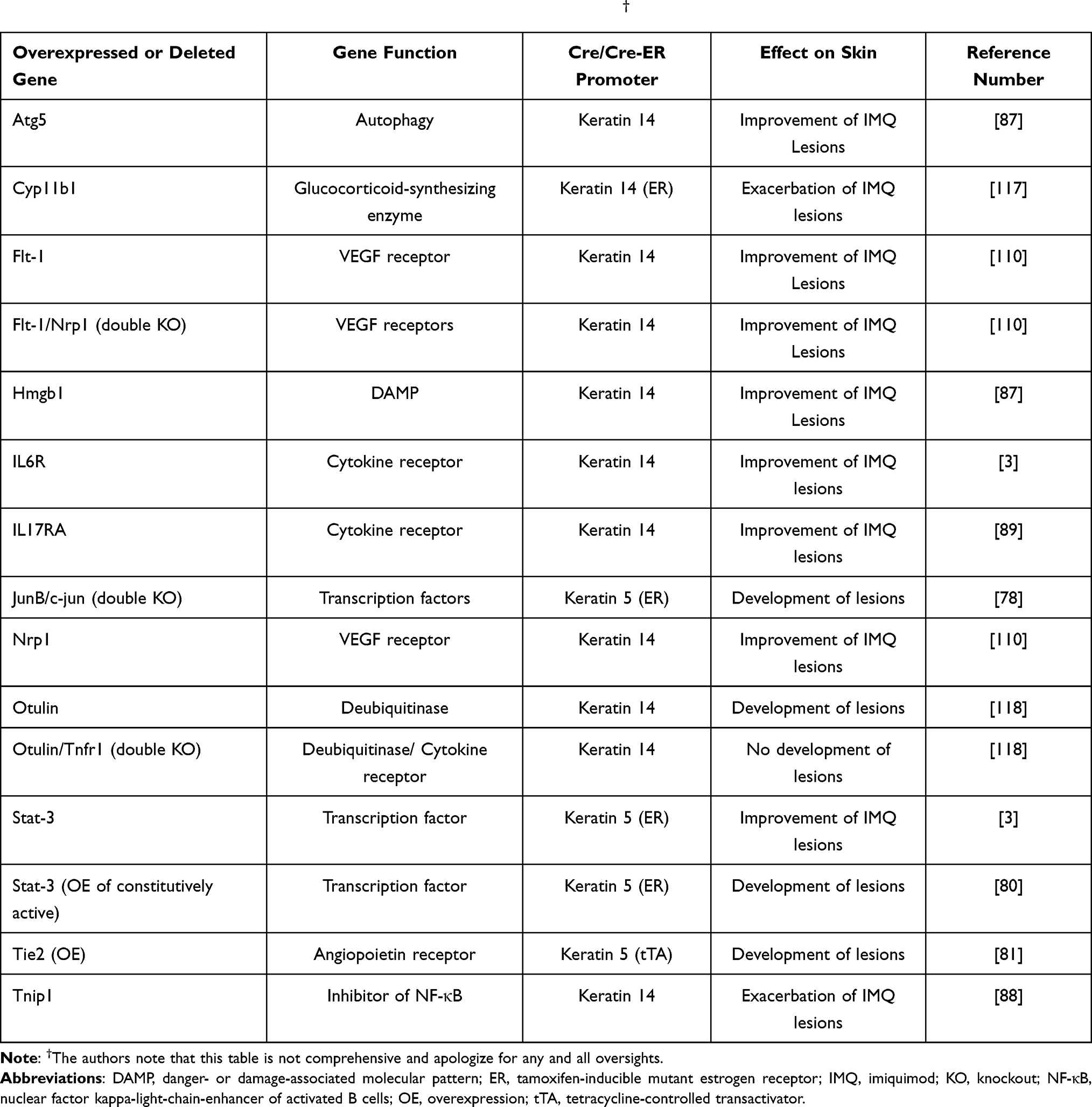 |
Table 1 Examples of the Role of Keratinocyte-Specific Gene Deletion or Autonomous Keratinocyte-Specific Overexpression in the Psoriasiform Phenotype Either with or Without Treatment with Imiquimod† |
The Imiquimod Mouse Model of Psoriasis
One mouse model of psoriasis that has proven quite useful for examining the involvement of different cell types in the development of skin lesions is that generated by topical imiquimod application.82 Imiquimod is an activator of pattern recognition receptors of the innate immune system, in particular toll-like receptor-7/8 (TLR7/8).83,84 Application of imiquimod to the shaved flank skin and/or ear of mice results in psoriasiform skin lesions that are at least partially dependent on cytokines, such as IL-17, which are key components of human psoriasis.82 Indeed, this medication is known to induce psoriasis in some susceptible individuals upon treatment of certain skin conditions, such as actinic keratosis, with imiquimod.82 Furthermore, the genetic changes in the skin elicited by imiquimod mimic to a large extent those observed in psoriatic lesions, particularly in mice of the C57BL/6 background,85 onto which many transgenic mouse models are bred. Therefore, imiquimod can be applied to transgenic mice, including cell/tissue-specific knockout or overexpressing mouse lines (either induced or constitutive), and their wild-type controls to determine the effect of a genetic manipulation in a particular cell type on the psoriasiform skin phenotype. However, it should be noted that, although the imiquimod is topically applied in this model, oral uptake through grooming is necessary for the complete development of the model. Thus, if the mice are prevented from ingesting imiquimod using Elizabethan collars, the developed skin lesions are less severe.86 This result suggests that systemic exposure to imiquimod is necessary for a maximal psoriasiform phenotype.
