")
Back to Archived Journals » Hypoxia » Volume 5
Update on hypoxia-inducible factors and hydroxylases in oxygen regulatory pathways: from physiology to therapeutics
Authors Ratcliffe P , Koivunen P, Myllyharju J , Ragoussis J, Bovée JV, Batinic-Haberle I, Vinatier C , Trichet V, Robriquet F, Oliver L, Gardie B
Received 30 November 2016
Accepted for publication 23 December 2016
Published 15 March 2017 Volume 2017:5 Pages 11—20
DOI https://doi.org/10.2147/HP.S127042
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Dörthe Katschinski
Peter Ratcliffe,1,2 Peppi Koivunen,3 Johanna Myllyharju,3 Jiannis Ragoussis,4 Judith VMG Bovée,5 Ines Batinic-Haberle,6 Claire Vinatier,7 Valérie Trichet,8 Florence Robriquet,9 Lisa Oliver,9 Betty Gardie9,10
1Target Discovery Institute, University of Oxford, 2The Francis Crick Institute, London, UK; 3Oulu Center for Cell-Matrix Research, Biocenter Oulu and Faculty of Biochemistry and Molecular Medicine, University of Oulu, Oulu, Finland; 4McGill University and Genome Quebec Innovation Centre, McGill University, Montreal, Canada; 5Department of Pathology, Leiden University Medical Center, Leiden, the Netherlands; 6Department of Radiation Oncology, Duke University School of Medicine, Durham, NC, USA; 7INSERM UMR 1229, Regenerative Medicine and Skeleton-RMeS, Team STEP, University of Nantes, UFR Odontology, 8UMR 1238 INSERM, Université de Nantes, Faculté de Médecine, 9CRCINA, INSERM, Université de Nantes, Nantes, 10Ecole Pratique des Hautes Etudes, PSL Research University, Paris, France
Abstract: The “Hypoxia Nantes 2016” organized its second conference dedicated to the field of hypoxia research. This conference focused on “the role of hypoxia under physiological conditions as well as in cancer” and took place in Nantes, France, in October 6–7, 2016. The main objective of this conference was to bring together a large group of scientists from different spheres of hypoxia. Recent advances were presented and discussed around different topics: genomics, physiology, musculoskeletal, stem cells, microenvironment and cancer, and oxidative stress. This review summarizes the major highlights of the meeting.
Keywords: hypoxia, genomics, lipid metabolism, musculoskeletal, cancer, oxidative stress
Introduction
Maintenance of oxygen homeostasis is a fundamental physiological challenge. Dysregulation of homeostasis with a consequent hypoxia is a component of many human diseases. Transcriptional response to hypoxia is mediated by hypoxia inducible factors (HIF1–3), the oxygen-sensitive signal being generated by a series of protein hydroxylases that catalyze prolyl and asparaginyl hydroxylation on specific residues in the regulatory HIF-α subunits (HIF1α, HIF-2α, and HIF-3α). Mammalian HIF-α subunits contain two hydroxylation sites called NODD (N-terminal Oxygen-dependant Degradation Domain) and CODD (C-terminal Oxygen-dependant Degradation Domain). In the presence of oxygen, prolyl hydroxylation by prolyl hydroxylase domain proteins (PHD1–4) directs HIF-α for proteasome destruction following binding by the product of the tumor suppressor gene von Hippel–Lindau (pVHL) and ubiquitination (Figure 1).1,2 In addition, asparaginyl hydroxylation by factor inhibiting HIF (FIH) blocks recruitment of HIF co-factors. The HIF hydroxylases belong to two distinct groups of Fe(II)- and 2-oxoglutarate (2-OG)-dependent dioxygenase, which split O2 and couple oxidation (hydroxylation) of HIF-α to oxidative decarboxylation of 2-OG to succinate and CO2. PHD2, in particular, has been described to play a dominant role in oxygen sensing.3,4 In the absence of oxygen, hydroxylases are inhibited, HIF-α factors are stabilized, enter the nucleus, and associate with HIF-β subunits (ie, aryl hydrocarbon receptor nuclear translocators ARNT1 or ARNT2) and cofactors (p300). These complexes form active transcription factors that bind hypoxia responsive elements (HREs) and drive the expressions of more than hundred target genes.5,6 These transcripts encode proteins that play a role in multiple biological pathways (cell survival, erythropoiesis, angiogenesis, metabolism, etc).7,8 The panel of genes expressed in different cell types is specific, and their regulation by different HIF isoforms is complex and still puzzling. In the meeting, Prof P Ratcliffe and Prof I Ragoussis presented recent pan-genomic studies that revealed some aspects of the hypoxia pathway complexity.
