")
Back to Journals » Psoriasis: Targets and Therapy » Volume 5
Update on the treatment of psoriasis and psoriatic arthritis – role of apremilast
Authors Forchhammer S, Ghoreschi K
Received 24 June 2015
Accepted for publication 6 August 2015
Published 7 September 2015 Volume 2015:5 Pages 117—124
DOI https://doi.org/10.2147/PTT.S69476
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Prof. Dr. Uwe Wollina
Stephan Forchhammer, Kamran Ghoreschi
Department of Dermatology, University Medical Center, Eberhard Karls University of Tübingen, Tübingen, Germany
Abstract: Psoriatic arthritis (PsA) is an inflammatory arthritis associated with psoriasis (PsO). The treatment of PsA can be challenging and includes non-steroidal anti-inflammatory drugs, synthetic disease modifying antirheumatic drugs, and biologicals. One novel oral compound that has been recently established for the treatment of PsO and PsA is apremilast, a small molecule PDE4 inhibitor. The inhibition of PDE4 results in increased intracellular cAMP levels and modulates the expression of inflammatory mediators critically involved in PsO and PsA pathogenesis like TNF, IL-12, IL-17, and IL-23. Apremilast received US Food and Drug Administration approval for the treatment of PsO and PsA in 2014 and received approval from the European Medicines Agency in early 2015. This article summarizes the pharmacology of apremilast, its efficacy and safety in clinical studies, and its potential position in modern PsO/PsA management.
Keywords: apremilast, phosphodiesterase-4-inhibitor, PsO, PsA, systemic therapy, efficacy, safety
Introduction
Psoriasis (PsO) is a chronic inflammatory disease of the skin and joints occurring in 3% of the population1,2 and approximately 20% – 30% of patients diagnosed with PsO develop psoriatic arthritis (PsA).3,4 PsO is currently considered an autoimmune inflammatory disease, mainly caused by the interplay between the innate and the adaptive immune system. Predominant cells of the psoriatic inflammation are T-cells, antigen presenting cells like dendritic cells and neutrophilic granulocytes. Their mediators are responsible for the proliferative response by keratinocytes and vascular endothelial cells.5,6 There is increasing evidence that besides Th1 cells, Th17 cells, a subset of CD4+ T-cells that preferentially produce IL-17, are the key players in PsO pathogenesis.7 IL-23 is an important cytokine that promotes the generation of pathogenic Th17 cell differentiation.8 Upon activation, Th1 and Th17 cells produce inflammatory cytokines like IL-2, IFN-γ (Th1-cells), IL-17A, IL-17F, IL-21, IL-22 (Th17-cells), and TNF (Th1- and Th17- cells).9–11 Similar to PsO pathogenesis, Th17 cells have been found to play an important role in joint inflammation in PsA.12
PsA is a separate clinical entity from rheumatoid arthritis. It is usually seronegative for rheumatoid factor, has a frequent involvement of distal joints, and has an asymmetric tendency. Moreover, PsA has a common spinal involvement and often goes along with enthesitis and dactylitis.13 Patients affected with PsA show a reduced quality of life and demonstrate a reduced physical function, as measured in lower scores of the Medical Outcome Survey Short Form 36 (SF-36) and higher scores of the Health Assessment Questionnaire (HAQ).14,15
For the management of PsA patients it is important to adjust treatment according to the prevailing clinical pattern and to pay attention to both skin and joint manifestations. There are patients presenting with predominant skin involvement and mild arthritis, who are successfully treated for PsA with non-steroidal anti-inflammatory drug (NSAID) treatment. Yet, a substantial percentage of patients with PsA (20%) develop a severe, mutilating, and debilitating form of arthritis.16,17
Treatment of PsA has fundamentally changed over the last years through development of new synthetic and biological agents. Depending on PsA activity and severity, type of joint involvement and extra-articular manifestations, therapy usually starts with NSAIDs. When PsA shows high activity or when NSAIDs are inefficient, therapy is escalated to synthetic disease modifying antirheumatic drugs (DMARDs) with methotrexate being the most common agent. Second line therapy is used when DMARDs show lack of efficacy or show toxicities. The second line DMARDs are currently represented by biologicals with TNF antagonists being the most popular biological DMARD.9,18 The introduction of TNF-neutralizing fusion proteins and antibodies was a breakthrough in the treatment of PsA and PsO.
