")
Back to Journals » The Application of Clinical Genetics » Volume 12
Update On The Clinical Perspectives And Care Of The Child With 47,XXY (Klinefelter Syndrome)
Authors Samango-Sprouse CA, Counts DR, Tran SL, Lasutschinkow PC, Porter GF, Gropman AL
Received 1 June 2019
Accepted for publication 20 September 2019
Published 23 October 2019 Volume 2019:12 Pages 191—202
DOI https://doi.org/10.2147/TACG.S180450
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Carole A Samango-Sprouse,1–3 Debra R Counts,4 Selena L Tran,3 Patricia C Lasutschinkow,3 Grace F Porter,3 Andrea L Gropman5,6
1Department of Pediatrics, George Washington University, Washington, DC, USA; 2Department of Human and Molecular Genetics, Florida International University, Miami, FL, USA; 3The Focus Foundation, Davidsonville, MD, USA; 4Pediatric Endocrinology, Sinai Hospital, Baltimore, MD, USA; 5Department of Neurology, George Washington University, Washington, DC, USA; 6Division of Neurogenetics and Developmental Pediatrics, Children’s National Medical Center, Washington, DC, USA
Correspondence: Carole A Samango-Sprouse
The Focus Foundation, 820 W. Central Ave. #190, Davidsonville, MD 21035, USA
Tel +1 443-223-7323
Fax +1 855-550-8696
Email [email protected]
Abstract: 47,XXY (Klinefelter syndrome [KS]) is the most common sex chromosomal aneuploidy (1:660), yet, despite this, only 25% of the males are ever diagnosed. Males with 47,XXY present with characteristic symptoms throughout their lifetime with typical physical and neurodevelopmental manifestations focused in growth, cognitive development, endocrine function, and reproduction. Studies have demonstrated that optimal outcomes are dependent on early detection combined with consistent and targeted neurodevelopmental treatment throughout the lifespan. During infancy and into the preschool years, individuals with 47,XXY commonly face deficits in growth and development in the areas of early hormonal, motor, speech, and behavioral development. As they transition into school, the primary neurodevelopmental concerns include language difficulty, executive dysfunction, behavior, and learning and reading deficits. Adults with 47,XXY often present with taller than average height, low levels of fertility, azoospermia, and elevated gonadotropin levels. These presentations may persist from early childhood through adulthood but can be mitigated by appropriate interventions. Early neurodevelopmental and hormonal treatment has been shown to have a minimizing effect on the physical and neurodevelopmental manifestations in individuals with 47,XXY. With innovative and current research studies, the features common to the neurodevelopmental profile of 47,XXY have been further expanded and defined. Further research is necessary to elucidate and understand the relationship between the brain, behavior, and the phenotypic profile of 47,XXY.
Keywords: 47, XXY, Klinefelter syndrome, neurodevelopment, hormonal treatment
Introduction
Currently estimated to occur in 1 in 660 males, 47,XXY (Klinefelter syndrome [KS]) is the most common sex chromosomal aneuploidy.1 This prevalence was described in a Denmark study that identified 163 cases of 47,XXY out of 76,526 prenatal examinations during 1970–2000.2 According to recent research, the incidence of 47,XXY is between 1:581 and 1:917 male births.3 Only 25% of this population are diagnosed during childhood or adulthood, only after presenting with developmental delay, behavioral disruptions, hypogonadism, gynecomastia, or low fertility.4 Even fewer receive an early diagnosis, as less than 10% of the boys with 47,XXY are diagnosed prenatally by amniocentesis or chorionic villus sampling (CVS).2 This may change as the use of quantitative fluorescent polymerase chain reaction (QR-PCR) and NIPT are becoming more accessible for the prenatal detection of chromosomal abnormalities.5,6
Although this disorder was initially discovered as a result of endocrinological deficiencies,4 the chromosomal complement was determined to be the result of an additive X chromosome.7 Since then, researchers have further identified deficiencies in the dental, musculoskeletal, and neurodevelopmental systems, and, more recently appreciated, brain dysmorphology.8 The evolution of this syndrome’s complexity and its multi-systemic involvement has resulted in greater understanding of the intimate relationship among hormonal development, neurodevelopmental performance, and health and well-being.9–12 Although these boys are not intellectually impaired, they often present with language-based learning difficulties, including deficits in expressive language, social language, and reading.3,12–14 Motor planning deficiencies contribute to delayed speech and language development, as well as significantly impacting on the development of balance and motor proficiency skills.15 These early delays are not isolated developmental disturbances but rather a reflection of this polygenic disorder and its complexity.
Although the profile of boys with 47,XXY varies contingent on several salient factors, recent research has expanded our understanding of the phenotype and presentation of medical issues resulting from the additive X chromosome. This paper presents recent research in order to provide an updated, comprehensive overview of the 47,XXY profile. The following topics will be discussed: etiology and genetics, detection and diagnosis, development and growth, language and behavior, and, lastly, associated medical complications.
Etiology And Genetics
47,XXY occurs as a result of a randomly occurring nondisjunction error that happens during meiotic division. The origin of the additional X chromosome is maternally derived in approximately 50% of the cases.16 Nondisjunction in paternally derived cases occur in the first meiotic division, while maternally derived cases can occur in meiosis I (24%), meiosis II (14%), or post-zygotically (8%). The process of nondisjunction is outlined in Figure 1. In the remaining 4% of maternally derived cases, the origin of nondisjunction is unknown.16
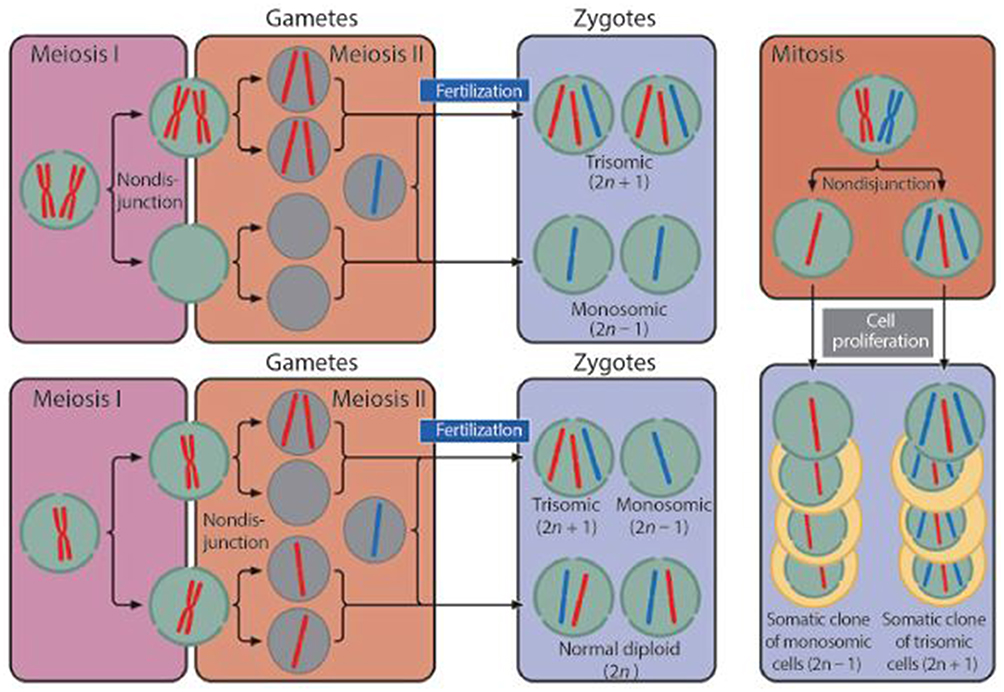 |
Figure 1 The process of nondisjunction.Note: Copyright |
Parental age has been thought to be a potential contributing factor to these nondisjunction events, as recent studies have suggested that mothers of advanced maternal age (AMA), over 35 years of age, have a statistically higher risk of giving birth to a child with 47,XXY.17 However, while a study by Thomas et al (2000)18 found that AMA was associated with errors in maternal meiosis I, AMA does not appear to be related to errors that occur during meiosis II.19 More recently, the use of non-invasive prenatal testing (NIPT) has resulted in an increasingly younger mean maternal age associated with X chromosome aneuploidy due to the spike in detection among young, low-risk mothers.6,18,20,21 Additionally, paternal age does not seem to play a role, even in paternally derived cases.18,20
In addition to researching the genetic etiology of 47,XXY, many studies have attempted to find a genetic mechanism that may explain the variability in the neurodevelopmental and behavioral phenotype observed in 47,XXY. While studies have not found phenotypic differences between maternally and paternally derived cases of 47,XXY, it is thought that some of the variability stems from a gene dosage effect due to the additional X chromosome. Another assumption is that the phenotype is caused by irregularities in gene transcription on the X chromosome, as most of the genes that escape X inactivation are on the short arm of this chromosome. However, the number of genes involved or sufficient in impacting the phenotype remains unknown. Some researchers have hypothesized that there may be two dysfunctional X-linked alleles caused by the same X chromosome being copied twice during a nondisjunction event that occurs during meiosis II or postzygotic division that does not occur during meiosis I. Thus, variability in the 47,XXY phenotype may be evident between maternally and paternally derived cases, though studies have not shown this. One of the more recent theories, however, is that the variability in phenotype is due to a significantly higher frequency of skewed X-inactivation reported in 47,XXY.22