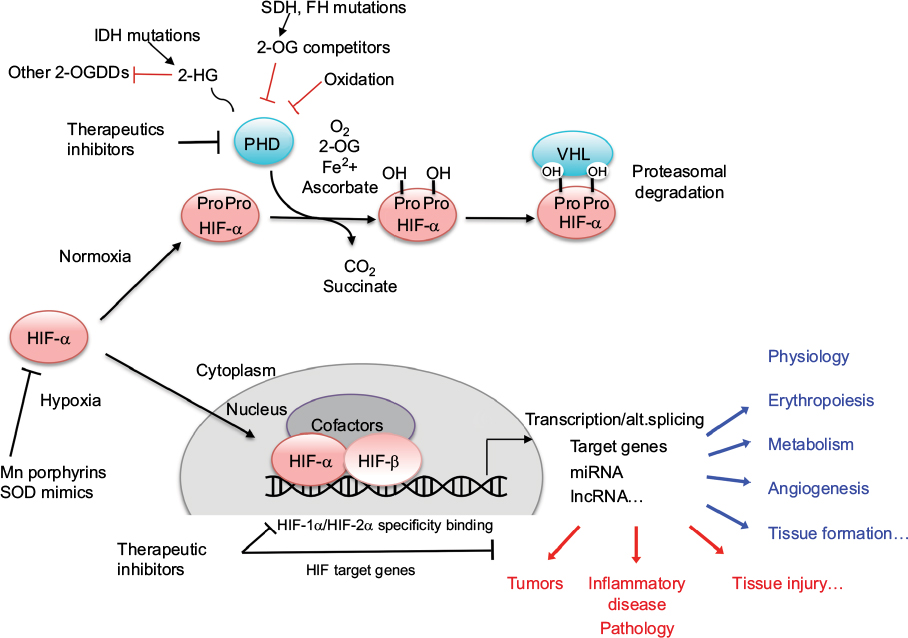 | Figure 1 Schematic representation of the hypoxia regulation pathway. Abbreviations: 2-OG, 2-oxoglutarate; 2-HG, 2-hydroxyglutarate; 2-OGDDs, 2-oxoglutarate-dependent dioxygenases; IDH, isocitrate dehydrogenase; FH, fumarate hydratase; SDH, succinate dehydrogenase; PHD, prolyl hydroxylase domain protein; VHL, von Hippel–Lindau; HIF, hypoxia inducible factor; alt.splicing, alternative splicing; SOD, superoxide dismutase. |
Complexity of the Hypoxia pathway
Complex HIF hydroxylase modulation and transcriptional architecture of HIF-1α versus HIF-2α binding
Prof P Ratcliffe provided an overview of the HIF hydroxylase pathway and considered opportunities and challenges in its therapeutic modulation, specifically whether it might be possible (and advantageous) to modulate specific components of the pathway under specific clinical settings. The evolution of the HIF prolyl hydroxylase pathway was outlined, highlighting the primitive PHD2/HIF-1α/VHL triad.9 Of particular relevance to the question of specific therapeutic modulation is the appearance of multiple HIF and PHD isoforms, through gene duplication events at the base of vertebrate evolution.10 This raises a question as to what extent these different isoforms have particular functions in the specialized oxygen delivery systems (the blood, vascular, and cardiopulmonary systems) of vertebrates, which are often the target of diseases. As a paradigm, the question of whether PHD inhibition could generate medically useful, isoform specific, activation of HIF-1α versus HIF-2α was considered.