Despite the vast advancement in PsA therapy over the past 15 years, there is still need for further improvement. Biological DMARDs have to be administered intravenously or subcutaneously and are very cost intensive for patients and the health system. Moreover, immunosuppressive biologicals show increased risks for infections and certain malignancies. Therefore, recent developments focus on oral DMARDs, which selectively silence inflammatory immune responses with acceptable safety profiles.9,19 One of these novel chemical compounds is apremilast, which has been recently approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) as second line treatment for PsA and PsO.
Apremilast – mode of action
Apremilast – (S)-N-{2-[1-(3-ethoxy-4-methoxyphenyl)-2-(methylsulfonyl)ethyl-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl}acetamide – is an orally available small molecule inhibitor of PDE4.20 Inhibition of PDE4 leads to reduced production of pro-inflammatory mediators like TNF, IFN-γ, IL-12/23p40, IL-23p19, IL-17A, and IL-22. On the other hand, apremilast has been reported to increase the production of IL-6 and IL-10.20 This is of interest, since IL-10 is a cytokine with potent anti-inflammatory properties, while IL-6 is a cytokine with pro- and anti-inflammatory features.21,22
Mechanistically, PDEs degrade intracellular cAMP levels by enzymatic hydrolysis. The cyclic nucleotide cAMP is an important intracellular secondary messenger. Extracellular inflammatory signals like chemokines, leukotrienes, and PGs bind to GPCRs, leading to activation of AC, which increases the production of cAMP.23 The secondary messenger cAMP then interacts with and activates effector proteins like PKA. On the one hand this kinase results in phosphorylation and activation of transcription factors like CREB and ATF-1. On the other hand PKA activation leads to inhibition of other transcription factors like NF-κB.24,25 In this way cAMP is involved in immune homeostasis by increasing inflammatory signals when cAMP levels are low and by reducing inflammatory signaling when cAMP levels are high (Figure 1).
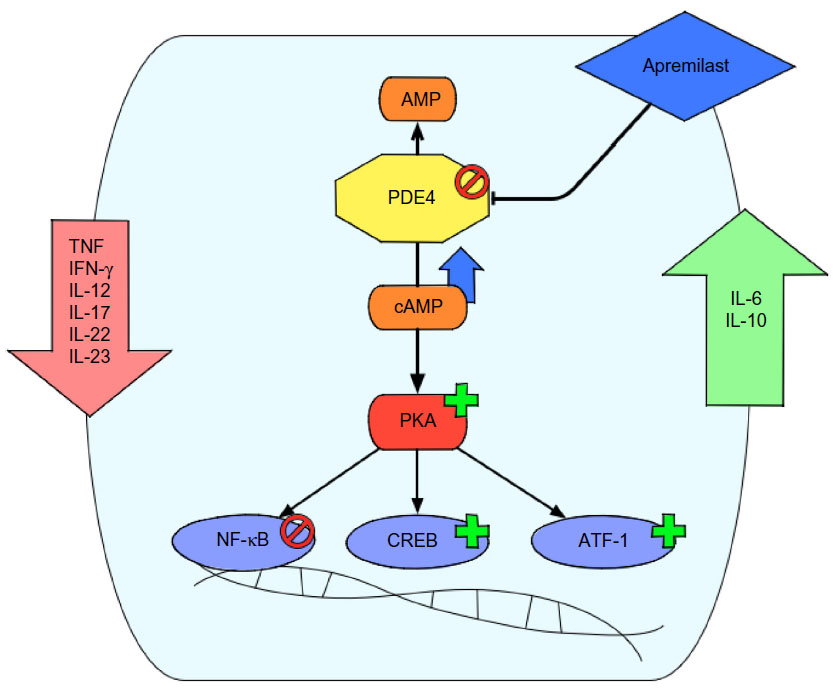 | Figure 1 Mode of action of apremilast. |
There are eleven individual families of PDEs described in mammalian species (PDE1–PDE11) with different selectivity to cAMP/cGMP hydrolyzation and distinct predominant cellular localization.26 PDE4 is specific for cAMP hydrolyzation and is the major PDE isoenzyme found in immune cells like monocytes, T-cells, and neutrophils.27 Apremilast is a selective inhibitor of PDE4, with significant inhibition observed only for the PDE4 isoenzymes encoded by four genes (PDE4A, PDE4B, PDE4C, and PDE4D). In a recent study, apremilast (10 μM) showed an average of approximately 95% inhibition of the PDE4 enzymes in vitro. There was no significant inhibition measured on other members of the PDE family.28 Originally, apremilast was developed as a thalidomide analog as both drugs have the phthalimide ring in common.29 Despite this relatedness, apremilast shows no binding to CRBN, the primary molecular target of thalidomide teratogenicity, as this binding is mediated through the glutarimide ring of thalidomide and apremilast does not contain a glutarimide ring.28,30
Apremilast treatment has been reported to elevate intracellular cAMP levels in human peripheral blood mononuclear cells (PBMCs) in response to PGE2.28 In vitro, treatment of PBMCs with apremilast inhibits LPS-induced TNF production and reduces CpG ODN-stimulated IFN-α production.28 Further, the production of cytokines like IL-2, IL-5, IL-10, IL-13, Il-17, TNF, GM-CSF, and IFN-γ by anti-CD3-stimulated T-cells was inhibited by apremilast.28 In contrast, the presence of apremilast significantly enhanced IL-10 and IL-6 production from LPS-stimulated PBMCs.20 The different regulation of IL-10 in T-cells compared to PBMCs by apremilast could be explained by the presence of antigen presenting cells when studying cytokine production in PBMCs. In an antigen-specific transgenic T- and B-cell clonal expansion mouse model, apremilast had no effect on the clonal expansion of T- or B-cells and had no effect on antibody responses.28