Detection And Diagnosis
Prenatal Diagnosis
Invasive procedures for prenatal testing, such as amniocentesis or chorionic villus sampling (CVS), are utilized as diagnostic options for mothers at high-risk due to AMA or a family history of genetic disorders. When electing the use of these tests, parents are most often concerned with identifying common autosomal abnormalities such as trisomies 13, 18, and 21 (Down Syndrome) without necessarily knowing that these methods also have the ability to prenatally detect sex chromosome aneuploidies (SCAs), leading to incidental diagnosis of SCAs, such as 47,XXY.6,21
The introduction of NIPT has also increased prenatal identification of 47,XXY.6,21 Conducted as early as 10 weeks gestation, NIPT isolates and analyzes cell-free fetal DNA (cffDNA) in maternal blood to identify chromosomal anomalies. This is now a screening option for all pregnant women, regardless of age, and is often considered favorably due to the noninvasive nature of the test. Several major societies have released consensus statements supporting the utilization of NIPT for detecting X and Y chromosomal variations, such as 47,XXY.23–26
Although NIPT is considered to be highly accurate (sensitivity of 91.0% and specificity of 99.6%), this testing method is not diagnostic. Additionally, current NIPT methods do not possess the capabilities to confirm the presence of more than one additive X. Positive NIPT results, therefore, must be confirmed via amniocentesis or CVS prenatally, or with chromosomal microarray (CMA) or karyotype testing after birth.21
However, despite these limitations of NIPT, parents of children with 47,XXY have reported favorably to the use of this screening tool in identifying those at-risk for the disorder, stating that the NIPT results gave them time to research and learn about the disorder, prepare early interventions, and understand their child prior to his birth.27 Thus, confirming the exact genotype for the X or Y aneuploidy during pregnancy provides opportunity for preparation and may shape parental expectations. This shaping of parental perspective may also better prepare families in cases where NIPT revealed 47,XXY during pregnancy, but later identified postnatally as 48,XXXY or 49,XXXXY.28
The emergence of the QR-PCR test has provided an additional noninvasive mechanism in which chromosomal aneuploidies, such as 47,XXY, can be detected prenatally. This assay yields results as quickly as a few hours after the sample collection, allowing for parents to be informed within 24 hrs of testing.5 In a study with 43,000 prenatal mothers, the karyotype of 41,019 cases (95%) were correctly identified; 65 of these cases were 47,XXY karyotypes.5
Postnatal Diagnosis
Many boys with 47,XXY remain undiagnosed throughout their lifetime as a result of nonspecific symptoms, a lack of dysmorphic appearance, and varied clinical presentation of the disorder. Although symptoms may appear as early as the newborn period, pediatric care providers rarely consider 47,XXY as an etiology for developmental dysfunction prior to adolescence. Postnatal diagnoses, therefore, often result from evident physical anomalies that may present in early childhood, including hypospadias, small phallus, or cryptorchidism.29–31
In infancy, many boys with 47,XXY exhibit early manifestations of developmental dyspraxia, as identifiable through speech and motor delays.10,32 They are often referred to physical therapy for motor delays prior to 12 months of age, and speech and language therapy for delayed speech skills by 18 to 24 months of age. Without early hormonal therapy (EHT), these boys may struggle with all aspects of motor planning in speech and motor domains throughout their lives.10,30,32
Although the onset of puberty in boys with 47,XXY may occur at the expected age, the testes in these boys do not reach the size observed in normal adults.33 This key feature often prompts medical providers into a postnatal diagnosis for boys who have remained undetected. The untreated population also displays eunuchoid body habitus, small testes, gynecomastia, as well as increased levels of gonadotropins, a follicle-stimulating and luteinizing hormone. Additional presentations found during adolescence include scoliosis and kyphosis, ligamentous laxity, wide hips, narrow shoulders, and truncal hypotonia.10,30 However, research has shown that exogenous testosterone supplementation, usually in the form of hormonal replacement therapy (HRT), in mid-to-late puberty will allow for full pubertal development and age-appropriate attainment of secondary sexual characteristics.34
In adulthood, males with 47,XXY exhibit symptoms of testosterone deficiency (small testes, low fertility), as well as social and behavioral anomalies, and executive dysfunction.9,12,35,36 Many males with 47,XXY are diagnosed in late adolescence or adulthood as a result of the presentation of low fertility or incomplete puberty. Providers who determine their patients to have delayed puberty or suspected infertility should conduct a chromosomal microarray to rule out 47,XXY as an etiological explanation for these features.
Development And Growth
Neurological Structure And Function
Our understanding of the interaction between brain development, neurocognition, and neurodevelopmental outcome in the 47,XXY child has expanded and benefited as a result of improved neuroimaging techniques of the brain.8,37 Neuroimaging studies on the 47,XXY population have described a reduction of brain volume in the caudate, frontal, and temporal regions of the brain.8 Anomalies in MRIs of the caudate and frontal brain are comparable to those of boys with attention-deficit hyperactivity disorder (ADHD), which may explain the executive function deficits often exhibited by boys with 47,XXY.8,36 Additional abnormalities noted in the caudate nucleus of the brain are believed to negatively impact speech and language development, as well as the mild oral motor dysfunction commonly seen in 47,XXY boys.8
Impairments have been found in the temporal lobes of untreated males with 47,XXY, which may lead to deficits in reading, social language, processing of verbal information, and facial recognition.1 A neuroimaging study of individuals with 47,XXY revealed decreased gray matter density in the insula region of the brain—a possible explanation for the social and emotional processing deficits faced by some boys with 47,XXY.38 The parietal lobe, however, appears normal in cortical volume and thickness, and this preservation is associated with typically enhanced spatial cognitive skills in individuals with 47,XXY.1,8,9
Selected studies have suggested that receiving HRT is associated with more normalized brain development.37,39 Patwardhan et al (2000) compared groups of treated and untreated individuals with 47,XXY and found that temporal gray matter volume was preserved in the treated group and diminished in the untreated group.37 EHT, ideally given between 4 months and 9 months of age, has been shown to normalize neurodevelopmental differences, while simultaneously having a beneficial impact on other features such as energy level, social engagement, and general well-being.10,11,34 Additional studies are warranted to further investigate the possible restorative impact of HRT during infancy and adolescence on the abnormal brain areas described in boys with 47,XXY.
