")
Back to Journals » Adolescent Health, Medicine and Therapeutics » Volume 8
Update on the management of systemic juvenile idiopathic arthritis and role of IL-1 and IL-6 inhibition
Received 5 April 2017
Accepted for publication 2 September 2017
Published 9 November 2017 Volume 2017:8 Pages 125—135
DOI https://doi.org/10.2147/AHMT.S109495
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Alastair Sutcliffe
Sriharsha Grevich, Susan Shenoi
Department of Rheumatology, Seattle Children’s Hospital, Seattle, WA, USA
Abstract: Systemic juvenile idiopathic arthritis (SJIA) is a disease marked with arthritis and several features of systemic inflammation including fevers, rashes, hepatosplenomegaly, lymphadenopathy, and serositis. The presentation can be variable and arthritis can be a later feature. Macrophage activation syndrome can be a life-threatening complication of this illness and requires early recognition and prompt therapy. Advancements in understanding the biology of SJIA have led to the development of cytokine-targeted therapies, mainly interleukin-1 (IL-1) and IL-6 inhibitors that have significantly improved outcomes. In this review, we provide an update on the advances in the understanding of SJIA biology and also the therapeutic options.
Keywords: systemic juvenile idiopathic arthritis, IL-1 inhibitors, IL-6 inhibitors, anakinra, tocilizumab, canakinumab, macrophage activation syndrome
Epidemiology and pathogenesis
Juvenile idiopathic arthritis (JIA) is a childhood (onset <16 years) chronic inflammatory disease of unclear etiology. International League of Associations for Rheumatology (ILAR) classifies JIA into seven different categories based on clinical presentation during the first 6 months of disease.1 Systemic juvenile idiopathic arthritis (SJIA) is uniquely characterized with a range of extra-articular symptoms and accounts for roughly 10% of JIA in North America and Europe and up to 25%–50% in Asian countries.2–4 There is no gender predominance in SJIA, and the disease can occur at any time during childhood with a peak age of onset between ages 1 and 5 years. The term adult-onset Still’s disease is used for those who develop the disease after 16 years and the incidence reported in a French study is 0.16 cases per 100,000 people.5
The underlying etiology for SJIA is thought to be multigenic, and both environmental and genetic factors are implicated. Unlike other categories of JIA, SJIA is thought to be an autoinflammatory disease and lacks characteristic autoantibodies, and dysregulated innate immunity is thought to play an important role.6 Given the increased incidence of SJIA compared to Still’s disease, infections encountered in childhood have been postulated as a possible trigger for SJIA, though no specific pathogens have been identified. Some studies also point to possible seasonal variation in disease onset, while this has been refuted in other studies.6,7 Natural killer cell dysfunction and increase in number of innate immune cells such as monocytes and neutrophils during SJIA flare have been reported.8,9 Several polymorphisms in promoter elements and genes coding for cytokines of the innate immune response have been reported.6,7 These include SNPs in both the interleukin-1 (IL-1) ligand and receptor cluster regions,10 IL-6 gene,11,12 tumor necrosis factor (TNF) α gene,13 IL-10 promoter polymorphisms,14,15 and polymorphism of the proinflammatory macrophage migration inhibitory factor (MIF) gene.16,17 In a Turkish study, MEFV gene mutations associated with familial Mediterranean fever were found in 14% of the 35 SJIA patients who were studied. Mutations in this gene cause activation of the IL-1β pathway and chronic inflammation.18
Though in the past the relationship between major histocompatibility complex (MHC) and SJIA has been uncertain, a study in 2015 identified HLA-DRB1*11 and variants of the MHC class II locus as risk factors for SJIA, thus implicating a role of adaptive immune system in the pathogenesis of SJIA.19 A genome-wide association study by the same group confirmed the MHC locus on chromosome 6 as a risk loci for SJIA and also identified a new risk locus on the short arm of chromosome 1 (1p36.32). In addition, the study found 23 other novel loci suggestive of association with SJIA and found no evidence of shared genetic risk factors between SJIA and other JIA categories.20
Clinical manifestations
Increased levels of several inflammatory cytokines and defective inhibitory responses likely explain the unique systemic inflammatory features of SJIA.7 ILAR classification, criteria for SJIA are shown in Table 1.
 | Table 1 ILAR SJIA classification criteria Note: Data from Petty et al.1 Abbreviations: ILAR, International League of Associations for Rheumatology; SJIA, systemic juvenile idiopathic arthritis; IgM, immunoglobulin M. |
It is important to note that these criteria were developed to identify clinically homogenous patients for research and not diagnostic purposes. Out of 528 patients with SJIA enrolled in the childhood arthritis and rheumatology research alliance (CARRA) registry between 2010 and 2013, 14.5% of patients did not meet the ILAR criteria, though the enrolling physician considered them to have SJIA.21 A Pennsylvania-based SJIA registry found that only 42 (30%) of the 136 patients diagnosed as having SJIA by the treating physician met the ILAR criteria. Of the remaining 94 patients, 51% did not meet the ILAR fever criteria though all of them had intermittent fever documented. This study highlights the heterogeneity of clinical presentation at disease onset. About 98% of patients had fever at presentation, but there was a significant variability in fever pattern, such as daily morning or evening fevers, twice daily fevers, intermittent fevers, and continuous fevers. About 88% of patients had arthritis, out of which 47% was polyarticular, 45% oligoarticular, and 8% monoarticular. About 81% had rash, 31% had lymphadenopathy, 10% had serositis (pericarditis), and even fewer patients had organomegaly.22
In addition to the clinical symptoms noted in ILAR criteria, children will often present with generalized malaise, myalgias, arthralgias, abdominal pain, and weight loss. Characteristic fever pattern in SJIA tends to be a high grade fever (39°C or higher) with a quotidian pattern (daily spikes with return to baseline or subnormal temperature). Fever can occur in conjunction with chills and is often accompanied by a characteristic “salmon-pink” colored or erythematous macular rash found commonly on the trunk and extremities. The rash is usually migratory/evanescent and shows a Koebner response. Histologically, analysis of the cutaneous rash in SJIA shows perivascular infiltration with neutrophils and monocytes and epithelial activation with the expression of myeloid related protein -8 (MRP-8) and MRP-14.23
Arthritis in SJIA most commonly involves knees, wrists, and ankles; however, arthritis of small joints of the hand and foot, elbow, shoulder, hip, temporomandibular joint, and cervical spine can also be seen.22 Bicipital synovial cysts can present as acute arm swelling in SJIA.7,24 Like other forms of JIA, synovial fluid can show neutrophil predominant leukocytosis. Radiographically soft tissue swelling and osteoporosis are common in SJIA, and subchondral irregularity and sclerosis followed by erosions can be seen within 2 years of disease onset. Later findings can include joint ankyloses, subluxation, and protrusio acetabuli.25
Reticuloendothelial system is frequently involved in SJIA with non-tender mobile lymphadenopathy caused by follicular hyperplasia. Rubbery or matted lymphadenopathy should raise concern for diagnoses such as lymphoma.26 Organomegaly such as splenomegaly and/or hepatomegaly is less common than lymphadenopathy.22