Using this model, there are multiple examples of knockout mouse models for which keratinocyte-specific deletion of a gene results in a modulation of the skin lesions developed in response to imiquimod. For instance, Wang et al87 generated a keratinocyte-specific (driven by the keratin 14 promoter) knockout of the gene encoding Atg5, which is important for autophagy. Loss of the Atg5 gene leads to amelioration of skin lesions and reduced epidermal thickening upon treatment with imiquimod.87 This beneficial effect of Atg5 loss appears to be at least in part due to decreased autosecretion of the DAMP high mobility group, box 1 (Hmgb1) from the knockout keratinocytes; indeed, creation of a keratinocyte-specific knockout of Hmgb1 also improves the imiquimod-induced skin phenotype.87 On the other hand, keratinocyte-specific knockout of the Tnip1 gene, which encodes a protein that regulates inflammatory signaling and has been linked to psoriasis, exacerbates imiquimod-induced skin lesion formation and epidermal thickening.88 These few examples (and others summarized in Table 1) do not by any means represent an exhaustive list of the mouse models that have been investigated using this model, especially since new such models are being identified constantly, but instead provide evidence of the importance of keratinocytes in the psoriasis-like phenotype produced by treatment of mice with imiquimod.
In some cases, the imiquimod model has been used to examine the importance of various cell types in skin lesion development using mouse models in which a gene was deleted specifically in different cell types. For example, tamoxifen-induced deletion of the gene encoding Stat-3 specifically in keratinocytes (under the control of the keratin 5 promoter) resulted in reduced inflammation compared to wild-type animals upon their treatment with imiquimod.3 In contrast, transgenic mouse models in which Stat-3 was knocked out in T cells by Cre recombinase under the control of two different T cell-specific promoters (Lck and Tcrδ) showed no decrease in imiquimod-induced inflammation, arguing that the key cell type in terms of promoting imiquimod-induced inflammation and ear and epidermal thickness is the keratinocyte. The overexpression of constitutively active Stat-3 under the control of the keratin 14 promoter had the opposite effect on the imiquimod-induced phenotype.3 Likewise, keratinocyte-specific ablation of the receptor for IL-6 (under the control of the keratin 14 promoter) resulted in a lesser imiquimod-elicited increase in inflammation and ear thickness,3 again suggesting a likely important role for keratinocytes in psoriasis. Furthermore, these results also argue that certain medications thought to target components of the immune system, such as certain cytokines, may also affect non-immune cells like keratinocytes.
Effects on Keratinocytes May Underlie Cytokine Involvement in Psoriasis
The idea that biologics targeting cytokines may function through effects on keratinocytes in addition to immune cells is supported by studies in which certain cytokine receptors are ablated in keratinocytes. As mentioned above, keratinocyte-specific knockout of the IL-6 receptor inhibits the imiquimod-induced skin phenotype.3 Similarly, mice lacking keratinocyte expression of IL-17 receptor A (IL-17RA, deleted using Cre driven by the keratin 14 promoter) also exhibit a blunted psoriasiform phenotype in response to imiquimod comparable to the response observed in mice with global knock out of IL-17RA.89 In contrast, loss of the IL17RA gene in T cells (under the control of the CD4 promoter) or myeloid cells (using LysM promoter-driven Cre) has essentially no effect on imiquimod-induced skin lesions, ear inflammation or epidermal thickness. Interestingly, the infiltration of neutrophils, but not monocytes, into the imiquimod-treated skin depends on keratinocyte IL-17R.89 Together, these results indicate a key role for keratinocytes in psoriasis pathogenesis. Furthermore, the correlation between decreased neutrophil influx and reduced psoriasiform lesion development suggests that neutrophils also play a key role in psoriasis pathogenesis. This idea has been supported by other studies, as reviewed previously.90 Thus, both the innate immune system and keratinocytes are important in the etiology of psoriasis, at least in mouse models of the disease.
Finally, it should be pointed out that keratinocytes comprise approximately 90% of the cells of the epidermis, with the remaining 10% consisting of melanocytes, Langerhans cells, and Merkel cells. Therefore, even if each keratinocyte produces only a small amount of a particular secreted protein, such as a cytokine, chemokine or other protein, the sheer number of keratinocytes will result in significant secretion of the molecule. Thus, keratinocytes should not necessarily be discounted as a source of some of the “immune cell factors” found to be upregulated in psoriasis. Indeed, it is thought that keratinocytes express the majority of the IFN-β observed in psoriatic skin.5 This principle is also illustrated by the work of Wang et al87 mentioned above. Although these investigators observed an improved imiquimod-induced psoriasiform phenotype with keratinocyte-specific ablation of Hmgb1, knockout of the Hmgb1 gene in dendritic cells (using Itgax promoter-driven Cre in Hmgb1 floxed mice), myeloid cells (Lyz2 promoter-driven Cre) or T cells (Lck promoter-driven Cre) produced no significant effect.87 Similarly, the work of Croxford et al91 and Wohn et al.92 support the idea that keratinocytes can be a source of significant amounts of a particular secreted protein. In the former report IL-17A was overexpressed from a single allele in keratinocytes under the control of the keratin 14 promoter; 100% of the mice rapidly develop skin lesions reminiscent of psoriasis, with acanthosis, agranulosis (loss of the stratum granulosum), hyper- and parakeratosis, infiltration of granulocytes, macrophages and effector T cells, and neutrophilic (Munro) skin abscesses, as well as signs of uveitis and arthritis,91 all of which are characteristic or associated with human psoriasis (reviewed in26,93). On the other hand, although overexpression of IL-17A from a single allele in dendritic cells (under the control of the CD11c promoter) also results in a psoriasiform phenotype, lesions require a much longer time to develop (onset at approximately 18 weeks of age) and less than half of the mice show skin lesions.92 If the amount of IL-17A is boosted by expressing from two alleles, 100% of the mice develop a phenotype starting at about 8 weeks of age.92 Although the potency of the CD11c versus the keratin 14 promoters may differ, it is tempting to speculate that greater IL-17A quantities are generated, even from a single allele, because of the large number of keratinocytes in the skin, to result in more rapid and highly penetrant development of the skin phenotype in these mice compared with mice overexpressing IL-17A in dendritic cells. In fact, serum levels of IL-17A support this idea, with control mice showing lower levels than single-allele CD11c, which were less than double-allele CD11c promoter mice;92 serum IL-17A levels of the mice with single-allele keratin 14 promoter-driven IL-17A expression were the highest.91 Although keratinocytes are not thought to express IL-17A naturally (although IL-17A expression/secretion has been observed in response to stimulation of the HaCaT keratinocyte cell line with the TLR9 agonist CpG94), they do express IL-17B, IL-17C and IL-17E (also known as IL-25). IL-17C is likely important in psoriasis, as IL-17C is able to stimulate Th17 cells to produce IL-17A (reviewed in95), and a neutralizing antibody to IL-17C has been reported to improve inflammation in an IL-23 ear injection mouse model of psoriasis.96 Importantly, IL-17C protein is found at much higher levels in psoriatic skin lesions than is IL-17A,97 and transgenic overexpression of IL-17C in mice leads to skin disease reminiscent of psoriasis.97 IL-17E (IL-25) is also an important cytokine in the disease: keratinocyte-specific deletion of the IL-25 gene inhibits the imiquimod-induced development of the psoriasis phenotype in knockout mice,98 suggesting that keratinocyte-derived IL-17/IL-25 is likely an important contributor to the disease process.