PDE4 inhibition has also been tested in preclinical models of PsO and PsA. In vivo studies using a PsO mouse model showed significant reduced epidermal thickness, reduced proliferation index, and recovery of psoriasiform histological features upon apremilast treatment.20 In a murine model of arthritis apremilast treatment blocked synovial inflammation, cartilage damage, and bone erosion.31
Efficacy of apremilast
Clinical efficacy of apremilast in PsA was studied in four randomized, placebo-controlled trials with open-label extension periods.32 This extensive Phase III clinical trial program is called the Psoriatic Arthritis Longterm Assessment of Clinical Efficacy (PALACE).32 For clinical efficacy assessment of apremilast in plaque-type PsO two randomized, placebo-controlled Phase III clinical studies with open-label extensions have been performed, ESTEEM 1 and ESTEEM 2.33
Recently, the first results of a Phase III clinical trial comparing apremilast to placebo and etanercept called the LIBERATE trial were presented at the 73rd Annual Meeting of the American Academy of Dermatology.34
Efficacy of apremilast in PsA
Efficacy data of apremilast in PsA are available from the PALACE 1, PALACE 2, and PALACE 3 studies. Those trials had a similar design and enrolled patients with active PsA defined by ≥3 swollen joints and ≥3 tender joints despite prior or current treatment with DMARDs (small-molecule and/or biologic). Different to PALACE 1 and PALACE 2, the PALACE 3 trial studied the efficacy of apremilast in patients with active PsA who had at least one psoriatic lesion with a body surface area (BSA) ≥3%.32,35,36 Overall there have been 1,493 patients randomized and treated either with placebo or apremilast 20 mg twice daily (BID) or 30 mg BID.33 Apremilast was either given as a monotherapy (34.8%) or in combination with stable doses of non-biological DMARDs (65.2%). Methotrexate (≤25 mg per week) was the most common co-medication of patients (54.5%).33 Primary efficacy endpoint was the proportion of patients meeting 20% improvement in modified American College of Rheumatology response criteria (ACR20) at week 16. Additional efficacy outcome measures included symptoms of PsA, physical function, enthesitis, dactylitis, and PsO.32 At week 16, significantly more patients receiving apremilast 20 mg BID (30.4%) or 30 mg BID (38.1%) achieved an ACR20 response compared to placebo treatment (19.0%) in the PALACE 1 trial.32 The pooled data of PALACE 1–3 trials after 16 weeks of treatment with apremilast 30 mg BID showed a significantly higher ACR20 response (37.0%) compared to placebo (18.8%).33 Patients with baseline enthesitis had a significantly higher change from the Maastricht Ankylosing Spondylitis Enthesitis Score when treated with apremilast 30 mg BID versus placebo. In patients with dactylitis, the mean change from baseline in dactylitis severity score was higher with apremilast versus placebo but did not reach statistical significance at week 24.32 After 24 weeks of treatment, the placebo-controlled phase ended and the remaining placebo patients were re-randomized to either apremilast 20 mg BID or apremilast 30 mg BID.32 The ACR20 response could be sustained among the patients who continued receiving treatment with apremilast through week 52. At week 52, 63.0% of patients who received apremilast 20 mg BID and 54.6% who received 30 mg BID achieved an ACR20 response (Table 1).37
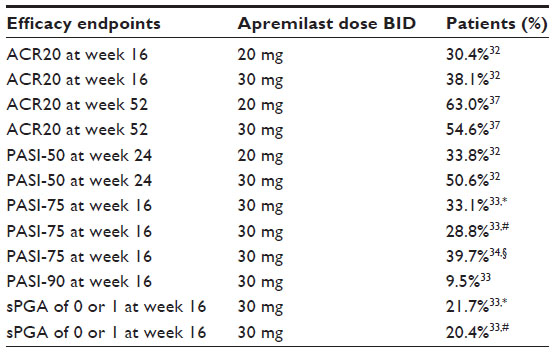 | Table 1 Efficacy of apremilast treatment |
Efficacy of apremilast in PsO
Data on the efficacy of apremilast on PsO were collected in trials for PsA and in PsO trials. The PALACE studies showed a reduction of the baseline Psoriasis Area and Severity Index (PASI) in patients with PsO affecting ≥3% of the BSA by at least 50% (PASI-50) in a significantly higher proportion of patients receiving apremilast 20 mg BID (33.8%) or apremilast 30 mg BID (50.6%) compared to placebo (18.5%) after 24 weeks.32 In the ESTEEM 1 and ESTEEM 2 trials, 1,257 patients with moderate to severe plaque PsO defined by a PASI score ≥12, a BSA ≥10%, and a static physician global assessment (sPGA) ≥3, who were candidates for systemic or phototherapy were included.33 Patients were treated with either apremilast 30 mg BID or placebo (2:1) for 16 weeks, from week 16 to week 32 every patient received apremilast 30 mg BID followed by a randomized reset phase from week 32 to week 52, depending on primary PASI response. The primary efficacy endpoint was a PASI-75 response after 16 weeks, the secondary efficacy endpoint was the proportion of patients reaching an sPGA of 0 (clear) or 1 (almost clear) after 16 weeks.