Endocrine Development
In typically developing infant males, testosterone levels peak between 2 and 4 months of age after rising steadily during the first month of life. Some studies have shown that infants with 47,XXY also experience normal or high normal testosterone levels.40,41 However, there are other studies that have demonstrated that this testosterone peak during infancy is reduced in males with 47,XXY.35,42 Thus, the data on the presence of hypoandrogenism in infants with 47,XXY are conflicting. However, studies over the last 10 years have suggested that supplementing this typical testosterone peak with EHT may potentially improve the neurodevelopment of infants with 47,XXY in multiple domains including behavior, motor, and speech.10,11,43,44 The biochemical abnormalities and clinical manifestations often seen in 47,XXY as well as appropriate interventions are described in Table 1. It is important to note that these are suggested guidelines, as there is currently no specific protocol for HRT in males with 47,XXY. These age-appropriate formulations and dosages have been extrapolated from the Endocrine Society’s Clinical Practice Guidelines for the treatment of hypogonadal men.45
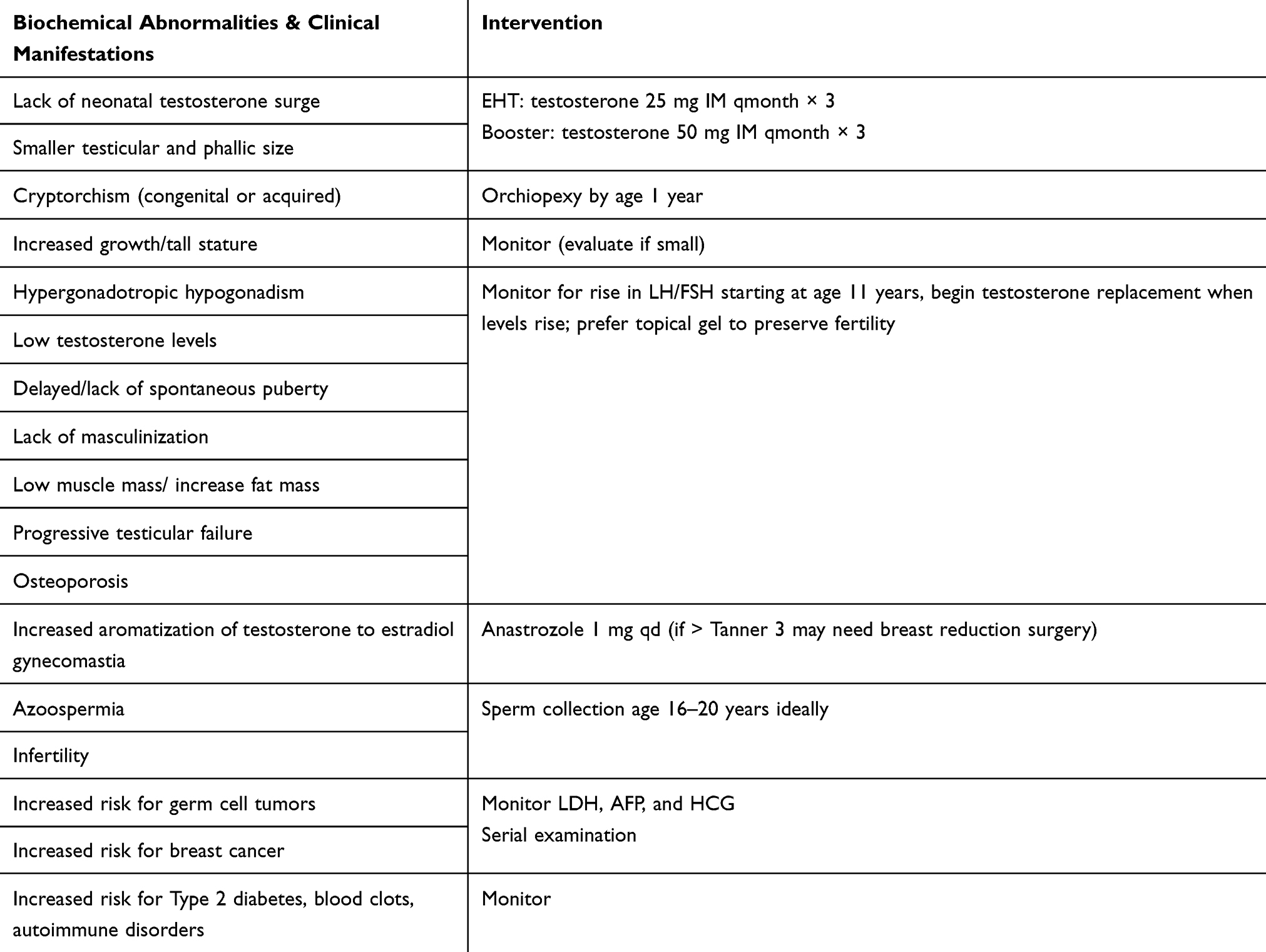 |
Table 1 Management Of Endocrinology Issues |
Previous research studies have shown that most boys with 47,XXY do not experience delayed puberty and progress the Tanner stages of puberty normally.46–48 However, it has been observed that testicular growth and volume for boys with 47,XXY does not exceed 4–5 mL.49,50 Additionally, while testosterone production in these boys during puberty is sufficient for the development of primary sexual characteristics, this development typically does not occur until 3–4 years later when compared to boys with 46,XY.51 Estrogen levels have been observed to be elevated early during puberty and remain slightly elevated when compared to controls, but these levels can also be normal.47,48,52
During mid-puberty, adolescents with 47,XXY experience a gradual increase in blood luteinizing hormone (LH). This is due to a compensatory feedback mechanism that maintains the Leydig cell function in males with 47,XXY. A surge in the follicle-stimulating hormone (FSH) has also been observed in males with 47,XXY at this stage, and this indicates progressive tubular damage, which leads to hypergonadotropic hormonal pattern by late puberty.
Due to a compensatory effort that the 47,XXY male body makes to maintain sufficient circulating testosterone level and a normal estrogen to testosterone ratio, males with 47,XXY may display multiple characteristics of hypogonadism once they reach pubertal age.33 This results in limited body hair, a low incidence of acne, and more feminized distributions of pubic hair. In addition, adolescents with 47,XXY can have increased fatigue, atypical fat distribution, and poor muscular development secondary to testosterone deficiency. Males with 47,XXY have also been found to have increased breast tissue, with rates as high as 50–75% as determined by palpation53 and 20% of the adolescents and adults presenting with gynecomastia.54 This is thought to be due to aromatase CYP19 being over-expressed, which causes increased aromatization of testosterone to estradiol.55 The gynecomastia may be minimized effectively with pharmacological intervention that includes an estrogen inhibitor.