Conclusions
Thus, accumulating data are pointing to important roles for both the immune system and keratinocytes in the etiology of psoriasis. Indeed, activated keratinocytes produce immune-stimulating molecules, and these immune cells secrete keratinocyte-activating factors, thereby establishing a “vicious cycle” to initiate and promote psoriasis (reviewed in93,99). Interrupting the cycle at the level of the immune system or the keratinocyte should therefore be effective for treating this disease. Immune-targeted therapies are common but have the disadvantage of side effects (e.g., potential issues with infection4), which are unlikely to arise with treatments that do not act to suppress the immune system; nevertheless, even those medications thought to target the immune system may also affect keratinocytes, such as the biologics inhibiting TNF-α, IL-6 and IL-17 (e.g.,3,71,89).
Examples of keratinocyte-produced immune-activating molecules are the anti-microbial peptides, which have been shown to serve as DAMPs to activate TLRs, like TLR2 and TLR4, and stimulate the production of cytokines/chemokines (e.g.,52,100–104), which in turn can recruit immune cells into the skin. While it may be inefficient to attempt to inhibit the action of individual anti-microbial peptides, a therapy to inhibit TLR activation by multiple DAMPs might be a potentially effective strategy for treating psoriasis. Phosphatidylglycerol has recently been shown to inhibit TLR2 and TLR4 activation in vitro by the anti-microbial peptides S100A9 and β-defensin-2,52 as well as microbial components like lipopolysaccharide.52,100 In addition, phosphatidylglycerol has been shown to ameliorate inflammation stimulated by the skin irritant, 12-O-tetradecanoylphorbol acetate and by imiquimod, as well as the imiquimod-induced development of skin lesions, in in vivo mouse models.52,105 Phosphatidylglycerol does not seem to act as a global immunosuppressant (since, for example, it inhibits viral infection of the lungs106,107). Therefore, since phosphatidylglycerol is a naturally occurring phospholipid with a presumably good safety profile, these results suggest the possibility of developing this lipid as a therapy for the treatment of psoriasis.
On the other hand, there are data to support the involvement in psoriasis of cell types/tissues in addition to the immune system and keratinocytes. Indeed, since the skin disorder classified as psoriasis likely has multiple etiologies, these diverse cell types may be differentially involved in the disease in various individuals, such that targeting of these different cell types may also be beneficial in psoriasis treatment. Thus, changes are observed in the dermal vasculature in psoriasis and evidence supports a potential role for blood vessels in this disease (reviewed in108). For example, expression of the well-known angiogenic molecule, vascular endothelial growth factor (VEGF) in keratinocytes (under the control of the keratin 14 promoter) has been shown to result in a phenotype characterized by acanthosis (epidermal hyperplasia), hyperkeratosis and parakeratosis, epidermal neutrophilic microabscesses and immune cell infiltration, similar to the changes characterizing psoriasis.109 (On the other hand, VEGF may have effects in keratinocytes in addition to its effects on endothelial cells: for example, mice with keratinocyte-specific deletion of the VEGF receptor, Flt-1 show amelioration of the psoriasiform phenotype induced by imiquimod.110) Finally, there is evidence for an involvement of the nervous system in psoriasis as well (reviewed in111). Thus, case reports show that damage to sensory neurons leading to loss of sensation results in unilateral remission of psoriatic plaques that return once nerve function is restored.112,113 In addition, blocking nerve transmission with a botulinum toxin can improve psoriasis area and severity index and physician global assessment scores in patients with recalcitrant plaque psoriasis.114
To summarize, then, psoriasis appears to be a complex inflammatory skin disease with participation of multiple cell types, tissues and systems in its pathogenesis. Therefore, it seems likely that optimal management of this disease will require targeting more than just a single cell or system for complete resolution. Such considerations are important since psoriatic individuals are often dissatisfied with current therapies115 because some may be ineffective and others, like the biologic agents, can be expensive and exhibit a spectrum of undesirable side effects, for example, increasing the risk of serious infections and possibly lymphoma.4,116 Therefore, the better the understanding of the mechanisms underlying psoriasis initiation and maintenance, as well as relapse, the more and better targets may be identified to develop therapies for psoriasis treatment.