Pericardial effusions and pericarditis are more common than myocarditis in SJIA. Cardiac tamponade, coronary artery abnormalities, and valvular disease have also been reported in SJIA. In an international study cohort of 25 patients with severe SJIA and pulmonary manifestations identified by Kimura et al, 64% of patients had pulmonary arterial hypertension, 28% had interstitial lung disease, and 20% had alveolar proteinosis or lipoid pneumonia.27 Compared to other forms of JIA, uveitis is quite rare in SJIA, occurring in <1% of patients.28
Patients with SJIA can follow variable disease courses. Monocyclic disease course is defined as a single episode of illness followed by clinical remission, whereas polycyclic course can present with episodes of active disease with intermittent periods of quiescence. Persistent disease course is often associated with progressive arthritis with or without systemic symptoms and results in significant morbidity. While the proportion of patients reported to follow each specific course has varied between studies (monocyclic 11%–40%, polycyclic 2.3%–34%, persistent 51%–66%), more than half the patients seem to follow the persistent disease course.29–31 Active disease at 6 months with arthritis, corticosteroid use, and, an elevated erythrocyte sedimentation rate (ESR) was predictive of a persistent disease course with longer times to achieve remission.29 Patients with early onset SJIA, defined as disease onset before 18 months were found to have a more aggressive disease with more common serositis, organomegaly, macrophage activation syndrome (MAS), growth failure, and destructive arthritis.32 Other predictors of destructive arthritis include persistent systemic symptoms, presence of polyarticular arthritis, hip arthritis, thrombocytosis, and corticosteroid use.33–35
SJIA is a diagnosis of exclusion, and since many of the presenting symptoms are non-specific, other etiologies such as malignancy, infectious causes, and other autoinflammatory conditions must be considered in the differential. SJIA is a clinical diagnosis made with supporting laboratory findings, and it is important to recognize as noted above that patients may not meet the ILAR classification criteria. Given that some patients may present with several systemic clinical features of SJIA but never develop arthritis, some have proposed renaming the diagnosis, possibly to Still’s disease as arthritis is not required for the diagnosis of adult-onset Still’s disease.36 With advancements in understanding of the pathogenesis of JIA, it is inevitable that the nomenclature and categorization may evolve in the future.37
Laboratory findings in SJIA are notable for non-specific markers of inflammation such as elevated C-reactive protein (CRP), ESR, white blood count (WBC), and platelet count, and, low hemoglobin and albumin.38 While not available in clinical practice, research has demonstrated that certain biomarkers may have a role in distinguishing SJIA from other non-SJIA causes of fever. S100 proteins including S100A12 secreted by activated phagocytes, and pro-inflammatory MRP-8 (S100A8) and MRP-14 (S100A9) have been reported to be higher in SJIA patients, especially with active disease compared to those with infection and malignancy.39–42
MAS
Though the exact incidence is unknown, about 10% of SJIA will develop MAS, a potentially life-threatening complication, and many more likely have subclinical MAS.43 The impaired immune response in MAS is similar to that seen in familial (primary) and secondary forms of hemophagocytic lymphohistiocytosis (HLH). In HLH, defective cytolytic pathways lead to the expansion of cytotoxic cells and hypersecretion of inflammatory cytokines.44 Similarly, MAS is characterized by the expansion of hemophagocytic T lymphocytes and macrophages leading to cytokine storm and ultimately multiorgan damage.45 Genetic variations seen in primary HLH that cause natural killer cell dysfunction and disrupt cytolysis such as mutations in genes UNC13D, PRF1 STXBP2, and RAB27 A have been reported in some cases of MAS.46–48 Defective interferon-γ, toll-like receptor 9 (TLR-9), IL-18, and IL-10 pathways have also been implicated in MAS.44
In an international multicenter study of 362 patients with SJIA-associated MAS, MAS was noted in 22% at SJIA onset. Clinical manifestations of MAS included fever (96%, often persistent rather than quotidian patterns), hepatomegaly (70%), splenomegaly (58%), and lymphadenopathy (51%). Central nervous system dysfunction ranging from seizures, lethargy, irritability, confusion, headache, and coma, occurred in 35%. Cardiac involvement occurred in 26% of patients with pericardial involvement being the most common. Pulmonary involvement with pleural effusions occurred in 22% of patients. Hemorrhagic symptoms ranged from petechiae, purpura, mucosal, or gastrointestinal bleeding in 20% and disseminated intravascular coagulation in 1%. Renal involvement ranging from renal failure, hematuria, and proteinuria was seen in 15%. It is important to note that MAS was present in almost 40% of patients who did not demonstrate tissue hemophagocytosis.49 Laboratory features of MAS include a drop in ESR, WBC, platelet counts, and fibrinogen levels with rising and extremely elevated ferritin levels, elevated liver enzymes, lactate dehydrogenase, triglycerides, D-dimer, and prolonged prothrombin time, and partial thromboplastin time. Soluble IL-2 receptor and soluble CD163 are also elevated in active MAS.38
In 2016, classification criteria for MAS complicating SJIA were established by an expert panel at a consensus conference with a sensitivity of 0.73 and a specificity of 0.99 in preliminary validation analysis (Table 2).50 It is important for the clinician to recognize that MAS is an evolving process and that the patient may not meet all the criteria at onset. The clinician should trend laboratory results closely to recognize MAS promptly so that treatment can be initiated in a timely manner. For example, the only abnormal laboratory parameters may be a trend of dropping platelets, but this change in the setting of rising hepatic enzymes should alert the physician to be more vigilant about MAS.
 | Table 2 MAS in SJIA classification criteria Note: Data from Ravelli et al.50 Abbreviations: MAS, macrophage activation syndrome; SJIA, systemic juvenile idiopathic arthritis. |
Treatment
Goals of treatment in SJIA are to provide symptomatic relief, control the underlying inflammatory process, and achieve and maintain sustained disease remission. To achieve tight disease control and to improve disease outcomes, there is a suggestion now to incorporate a treat-to-target approach in the management of JIA.51 The already defined descriptions for various disease states including inactive disease, minimal disease activity, or parent- or child-acceptable symptom states can be used in guiding the treat-to-target approach.51–53 Clinically inactive disease is defined as having no joints with active arthritis, no systemic symptoms attributable to JIA including fever, rash, serositis, splenomegaly, or generalized lymphadenopathy, no active uveitis, no elevation in ESR or CRP attributable to JIA, physician’s global assessment of disease activity of zero, and <15 minutes of morning stiffness.54
Based on the advancements in understanding of SJIA pathogenesis, especially the role of IL-1 and IL-6, biologic therapies targeting these specific cytokines have been developed in recent years. Of these biologics, anti-IL-1 agent canakinumab and anti-IL-6 agent tocilizumab are US food and drug administration (FDA) approved for SJIA in children ≥2 years, while anti-IL-1 agents anakinra and rilonacept are not FDA approved. Table 3 summarizes the available therapeutic options for SJIA.