Although the differential regulation of HIF-α isoforms is not completely understood, they demonstrate differential dependence on each of two prolyl hydroxylation sites in the N-terminal and C-terminal portions of their oxygen-dependent degradation domains (NODD and CODD).11 Thus, inhibitors that operate differentially on the hydroxylation of each of these sites might impart at least partial HIF-α isoform selectivity. Initial evidence that this might be possible has been provided by the action of human mutations in PHD2, some of which generate striking differences in hydroxylation of HIF-1α NODD versus CODD.12 The feasibility of generating relatively specific inhibitors is also supported by differences in the binding of human NODD and CODD to PHD212 and from the ability of some, but not all, PHD inhibitors to displace CODD substrate as well as 2-OG from PHD2.13 All the PHDs, and ~60–70 other human dioxygenases in this family, use 2-OG as co-substrate; hence, inhibitors that simply act as 2-OG analogs are unlikely to be highly specific.14 Thus, it was argued that greater efforts would be required to develop substrate specific inhibitors and that at least some selectivity would likely be feasible.
In light of this, recent work on the functional differentiation of HIF-1α versus HIF-2α was considered from two contrasting perspectives: 1) transcriptional biology, as assessed by pan-genomic assays of the HIF transcriptional response using ChIPseq and RNAseq analyses, and 2) the integrated physiology of hypoxia, assessed in recombinant mice bearing inactivating alleles of HIF-1α and HIF-2α.
As an example of HIF-α isoform-specific actions on integrated physiology, recent work on erythropoiesis and ventilator sensitivity to hypoxia was considered.15,16 Increased erythropoiesis and enhanced ventilator sensitivity to hypoxia are key physiological components of the process of acute acclimatization. Direct comparison of conditional inactivation of HIF-1α with HIF-2α in adult life (as a surrogate for potential actions of specific pharmacological intervention) reveals marked specificity for the HIF-2α isoforms, at least under the conditions of experimental testing.15,16 This is consistent with, and likely in part driven by, contrasting patterns of expression. HIF-2α is expressed at much higher levels in the renal erythropoietin-producing fibroblasts and in the type 1 cells of the carotid body.17,18 Thus, it might be concluded that specific activation of HIF-2α would be of value in therapeutic strategies that aimed to augment these responses (ie, in the correction of anemia or in the stimulation of ventilation).
As a counterpoise to this work, recent insights into the transcriptional biology of HIF were then considered from the perspective of pan-genomic studies that contrast patterns of HIF1α and HIF-2α at transcriptional target loci. These studies have defined distinct, though partially overlapping, sets of DNA binding sites for HIF-1α and HIF-2α.19 Depending on the level of analytical stringency applied, each isoform binds at hundreds to a few thousand sites, at levels sufficient to have an effect on transcription. No differences for HIF-1α versus HIF-2α are seen in consensus binding sequences. However, marked differences are observed across the genome in the distribution of binding at >106 potential hypoxia response elements in open chromatin. Gene ontology programs define differences in the functional pathways associated with gene loci that preferentially bind HIF-1α or HIF-2α, but these are generally not tightly demarcated; rather moderate pathway bias is observed in one or other isoform.20 Interestingly, significant biases are also observed in the type of transcriptional target, with HIF-2α showing greater enrichment than HIF-1α at loci associated with long non-coding RNAs.21 However, the most striking differences are observed in the distance between the HIF-α binding sites and gene promoters.20,22 HIF-1α is much more frequently bound close to promoters, with HIF-2α being more commonly bound at a distance from promoters. Moreover, even when both the HIF-α isoforms bound close to a promoter, functional studies reveal that it is generally HIF-1α that mediates the transcriptional response.23 Thus, when considered both are considered at the level of transcriptional biology and in the setting of integrated physiology, HIF-2α manifests features consistent with it being the “modern” isoform. HIF-2α mediates several of the adaptive responses to hypoxia that are specific to higher animals; it binds and functions at greater distances from its target genes, its expression is more cell-type specific, and it exhibits more frequent interactions with non-coding RNA networks. However, none of these properties is absolutely specific.