Acknowledgments
LIO-L was supported by National Institutes of Health/National Institute on Aging award #AG067577. WBB was supported as a VA Research Career Scientist (#BX005691).
Disclosure
LIO-L and VC report no conflicts of interest in this work. WBB is an inventor of an Augusta University patent (8,808,715) for the use of phosphatidylglycerol to modulate keratinocyte function.
References
1. Wikramanayake TC, Stojadinovic O, Tomic-Canic M. Epidermal differentiation in barrier maintenance and wound healing. Adv Wound Care. 2014;3(3):272–280. doi:10.1089/wound.2013.0503
2. Madison KC. Barrier function of the skin: “la raison d’etre” of the epidermis. J Invest Dermatol. 2003;121:231–241. doi:10.1046/j.1523-1747.2003.12359.x
3. Ravipati A, Nolan S, Alphonse M, et al. IL-6R/signal transducer and activator of transcription 3 signaling in keratinocytes rather than in T cells induces psoriasis-like dermatitis in Mice. J Invest Dermatol. 2021;142(4):1126–1135.e4. doi:10.1016/j.jid.2021.09.012
4. Singh R, Koppu S, Perche PO, Feldman SR. The cytokine mediated molecular pathophysiology of psoriasis and its clinical implications. Int J Mol Sci. 2021;22:23. doi:10.3390/ijms222312793
5. Takahashi T, Yamasaki K. Psoriasis and antimicrobial peptides. Int J Mol Sci. 2020;21(18):6791. doi:10.3390/ijms21186791
6. Dopytalska K, Ciechanowicz P, Wiszniewski K, Szymanska E, Walecka I. The role of epigenetic factors in psoriasis. Int J Mol Sci. 2021;22:17. doi:10.3390/ijms22179294
7. Lowes MA, Suarez-Farinas M, Krueger JG. Immunology of psoriasis. Annu Rev Immunol. 2014;32:227–255. doi:10.1146/annurev-immunol-032713-120225
8. Weinstein GD, McCullough JL, Ross P. Cell proliferation in normal epidermis. J Invest Dermatol. 1984;82(6):623–628.
9. Orsmond A, Bereza-Malcolm L, Lynch T, March L, Xue M. Skin barrier dysregulation in psoriasis. Int J Mol Sci. 2021;22:19. doi:10.3390/ijms221910841
10. Zhang J, Wu J, Sun M, et al. Phospholipase C epsilon mediates cytokine cascade induced by acute disruption of epidermal permeability barrier in mice. Biochem Biophys Rep. 2020;24:100869. doi:10.1016/j.bbrep.2020.100869
11. Ye L, Lv C, Man G, Song S, Elias PM, Man MQ. Abnormal epidermal barrier recovery in uninvolved skin supports the notion of an epidermal pathogenesis of psoriasis. J Invest Dermatol. 2014;134(11):2843–2846. doi:10.1038/jid.2014.205
12. Voorhees JJ. Pathophysiology of psoriasis. Annu Rev Med. 1977;28:467–473. doi:10.1146/annurev.me.28.020177.002343
13. Cai Y, Fleming C, Yan J. New insights of T cells in the pathogenesis of psoriasis. Cell Mol Immunol. 2012;9(4):302–309. doi:10.1038/cmi.2012.15
14. Lande R, Botti E, Jandus C, et al. The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. Nat Commun. 2014;5:5621. doi:10.1038/ncomms6621
15. Morizane S, Yamasaki K, Muhleisen B, et al. Cathelicidin antimicrobial peptide LL-37 in psoriasis enables keratinocyte reactivity against TLR9 ligands. J Invest Dermatol. 2012;132(1):135–143. doi:10.1038/jid.2011.259
16. Ran D, Cai M, Zhang X. Genetics of psoriasis: a basis for precision medicine. Precision Clin Med. 2019;2:120–130. doi:10.1093/pcmedi/pbz011
17. Capon F. The genetic basis of psoriasis. Int J Mol Sci. 2017;18:12. doi:10.3390/ijms18122526
18. Gran F, Kerstan A, Serfling E, Goebeler M, Muhammad K. Current developments in the immunology of psoriasis. Yale J Biol Med. 2020;93(1):97–110.