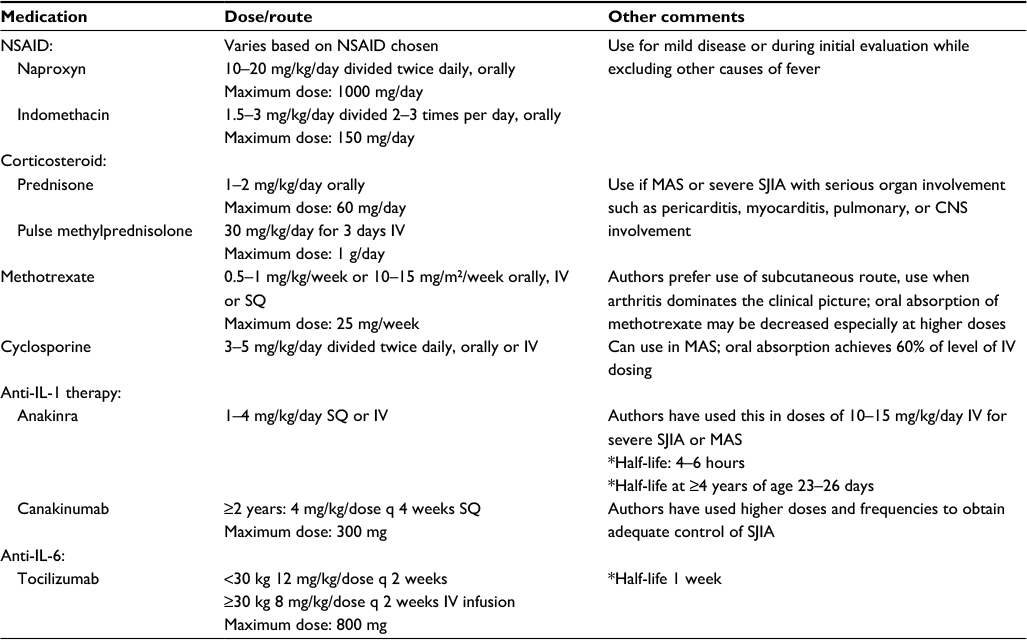 | Table 3 Therapeutic agents commonly used for SJIA treatment Notes: *Source package insert. Reprinted from The Journal of Pediatrics, 177, Shenoi S, Wallace CA, Diagnosis and treatment of systemic juvenile idiopathic arthritis, 19–26, Copyright © 2016, with permission from Elsevier.57 Abbreviations: NSAID, nonsteroidal anti-inflammatory drug; SQ, subcutaneous; IV, intravenous; CNS, central nervous system; MAS, macrophage activation syndrome; q, every; SJIA, systemic juvenile idiopathic arthritis. |
Different cytokine clusters were identified in SJIA compared to oligo and polyarticular JIA categories using multiplex immunoassays and cluster analysis, suggesting that the immune mechanisms in SJIA are different from other categories of JIA.55 The differences in pathogenesis seen in SJIA contribute to the difference in clinical responses to various immunosuppressive agents compared to other categories of JIA. While nonsteroidal anti-inflammatory agents (NSAIDs) can provide some symptomatic relief as antipyretics and analgesics, they usually do not fully control arthritis or systemic symptoms. Oral or intravenous glucocorticoids are sometimes used as first-line therapy depending on severity; however, given the significant side effect profile of steroids, they are not ideal for long-term management. While methotrexate is a commonly used disease-modifying antirheumatic drug (DMARD) in the treatment of JIA, a randomized, placebo-controlled trial using low-dose methotrexate (15–20 mg/m2) did not demonstrate significant improvement in arthritis or systemic features in SJIA compared to those with extended oligo-articular arthritis.56
Treatment with TNF inhibitors such as etanercept, adalimumab, and infliximab is considered to be less effective in SJIA compared to other JIA categories.57 An Argentinian study of 45 SJIA patients on anti-TNF therapy (etanercept, infliximab, and adalimumab) demonstrated remission in 24% after an average of 26 months of therapy, but half of these patients flared within a year.58 In a US survey-based study of 82 patients with refractory SJIA (refractory to NSAIDs, steroids, and methotrexate) treated with etanercept, one third of the patients showed an excellent response, but more than half the patients had fair or poor response and disease flares were common.59 Abatacept, a fusion protein consisting of CTLA-4 linked to Fc portion of human IgG1, is a selective T-cell activation inhibitor and is FDA approved for polyarticular course JIA. Thirty-seven patients with SJIA without systemic features were included in a randomized, placebo-controlled withdrawal trial of abatacept with inadequate response to at least one DMARD. At the end of the 4-month open-label treatment with abatacept, 65% of the 37 SJIA patients showed American College of Rheumatology (ACR) 30 response. About 16% of the 32 patients included in the open-label extension of up to 1.5 years showed an ACR 90 response.60,61 Therapies that have been tried for SJIA with treatment failure to commonly used biologics include cyclophosphamide, rituximab, thalidomide, tacrolimus, plasmapheresis, and autologous stem cell transplantation.57
The ACR recommendations for SJIA treatment were updated in 2013 to incorporate the anti-IL-1 and anti-IL-6 biologic therapies.62 They categorized SJIA treatment recommendations based on three different SJIA phenotypes. For SJIA patients with active systemic features and varying degrees of synovitis, with a physician global assessment score (medical doctor [MD] global) of 5 irrespective of active joint count (AJC), they recommend initial therapy with anakinra or glucocorticoids. However, glucocorticoid monotherapy should not exceed more than 2 weeks, if children have continued disease activity with MD global score over 5 and a biologic agent such as anakinra, tocilizumab, or canakinumab should be added. NSAID monotherapy can be considered in children with MD global of <5; however, if they continue to have active disease after 1 month, additional therapy is warranted. If patients continue to have active disease after 1 month of anakinra, they recommend changing to other biologic agent if systemic features persist or considering DMARD agents such as methotrexate or leflunomide if they have an AJC >0 and improvement in systemic features. TNFα inhibitor was recommended for patients with AJC >4 after a trial of an IL-1 or IL- 6. Abatacept (CTLA4-Ig) and calcineurin inhibitors can be considered in patients with persistent disease activity after trialing IL-1 and IL-6 inhibitors. Rilonacept was considered an inappropriate initial therapy for children with systemic features, and its use as a second-line agent is uncertain.