One of the features of 47,XXY that often leads to a postnatal diagnosis is the decrease in penile length and testicular size throughout the lifespan. Significantly smaller sizes were reported by Ross et al (2005) in infant males with 47,XXY between 1 and 23 months of age.35 In addition, pre-pubertal males with 47,XXY have been found to present with borderline small testes, although the development of the Leydig cells and seminiferous tubules did not appear to be abnormal.35 While primary spermatogonia is typically present in the early childhood of boys with 47,XXY, they have been found to be reduced when compared to 46,XY males.56 At the onset of puberty, there is an increase in germ cell attrition that results in primary spermatogonia becoming absent and the tubules only containing Sertoli cells. While Inhibin B levels are within normal limits prior to puberty, apoptosis occurs during puberty resulting in dramatically low levels of Inhibin B levels in adult men with 47,XXY.57
In adult males with 47,XXY, Christiansen et al (2003) found that the elevations in LH and FSH reflect the sex steroids having an insufficient amount of feedback inhibition, in addition to testicular dysfunction.57 Some elements of function remain intact with normal suppression of LH and FSH to exogenous testosterone and a normal response of the pituitary gland to gonadotropin-releasing hormones. Even when levels of serum testosterone are within normal limits, serum gonadotropins are elevated, which is typically taken as a sign of compensated hypogonadotropic hypogonadism.
Some adult males with 47,XXY present with a smaller prostate, as well as continued smaller testicular size. While the testes of typical male adults are on average 3.5 centimeters in diameter, the adult testes seen in 47,XXY are typically no more than 2 centimeters. In addition to the difference in size of the testes, they are also typically firmer than normal due to germinal atresia. The seminiferous tubules in men with 47,XXY present with hyalinization, fibrosis, and decreased amounts of Leydig cells, which lack the typical Reinke crystalloids. While Sertoli cells appear not to be impacted, germ cells in 47,XXY are greatly decreased. Along with the decreased presence of Leydig and germ cells, testicular biopsies in 47,XXY have also identified thickened tubular member basement membranes and sclerosis of the germ cell compartment often resulting in azoospermic infertility or severe oligospermia.58
Although all men with 47,XXY were originally thought to be infertile, research in the last 15 years have found that sperm can be recovered in some men either during a testicular biopsy or in the ejaculate. However, the latter is much less common, and documented success rates of extraction have been varied. A study of 36 Korean men with 47,XXY found that only 27% had mature sperm, with higher rates of successful sperm retrieval in mosaic (46,XY/47,XXY) men.59 However, a larger study from Vernaeve et al (2004) documented that sperm were recovered in 48% (24) of the non-mosaic men studied.60 More recently, a study by Franik et al (2016) found that retrieval rates using testicular sperm extraction (TESE) for males between the ages of 16 and 30 were around 40–70%, while retrieval rates for adolescents younger than 16 were between 0% and 20%.61 More research is needed to identify the cause of the variability in successful sperm retrieval.
Successful fertilization rates after sperm retrieval has been obtained are thought to be around 50%62 with no differences found in fertilization rates between fresh and cryopreserved sperm.63 Thus, extraction of sperm need not correspond with the reproductive cycle of the partner to result in fertilization. Due to the fertilization rates, it is recommended that couples be offered access to donor sperm concomitantly to the male partner’s sperm in case fertilization is not successful. In 2003, Staessen et al completed a study in which intracytoplasmic sperm injection (ICSI) was performed in 20 couples, where the partner had 47,XXY.64 Eight pregnancies resulted from 32 cycles of ICSI, in which five were live births.64 Genetic diagnosis of the embryos prior to implantation demonstrated that 54% of the embryos were cytogenetically normal compared to the center’s average of 77%. In addition, of the embryos, sex chromosome aneuploidy occurred twice as much in the couples where the male was 47,XXY. While prenatal genetic diagnosis should be offered to couples in which the male partner has 47,XXY due to these findings, all live born offspring to date have been normal.63–65
Growth
Growth rates are variable in the boys with 47,XXY, as some boys are above the 75th height percentile prior to 12 months, and others remain within the normal range under 3 years of age. However, the average height of this population increases with age and is only mildly impacted by parental height. Rarely do boys with 47,XXY remain below the 25th height percentile. Their average height increases to the 30th percentile before age 2 and further height increases between 5 and 8 years of age until the individual reaches the 75th to 90th percentile by 18 years of age.66
Adults with 47,XXY present with increased leg length, decreased body ratio, as well as taller than average stature, with a mean height of 190.5 centimeters (cm).67 The tall stature that typically manifests in 47,XXY likely results from the three copies of X and Y chromosome height determining gene: SHOX. Delayed epiphyseal fusion secondary to testosterone deficiency may also explain tall stature in these boys.68 Although the bone age of boys with 47,XXY is typically qualified as clinically normal, osteoporosis, coupled with hypotonia, that is commonly seen in the untreated population can lead to further orthopedic complications.69
Untreated boys with 47,XXY often develop truncal obesity in adolescence secondary to the hypogonadism typically associated with the disorder. HRT serves as a highly effective method of weight management when supplemented with a balanced and nutritional diet, as well as a well-rounded exercise regimen that includes a combination of aerobic exercise, muscle strength, and endurance training. At the first indicators of delayed puberty, HRT may be helpful in increasing muscle development, develop secondary sexual characteristics, and improve bone health.70,71
Feeding Difficulties
Feeding disruptions serve as infantile indications of the oral motor-based dysfunction that are more common in the child with 47,XXY. In a large cohort of 171 prenatally diagnosed infants with 47,XXY, 47.95% presented with feeding difficulties (eg, sucking/swallowing difficulties or latching on for breastfeeding).12 These feeding difficulties typically resolve within the first three months of life. The assistance of a lactation specialist, pediatric occupational therapist, or speech and language pathologist can be quite beneficial in diminishing these delays, particularly for primigravida mothers. The developmental periods at which these difficulties present in individuals with 47,XXY, as well as appropriate management for these concerns are detailed in Table 2.