19. Nestle FO, Conrad C, Tun-Kyi A, et al. Plasmacytoid predendritic cells initiate psoriasis through interferon-alpha production. J Exp Med. 2005;202(1):135–143. doi:10.1084/jem.20050500
20. Kulig P, Musiol S, Freiberger SN, et al. IL-12 protects from psoriasiform skin inflammation. Nat Commun. 2016;7:13466. doi:10.1038/ncomms13466
21. Nograles KE, Zaba LC, Guttman-Yassky E, et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br J Dermatol. 2008;159(5):1092–1102. doi:10.1111/j.1365-2133.2008.08769.x
22. Yost J, Gudjonsson JE. The role of TNF inhibitors in psoriasis therapy: new implications for associated comorbidities. F1000 Med Rep. 2009;1. doi:10.3410/M1-30
23. Brotas AM, Cunha JM, Lago EH, Machado CC, Carneiro SC. Tumor necrosis factor-alpha and the cytokine network in psoriasis. An Bras Dermatol. 2012;87(5):
24. Morizane S, Gallo RL. Antimicrobial peptides in the pathogenesis of psoriasis. J Dermatol. 2012;39(3):225–230. doi:10.1111/j.1346-8138.2011.01483.x
25. Ghoreschi K, Weigert C, Röcken M. Immunopathogenesis and role of T cells in psoriasis. Clin Dermatol. 2007;25:574–580. doi:10.1016/j.clindermatol.2007.08.012
26. Fotiadou C, Lazaridou E. Psoriasis and uveitis: links and risks. Psoriasis. 2019;9:91–96. doi:10.2147/PTT.S179182
27. Vicic M, Kastelan M, Brajac I, Sotosek V, Massari LP. Current Concepts of Psoriasis Immunopathogenesis. Int J Mol Sci. 2021;22:21. doi:10.3390/ijms222111574
28. Benhadou F, Mintoff D, Del Marmol V. Psoriasis: keratinocytes or immune cells - which is the trigger? Dermatology. 2019;235(2):91–100. doi:10.1159/000495291
29. Piipponen M, Li D, Landen NX. The immune functions of keratinocytes in skin wound healing. Int J Mol Sci. 2020;21:22. doi:10.3390/ijms21228790
30. de Cid R, Riveira-Munoz E, Zeeuwen PL, et al. Deletion of the late cornified envelope LCE3B and LCE3C genes as a susceptibility factor for psoriasis. Nat Genet. 2009;41(2):211–215. doi:10.1038/ng.313
31. Schmuth M, Blunder S, Dubrac S, Gruber R, Moosbrugger-Martinz V. Epidermal barrier in hereditary ichthyoses, atopic dermatitis, and psoriasis. J Dtsch Dermatol Ges. 2015;13(11):1119–1123. doi:10.1111/ddg.12827
32. Garg A, Chren MM, Sands LP, et al. Psychological stress perturbs epidermal permeability barrier homeostasis: implications for the pathogenesis of stress-associated skin disorders. Arch Dermatol. 2001;137(1):53–59. doi:10.1001/archderm.137.1.53
33. Sano S. Psoriasis as a barrier disease. Dermatol Sin. 2015;33:64–69. doi:10.1016/j.dsi.2015.04.010
34. Yamamoto H, Hattori M, Chamulitrat W, Ohno Y, Kihara A. Skin permeability barrier formation by the ichthyosis-causative gene FATP4 through formation of the barrier lipid omega-O-acylceramide. Proc Natl Acad Sci U S A. 2020;117(6):2914–2922. doi:10.1073/pnas.1917525117
35. Cameron DJ, Tong Z, Yang Z, et al. Essential role of Elovl4 in very long chain fatty acid synthesis, skin permeability barrier function, and neonatal survival. Int J Biol Sci. 2007;3(2):111–119. doi:10.7150/ijbs.3.111
36. Knox S, O’Boyle NM. Skin lipids in health and disease: a review. Chem Phys Lipids. 2021;236:105055. doi:10.1016/j.chemphyslip.2021.105055
37. Luczaj W, Wronski A, Domingues P, Domingues MR, Skrzydlewska E. Lipidomic analysis reveals specific differences between fibroblast and keratinocyte ceramide profile of patients with psoriasis vulgaris. Molecules. 2020;25(3):630. doi:10.3390/molecules25030630
38. Zeng C, Wen B, Hou G, et al. Lipidomics profiling reveals the role of glycerophospholipid metabolism in psoriasis. Gigascience. 2017;6(10):1–11. doi:10.1093/gigascience/gix087
39. Ali G, Elsayed AK, Nandakumar M, et al. Keratinocytes derived from patient-specific induced pluripotent stem cells recapitulate the genetic signature of psoriasis disease. Stem Cells Dev. 2020;29(7):383–400. doi:10.1089/scd.2019.0150
40. Chen J, Zeng F, Forrester SJ, Eguchi S, Zhang MZ, Harris RC. Expression and function of the epidermal growth factor receptor in physiology and disease. Physiol Rev. 2016;96(3):1025–1069. doi:10.1152/physrev.00030.2015
41. Zeng C, Tsoi LC, Gudjonsson JE. Dysregulated epigenetic modifications in psoriasis. Exp Dermatol. 2021;30(8):1156–1166. doi:10.1111/exd.14332
42. Bikle DD, Xie Z, Tu CL. Calcium regulation of keratinocyte differentiation. Expert Rev Endocrinol Metab. 2012;7(4):461–472. doi:10.1586/eem.12.34
43. Menon GK, Grayson S, Elias PM. Ionic calcium reservoirs in mammalian epidermis: ultrastructural localization by ion-capture cytochemistry. J Invest Dermatol. 1985;84:508–512. doi:10.1111/1523-1747.ep12273485
44. Menon GK, Elias PM. Ultrastructural localization of calcium in psoriatic and normal human epidermis. Arch Dermatol. 1991;127:57–63. doi:10.1001/archderm.1991.01680010067010
45. Tu C-L, Chang W, Xie Z, Bikle DD. Inactivation of the calcium sensing receptor inhibits E-cadherin-mediated cell-cell adhesion and calcium-induced differentiation in human epidermal keratinocytes. J Biol Chem. 2008;283:3519–3528. doi:10.1074/jbc.M708318200
46. Tu CL, Crumrine DA, Man MQ, et al. Ablation of the calcium-sensing receptor in keratinocytes impairs epidermal differentiation and barrier function. J Invest Dermatol. 2012;132(10):2350–2359. doi:10.1038/jid.2012.159