Patients included in this trial had a mean PASI score of 19.07, 70% had an sPGA score of 3 (moderate) and 29.8% had an sPGA score of 4 (severe). Eighteen percent of patients in the ESTEEM 1 and ESTEEM 2 trial also suffered from PsA.33 After 16 weeks of treatment with apremilast 30 mg BID, 33.1% of patients in the ESTEEM 1 trial had a PASI-score improvement of 75% compared to baseline (5.3% with placebo treatment). In the ESTEEM 2 trial, 28.8% of patients reached a PASI-75 response after 16 weeks compared to 5.8% following placebo treatment. An sPGA score of clear or almost clear was reached by 21.7% of patients in the ESTEEM 1 trial (3.9% with placebo) and by 20.4% of patients in the ESTEEM 2 trial (4.4% with placebo). Seventy-nine out of 835 patients (9.5%) treated with apremilast 30 mg BID in both trials had a 90% improvement in PASI score (PASI-90) after 16 weeks (Table 1).33
Beside PASI and sPGA response, patients treated with apremilast in the ESTEEM 1 and ESTEEM 2 trial had an improvement in nail PsO, scalp PsO, and pruritus.33
In the LIBERATE (EvaLuatIon from a PlaceBo-controllEd Study of ORal ApremilasT and Etanercept in Plaque Psoriasis) Phase IIIb clinical trial to measure the efficacy and safety of apremilast, etanercept, and placebo in patients with moderate to severe plaque PsO, 250 subjects who had no prior exposure to biological agents were randomized 1:1:1 to receive either apremilast 30 mg BID, etanercept 50 mg subcutaneously once weekly or placebo for 16 weeks. After week 16 all patients received apremilast 30 mg BID through week 104.34 The primary endpoint was to evaluate efficacy and safety of apremilast 30 mg BID compared to placebo at week 16. The study was not designed to directly compare apremilast to etanercept treatment.34
After 16 weeks, 33 out of 83 (40%) patients receiving apremilast 30 mg BID showed a PASI-75 response, compared to ten out of 84 (12%) patients receiving placebo treatment and 40 out of 84 (48%) patients receiving etanercept 50 mg subcutaneously once weekly.34
Safety and tolerability of PDE4 inhibition
In a pooled analysis, 1,493 patients receiving either placebo (n=495), apremilast 20 mg BID (n=501) or apremilast 30 mg BID (n=497) in the PALACE 1, 2, and 3 clinical trials were included in a safety population to assess safety and tolerability of apremilast treatment.38 After 24 weeks, all patients receiving placebo were re-randomized to either 20 mg BID or 30 mg BID apremilast. The apremilast exposure in this pooled analysis included 720 patients exposed to apremilast 20 mg BID (766.4 patient years) and 721 patients exposed to apremilast 30 mg BID (769.0 patient years).38 The most common adverse events (AEs) in these studies were diarrhea (14.3%), nausea (12.6%), urinary tract infections (10.3%), headache (10.1%), and nasopharyngitis (7.4%) (Table 2).33 In the PALACE 1 clinical trial, most AEs occurred during the first 24 weeks of treatment, with most AEs (over 90%) being mild to moderate in severity and discontinuation rates because of AEs were under 10%.37 Diarrhea and nausea occurred most often during the first 2 weeks of treatment and usually resolved within 4 weeks despite continued treatment and without medical intervention.37 From week 0 to 52, 6.1% of patients in both treatment arms (apremilast 20 mg BID and apremilast 30 mg BID) reported severe AEs (SAEs) while 3.6% of patients treated with placebo reported an SAE from week 0 to 24. With the exception of myocardial infarction, which was reported in two patients receiving apremilast 20 mg BID, no individual SAE was reported more than once per treatment group.37