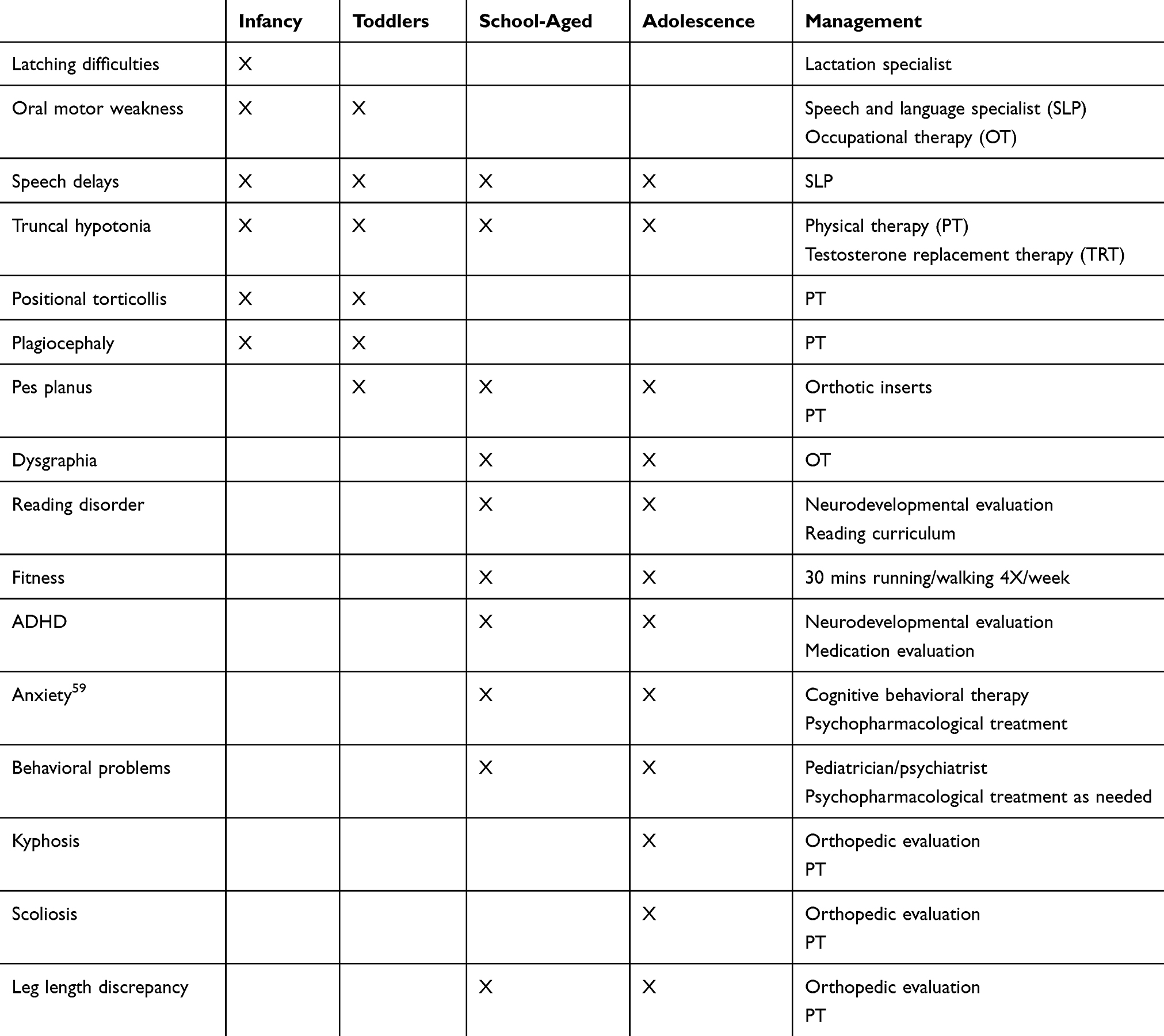 |
Table 2 Management Of Neurodevelopmental Concerns Throughout The Lifespan |
Motor Planning
The 47,XXY population presents with increased incidences of truncal hypotonia and motor planning difficulties before one year of age.10,32 As a result, these boys often develop positional torticollis combined with plagiocephaly (Table 2). Therefore, prone development, commonly referred to as “tummy time,” must be strongly encouraged in order to prevent the development of positional torticollis secondary to inactivity and truncal weakness.
Speech delay and deficiencies are typical of the child with 47,XXY, manifesting as early as 6 months of age (Table 2). Typically, these boys are described as “quiet babies,” as they exhibit decreased vocalizations during infancy, secondary to delayed phonemic development, limited variegated sounds, and deficiencies in sound imitation. These delays are further characterized by reduced reciprocal synchrony with primary caregivers, thus limiting social interaction.10,13,30,32,66 Infants with 47,XXY may appear expressionless as a result of oral facial hypotonia. These motor planning deficits influence all other factors of the 47,XXY phenotype and may provide additional explanation for disruptions in the behavior of these boys. These skill deficits are further evidenced with age, as complex, multi-faceted motor movements evolve throughout childhood and adolescence. Although these neurodevelopmental challenges were present during infancy and toddler years, the deficiencies associated with 47,XXY become more evident over time.
Language And Behavior
The variability of the profile of 47,XXY must be considered when discussing the behaviors and social communication demonstrated in this population. While some boys exhibit difficulties with conflict resolution, peer interaction, or social fluency in social environments, others demonstrate little to no behavioral problems. Boys with a prenatal 47,XXY diagnosis present with fewer problems in general, but require consistent support and oversight to be successful.30 Those children who receive EHT in infancy have fewer problems than their untreated and postnatally diagnosed counterparts.11,72 Frustration stemming from the low expressive capabilities associated with 47,XXY may further explain some of the behavioral problems displayed by these untreated boys.30,71 Since EHT may increase frontal lobe function and expressive ability,8,37 the administration of EHT may minimize such behavioral difficulties exhibited in these boys.10 Further research has also found EHT to decrease autistic traits, aggression, and affective problems, as well as improve social communication and social cognition.11
Research has found individuals with 47,XXY to be at an increased risk for social communication deficits, increased anxiety, autistic traits, ADHD symptoms, and emotional dysregulation (Table 2).73–76 The social emotional profile of 47,XXY is variable, but these boys typically present as timid, and are often susceptible to heightened anxiety secondary to social language difficulties, particularly when hormonal replacement therapy has not been initiated in a timely manner. Due to the prevalence of language-based learning disorders in this population, boys with 47,XXY may have difficulty identifying and verbalizing their emotions in comparison with their neurotypical peers.73,77 These deficits have been shown to be reduced with the implementation of early intervention services and biological treatment.
While some studies have reported increased incidences of autistic-like traits in boys with 47,XXY,73,74 the social deficits described are likely to have been compounded by timing of diagnosis, as well as untreated androgen deficiency. These social language deficiencies have shown to be significantly improved with prenatal diagnosis, the administration of EHT in infancy, HRT in adolescence, and early intervention.10,11,34,36,72 Social language deficits and anxiety may also be mitigated by timing of diagnosis of 47,XXY.32,72
Research studies have found increased incidences of ADHD symptoms, with manifestations reported in 41–63% of the boys with 47,XXY.75,76 The results of these studies, however, may be confounded, as familial history of ADHD, dyslexia, and other learning difficulties was not accounted for in the data segregation; therefore, such results should be considered with caution. Consequently, the incidence of 47,XXY and ADHD has not been addressed comprehensively and is subsequently, not well understood. However, boys with 47,XXY should be monitored for ADHD throughout childhood and into young adulthood. The etiology of the behavioral variability in 47,XXY is not well understood yet, but critical factors may include early biological treatment, timing of diagnosis, family history, skewed X inactivation, or a synergy among these factors.
Associated Medical Complications
Musculoskeletal
The musculoskeletal profile of boys with 47,XXY features several mild to complex abnormalities, including positional torticollis, mild midface underdevelopment, scoliosis, kyphosis, pes planus, ligamentous laxity, fifth toe anomalies, fifth finger clinodactyly, pectus excavatum or carinatum.30 The identification of any of these anomalies, specifically positional torticollis, requires immediate referral to pediatric physical therapy to manage and prevent secondary symptoms (such as plagiocephaly), and encourage appropriate motor development.
Immunologic Disorders
Current research suggests an increased incidence of pulmonary diseases in the 47,XXY population, including bronchitis, asthma, and emphysema.78,79 Anecdotal information reports that asthma can be exacerbated in boys with 47,XXY if pulmonary issues are present in the family history. If repeat upper respiratory infections, ear infections, pneumonia, or any incidence of bronchiolitis present, an immunology evaluation can be very helpful for appropriate medical management.