47. Tu CL, Bikle DD. Role of the calcium-sensing receptor in calcium regulation of epidermal differentiation and function. Best Pract Res Clin Endocrinol Metab. 2013;27(3):415–427. doi:10.1016/j.beem.2013.03.002
48. Bollag WB. Down-regulated calcium-sensing receptor in keratinocytes and skin from aged mice and humans impairs function. J Invest Dermatol. 2021;141(11):2558–2561. doi:10.1016/j.jid.2021.04.005
49. Karvonen S-J, Korkiamäki T, Ylä-Outinen H, et al. Psoriasis and altered calcium metabolism: downregulated capacitative calcium influx and defective calcium-mediated cell signaling in cultured psoriatic keratinocytes. J Invest Dermatol. 2000;114:693–700. doi:10.1046/j.1523-1747.2000.00926.x
50. Leuner K, Kraus M, Woelfle U, et al. Reduced TRPC channel expression in psoriatic keratinocytes is associated with impaired differentiation and enhanced proliferation. PLoS One. 2011;6(2):e14716. doi:10.1371/journal.pone.0014716
51. Erridge C. Endogenous ligands of TLR2 and TLR4: agonists or assistants? J Leukoc Biol. 2010;87(6):989–999. doi:10.1189/jlb.1209775
52. Choudhary V, Uaratanawong R, Patel RR, et al. Phosphatidylglycerol inhibits toll-like receptor-mediated inflammation by danger-associated molecular patterns. J Invest Dermatol. 2019;139(4):868–877. doi:10.1016/j.jid.2018.10.021
53. Bowcock AM, Shannon W, Du F, et al. Insights into psoriasis and other inflammatory diseases from large-scale gene expression studies. Hum Mol Genet. 2001;10(17):1793–1805. doi:10.1093/hmg/10.17.1793
54. Nakajima K, Kanda T, Takaishi M, et al. Distinct roles of IL-23 and IL-17 in the development of psoriasis-like lesions in a mouse model. J Immunol. 2011;186(7):4481–4489. doi:10.4049/jimmunol.1000148
55. Racz E, Prens EP, Kurek D, et al. Effective treatment of psoriasis with narrow-band UVB phototherapy is linked to suppression of the IFN and Th17 pathways. J Invest Dermatol. 2011;131(7):1547–1558. doi:10.1038/jid.2011.53
56. Waite JC, Skokos D. Th17 response and inflammatory autoimmune diseases. Int J Inflam. 2012;2012:819467. doi:10.1155/2012/819467
57. Schonthaler HB, Guinea-Viniegra J, Wculek SK, et al. S100A8-S100A9 protein complex mediates psoriasis by regulating the expression of complement factor C3. Immunity. 2013;39(6):1171–1181. doi:10.1016/j.immuni.2013.11.011
58. Nograles KE, Davidovici B, Krueger JG. New insights in the immunologic basis of psoriasis. Semin Cutan Med Surg. 2010;29(1):3–9. doi:10.1016/j.sder.2010.03.001
59. Nakajima K, Terao M, Takaishi M, et al. Barrier abnormality due to ceramide deficiency leads to psoriasiform inflammation in a mouse model. J Invest Dermatol. 2013;133(11):2555–2565. doi:10.1038/jid.2013.199
60. Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009;361(5):496–509. doi:10.1056/NEJMra0804595
61. Jansen PA, Rodijk-Olthuis D, Hollox EJ, et al. Beta-defensin-2 protein is a serum biomarker for disease activity in psoriasis and reaches biologically relevant concentrations in lesional skin. PLoS One. 2009;4(3):e4725. doi:10.1371/journal.pone.0004725
62. Gu X, Nylander E, Coates PJ, Fahraeus R, Nylander K. Correlation between reversal of DNA methylation and clinical symptoms in psoriatic epidermis following narrow-band UVB phototherapy. J Invest Dermatol. 2015;135(8):2077–2083. doi:10.1038/jid.2015.128
63. Zhou F, Wang W, Shen C, et al. Epigenome-wide association analysis identified nine skin DNA methylation loci for psoriasis. J Invest Dermatol. 2016;136(4):779–787. doi:10.1016/j.jid.2015.12.029
64. Chandra A, Senapati S, Roy S, Chatterjee G, Chatterjee R. Epigenome-wide DNA methylation regulates cardinal pathological features of psoriasis. Clin Epigenetics. 2018;10(1):108. doi:10.1186/s13148-018-0541-9
65. Wang X, Liu X, Liu N, Chen H. Prediction of crucial epigenetically associated, differentially expressed genes by integrated bioinformatics analysis and the identification of S100A9 as a novel biomarker in psoriasis. Int J Mol Med. 2020;45(1):93–102. doi:10.3892/ijmm.2019.4392
66. Hollox EJ, Huffmeier U, Zeeuwen PL, et al. Psoriasis is associated with increased beta-defensin genomic copy number. Nat Genet. 2008;40(1):23–25. doi:10.1038/ng.2007.48
67. Ni X, Lai Y. Keratinocyte: a trigger or an executor of psoriasis? J Leukoc Biol. 2020;108(2):485–491. doi:10.1002/JLB.5MR0120-439R
68. Qiao P, Guo W, Ke Y, et al. Mechanical stretch exacerbates psoriasis by stimulating keratinocyte proliferation and cytokine production. J Invest Dermatol. 2019;139(7):1470–1479. doi:10.1016/j.jid.2018.12.019
69. Okamoto T, Ogawa Y, Kinoshita M, et al. Mechanical stretch-induced ATP release from keratinocytes triggers Koebner phenomenon in psoriasis. J Dermatol Sci. 2021;103(1):60–62. doi:10.1016/j.jdermsci.2021.06.001
70. Ferrari D, Casciano F, Secchiero P, Reali E. Purinergic signaling and inflammasome activation in psoriasis pathogenesis. Int J Mol Sci. 2021;22(17):9449. doi:10.3390/ijms22179449
71. Kennedy-Crispin M, Billick E, Mitsui H, et al. Human keratinocytes’ response to injury upregulates CCL20 and other genes linking innate and adaptive immunity. J Invest Dermatol. 2012;132(1):105–113. doi:10.1038/jid.2011.262