 | Table 2 Safety and tolerability of apremilast treatment – most common adverse events |
During the 52-week period there were no cases of lymphoma, de novo tuberculosis or tuberculosis reactivations reported, while there has been one squamous cell carcinoma of the skin identified in this period.37 One death occurred in the placebo-controlled phase in a 52-year old woman receiving apremilast 20 mg BID and methotrexate due to multi-organ failure secondary to pre-existing vitamin B12 deficiency and was not considered to be study drug related by the investigator.37
There were no clinically meaningful effects on laboratory measurements detected during the first 52 weeks of the trial with no marked abnormalities in leukocytes, neutrophils, or platelets and only in one or two patients have there been marked increases in ALT (n=2) or creatinine (n=1), or marked decreases in hemoglobin (n=2).37,38
Preclinical trials showed that PDE4B deficient mice were leaner, had lower fat pad weights, smaller adipocytes, and decreased serum leptin levels compared to wild type mice.39 In the PALACE 1 trial, weight decrease was reported as an AE in 1.6% of patients receiving apremilast 20 mg BID and 2.0% of patients receiving apremilast 30 mg BID during the apremilast exposure period.40 Weight loss, that was not considered an AE, was observed in a larger proportion of patients, as after 52 weeks weight loss greater than 5% was observed in 15.8% of patients receiving apremilast 20 mg BID and in 17.2% of patients receiving apremilast 30 mg BID. Patients treated with apremilast 30 mg BID had a mean weight loss of −1.79 kg after 52 weeks (0.91 kg with apremilast 20 mg BID treatment).37 There was no association between weight loss and gastrointestinal AEs such as diarrhea or nausea and vomiting.40
Besides anti-inflammatory effects, PDE4 inhibitors are known to mediate behavioral changes in the animal model.28 In the conscious ferret model, mild behavioral changes like flattened posture, lip licking, and backward walking were observed at doses under 10 mg/kg apremilast. Higher doses led to marked emesis along with pronounced behavioral changes in these animals.28 During the placebo-controlled phase of the clinical trials, 1.2% (14/1,184) of patients treated with apremilast compared to 0.5% (2/418) of patients treated with placebo reported depressive mood or depression. None of these depressions was classified as severe or led to discontinuation of the study.33
Patient focused perspectives
PsO and PsA go along with severe disease-related limitations in quality of life. PsO patients indicated itching, scales, and flaking as most bothersome, while joint pain was reported by 89% of patients with PsA.41 Patients with more than four affected joints answered “much difficulty” or “unable to do” for many daily life tasks like bending down to pick up clothing from the floor (26%), walking outdoors on flat ground (18%), dressing themselves (15%), getting in and out of bed or the car (15%), washing and drying their body (12%), turning faucets on and off (8%), and lifting a full cup or glass to their mouth (7%).41
In the PALACE 1 clinical trial, at week 16, apremilast treatment was associated with significantly greater reductions (improvements) in the HAQ-Disability Index (HAQ-DI) compared with placebo.32 The HAQ-DI scores were maintained over 52 weeks with mean reductions in HAQ-DI score of −0.37 with apremilast 20 mg BID and −0.32 with apremilast 30 mg BID.37 A significant improvement was also measured in physical functions by the 36-Item Short-Form Health Survey v2 Physical Function domain, in health-related quality of life (SF-36v physical component summary and Functional Assessment of Chronic Illness Therapy-Fatigue) and in patient assessment of pain.33,37
With pruritus being one of the major problems of PsO, apremilast treatment led to a reduction of −31.5 points (ESTEEM 1) and −33.5 points (ESTEEM 2) in the visual analog scale after 16 weeks compared to an improvement of −7.3 points (ESTEEM 1) and −12.2 points (ESTEEM 2) in the visual analog scale when treated with placebo.33