Life Expectancy
Although longitudinal research is limited due to decreased identification of males with 47,XXY, several current research studies indicate mildly increased mortality risks in this population as a result of cardiovascular, hemostatic, or metabolic complications.69,78 Previous research has also indicated a possible increased risk for breast cancer, non-Hodgkin lymphoma, and lung cancer in the 47,XXY population.80 Further research is required to better understand the interaction of the disorder with a variety of factors including mortality risk, socioeconomic status (SES), family history, timing of diagnosis, and administration of hormonal replacement therapy.
Copy Number Variants (CNVs)
While typically intellectually intact, the variable presentation of clinical features in 47,XXY can be impacted by additional factors, including prematurity, brain malformations, family history of learning disabilities, or, potentially, CNVs. A recent research publication described increased incidences of CNVs in association with 47,XXY.81 Further research regarding the influence of a CNV in the neurodevelopmental profile may expand our understanding of its potentially modulating effect on the variable phenotype of 47,XXY.
Conclusion And Future Research
47,XXY (KS) is the most common sex chromosomal variation, occurring equally across races, ethnicity, and SES.2,12 However, it continues to be rarely diagnosed and largely misunderstood. Individuals with 47,XXY may have multiple systems compromised with medical disorders ranging from asthma, ADHD, motor planning deficits, low fertility, and social language challenges. The variability of these symptoms is mitigated by several factors, including family history, timing of diagnosis, access to early intervention, and biological treatment. HRT may improve neurodevelopmental outcome during prepubertal years in boys with 47,XXY. This novel finding suggests that there is a biological treatment that may be both impactful and easily delivered to an extremely vulnerable population of males throughout the world.
Future research is warranted to expand our understanding of the endocrinological impact of HRT at various stages in life in males with 47,XXY, specifically in school-age, prepubescent children since previous research has shown that boys with 47,XXY enter puberty at the expected age but may experience a delay of primary and secondary sexual features. There is a need, therefore, to explore the impact of HRT timing, as research in this age population is limited. Additionally, more research is needed to determine if adverse effects of HRT administration in infants and adolescents with 47,XXY exist, if any, as none have previously been reported. Further investigation of the restorative and beneficial impact of early diagnoses and biological treatment on the neurodevelopmental profile of these boys will generate increased access to knowledge of 47,XXY for genetic counselors, pediatric specialists, and primary care providers. Current literature suggests an extraordinary need for increased detection, early treatment, and multidisciplinary care to boys with 47,XXY in order to optimize neurodevelopmental outcome, health, and well-being.
Abbreviations
AMA, advanced maternal age; cffDNA, cell-free fetal DNA; CMA, chromosomal microarray; CVS, chorionic villus sampling; EHT, early hormonal therapy; FSH, follicle-stimulating hormone; HRT, hormonal replacement therapy; ICSI, intracytoplasmic sperm injection; KS, Klinefelter Syndrome; LH, luteinizing hormone; NIPT, non-invasive prenatal testing; TESE, testicular sperm extraction.
Acknowledgment
The authors would like to thank The Focus Foundation team, especially Teresa Sadeghin, for their contributions to this article.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Savic I. Advances in research on the neurological and neuropsychiatric phenotype of Klinefelter syndrome. Curr Opin Neurol. 2012;25(2):138–143. doi:10.1097/WCO.0b013e32835181a0
2. Bojesen A, Juul S, Gravholt CH. Prenatal and postnatal prevalence of Klinefelter syndrome: a national registry study. J Clin Endocrinol Metab. 2003;88(2):622–626.
3. Samango-Sprouse CA, Kirkizlar E, Hall MP, et al. Incidence of X and Y chromosomal 347 aneuploidy in a large child bearing population. PLoS One. 2016;11:8. doi:10.1371/journal.pone.0161045
4. Klinefelter HF
5. Cirigliano V, Voglino G, Ordonez E. Rapid prenatal diagnosis of common chromosome aneuploidies by QF-PCR, results of 9 years of clinical experience. Prenatal Diagn. 2009;29(1):40–49. doi:10.1002/pd.2192
6. Howard-Bath A, Poulton A, Halliday J, et al. Population-based trends in the prenatal diagnosis of sex chromosome aneuploidy before and after non-invasive prenatal testing. Prenat Diagn. 2018;38(13):1062–1068. doi:10.1002/pd.5363
7. Jacobs PA, Strong JA. A case of human intersexuality having a possible XYY sex-determining mechanism. Nature. 1959;183(4657):302–303. doi:10.1038/183302a0
8. Giedd JN, Clasen LS, Wallace GL, et al. XXY (Klinefelter syndrome): a pediatric quantitative brain magnetic resonance imaging case-control study. Pediatr. 2007;119(1):e232–e240. doi:10.1542/peds.2005-2969
9. Samango-Sprouse C, Law P. The neurocognitive profile of the young child with XXY. Eur J Hum Genet. 2001;9(Suppl 1):193.
10. Samango-Sprouse CA, Sadeghin T, Mitchell FL, et al. Positive effects of short course androgen therapy on the neurodevelopmental outcome in boys with 47,XXY syndrome at 36 and 72 months of age. Am J Med Genet Pt A. 2013;161(3):501–508. doi:10.1002/ajmg.a.35769
11. Samango-Sprouse C, Stapleton EJ, Lawson P, et al. Positive effects of early androgen therapy on the behavioral phenotype of boys with 47,XXY. Am J Med Genet C Semin Med Genet. 2015;169(2):150–157. doi:10.1002/ajmg.c.31437
12. Samango-Sprouse C, Gropman AL, X & Y chromosomal variations: hormones, brain development, and neurodevelopmental performance. Colloquium Series Developing Brain. 2017;5(2):i–122. Morgan & Claypool Life Sciences. doi:10.4199/C00134ED1V01Y201506DBR013
13. Graham JM, Bashir AS, Stark RA, et al. Oral and written language abilities of XXY boys: implications for anticipatory guidance. Pediatr. 1988;81(6):795–806.
14. Re L, Birkhoff JM. The 47, XYY syndrome, 50 years of certainties and doubts: a systematic review. Aggress Violent Behav. 2015;22:9–17. doi:10.1016/j.avb.2015.02.003
15. Gropman A, Samango-Sprouse CA. Neurocognitive variance and neurological underpinnings of the X and Y chromosomal variations. Am J Med Genet Pt C Semin Med Genet. 2013;163(1):35–43. doi:10.1002/ajmg.c.31352
16. Hall H, Hunt P, Hassold T. Meiosis and sex chromosome aneuploidy: how meiotic errors cause aneuploidy; how aneuploidy causes meiotic errors. Curr Opin Genet Dev. 2006;16(3):323–329. doi:10.1016/j.gde.2006.04.011
17. Morris JK, Alberman E, Scott C, et al. Is the prevalence of Klinefelter syndrome increasing? Eur J Hum Genet. 2008;16(2):163–170. doi:10.1210/jc.2009-2354
18. Thomas NS, Collins AR, Hassold TJ, Jacobs PA. A reinvestigation of non-disjunction resulting in 47,XXY males of paternal origin. Eur J Hum Genet. 2000;8(10):805–808. doi:10.1038/sj.ejhg.5200531
19. Hassold TJ, Sherman SL, Pettay D, et al. XY chromosome nondisjunction in man is associated with diminished recombination in the pseudoautosomal region. Am J Hum Genet Pt A. 1991;49(2):253–260.