In the discussion, it was speculated that this lack of clear organization was a fundamental reflection of Darwinian evolution, in which mutations are generated agnostically and alter specific biochemical processes, but selection is applied to the performance of integrated physiological systems. This process runs counter to the aim of dissecting highly specific drug targets out of complex systems. The conclusions were that the development of more specific HIF hydroxylase inhibitors, which target components of the HIF pathway with moderate levels of selectivity, would be feasible. Nevertheless, accurate predictions about what might or might not be useful in a specific medicinal situation remain difficult. The development of multiple inhibitors with diverse kinetic and chemical properties, coupled careful testing in physiological models, and (ideally) human experimental medicine studies, would be an ideal approach to this new area of medicine development.
Complex expression of genes in hypoxia identified by RNAseq
A major part of the adaptive response to hypoxia is mediated through transcriptional regulation, with the HIF transcription factors taking central stage. As presented by Prof I Ragoussis, the detailed study of the transcriptional response is conditional upon developing and applying genomic technologies that allow the global characterization of a wide universe of transcripts in the cell. Methodological developments utilizing the power of short read next generation sequencing technology have been applied, which allow the analysis of long and short transcripts following the removal of the highly abundant ribosomal RNA, while also retaining information on transcriptional orientation. Thus, for the first time, it was possible to identify global transcriptional changes under hypoxia in all classes of coding and non-coding RNAs, including small RNAs.21 As a result, our understanding of the transcriptional response to hypoxia is greatly enhanced and now covers the entire spectrum of RNAs, coding and non-coding, regulatory, microRNAs, t-RNAs, small nuclear RNAs, and others. In addition, through the integration of ChIP data produced using antibodies against HIF-1α, HIF-2α, HIF-1β, and PolII as well as the histones H3K4Me3 and H3K4me1, it was possible to determine that poised PolII is localized at the promoters of both coding and non-coding transcripts, the increased transcription of which is associated with HIF binding.21 More recently, this binding has been determined to take place at pre-existing promoter, promoter–enhancer, and enhancer–enhancer interactions using chromatin conformation assays.23 The role of long non-coding RNAs in hypoxia response is now the focus of intense research.24 Typical examples include HIF-2α regulated lncRNA NEAT1 that leads to the identification of paraspeckle formation as a response to hypoxia25 in breast cancer cells, whereas H19 is found to play a role in cell cycle in hypoxic HUVEC cells.26
There is good evidence that alternative splicing contributes to a substantial part of the hypoxic response and is also directly regulated by HIF and can lead to the production of non-coding mRNA isoforms implicated in upregulated genes.27–30 Sena et al27 observed a dichotomy between high frequency of exon inclusion in hypoxia up-regulated genes, whereas exon exclusion was found predominant in downregulated genes using HEP3B cells. Following that, Memon et al29 observed that there is a major switch to non-coding isoforms in hypoxic HTCC116 human colon carcinoma cells, which was recapitulated in a large set of colorectal cancer samples from TCGA. More work is needed in a wider range of tissues and cancers in order to confirm the observations related to HIF’s role in splicing, the trends related to up- or down-regulated transcripts and any pathway-specific splicing effects, but it is clear that alternative splicing plays a major role in the hypoxia response.
In terms of technological developments, inferring alternatively spliced isoforms of genes from short read data through statistical assignment of the most probable combination of exons is still computationally challenging and not very accurate. New, single molecule sequencing technologies are now enabling the direct sequencing of complete cDNA or even RNA molecules and their application31,32 has the potential to allow the characterization of the hypoxia transcriptome at even higher resolution.
Hypoxia pathway in physiology and therapies
In the meeting, sessions were dedicated to the physiological aspects of hypoxia. In fact, the physiological reaction to low oxygen induces adaptation and is essential to cell survival. As described earlier, the HIFs induce the expression of hundred of genes that play roles in many biological pathways.