In the Dermatology Life Quality Index score where 0 is the best and 30 is the worst value of daily life quality, apremilast treatment of 30 mg BID showed a mean improvement of −6.6 points (ESTEEM 1) and −6.7 points (ESTEEM 2) compared to −2.1 points (ESTEEM 1) and −2.8 points (ESTEEM 2) upon placebo treatment. These improvements could be maintained until week 52.33
There were also statistically significant improvements seen in the 36-Item Short-Form Health Survey v2 mental component score and in “Work Limitations Questionnaire (WLQ-25) Index” upon apremilast treatment compared to placebo.33
Apremilast’s position in PsO therapy
Apremilast is an orally available PDE4 inhibitor with good efficacy in PsA and PsO. The safety profile of apremilast within a treatment period of 52 weeks seems to be favorable with no increased rates of laboratory abnormalities, severe infections or malignancies. Rare AEs reported in some of the patients are weight loss and depression.33 These side effects seem to be compound-specific. Weight loss should be closely monitored in patients treated with apremilast, underweight patients should only be treated with caution. However, weight loss may be a beneficial side effect in selected patients with adiposity, which is a strong risk factor for incident PsO and obesity-related disorders like diabetes mellitus type 2, hyperlipidemia, hypertension, and ischemic heart disease.42 However, for a concluding statement on apremilast safety profile additional data from long-term extension phases of clinical trials, Phase IV studies, and registry data are needed. The manufacturer does not recommend performing pre-examinations for tuberculosis, or laboratory monitoring during therapy with apremilast.33 However, it is up to the dermatologist and the rheumatologist to evaluate the need for laboratory testing. Current therapy guidelines recommend PsA treatment with NSAIDs, followed by methotrexate treatment or other orally available DMARDs like leflunomide or sulfasalazine when clinical progression is observed. If these DMARDs are contraindicated, show toxicity or lack of efficacy, second line therapy with biological agents like etanercept or other TNF-blocking drugs, with or without DMARD co-medication is indicated.18 In patients with PsA, apremilast could find its place early in the treatment ladder due to its favorable safety profile and its clinical response to signs and symptoms of PsA.
Compared to biological agents, apremilast seems not to increase the risk of infections and malignancies and has the advantage of an oral route of application, but head to head studies are helpful to evaluate its clinical efficacy in PsA. Moreover, efficacy and safety trials of combination therapies of apremilast and biological DMARDs are needed, since this approach may be of benefit in difficult to treat patients or in patients with AEs or contraindications to methotrexate.
With approximately one out of three patients reaching a PASI-75 response and one out of ten (9.5%) reaching a PASI-90 response after 16 weeks of treatment, apremilast will probably stay a second line drug for PsA patients with major skin involvement. With newly introduced biological agents like secukinumab, an IL-17A antibody, approximately 70%–75% of patients achieve a PASI-90 response after 16 weeks of treatment.43 Other second generation biologicals targeting the Th17 axis like antibodies neutralizing IL-23p19 are under clinical investigation.
Taken together, apremilast is a modern anti-psoriatic oral compound with a favorable safety profile, which primarily will be used in patients with PsO and PsA who do not respond adequately to methotrexate or other oral anti-psoriatics.
Disclosure
Stephan Forchhammer has been an investigator for Almirall, Biogen Idec, Celgene, Delenex Therapeutics, Eli Lilly and Company, Galderma SA, Janssen-Cilag, MSD Sharp and Dohme, Novartis Pharmaceuticals, and Pfizer. Kamran Ghoreschi has been a consultant, lecturer or investigator for Abbvie, Almirall, Biogen Idec, Celgene, Delenex Therapeutics, Eli Lilly and Company, Galderma SA, Janssen-Cilag, MSD Sharp and Dohme, Novartis Pharmaceuticals, Pfizer, and the Schering-Plough Research Institute and received research grants from Fumapharm AG (now Biogen Idec) and Pfizer.