For children with active synovitis without active systemic features, initial therapy with methotrexate or leflunomide is recommended if the AJC >4, intraarticular glucocorticoid injection if AJC ≤4 and NSAID monotherapy can be trialed for a maximum of 1 month for AJC ≤4. Anakinra can be used in children with persistent synovitis that fail intraarticular injection, NSAID monotherapy, or methotrexate or leflunomide therapy based on the AJC. Abatacept, TNFα inhibitor, or tocilizumab can be considered in children with active synovitis that fail methotrexate, leflunomide, or anakinra. Initiation of therapy with rilonacept was considered uncertain regardless of the joint count.
For children with SJIA with features of, MAS they note that the recommended treatment options are not mutually exclusive and clinical situation may warrant the initiation of multiple medications at the same time. Initial therapeutic options for these children may include anakinra, calcineurin inhibitor, and/or systemic glucocorticoid therapy; however, glucocorticoid monotherapy should not be continued for >2 weeks if MAS features persist.62 Given that MAS can be life threatening, it is important to recognize and treat promptly to prevent multisystem organ failure and death. There are no controlled studies on MAS treatment, and management of this condition is based on anecdotal experience. Typically in clinical practice anakinra is commonly used given its quick onset of action and short half-life. High-dose intravenous methylprednisolone and cyclosporine are often used in conjunction with anakinra based on the severity of presentation. In-depth discussion on MAS treatment is outside the scope of this article, and readers are encouraged to refer to specific literature for a detailed review.45,63
IL-1 inhibitors
IL-1 refers to the two pro-inflammatory cytokines IL-1α and IL-1β, which mediate their downstream effects by binding to the IL-1 receptor (IL1R1). IL-1 receptor antagonist, IL-1Ra, is an endogenous IL-1 inhibitor that works as a competitive inhibitor for the binding of IL-1 to IL1R1.64 IL-1 plays a role in both the innate and adaptive immune mechanisms that contribute to the theorized biphasic model of SJIA. In the early phase, the innate immune activity of IL-1 is thought to cause symptoms such as fever, rash, and early synovitis. Eventually, IL-1 is thought to affect adaptive regulatory mechanisms by promoting T-cell differentiation into the pro-inflammatory TH17 cells and by inhibiting T-reg cell activity. Autoimmune T-cell mechanisms are thought to primarily drive the chronic arthritis of SJIA in this late phase.65
Anakinra
Anakinra is a recombinant IL-1 receptor antagonist effective in inhibiting both IL-1α and IL-1β binding to IL-R1.57 The first report of its effectiveness in both clinical and laboratory parameters in two patients with SJIA resistant to several immunosuppressant (corticosteroids, methotrexate, anti-TNFα agents, and cyclosporine), both with severe disease and one with MAS, was published in 2004.66 Pascual et al treated nine refractory SJIA patients with anakinra and obtained complete disease remission in seven patients.67 Anakinra may be less effective in treating arthritis compared to systemic features of SJIA.68 A randomized, placebo-controlled trial of anakinra, called the ANAJIS trial with 12 SJIA patients in each group, showed that ACR pediatric 30 response was achieved by eight out of 12 patients in the anakinra group versus only one patient in the placebo group at 1 month. Interestingly, by 12 months, there was a lack of sustained response in several patients, and this was thought to be due to enrollment of patients with diffuse polyarthritis as the main feature, for which anakinra is reported to be less effective; using lower dose of anakinra in the study (2 mg/kg subcutaneous injection daily with a maximum dose of 100 mg) and enrolling refractory SJIA patients in the study.69
Early treatment with IL-1 blockade may take advantage of a “window of opportunity” and alter the course of disease to prevent the chronic destructive arthritis seen in the later phase of SJIA. In patients who received anakinra as part of their initial therapy (N=46, 10 patients on anakinra monotherapy, while other received corticosteroids and/or other DMARDs in addition to anakinra), 60% achieved a complete response and at 6-month follow-up 89% were arthritis free.70 Vastert et al studied 20 new onset SJIA patients using anakinra as the initial therapy and showed that by 3 months, 85% achieved an ACR pediatric 90 response with 15/20 patients responding to anakinra monotherapy. Of these 15 patients, 13 had inactive disease by 1 year and 73% who achieved the ACR pediatric 90 response at 3 months were able to stop anakinra.71 What percent of these patients would have had monophasic SJIA regardless of anakinra therapy is unclear in both these studies. Reported side effects with anakinra include injection site pain and erythema, leukopenia, infections, and increase in serum transaminases.67,69,70
Treatment with anakinra has been shown to be effective in controlling SJIA-related MAS, even in setting of inadequate response to other immunosuppressive agents such as corticosteroids, cyclosporine, intravenous immunoglobulin, and etoposide.72–74 On the other hand, however, in the study by Nigrovic et al,70 five episodes of MAS occurred in four patients while receiving anakinra (1–2 mg/kg/day). One of the cases occurred when a patient abruptly discontinued corticosteroids and another in the setting of Epstein–Barr virus infection. Dose escalation of anakinra and in some cases addition of cyclosporine and corticosteroids seemed to help the episodes of MAS, and permanent discontinuation of anakinra was not necessary in any of the cases. They suggest that anakinra at a dose of 1–2 mg/kg/day may not always be sufficient to control MAS in SJIA.70
Canakinumab
Canakinumab is human monoclonal antibody that selectively binds IL-1β, thus preventing it from binding to the IL-1 receptor. In a Phase II multicenter dose-escalation study, 60% of 23 children with active SJIA achieved an ACR pediatric 50 response in 15 days of first canakinumab dose. Response was sustained by several patients, and steroids were tapered by at least 50% by 5 months. Similar to what was seen in studies with anakinra, responders seem to have fewer AJCs and higher white blood cell count at baseline. Common adverse events were mild to moderate in nature and included gastrointestinal symptoms such as abdominal pain, vomiting, diarrhea, and infections.75