Role on glucose and lipid metabolism
Prof P Koivunen presented HIF target genes and use of mouse models to study the connection of HIF with glucose and lipid metabolism. In agreement with a key role of hypoxia in lipid metabolism, genetic inhibition of Phd2 has recently been shown to reduce body weight and the amount of white adipose tissue (WAT) in mice.33 These mice hypomorphic for Phd2 also had smaller adipocytes, less WAT inflammation, and better glucose tolerance, and did not develop insulin resistance when fed with high-fat diet (HFD) or aged.33 Moreover, they had lowered serum cholesterol levels and an improved high density lipoprotein (HDL)/low density lipoprotein (LDL) + very low density lipoprotein (VLDL) cholesterol ratio and were protected against steatohepatitis.33 The molecular-level determinants of the phenotype were upregulation of the HIF-1α target Glut1 and Glut4 mRNAs, several enzymes of glycolysis and Pdk1 mRNA in several tissues, downregulation of some inflammatory mRNAs in WAT and upregulation of the insulin sensitivity increasing, HIF2α target Irs2 mRNA and the concomitant downregulation of the lipogenesis regulating Srebp1c mRNA and its downstream targets Fas, Acca, and Scd1 in the liver.33,34 Mice deficient for Phd2 in adipose tissue also showed resistance to HFD-induced obesity and had a better glucose tolerance.35 In another study, knockout of Phd2 in adipocytes blunted lipolysis and increased intracellular lipid storages, therefore reducing ectopic lipid deposition.36 When wild-type mice were treated with a pharmacological PHD inhibitor FG-4497, obesity and metabolic syndrome opposing phenotype was observed.33 In support of these data, clinical trials with two different PHD inhibitors, FG-4592 (Roxadustat) and GSK1278863, for the treatment of anemia and peripheral vascular disease, respectively, report lowered serum cholesterol levels and an improved HDL/LDL profile with the subjects treated with the inhibitors.37,38 Dyslipidemia and metabolic syndrome predispose to atherosclerosis. Recent data from mice associate inhibition of PHDs 1 and 2 with protection against atherosclerosis.34,39 The mechanisms involved were HIF-mediated modifications to metabolism including that for cholesterol, reduced inflammation, and beneficial alterations to the immune system.34,39 Although metabolic syndrome and the connected diseases are highly associated with life style only a minority of patients succeed in its improvement and end up in medication. Recent data provide evidence for justification to explore PHD inhibitors and activation of the endogenous hypoxia response pathway, to treat obesity, metabolic dysfunction, and atherosclerosis.
Role on musculoskeletal system
Prof J Myllyharju presented another example of the physiological role of the hypoxia pathway, related to the regulation of extracellular matrix homeostasis.40 The presentation focused on the roles of collagen prolyl 4-hydroxylases (C-P4Hs) and lysyl oxidase (LOX) in the musculoskeletal system. Indeed, HIF induces the expression of several key enzymes such as CP4Hs, lysyl hydroxylases, and LOX that are required for proper collagen synthesis and assembly, and hence correct structure and function of connective tissues.41,42 Remodeling of extracellular matrix by these collagen-modifying enzymes has recently been shown to be important in hypoxic cancer metastasis.43,44 C-P4Hs are essential enzymes for collagen synthesis as the modifications catalyzed by these enzymes are required for thermal stability of collagens at body temperature. The C-P4H family consists of three isoenzymes, C-P4Hs I and II being the major forms.41 CP4H-I is expressed ubiquitously, whereas C-P4H-II has a more restricted expression pattern, being a prominent form in for example chondrocytes and osteoblasts.41 It has been shown that HIF-1α-regulated induction of C-P4Hs is necessary for sufficient collagen production in a hypoxic tissue environment such as, for example, in the cartilage chondrocytes.45 Studies with gene-modified mice lines have shown that c-P4h-1−/−mice die during early embryogenesis because of nonfunctional assembly of collagen IV into basement membranes.46 Reduced C-P4H activity in c-P4h-1+/−/ c-P4h-2−/−double mutant mice leads to several connective tissue abnormalities. These include abnormal development of cartilage and bone resulting in structural and biomechanical impairment of these tissues and chondrodysplasia.47 Similar, but much milder abnormalities are present in the c-P4h-2−/−single mutant mice.47 As no signs of uncompensated endoplasmic reticulum stress were observed in these mice, the main cause of the chondrodysplasia is likely to be aberrant mechanosensing and signaling cues mediated by the abnormal