References
Kurd SK, Gelfand JM. The prevalence of previously diagnosed and undiagnosed psoriasis in US adults: results from NHANES 2003–2004. J Am Acad Dermatol. 2009;60(2):218–224. | |
Parisi R, Symmons DP, Griffiths CE, Ashcroft DM; Identification and Management of Psoriasis and Associated ComorbidiTy (IMPACT) project team. Global epidemiology of psoriasis: a systematic review of incidence and prevalence. J Invest Dermatol. 2013;133(2):377–385. | |
Gladman DD. Psoriatic arthritis from Wright’s era until today. J Rheumatol Suppl. 2009;83:4–8. | |
Reich K, Krüger K, Mössner R, Augustin M. Epidemiology and clinical pattern of psoriatic arthritis in Germany: a prospective interdisciplinary epidemiological study of 1511 patients with plaque-type psoriasis. Br J Dermatol. 2009;160(5):1040–1047. | |
Boehncke WH, Schön MP. Psoriasis. Lancet. Epub 2015 May 26. | |
Ghoreschi K, Weigert C, Röcken M. Immunopathogenesis and role of T cells in psoriasis. Clin Dermatol. 2007;25(6):574–580. | |
Ghoreschi K, Laurence A, Yang X, Hirahara K, O’Shea JJ. T helper 17 cell heterogeneity and pathogenicity in autoimmune disease. Trends Immunol. 2011;32(9):395–401. | |
Ghoreschi K, Laurence A, Yang X, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-b signalling. Nature. 2010; 467(7318):967–971. | |
Belge K, Brück J, Ghoreschi K. Advances in treating psoriasis. F1000Prime Rep. 2014;6:4. | |
Baliwag J, Barnes DH, Johnston A. Cytokines in psoriasis. Cytokine. 2015;73(2):342–350. | |
Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009;361(5):496–509. | |
Leipe J, Grunke M, Dechant C, et al. Role of Th17 cells in human autoimmune arthritis. Arthritis Rheum. 2010;62(10):2876–2885. | |
Gladman DD. Clinical aspects of the spondyloarthropathies. Am J Med Sci. 1998;316(4):234–238. | |
Husted JA, Gladman DD, Farewell VT, Cook RJ. Health-related quality of life of patients with psoriatic arthritis: a comparison with patients with rheumatoid arthritis. Arthritis Rheum. 2001;45(2):151–158. | |
Husted JA, Tom BD, Farewell VT, Schentag CT, Gladman DD. A longitudinal study of the effect of disease activity and clinical damage on physical function over the course of psoriatic arthritis: Does the effect change over time? Arthritis Rheum. 2007;56(3):840–849. | |
Gladman DD, Shuckett R, Russell ML, Thorne JC, Schachter RK. Psoriatic arthritis (PSA) – an analysis of 220 patients. Q J Med. 1987; 62(238):127–141. | |
Huynh D, Kavanaugh A. Psoriatic arthritis: current therapy and future approaches. Rheumatology (Oxford). 2015;54(1):20–28. | |
Gossec L, Smolen JS, Gaujoux-Viala C, et al. European League Against Rheumatism recommendations for the management of psoriatic arthritis with pharmacological therapies. European League Against Rheumatism. Ann Rheum Dis. 2012;71(1):4–12. | |
Ghoreschi K, Gadina M. Jakpot! New small molecules in autoimmune and inflammatory diseases. Exp Dermatol. 2014;23(1):7–11. | |
Schafer PH, Parton A, Gandhi AK, et al. Apremilast, a cAMP phosphodiesterase-4 inhibitor, demonstrates anti-inflammatory activity in vitro and in a model of psoriasis. Br J Pharmacol. 2010;159(4):842–855. | |
de Waal Malefyt R, Abrams J, Bennett B, Figdor CG, de Vries JE. Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med. 1991;174(5):1209–1220. | |
Tilg H, Trehu E, Atkins MB, Dinarello CA, Mier JW. Interleukin-6 (IL-6) as an anti-inflammatory cytokine: induction of circulating IL-1 receptor antagonist and soluble tumor necrosis factor receptor p55. Blood. 1994;83(1):113–118. | |
Serezani CH, Ballinger MN, Aronoff DM, Peters-Golden M. Cyclic AMP: master regulator of innate immune cell function. Am J Respir Cell Mol Biol. 2008;39(2):127–132. | |
Zambon AC, Zhang L, Minovitsky S, et al. Gene expression patterns define key transcriptional events in cell-cycle regulation by cAMP and protein kinase A. Proc Natl Acad Sci U S A. 2005;102(24):8561–8566. | |
Ollivier V, Parry GC, Cobb RR, de Prost D, Mackman N. Elevated cyclic AMP inhibits NF-kappaB-mediated transcription in human monocytic cells and endothelial cells. J Biol Chem. 1996;271(34):20828–20835. | |