Two randomized Phase III trials conducted by Ruperto et al76 also demonstrated efficacy of canakinumab in active SJIA. In trial 1, 36 of 43 (84%) of patients who received single dose of canakinumab (4 mg/kg) versus 4 of 41 (10%) who received placebo achieved an ACR pediatric 30 response by day 15. Fourteen patients in the canakinumab group were also considered to have clinically inactive disease at this point. Trial 2 was a two-part withdrawal design in which 177 patients were treated with canakinumab in the open-label phase (12–32 weeks) and those who had a response (at least ACR pediatric 30) and tolerated glucocorticoid tapering (or were not on glucocorticoids) were enrolled in the randomized, placebo-controlled withdrawal phase of the study. Patients who did not show a response, who could not tolerate the steroid wean, and who flared entered an open-label extension. Of the 128 patients on steroids, 45% were able to undergo tapering and 33% were able to discontinue steroids completely. A total of 100 patients were eligible for the withdrawal phase of the study, and within this group, 74% of patients who continued to receive canakinumab had no flare versus 25% in the placebo group, suggesting a relative risk reduction of 64% in regard to SJIA flares. After 37 patients flared, withdrawal phase was discontinued, and all patients were enrolled in the open-label extension. By the end of the withdrawal phase, 62% in canakinumab group had inactive disease versus 34% in the placebo group. There were seven cases of MAS, one occurring in canakinumab and placebo group each in trial 1, four cases in canakinumab group in trial 2, and one case in the placebo group in the withdrawal phase. Two of the seven cases with MAS resulted in death with one case having concurrent urosepsis and another with adenoviral gastroenteritis. Other reported adverse events in the study included non-opportunistic infections, neutropenia, thrombocytopenia, and aminotransferase elevation. It was concluded that the mortality rate in these two randomized trials was consistent with the mortality rate associated with SJIA.76 After the second MAS case in the above Phase III study, an MAS adjudication committee (MASAC) was convened to study canakinumab’s role in the development of MAS. They found that most of the canakinumab-treated patients had well-controlled SJIA at the time of MAS with a median of 292 days from canakinumab treatment to MAS. There was no significant difference of probable MAS events (predefined by criteria) between the placebo and canakinumab groups. They concluded that while MAS can occur in well-controlled SJIA patients, canakinumab does not contribute significantly to the MAS risk or its clinical features and found that infections were the most common trigger for MAS.77
Rilonacept
Rilonacept, also known as IL-1 trap, is a soluble decoy receptor fusion protein consisting of IL-1 receptor type 1 and IL-1 receptor accessory protein combined with the Fc portion of human IgG1. It blocks IL-1 signaling by binding to both cytokines (IL-1α and IL-1β).78 Rilonacept is not commonly used for the treatment of SJIA in clinical practice and a recent meta-analysis of randomized clinical trials of biologic agents in SJIA demonstrated better efficacy of canakinumab and tocilizumab compared to rilonacept.79
IL-6 inhibitor: tocilizumab
Serum IL-6 levels are elevated during active SJIA and correlate with the severity of joint involvement and thrombocytosis, indicating its role in pathogenesis of SJIA.80 Tocilizumab is a monoclonal humanized anti IL-6 receptor antibody. In a randomized, placebo-controlled, withdrawal Phase III trial in 56 SJIA patients, 91% of patients achieved ACR pediatric 30 response by the end of a 6-week open-label lead in phase. In the double-blind phase, 80% in the tocilizumab group versus 17% in the placebo group maintained an ACR pediatric 30 response and CRP <15 mg/L. Liver transaminases seemed to increase during the early phase of treatment with tocilizumab (first 3–6 months) and subside as treatment continued.81 A multicenter randomized trial of 112 active SJIA patients showed that 85% in the tocilizumab group versus 24% in the placebo group achieved the primary endpoint of ACR pediatric 30 score at the end of the 12-week double-blind phase. At the end of the 52 weeks, 59% of the patients had 90% improvement, 48% had no active arthritis, and 52% discontinued oral glucocorticoids. Adverse events included elevation of aminotransferase levels, infection, neutropenia, and three cases of MAS (one in setting of varicella infection).82
In a Japanese surveillance registry of SJIA patients treated with tocilizumab, three events of definitive MAS in three patients and 12 events of probable MAS in 11 patients were noted. In this study, 3.6% of 394 SJIA patients treated with tocilizumab developed MAS. Majority of the patients in the Japanese study resumed tocilizumab after improvement of MAS and the authors suggest that it is unlikely that tocilizumab induces MAS.83 While treating with tocilizumab, it is important to note that CRP production by the liver is IL-6 dependent and hence CRP cannot be used as a reliable indicator of inflammation associated with SJIA. It has been shown that treatment with tocilizumab can mask the symptoms of SJIA-associated MAS by having normal CRP levels and only minimal increase in ferritin levels.84 Reports of MAS in SJIA while undergoing treatment with IL-1 or IL-6 inhibitors as noted above suggest that perhaps single cytokine blockade may not be sufficient to prevent MAS and other cytokines may be playing a role.77
Outcomes and future direction
SJIA is a heterogeneous disease with variability in presenting symptoms and disease course. MAS remains the most significant cause of mortality in SJIA with a mortality rate of 8% with one third of the patients requiring intensive care.49 In a study of 962 cases with SJIA who were followed for a mean of 8 years, the standardized mortality ratio reported was 1.8; however, over the years, the outcomes have improved, likely attributable to the advances in therapies.85
With the number of treatment options now available for SJIA, there is a wide variability in treatment approaches among practitioners, and the ideal treatment approach is unknown and also likely dependent on the features and severity of each individual case of SJIA. To study the comparative effectiveness of the diverse therapeutic options in an observational setting, CARRA developed four standardized consensus treatment plans (CTPs) – glucocorticoid alone, methotrexate ± glucocorticoid, IL-1 inhibitor ± glucocorticoid, and IL-6 inhibitor ± glucocorticoid.86 A pilot study of 30 untreated SJIA patients from 13 different sites demonstrated the feasibility of using these four CTPs in a larger observational registry. In the pilot study, 37% of patients in the biologic group versus none in the nonbiologic group seemed to achieve the study outcome of clinically inactive disease off glucocorticoids at 9 months.87 Enrollment is currently underway for the First-line Options for SJIA Treatment (FROST) study designed to implement these CTPs at CARRA registry sites to assess their comparative effectiveness in clinical practice. Studies of newer therapies in SJIA such as sarilumab (monoclonal antibody against IL-6 receptor) and tofacitinib (inhibitor janus kinase 1 and 3) are in the pipeline (interested readers can refer to https://clinicaltrials.gov for further details). Future directions for research would include identifying both biomarkers for diagnosis of SJIA and prediction for flares of existing SJIA. There remains a subset of patients who seem to be refractory to current therapy, and continued advancements in understanding the pathogenesis of SJIA will hopefully help pave the way toward discovery of newer therapies targeted toward different cytokines/molecules.