20. Samango-Sprouse C, Banjevic M, Ryan A, et al. SNP-based non-invasive prenatal testing detects sex chromosome aneuploidies with high accuracy. Prenat Diagn. 2013;33(7):643–649. doi:10.1002/pd.4159
21. Samango-Sprouse C, Keen C, Sadeghin T, Gropman A. The benefits and limitations of cell-free DNA screening for 47,XXY (Klinefelter syndrome). Prenat Diagn. 2017;37(5):497–501. doi:10.1002/pd.5044
22. Iitsuka Y, Bock A, Nguyen DD, et al. Evidence of skewed X-chromosome inactivation in 47,XXY and 48,XXYY Klinefelter patients. Am J Med Genet Pt A. 2001;98(1):25–31. doi:10.1002/1096-8628(20010101)98:1<25::AID-AJMG1015>3.0.CO;2-X
23. Gregg AR, Gross SJ, Best RG, et al. ACMG statement on noninvasive prenatal screening for fetal aneuploidy. Genet Med. 2013;15(5):395–398. doi:10.1038/gim.2013.29
24. Practice Bulletin No. 163: Screening for Fetal Aneuploidy. Obstet Gynecol. 2016;127(5):e123–137. doi:10.1097/aog.0000000000001406.
25. Gregg AR, Skotko BG, Benkendorf JL, et al. Noninvasive prenatal screening for fetal aneuploidy, 2016 update: a position statement of the American College of Medical Genetics and Genomics. Genet Med. 2016;18(10):1056–1065. doi:10.1038/gim.2016.97
26. NSGC Headquarters. Prenatal cell-free DNA screening: position statement. Available from: https://www.nsgc.org/p/bl/et/blogaid=805. Published October 11, 2016. Updated April 12, 2018.Accessed October 17, 2019.
27. Samango-Sprouse CA, Porter GF, Lasutschinkow P, et al. Investigating the impact of early diagnosis and non-invasive prenatal testing (NIPT): knowledge, attitudes, and experiences of 55 parents of children with sex chromosome aneuploidies (SCAs). In press 2019.
28. Ramdaney A, Hoskovec J, Harkenrider J, Soto E, Murphy L. Clinical experience with sex chromosome aneuploidies detected by noninvasive prenatal testing (NIPT): accuracy and patient decision-making. Prenat Diagn. 2018;38(11):841–848. doi:10.1002/pd.5339
29. Abramsky L, Chapple J. 47,XXY (Klinefelter syndrome) and 47,XYY: estimated rates of and indicated for postnatal diagnosis with implications for prenatal counseling. Prenat Diag. 1997;17:363–368. doi:10.1002/(SICI)1097-0223(199704)17:4<363::AID-PD79>3.0.CO;2-O
30. Simpson JL, de la Cruz FF, Swerdloff RS, et al. Klinefelter syndrome: expanding the phenotype and identifying new research directions. Genet Med. 2003;5(6):460–468.
31. Aksglaede L, Link K, Giwercman A, et al. 47,XXY Klinefelter syndrome: clinical characteristics and age-specific recommendations for medical management. Am J Med Genet C. 2013;163C(1):55–63. doi:10.1002/ajmg.c.31349
32. Samango-Sprouse C, Rogol A. The hidden disability and a prototype for an infantile presentation of developmental dyspraxia (IDD). Infants Young Child. 2002;15:11–118. doi:10.1097/00001163-200207000-00004
33. Radicioni AF, De Marco E, Gianfrilli D, et al. Strategies and advantages of early diagnosis in Klinefelter’s syndrome. Mol Hum Reprod. 2010;16(6):434–440. doi:10.1093/molehr/gaq027
34. Mehta A, Paduch DA. Klinefelter syndrome: an argument for early aggressive hormonal and fertility management. Fertil Steril. 2012;98(2):274–283. doi:10.1016/j.fertnstert.2012.06.001
35. Ross JL, Samango-Sprouse C, Lahlou N, et al. Early androgen deficiency in infants and young boys with 47,XXY Klinefelter syndrome. Horm Res. 2005;64:39–45. doi:10.1159/000087313
36. Van Rijn S, Swaab H. Executive dysfunction and the relation with behavioral problems in children with 47,XXY and 47, XXX. Genes Brain Behav. 2015;14(2):200–208. doi:10.1111/gbb.12203
37. Patwardhan AJ, Eliez S, Bender B, et al. Brain morphology in Klinefelter syndrome: extra X chromosome and testosterone supplementation. Neurology. 2000;54(12):2218–2223. doi:10.1212/wnl.54.12.2218
38. Nagai M, Kishi K, Kato S. Insular cortex and neuropsychiatric disorders: a review of recent literature. Eur Psychiatry. 2007;22(6):387–394. doi:10.1016/j.eurpsy.2007.02.006
39. Ross JL, Kushner H, Kowal K, et al. Androgen treatment effects on motor function, cognition, and behavior in boys with Klinefelter syndrome. Pediatr. 2017;185:193–199. doi:10.1016/j.jpeds.2017.02.036
40. Aksglaede L, Petersen JH, Main KM, Skakkebaek NE, Juul A. High normal testosterone levels in infants with non-mosaic Klinefelter’s syndrome. Eur J Endocrinol. 2007;157(3):345–350. doi:10.1530/EJE-07-0310
41. Cabrol S, Ross JL, Fennoy I, et al. Assessment of Leydig and Sertoli cell functions in infants with nonmosaic Klinefelter syndrome: insulin-like peptide 3 levels are normal and positively correlated with LH levels. J Clin Endocrinol Metab. 2001;96(4):E746–E753. doi:10.1210/jc.2010-2103
42. Lahlou N, Fennoy I, Carel JC, et al. Inhibin B and anti-Mullerian hormone, but not testosterone levels, are normal in infants with nonmosaic Klinefelter syndrome. J Clin Endocrinol Metab. 2004;89(4):1864–1868. doi:10.1210/jc.2003-031624
43. Ross JL, Roeltgen DP, Stefanatos G, et al. Cognitive and motor development during childhood in boys with Klinefelter syndrome. Am J Med Genet Pt A. 2008;146(6):708–719. doi:10.1002/ajmg.a.32232
44. Samango-Sprouse CA, Stapleton E, Chea S, et al. International investigation of neurocognitive and behavioral phenotype in 47,XXY (Klinefelter syndrome): predicting individual differences. Am J Med Genet Pt A. 2018;176(4):877–885. doi:10.1002/ajmg.a.38621
45. Bhasin S, Cunningham GR, Hayes FJ, et al. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010;95(6):2536–2559. doi:10.1210/jc.2009-2354
46. Topper E, Dickerman Z, Prager-Lewin R, et al. Puberty in 24 patients with Klinefelter syndrome. Eur J Pediatr. 1982;139(1):8–12. doi:10.1007/bf00442070
47. Salbenblatt JA, Bender BG, Puck MH, et al. Pituitary-gonadal function in Klinefelter syndrome before and during puberty. Pediatr Res. 1985;19(1):82–86. doi:10.1203/00006450-198501000-00022
48. Wikström AM, Dunkel L, Wickman S, et al. Are adolescent boys with Klinefelter syndrome androgen deficient? A longitudinal study of Finnish 47, XXY boys. Pediatr Res. 2006;59(6):854–859. doi:10.1203/01.pdr.0000219386.31398.c3
49. Aksglaede L, Molgaard C, Skakkebæk NE, Juul A. Normal bone mineral content but unfavourable muscle/fat ratio in Klinefelter syndrome. Arch Dis Child. 2008;93(1):30–34. doi:10.1136/adc.2007.120675
50. Wikström AM, Raivio T, Hadziselimovic F, et al. Klinefelter syndrome in adolescence: onset of puberty is associated with accelerated germ cell depletion. J Clin Endocrinol Metab. 2004;89(5):2263–2270. doi:10.1210/jc.2003-031725
51. Hall S, Marteau TM, Limbert C, et al. Counselling following the prenatal diagnosis of Klinefelter syndrome: comparisons between geneticists and obstetricians in five European countries. Public Health Genomics. 2001;4(4):233–238. doi:10.1159/000064198
52. Chang S, Skakkebæk A, Gravholt CH. Klinefelter syndrome and medical treatment: hypogonadism and beyond. Hormones. 2015;14(4):531–548. doi:10.14310/horm.2002.1622
53. Raboch J, Mellan J, Starka L. Klinefelter’s syndrome: sexual development and activity. Arch Sex Behav. 1979;8(4):333–339. doi:10.1007/bf01541877