72. Larsen SB, Cowley CJ, Sajjath SM, et al. Establishment, maintenance, and recall of inflammatory memory. Cell Stem Cell. 2021;28(10):1758–1774 e1758. doi:10.1016/j.stem.2021.07.001
73. Gudjonsson JE, Johnston A, Dyson M, Valdimarsson H, Elder JT. Mouse models of psoriasis. J Invest Dermatol. 2007;127:1292–1308. doi:10.1038/sj.jid.5700807
74. Schon MP. Animal models of psoriasis–What can we learn from them? J Invest Dermatol. 1999;112:405–410. doi:10.1046/j.1523-1747.1999.00538.x
75. Gangwar RS, Gudjonsson JE, Ward NL. Mouse models of psoriasis: a comprehensive review. J Invest Dermatol. 2021;142(3 Pt B):884–897. doi:10.1016/j.jid.2021.06.019
76. Schon MP, Manzke V, Erpenbeck L. Animal models of psoriasis-highlights and drawbacks. J Allergy Clin Immunol. 2021;147(2):439–455. doi:10.1016/j.jaci.2020.04.034
77. Cook PW, Piepkorn M, Clegg CH, et al. Transgenic expression of the human amphiregulin gene induces a psoriasis-like phenotype. J Clin Invest. 1997;100:2286–2294. doi:10.1172/JCI119766
78. Zenz R, Efer R, Kenner L, et al. Psoriasis-like skin disease and arthritis caused by inducible epidermal deletion of Jun proteins. Nature. 2005;437:369–375. doi:10.1038/nature03963
79. Gudjonsson JE, Elder JT. Mouse models: psoriasis: an epidermal disease after all? Eur J Hum Genet. 2006;14(1):2–4. doi:10.1038/sj.ejhg.5201543
80. Sano S, Chan KS, Carbajal S, et al. Stat3 links activated keratinocytes and immunocytes required for development of psoriasis in a novel transgenic mouse model. Nat Med. 2005;11(1):43–49. doi:10.1038/nm1162
81. Wolfram JA, Diaconu D, Hatala DA, et al. Keratinocyte but not endothelial cell-specific overexpression of Tie2 leads to the development of psoriasis. Am J Pathol. 2009;174(4):1443–1458. doi:10.2353/ajpath.2009.080858
82. van der Fits L, Mourits S, Voerman JS, et al. Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol. 2009;182(9):5836–5845. doi:10.4049/jimmunol.0802999
83. Flutter B, Nestle FO. TLRs to cytokines: mechanistic insights from the imiquimod mouse model of psoriasis. Eur J Immunol. 2013;43(12):3138–3146. doi:10.1002/eji.201343801
84. Schon MP, Schon M. TLR7 and TLR8 as targets in cancer therapy. Oncogene. 2008;27(2):190–199. doi:10.1038/sj.onc.1210913
85. Swindell WR, Johnston A, Carbajal S, et al. Genome-wide expression profiling of five mouse models identifies similarities and differences with human psoriasis. PLoS One. 2011;6(4):e18266. doi:10.1371/journal.pone.0018266
86. Grine L, Steeland S, Van Ryckeghem S, et al. Topical imiquimod yields systemic effects due to unintended oral uptake. Sci Rep. 2016;6:20134. doi:10.1038/srep20134
87. Wang Z, Zhou H, Zheng H, et al. Autophagy-based unconventional secretion of HMGB1 by keratinocytes plays a pivotal role in psoriatic skin inflammation. Autophagy. 2021;17(2):529–552. doi:10.1080/15548627.2020.1725381
88. Ippagunta SK, Gangwar R, Finkelstein D, et al. Keratinocytes contribute intrinsically to psoriasis upon loss of Tnip1 function. Proc Natl Acad Sci U S A. 2016;113(41):E6162–E6171. doi:10.1073/pnas.1606996113
89. Moos S, Mohebiany AN, Waisman A, Kurschus FC. Imiquimod-induced psoriasis in mice depends on the IL-17 signaling of keratinocytes. J Invest Dermatol. 2019;139(5):1110–1117. doi:10.1016/j.jid.2019.01.006
90. Nakabo S, Romo-Tena J, Kaplan MJ. Neutrophils as drivers of immune dysregulation in autoimmune diseases with skin manifestations. J Invest Dermatol. 2021;142(3 Pt B):823–833. doi:10.1016/j.jid.2021.04.014
91. Croxford AL, Karbach S, Kurschus FC, et al. IL-6 regulates neutrophil microabscess formation in IL-17A-driven psoriasiform lesions. J Invest Dermatol. 2014;134(3):728–735. doi:10.1038/jid.2013.404
92. Wohn C, Brand A, van Ettinger K, et al. Gradual development of psoriatic skin lesions by constitutive low-level expression of IL-17A. Cell Immunol. 2016;308:57–65. doi:10.1016/j.cellimm.2015.11.006
93. Yamanaka K, Yamamoto O, Honda T. Pathophysiology of psoriasis: a review. J Dermatol. 2021;48(6):722–731. doi:10.1111/1346-8138.15913
94. Li ZJ, Choi DK, Sohn KC, et al. Induction of Interleukin-22 (IL-22) production in CD4(+) T cells by IL-17A secreted from CpG-stimulated keratinocytes. Ann Dermatol. 2016;28(5):579–585. doi:10.5021/ad.2016.28.5.579
95. Furue M, Furue K, Tsuji G, Nakahara T. Interleukin-17A and keratinocytes in psoriasis. Int J Mol Sci. 2020;21(4):1275. doi:10.3390/ijms21041275
96. Vandeghinste N, Klattig J, Jagerschmidt C, et al. Neutralization of IL-17C reduces skin inflammation in mouse models of psoriasis and atopic dermatitis. J Invest Dermatol. 2018;138(7):1555–1563. doi:10.1016/j.jid.2018.01.036
97. Johnston A, Fritz Y, Dawes SM, et al. Keratinocyte overexpression of IL-17C promotes psoriasiform skin inflammation. J Immunol. 2013;190(5):2252–2262. doi:10.4049/jimmunol.1201505
98. Xu M, Lu H, Lee YH, et al. An interleukin-25-mediated autoregulatory circuit in keratinocytes plays a pivotal role in psoriatic skin inflammation. Immunity. 2018;48(4):787–798 e784. doi:10.1016/j.immuni.2018.03.019
99. Helwa I, Gulotto M, Bollag WB. Keratinocytes in psoriasis: key players in the disease process. In: Smith P, Johnson N, editors. Psoriasis Types, Triggers and Treatment Strategies. Hauppauge, NY: Nova Science Publishers; 2013.