Disclosure
Fellowship salary for Sriharsha Grevich is funded by Pfizer Medical Academic Partnerships grant. Susan Shenoi has served on speakers’ bureau and advisory board of Novartis. The authors report no other conflicts of interest in this work.
References
Petty RE, Southwood TR, Manners P, et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol. 2004;31(2):390–392. | ||
Sawhney S, Magalhaes CS. Paediatric rheumatology – a global perspective. Best Pract Res Clin Rheumatol. 2006;20(2):201–221. | ||
Seth V, Kabra SK, Semwal OP, Jain Y. Clinico-immunological profile in juvenile rheumatoid arthritis--an Indian experience. Indian J Pediatr. 1996;63(3):293–300. | ||
Fujikawa S, Okuni M. Clinical analysis of 570 cases with juvenile rheumatoid arthritis: results of a nationwide retrospective survey in Japan. Acta Paediatr Jpn. 1997;39(2):245–249. | ||
Magadur-Joly G, Billaud E, Barrier JH, et al. Epidemiology of adult Still’s disease: estimate of the incidence by a retrospective study in west France. Ann Rheum Dis. 1995;54(7):587–590. | ||
Mellins ED, Macaubas C, Grom AA. Pathogenesis of systemic juvenile idiopathic arthritis: some answers, more questions. Nat Rev Rheumatol. 2011;7(7):416–426. | ||
De Benedetti F, Schneider R. Systemic juvenile idiopathic arthritis. In: Petty R, Laxer R, Lindsley C, Wedderburn L, editors. Textbook of Pediatric Rheumatology. Philadelphia: Elsevier; 2016:205–216. | ||
Macaubas C, Nguyen K, Deshpande C, et al. Distribution of circulating cells in systemic juvenile idiopathic arthritis across disease activity states. Clin Immunol. 2010;134(2):206–216. | ||
Villanueva J, Lee S, Giannini EH, et al. Natural killer cell dysfunction is a distinguishing feature of systemic onset juvenile rheumatoid arthritis and macrophage activation syndrome. Arthritis Res Ther. 2005; 7(1):R30–R37. | ||
Stock CJ, Ogilvie EM, Samuel JM, Fife M, Lewis CM, Woo P. Comprehensive association study of genetic variants in the IL-1 gene family in systemic juvenile idiopathic arthritis. Genes Immun. 2008; 9(4):349–357. | ||
Fishman D, Faulds G, Jeffery R, et al. The effect of novel polymorphisms in the interleukin-6 (IL-6) gene on IL-6 transcription and plasma IL-6 levels, and an association with systemic-onset juvenile chronic arthritis. J Clin Invest. 1998;102(7):1369–1376. | ||
Ogilvie EM, Fife MS, Thompson SD, et al. The -174G allele of the interleukin-6 gene confers susceptibility to systemic arthritis in children: a multicenter study using simplex and multiplex juvenile idiopathic arthritis families. Arthritis Rheum. 2003;48(11):3202–3206. | ||
Date Y, Seki N, Kamizono S, et al. Identification of a genetic risk factor for systemic juvenile rheumatoid arthritis in the 5’-flanking region of the TNFalpha gene and HLA genes. Arthritis Rheum. 1999;42(12):2577–2582. | ||
Moller JC, Paul D, Ganser G, et al. IL10 promoter polymorphisms are associated with systemic onset juvenile idiopathic arthritis (SoJIA). Clin Exp Rheumatol. 2010;28(6):912–918. | ||
Fife MS, Gutierrez A, Ogilvie EM, et al. Novel IL10 gene family associations with systemic juvenile idiopathic arthritis. Arthritis Res Ther. 2006;8(5):R148. | ||
Donn RP, Shelley E, Ollier WE, Thomson W; British Paediatric Rheumatology Study Group. A novel 5’-flanking region polymorphism of macrophage migration inhibitory factor is associated with systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2001;44(8):1782–1785. | ||
De Benedetti F, Meazza C, Vivarelli M, et al. Functional and prognostic relevance of the -173 polymorphism of the macrophage migration inhibitory factor gene in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2003;48(5):1398–1407. | ||
Ayaz NA, Ozen S, Bilginer Y, et al. MEFV mutations in systemic onset juvenile idiopathic arthritis. Rheumatology (Oxford). 2009;48(1):23–25. | ||
Ombrello MJ, Remmers EF, Tachmazidou I, et al. HLA-DRB1*11 and variants of the MHC class II locus are strong risk factors for systemic juvenile idiopathic arthritis. Proc Natl Acad Sci U S A. 2015;112(52):15970–15975. | ||
Ombrello MJ, Arthur VL, Remmers EF, et al. Genetic architecture distinguishes systemic juvenile idiopathic arthritis from other forms of juvenile idiopathic arthritis: clinical and therapeutic implications. Ann Rheum Dis. 2017;76(5):906–913. | ||
Janow G, Schanberg LE, Setoguchi S, et al. The systemic juvenile idiopathic arthritis cohort of the childhood arthritis and rheumatology research alliance registry: 2010-2013. J Rheumatol. 2016; 43(9):1755–1762. | ||
Behrens EM, Beukelman T, Gallo L, et al. Evaluation of the presentation of systemic onset juvenile rheumatoid arthritis: data from the Pennsylvania Systemic Onset Juvenile Arthritis Registry (PASOJAR). J Rheumatol. 2008;35(2):343–348. | ||
Frosch M, Metze D, Foell D, et al. Early activation of cutaneous vessels and epithelial cells is characteristic of acute systemic onset juvenile idiopathic arthritis. Exp Dermatol. 2005;14(4):259–265. | ||
Bloom BJ, Tucker LB, Miller LC, McCauley RG, Schaller JG. Bicipital synovial cysts in juvenile rheumatoid arthritis: clinical description and sonographic correlation. J Rheumatol. 1995;22(10):1953–1955. | ||
Lang BA, Schneider R, Reilly BJ, Silverman ED, Laxer RM. Radiologic features of systemic onset juvenile rheumatoid arthritis. J Rheumatol. 1995;22(1):168–173. | ||
Munro R, Porter DR, Sturrock RD. Lymphadenopathy in a patient with systemic onset juvenile chronic arthritis. Ann Rheum Dis. 1998;57(9):513–517. | ||
Kimura Y, Weiss JE, Haroldson KL, et al. Pulmonary hypertension and other potentially fatal pulmonary complications in systemic juvenile idiopathic arthritis. Arthritis Care Res (Hoboken). 2013;65(5):745–752. | ||
Saurenmann RK, Levin AV, Feldman BM, et al. Prevalence, risk factors, and outcome of uveitis in juvenile idiopathic arthritis: a long-term followup study. Arthritis Rheum. 2007;56(2):647–657. | ||