54. Becker KL. Clinical and therapeutic experiences with Klinefelter’s syndrome. Fertil Steril. 1972;23(8):568–578.
55. Wosnitzer MS, Paduch DA. Endocrinological issues and hormonal manipulation in children and men with Klinefelter syndrome. Am J Med Genet C Semin Med Genet. 2013;163C:16–26. doi:10.1002/ajmg.c.31350
56. Ferguson-Smith M, Mack WS, Ellis PM, et al. Paternal age and the source of the X chromosomes in XXY Klinefelter syndrome. Lancet. 1964;283(7323):41–46. doi:10.1016/S0140-6736(64)92198-1
57. Christiansen P, Andersson AM, Skakkebaek NE. Longitudinal studies of inhibin B levels in boys and young adults with Klinefelter syndrome. J Clin Endocrinol Metab. 2003;88(2):888–891. doi:10.1210/jc.2002-021379
58. Wiesner G, Everman D, Cassidy S. Constitutional chromosome disorders in adults. In: King R, Rotter J, Motulsky A, editors. The Genetic Basis of Common Diseases.
59. Seo JT, Park YS, Lee JS. Successful testicular sperm extraction in Korean Klinefelter syndrome. Urol. 2004;64(6):1208–1211. doi:10.1016/j.urology.2004.07.013
60. Vernaeve V, Staessen C, Verheyen G, et al. Can biological or clinical parameters predict testicular sperm recovery in 47,XXY Klinefelter’s syndrome patients? Hum Reprod. 2004;19(5):1135–1139. doi:10.1093/humrep/deh253
61. Franik YS, Hoeijmakers K, D’Hauwers DDM, et al. Klinefelter syndrome and fertility: sperm preservation should not be offered to children with Klinefelter syndrome. Hum Reprod. 2016;31(9):1951–1959. doi:10.1093/humrep/dew179
62. Ferlin A, Gavolla A, Forester C. Chromosome abnormalities in sperm of individuals with constitutional sex chromosome abnormalities. Cytogenet Genome Res. 2005;111(3–4):310–316. doi:10.1159/000086905
63. Friedler S, Raziel A, Strassburger D, et al. Outcome of ICSI using fresh and cryopreserved-thawed testicular spermatozoa in patients with non-mosaic Klinefelter’s syndrome. Hum Reprod. 2001;16(12):2616–2620. doi:10.1093/humrep/16.12.2616
64. Staessen C, Tournaye H, Van Assche E, et al. PGD in 47,XXY Klinefelter’s syndrome patients. Hum Reprod Update. 2003;9(4):319–330. doi:10.1093/humupd/dmg029
65. Ron-El R, Strassburger D, Gelman-Kohan S, et al. A 47,XXY fetus conceived after ICSI of spermatozoa from a patient with non-mosaic Klinefelter’s syndrome: case report. Hum Reprod. 2000;15(8):1804–1806. doi:10.1093/humrep/15.8.1804
66. Visootsak J, Graham JM
67. Bandmann HJ, Breit R, Perwein E, eds. Klinefelter’s Syndrome. Springer Science & Business Media; 2012.
68. Fideleff HL, Boquete HR, Suárez MG, et al. Burden of growth hormone deficiency and excess in children. Prog Mol Biol Transl Sci. 2016;138:143–166. doi:10.1016/bs.pmbts.2015.10.009
69. Bojesen A, Birkebaek N, Kristensen K, et al. Bone mineral density in Klinefelter syndrome is reduced and primarily determined by muscle strength and resorptive markers, but not directly by testosterone. Osteoporos Int. 2011;22(5):1441–1450. doi:10.1007/s00198-010-1354-7
70. Mehta A, Bolyakov A, Roosma J, et al. Successful testicular sperm retrieval in adolescents with Klinefelter syndrome treated with at least 1 year of topical testosterone and aromatase inhibitor. Fertil Steril. 2013;100(4):970–974. doi:10.1016/j.fertnstert.2013.06.010
71. Stagi S, Di Tommaso M, Manoni C, et al. Bone mineral status in children and adolescents with Klinefelter syndrome. Int J Endocrinol. 2016;(4):1–9. doi:10.1155/2016/3032759
72. Samango-Sprouse CA, Lasutschinkow P, Powell S, et al. The incidence of anxiety symptoms in boys with 47,XXY (Klinefelter syndrome) and the possible impact of timing of diagnosis and hormonal replacement therapy. Am J Med Genet Pt A. 2019;179(3):423–428. doi:10.1002/ajmg.a.61038
73. Van Rijn S, Swaab H, Aleman A, et al. X Chromosomal effects on social cognitive processing and emotion regulation: a study with Klinefelter men (47,XXY). Schizophr Res. 2006;84(2):194–203. doi:10.1016/j.schres.2006.02.020
74. Van Rijn S, Swaab H, Aleman A, et al. Social behavior and autism traits in a sex chromosomal disorder: Klinefelter (47,XXY) syndrome. J Autism Dev Disord. 2008;38(9):1034–1041. doi:10.1007/s10803-008-0542-1
75. Bruining H, Swaab H, Kas M, et al. Psychiatric characteristics in a self-selected sample of boys with Klinefelter syndrome. Pediatr. 2009;123(5):e865–e870. doi:10.1542/peds.2008-1954
76. Ross JL, Roeltgen DP, Kushner H, et al. Behavioral and social phenotypes in boys with 47,XYY syndrome or 47,XXY Klinefelter syndrome. Pediatr. 2012;129(4):769–778. doi:10.1542/peds.2011-0719
77. Visootsak J, Graham JM. Social function in multiple X and Y chromosome disorders: XXY, XYY, XXYY, XXXY. Dev Disabil Res Rev. 2009;15(4):328–332. doi:10.1002/ddrr.76
78. Bojesen A, Juul S, Birkebæk N, et al. Increased mortality in Klinefelter syndrome. J Clin Endocrinol Metab. 2004;89(8):3830–3834. doi:10.1210/jc.2004-0777
79. Seminog OO, Seminog AB, Yeates D, et al. Associations between Klinefelter’s syndrome and autoimmune diseases: English national record linkage studies. Autoimmunity. 2015;48(2):125–128. doi:10.3109/08916934.2014.968918
80. Swerdlow AJ, Shoemaker MJ, Higgins CD, et al. Cancer incidence and mortality in men with Klinefelter syndrome: a cohort study. J Natl Cancer Inst. 2005;97:1204–1210. doi:10.1093/jnci/dji240
81. Rocca MS, Pecile V, Cleva L, et al. The Klinefelter syndrome associated with high recurrence of copy number variations on the X chromosome with a potential role in the clinical phenotype. Andrology. 2016;4(2):328–334. doi:10.1111/andr.12146
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.