100. Choudhary V, Griffith S, Chen X, Bollag WB. Pathogen-associated molecular pattern-induced TLR2 and TLR4 activation increases keratinocyte production of inflammatory mediators and is inhibited by phosphatidylglycerol. Mol Pharmacol. 2020;97(5):324–335. doi:10.1124/mol.119.118166
101. Kandasamy P, Numata M, Berry KZ, et al. Structural analogs of pulmonary surfactant phosphatidylglycerol inhibit toll-like receptor 2 and 4 signaling. J Lipid Res. 2016;57(6):993–1005. doi:10.1194/jlr.M065201
102. Kandasamy P, Zarini S, Chan ED, Leslie CC, Murphy RC, Voelker DR. Pulmonary surfactant phosphatidylglycerol inhibits Mycoplasma pneumoniae-stimulated eicosanoid production from human and mouse macrophages. J Biol Chem. 2011;286(10):7841–7853. doi:10.1074/jbc.M110.170241
103. Kuronuma K, Mitsuzawa H, Takeda K, et al. Anionic pulmonary surfactant phospholipids inhibit inflammatory responses from alveolar macrophages and U937 cells by binding the lipopolysaccharide-interacting proteins CD14 and MD-2. J Biol Chem. 2009;284(38):25488–25500. doi:10.1074/jbc.M109.040832
104. Numata M, Chu HW, Dakhama A, Voelker DR. Pulmonary surfactant phosphatidylglycerol inhibits respiratory syncytial virus-induced inflammation and infection. Proc Natl Acad Sci U S A. 2010;107(1):320–325. doi:10.1073/pnas.0909361107
105. Xie D, Choudhary V, Seremwe M, et al. Soy phosphatidylglycerol reduces inflammation in a contact irritant ear edema mouse model in vivo. J Pharmacol Exp Ther. 2018;366(1):1–8. doi:10.1124/jpet.117.244756
106. Numata M, Kandasamy P, Nagashima Y, et al. Phosphatidylglycerol suppresses influenza A virus infection. Am J Respir Cell Mol Biol. 2012;46(4):479–487. doi:10.1165/rcmb.2011-0194OC
107. Numata M, Nagashima Y, Moore ML, et al. Phosphatidylglycerol provides short-term prophylaxis against respiratory syncytial virus infection. J Lipid Res. 2013;54(8):2133–2143. doi:10.1194/jlr.M037077
108. Heidenreich R, Rocken M, Ghoreschi K. Angiogenesis drives psoriasis pathogenesis. Int J Exp Pathol. 2009;90(3):232–248. doi:10.1111/j.1365-2613.2009.00669.x
109. Xia YP, Li B, Hylton D, Detmar M, Yancopoulos GD, Rudge JS. Transgenic delivery of VEGF to mouse skin leads to an inflammatory condition resembling human psoriasis. Blood. 2003;102(1):161–168. doi:10.1182/blood-2002-12-3793
110. Benhadou F, Glitzner E, Brisebarre A, et al. Epidermal autonomous VEGFA/Flt1/Nrp1 functions mediate psoriasis-like disease. Sci Adv. 2020;6(2):eaax5849. doi:10.1126/sciadv.aax5849
111. Zhang X, He Y. The role of nociceptive neurons in the pathogenesis of psoriasis. Front Immunol. 2020;11:1984. doi:10.3389/fimmu.2020.01984
112. Dewing SB. Remission of psoriasis associated with cutaneous nerve section. Arch Dermatol. 1971;104(2):220–221. doi:10.1001/archderm.1971.04000200108024
113. Joseph T, Kurian J, Warwick DJ, Friedmann PS. Unilateral remission of psoriasis following traumatic nerve palsy. Br J Dermatol. 2005;152(1):185–186.
114. Aschenbeck KA, Hordinsky MK, Kennedy WR, et al. Neuromodulatory treatment of recalcitrant plaque psoriasis with onabotulinumtoxinA. J Am Acad Dermatol. 2018;79(6):1156–1159. doi:10.1016/j.jaad.2018.07.058
115. Armstrong AW, Robertson AD, Wu J, Schupp C, Lebwohl MG. Undertreatment, treatment trends, and treatment dissatisfaction among patients with psoriasis and psoriatic arthritis in the United States: findings from the National Psoriasis Foundation surveys, 2003–2011. JAMA Dermatol. 2013;149(10):1180–1185. doi:10.1001/jamadermatol.2013.5264
116. Sivamani RK, Correa G, Ono Y, Bowen MP, Raychaudhuri SP, Maverakis E. Biological therapy of psoriasis. Indian J Dermatol. 2010;55(2):161–170. doi:10.4103/0019-5154.62754
117. Phan TS, Schink L, Mann J, et al. Keratinocytes control skin immune homeostasis through de novo-synthesized glucocorticoids. Sci Adv. 2021;7(5). doi:10.1126/sciadv.abe0337
118. Schunke H, Gobel U, Dikic I, Pasparakis M. OTULIN inhibits RIPK1-mediated keratinocyte necroptosis to prevent skin inflammation in mice. Nat Commun. 2021;12(1):5912. doi:10.1038/s41467-021-25945-1
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.