Singh-Grewal D, Schneider R, Bayer N, Feldman BM. Predictors of disease course and remission in systemic juvenile idiopathic arthritis: significance of early clinical and laboratory features. Arthritis Rheum. 2006;54(5):1595–1601. | ||
Lomater C, Gerloni V, Gattinara M, Mazzotti J, Cimaz R, Fantini F. Systemic onset juvenile idiopathic arthritis: a retrospective study of 80 consecutive patients followed for 10 years. J Rheumatol. 2000;27(2):491–496. | ||
Fantini F, Gerloni V, Gattinara M, Cimaz R, Arnoldi C, Lupi E. Remission in juvenile chronic arthritis: a cohort study of 683 consecutive cases with a mean 10 year followup. J Rheumatol. 2003;30(3):579–584. | ||
Russo RA, Katsicas MM. Patients with very early-onset systemic juvenile idiopathic arthritis exhibit more inflammatory features and a worse outcome. J Rheumatol. 2013;40(3):329–334. | ||
Schneider R, Lang BA, Reilly BJ, et al. Prognostic indicators of joint destruction in systemic-onset juvenile rheumatoid arthritis. J Pediatr. 1992;120(2 Pt 1):200–205. | ||
Modesto C, Woo P, Garcia-Consuegra J, et al. Systemic onset juvenile chronic arthritis, polyarticular pattern and hip involvement as markers for a bad prognosis. Clin Exp Rheumatol. 2001;19(2):211–217. | ||
Spiegel LR, Schneider R, Lang BA, et al. Early predictors of poor functional outcome in systemic-onset juvenile rheumatoid arthritis: a multicenter cohort study. Arthritis Rheum. 2000;43(11):2402–2409. | ||
Martini A. It is time to rethink juvenile idiopathic arthritis classification and nomenclature. Ann Rheum Dis. 2012;71(9):1437–1439. | ||
Prakken B, Albani S, Martini A. Juvenile idiopathic arthritis. Lancet. 2011;377(9783):2138–2149. | ||
Cimaz R. Systemic-onset juvenile idiopathic arthritis. Autoimmun Rev. 2016;15(9):931–934. | ||
Wittkowski H, Frosch M, Wulffraat N, et al. S100A12 is a novel molecular marker differentiating systemic-onset juvenile idiopathic arthritis from other causes of fever of unknown origin. Arthritis Rheum. 2008;58(12):3924–3931. | ||
Shenoi S, Ou JN, Ni C, et al. Comparison of biomarkers for systemic juvenile idiopathic arthritis. Pediatr Res. 2015;78(5):554–559. | ||
Frosch M, Ahlmann M, Vogl T, et al. The myeloid-related proteins 8 and 14 complex, a novel ligand of toll-like receptor 4, and interleukin-1beta form a positive feedback mechanism in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2009;60(3):883–891. | ||
Ling XB, Park JL, Carroll T, et al. Plasma profiles in active systemic juvenile idiopathic arthritis: biomarkers and biological implications. Proteomics. 2010;10(24):4415–4430. | ||
Behrens EM, Beukelman T, Paessler M, Cron RQ. Occult macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis. J Rheumatol. 2007;34(5):1133–1138. | ||
Ravelli A, Davi S, Minoia F, Martini A, Cron RQ. Macrophage activation syndrome. Hematol Oncol Clin North Am. 2015;29(5):927–941. | ||
Grom AA, Horne A, De Benedetti F. Macrophage activation syndrome in the era of biologic therapy. Nat Rev Rheumatol. 2016;12(5):259–268. | ||
Zhang K, Biroschak J, Glass DN, et al. Macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis is associated with MUNC13-4 polymorphisms. Arthritis Rheum. 2008;58(9):2892–2896. | ||
Vastert SJ, van Wijk R, D’Urbano LE, et al. Mutations in the perforin gene can be linked to macrophage activation syndrome in patients with systemic onset juvenile idiopathic arthritis. Rheumatology (Oxford). 2010;49(3):441–449. | ||
Zhang M, Behrens EM, Atkinson TP, Shakoory B, Grom AA, Cron RQ. Genetic defects in cytolysis in macrophage activation syndrome. Curr Rheumatol Rep. 2014;16(9):439. | ||
Minoia F, Davi S, Horne A, et al. Clinical features, treatment, and outcome of macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a multinational, multicenter study of 362 patients. Arthritis Rheumatol. 2014;66(11):3160–3169. | ||
Ravelli A, Minoia F, Davi S, et al. 2016 Classification criteria for macrophage activation syndrome complicating systemic juvenile idiopathic arthritis: a European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Ann Rheum Dis. 2016;75(3):481–489. | ||
Consolaro A, Negro G, Lanni S, Solari N, Martini A, Ravelli A. Toward a treat-to-target approach in the management of juvenile idiopathic arthritis. Clin Exp Rheumatol. 2012;30(4 Suppl 73):S157–S162. | ||
Magni-Manzoni S, Ruperto N, Pistorio A, et al. Development and validation of a preliminary definition of minimal disease activity in patients with juvenile idiopathic arthritis. Arthritis Rheum. 2008;59(8):1120–1127. | ||
Filocamo G, Consolaro A, Schiappapietra B, et al. Parent and child acceptable symptom state in juvenile idiopathic arthritis. J Rheumatol. 2012;39(4):856–863. | ||
Wallace CA, Giannini EH, Huang B, et al. American College of Rheumatology provisional criteria for defining clinical inactive disease in select categories of juvenile idiopathic arthritis. Arthritis Care Res (Hoboken). 2011;63(7):929–936. | ||
van den Ham HJ, de Jager W, Bijlsma JW, Prakken BJ, de Boer RJ. Differential cytokine profiles in juvenile idiopathic arthritis subtypes revealed by cluster analysis. Rheumatology (Oxford). 2009;48(8):899–905. | ||
Woo P, Southwood TR, Prieur AM, et al. Randomized, placebo-controlled, crossover trial of low-dose oral methotrexate in children with extended oligoarticular or systemic arthritis. Arthritis Rheum. 2000;43(8):1849–1857. | ||
Shenoi S, Wallace CA. Diagnosis and treatment of systemic juvenile idiopathic arthritis. J Pediatr. 2016;177:19–26. | ||
Russo RA, Katsicas MM. Clinical remission in patients with systemic juvenile idiopathic arthritis treated with anti-tumor necrosis factor agents. J Rheumatol. 2009;36(5):1078–1082. | ||
Kimura Y, Pinho P, Walco G, et al. Etanercept treatment in patients with refractory systemic onset juvenile rheumatoid arthritis. J Rheumatol. 2005;32(5):935–942. | ||