extracellular matrix.47 Another collagen-modifying enzyme highly induced by HIF is LOX that catalyzes crosslink formation in collagen fibrils and elastin and affects the maturation, turnover, and stiffness of connective tissue.42 LOX is the major isoenzyme of the LOX family.42 It has been shown that lack of LOX interferes with the development of several connective tissues resulting in dysfunction of the cardiovascular and respiratory systems, aortic aneurysms, and perinatal lethality in mouse.48,49 Interestingly, it has been shown recently that muscle composition is regulated by a LOX–TGFβ feedback loop. Lack of LOX was found to lead to excess expression of TGFβ that disrupts the balance between the amounts of myofibers and that of muscle connective tissue.50 This results in short and small muscles with reduced amount of myofibers and abnormal muscle patterning.50 Remarkably, this abnormal muscle development could be rescued by TGFβ inhibition.50 The regulatory effect of LOX on TGFβ signaling may be explained by either direct interaction between LOX and TGFβ or its receptor and/or increased susceptibility of insufficiently crosslinked collagens and elastin to proteolysis, which could result in elevated liberation of TGFβ from the extracellular matrix reservoir of the Lox−/− tissue.50
Hypoxia pathway in pathology and therapies
The meeting dedicated a session to the pathological aspect of hypoxia. The oxygen-sensing pathway may be dysregulated with a consequent constitutive activation of HIF transcription factors and target genes expression as observed, for example, in many types of tumors. The hypoxia pathway participates and sometimes initiates progression of a vast majority of tumors. The causes of HIF stabilization are wide-ranging, from a chronic hypoxic microenvironment to mutations in direct key regulator genes. The causes may also be indirect with the inhibition of hydroxylases by the production of 2-OG mimetics (following mutations in metabolic enzymes) or by oxidation (modification of the redox status, oxidation of co-factors, etc).
Example of chondrosarcoma
Prof J Bovée presented chondrosarcomas as an example of tumors linked to hypoxia. Chondrosarcoma is the second most frequent primary bone malignancy, predominantly affecting adults. They usually arise from their benign precursor lesions: 1) peripheral chondrosarcoma arising in osteochondroma (at the surface of bone, with mutations in exostosin genes EXT1 or EXT2 encoding glycosyltransferases involved in heparan sulphate biosynthesis) and 2) central chondrosarcoma arising in enchondroma (in the medulla of bone, with mutations in isocitrate dehydrogenase genes IDH1 or IDH2). The prognosis is strongly correlated with histological grading. Grade I chondrosarcoma, now reclassified as an atypical cartilaginous tumor, is poorly vascularized, behaves locally aggressive, but typically does not metastasize. High-grade chondrosarcomas (grades II and III) have an increased vascularity and increased metastases corresponding to poor patient survival. Surgery is the mainstay of treatment. If the tumor location is non-resectable or metastatic, there is still no curative treatment.51,52 Chondrosarcoma is notorious for its primary resistance to conventional chemo- and radiotherapy. 51,53 A multistep genetic model has been devised: while specific mutations (IDH, EXT) cause the benign precursor lesions, high-grade chondrosarcomas have complex karyotypes54 with many additional mutations.50 Since cartilaginous tissue as well as chondrosarcomas has a hypoxic microenvironment,55 it has been hypothesized that hypoxia is involved in chondrosarcoma aggressiveness, and this is supported by the increased microvessel density, vascular endothelial growth factor (VEGF), and HIF-1α levels in high-grade chondrosarcoma as compared to low grade.56,57 Interestingly, mutations in the IDH genes are found in up to 80% of enchondromas and ~50% of chondrosarcomas. IDH is involved in the tricarboxylic acid cycle. Mutations in IDH lead to the formation of a neoenzyme that catalyzes the reduction of 2-OG to D-2-hydroxyglutarate (D2HG),58 which is considered an oncometabolite and inhibits some α ketoglutarate dependent oxygenases. Regarding the effect of D2HG on prolyl hydroxylase and HIF pathway, mutations in IDH were first shown to associate with stabilization of HIF-1α,59 but later it was shown that D2HG produced by these mutations is not a potent PHD inhibitor.60,61 An inhibitory effect of D2HG has been demonstrated on the dioxygenase ten-elevan-translocation-2 (TET2),62 a dioxygenase that catalyzes hydroxylation of methylated DNA. This results in inhibition of DNA demethylation, causing hypermethylation,62,63 and altered histone60 that impacts tumor progression.