Francis SH, Blount MA, Corbin JD. Mammalian cyclic nucleotide phosphodiesterases: molecular mechanisms and physiological functions. Physiol Rev. 2011;91(2):651–690. | |
Houslay MD, Schafer P, Zhang KY. Keynote review: phosphodiesterase-4 as a therapeutic target. Drug Discov Today. 2005;10(22):1503–1519. | |
Schafer PH, Parton A, Capone L, et al. Apremilast is a selective PDE4 inhibitor with regulatory effects on innate immunity. Cell Signal. 2014; 26(9):2016–2029. | |
Molostvov G, Morris A, Rose P, Basu S, Muller G. The effects of selective cytokine inhibitory drugs (CC-10004 and CC-1088) on VEGF and IL-6 expression and apoptosis in myeloma and endothelial cell co-cultures. Br J Haematol. 2004;124(3):366–375. | |
Ito T, Ando H, Suzuki T, et al. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327(5971):1345–1350. | |
McCann FE, Palfreeman AC, Andrews M, et al. Apremilast, a novel PDE4 inhibitor, inhibits spontaneous production of tumour necrosis factor-alpha from human rheumatoid synovial cells and ameliorates experimental arthritis. Arthritis Res Ther. 2010;12(3):R107. | |
Kavanaugh A, Mease PJ, Gomez-Reino JJ, et al. Treatment of psoriatic arthritis in a phase 3 randomised, placebo-controlled trial with apremilast, an oral phosphodiesterase 4 inhibitor. Ann Rheum Dis. 2014;73(6):1020–1026. | |
Otezla® (apremilast) tablets for oral use [prescribing information]. Summit, NJ: Celgene Corporation; 2014. Available from http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/003746/WC500182627.pdf. Accessed August 26, 2015. | |
Celgene.com [homepage on the Internet]. Positive Results from Phase III Study Evaluating Oral OTEZLA® (Apremilast) or Injectable Etanercept versus Placebo in Patients with Moderate to Severe Plaque Psoriasis Presented at AAD. Celgene Corporation; 2015 [updated Mar 20, 2015; cited May 17, 2015]. Available from: http://ir.celgene.com/releasedetail.cfm?releaseid=902701. Accessed May 17, 2015. | |
Cutolo M, Myerson GE, Fleischmann RM, et al. Long-Term (52-Week) Results Of a Phase 3, Randomized, Controlled Trial Of Apremilast, An Oral Phosphodiesterase 4 Inhibitor, In Patients With Psoriatic Arthritis (PALACE 2). [abstract]. Arthritis Rheum. 2013;65(Suppl 10):815. | |
Edwards CJ, Blanco FJ, Crowley J, et al. Long-term 52-week results of PALACE 3, a Phase III, randomized, controlled trial of apremilast, an oral phosphodiesterase 4 inhibitor, in patients with psoriatic arthritis and current skin involvement [abstract 212]. Rheumatology. 2014;53 Suppl 1:i138. | |
Kavanaugh A, Mease PJ, Gomez-Reino JJ, et al. Longterm (52-week) results of a phase III randomized, controlled trial of apremilast in patients with psoriatic arthritis. J Rheumatol. 2015;42(3):479–488. | |
Mease PJ, Gladman DD, Kavanaugh A, et al. Long-Term Safety and Tolerability Of Apremilast, An Oral Phosphodiesterase 4 Inhibitor, In Patients With Psoriatic Arthritis: Pooled Safety Analysis Of Three Phase 3, Randomized, Controlled Trials. [abstract]. Arthritis Rheum. 2013;65(Suppl 10):310. | |
Zhang R, Maratos-Flier E, Flier JS. Reduced adiposity and high-fat diet-induced adipose inflammation in mice deficient for phosphodi-esterase 4B. Endocrinology. 2009;150(7):3076–3082. | |
Mease PJ, Gladman DD, Kavanaugh A, et al. Change in weight from baseline during the PALACE clinical trial program with apremilast, an oral phosphodiesterase 4 inhibitor: Pooled results from 3 phase 3, randomized, controlled trials. [abstract]. Ann Rheum Dis. 2014;73 (Suppl 2):1055. | |
Lebwohl MG, Bachelez H, Barker J, et al. Patient perspectives in the management of psoriasis: results from the population-based Multinational Assessment of Psoriasis and Psoriatic Arthritis Survey. J Am Acad Dermatol. 2014;70(5):871–881. | |
Setty AR, Curhan G, Choi HK. Obesity, waist circumference, weight change, and the risk of psoriasis in women: Nurses’ Health Study II. Arch Intern Med. 2007;167(15):1670–1675. | |
Richard G, Langley MD, Boni E, et al. Secukinumab in Plaque Psoriasis–Results of Two Phase 3 Trials. N Engl J Med. 2014;371(4):326–338. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.