Ruperto N, Lovell DJ, Quartier P, et al. Abatacept in children with juvenile idiopathic arthritis: a randomised, double-blind, placebo-controlled withdrawal trial. Lancet. 2008;372(9636):383–391. | ||
Ruperto N, Lovell DJ, Quartier P, et al. Long-term safety and efficacy of abatacept in children with juvenile idiopathic arthritis. Arthritis Rheum. 2010;62(6):1792–1802. | ||
Ringold S, Weiss PF, Beukelman T, et al. 2013 update of the 2011 American College of Rheumatology recommendations for the treatment of juvenile idiopathic arthritis: recommendations for the medical therapy of children with systemic juvenile idiopathic arthritis and tuberculosis screening among children receiving biologic medications. Arthritis Rheum. 2013;65(10):2499–2512. | ||
Boom V, Anton J, Lahdenne P, et al. Evidence-based diagnosis and treatment of macrophage activation syndrome in systemic juvenile idiopathic arthritis. Pediatr Rheumatol Online J. 2015;13:55. | ||
Gabay C, Lamacchia C, Palmer G. IL-1 pathways in inflammation and human diseases. Nat Rev Rheumatol. 2010;6(4):232–241. | ||
Nigrovic PA. Review: is there a window of opportunity for treatment of systemic juvenile idiopathic arthritis? Arthritis Rheumatol. 2014;66(6):1405–1413. | ||
Verbsky JW, White AJ. Effective use of the recombinant interleukin 1 receptor antagonist anakinra in therapy resistant systemic onset juvenile rheumatoid arthritis. J Rheumatol. 2004;31(10):2071–2075. | ||
Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med. 2005;201(9):1479–1486. | ||
Macaubas C, Wong E, Zhang Y, et al. Altered signaling in systemic juvenile idiopathic arthritis monocytes. Clin Immunol. 2016;163:66–74. | ||
Quartier P, Allantaz F, Cimaz R, et al. A multicentre, randomised, double-blind, placebo-controlled trial with the interleukin-1 receptor antagonist anakinra in patients with systemic-onset juvenile idiopathic arthritis (ANAJIS trial). Ann Rheum Dis. 2011;70(5):747–754. | ||
Nigrovic PA, Mannion M, Prince FH, et al. Anakinra as first-line disease-modifying therapy in systemic juvenile idiopathic arthritis: report of forty-six patients from an international multicenter series. Arthritis Rheum. 2011;63(2):545–555. | ||
Vastert SJ, de Jager W, Noordman BJ, et al. Effectiveness of first-line treatment with recombinant interleukin-1 receptor antagonist in steroid-naive patients with new-onset systemic juvenile idiopathic arthritis: results of a prospective cohort study. Arthritis Rheumatol. 2014;66(4):1034–1043. | ||
Miettunen PM, Narendran A, Jayanthan A, Behrens EM, Cron RQ. Successful treatment of severe paediatric rheumatic disease-associated macrophage activation syndrome with interleukin-1 inhibition following conventional immunosuppressive therapy: case series with 12 patients. Rheumatology (Oxford). 2011;50(2):417–419. | ||
Kahn PJ, Cron RQ. Higher-dose anakinra is effective in a case of medically refractory macrophage activation syndrome. J Rheumatol. 2013;40(5):743–744. | ||
Kelly A, Ramanan AV. A case of macrophage activation syndrome successfully treated with anakinra. Nat Clin Pract Rheumatol. 2008; 4(11):615–620. | ||
Ruperto N, Quartier P, Wulffraat N, et al. A phase II, multicenter, open-label study evaluating dosing and preliminary safety and efficacy of canakinumab in systemic juvenile idiopathic arthritis with active systemic features. Arthritis Rheum. 2012;64(2):557–567. | ||
Ruperto N, Brunner HI, Quartier P, et al. Two randomized trials of canakinumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367(25):2396–2406. | ||
Grom AA, Ilowite NT, Pascual V, et al. Rate and clinical presentation of macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis treated with canakinumab. Arthritis Rheumatol. 2016;68(1):218–228. | ||
Economides AN, Carpenter LR, Rudge JS, et al. Cytokine traps: multi-component, high-affinity blockers of cytokine action. Nat Med. 2003;9(1):47–52. | ||
Tarp S, Amarilyo G, Foeldvari I, et al. Efficacy and safety of biological agents for systemic juvenile idiopathic arthritis: a systematic review and meta-analysis of randomized trials. Rheumatology (Oxford). 2016;55(4):669–679. | ||
de Benedetti F, Massa M, Robbioni P, Ravelli A, Burgio GR, Martini A. Correlation of serum interleukin-6 levels with joint involvement and thrombocytosis in systemic juvenile rheumatoid arthritis. Arthritis Rheum. 1991;34(9):1158–1163. | ||
Yokota S, Imagawa T, Mori M, et al. Efficacy and safety of tocilizumab in patients with systemic-onset juvenile idiopathic arthritis: a randomised, double-blind, placebo-controlled, withdrawal phase III trial. Lancet. 2008;371(9617):998–1006. | ||
De Benedetti F, Brunner HI, Ruperto N, et al. Randomized trial of tocilizumab in systemic juvenile idiopathic arthritis. N Engl J Med. 2012;367(25):2385–2395. | ||
Yokota S, Itoh Y, Morio T, Sumitomo N, Daimaru K, Minota S. Macrophage activation syndrome in patients with systemic juvenile idiopathic arthritis under treatment with tocilizumab. J Rheumatol. 2015;42(4):712–722. | ||
Shimizu M, Nakagishi Y, Kasai K, et al. Tocilizumab masks the clinical symptoms of systemic juvenile idiopathic arthritis-associated macrophage activation syndrome: the diagnostic significance of interleukin-18 and interleukin-6. Cytokine. 2012;58(2):287–294. | ||
Hashkes PJ, Wright BM, Lauer MS, et al. Mortality outcomes in pediatric rheumatology in the US. Arthritis Rheum. 2010;62(2):599–608. | ||
DeWitt EM, Kimura Y, Beukelman T, et al. Consensus treatment plans for new-onset systemic juvenile idiopathic arthritis. Arthritis Care Res (Hoboken). 2012;64(7):1001–1010. | ||
Kimura Y BT, Morgan-DeWitt E, Mieszkalski KL, et al. Results from the childhood arthritis and rheumatology research alliance systemic JIA consensus treatment plans pilot study [abstract]. Arthritis Rheumatol. 2015;67(Suppl 10):abstract 959. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.