Example of Mn porphyrin-based redox-active drugs as anticancer therapeutics
Some aspects of anticancer therapies have been presented during the meeting. Notably, Prof I Batinic-Haberle focused on manganese (Mn) porphyrin-based superoxide dismutase (SOD) mimics as anticancer therapeutics. Porphyrins are molecules that comprise the active site of numerous key enzymes (hemoglobin, myoglobin, prolyl hydroxylase, cyt P450 oxidases, nitric oxide synthases, oxygenases, etc). These enzymes contain macrocyclic protoporphyrin ring that encapsulates iron and affords extreme stability to iron complex.
Batinic-Haberle, Spasojevic, Tovmasyan, and other collaborators have developed very stable and redox-active manganese porphyrins (MnPs) as powerful mimics of superoxide dismutase (SOD) family of enzymes.64–66 They have mimicked the nature in its use of iron protoporphyrin as active site of different proteins to run major metabolic functions such as oxygen transport and detoxification among those. Yet, instead of protoporphyrin bound to iron, MnP-based SOD mimics have N-substituted pyridylporphyrins bound to manganese. MnPs exhibit remarkable ability to protect normal tissue from radiation (RT)-induced damage, such as brain, salivary glands, mouth mucosa, colon, eye, prostate, hematopoietic stem cells, and lung. Moreover, MnPs are powerful tumor radio-and chemosensitizers as demonstrated with lymphoma and tumors of brain, breast, head and neck, ovary, and prostate. Based on differential properties of MnPs to protect normal while killing cancerous tissues, two lead compounds, MnTE-2-PyP5+ and MnTnBuOE-2-PyP5+, progressed to Phase I/II clinical trials, the latter at Duke University.
MnPs as mimics of superoxide family of enzymes also react with numerous other reactive species (RS) such as peroxynitrite (ONOO−), nitric oxide (.NO), and hypochlorite (ClO−) (reviewed in64–66). The actions of MnPs are also H2O2-driven and are frequently coupled with cellular reductants such as ascorbate, glutathione (GSH), and protein thiols. Although they have insignificant catalase-like activity, MnPs possess GSH peroxidase and thiol oxidase-like activities. The magnitude of all actions of MnPs thus far studied, including SOD-like activity, parallels the magnitude of their therapeutic efficacies and is dependent upon the metal-centered reduction potential of MnIIIP/MnIIP redox couple; in turn the magnitude of those actions parallels each other. In addition to reacting with different RS, MnPs inhibit activities of several transcription factors, such as HIF-1α, NF-κB, AP-1, SP-1, and Nrf2, thereby affecting cellular apoptotic and proliferative pathways.64–66 The nature of such interactions has mostly been explored with respect to NF-κB and has been ascribed to MnP-catalyzed and H2O2/GSH-driven oxidation/S-glutathionylation of thiols in its p50 and p65 subunits. Preliminary data also indicate the major role of Nrf2 in the actions of MnPs.
MnPs exhibit differential effects to normal and tumor tissues presumably by impacting NFκB/HIF-1α/Nrf2 pathways.64–66 The HIF and NF-κB pathways are closely related. Hypoxia contributes to the development of inflammation, at least in part through the activation and/or potentiation of NF-κB, a master regulator of genes involved in innate immunity, inflammation, and apoptosis.67,68 The differential effects of MnPs on these pathways arise from different redox environments and diverse accumulation of MnPs in those tissues. In normal cells/tissues, MnPs reduce inflammation. For example, in a rat pulmonary radioprotection study, MnPs suppressed RT-induced lung inflammation through the inhibition of HIF-1α activation and downregulation of its target gene VEGF, which seemed to be controlled by NF-κB.69,70
The cancer cell, though, relative to the normal cell, has excessive levels of endogenous H2O2, and thereby is often under oxidative stress and sensitive to any further increase in oxidative stress. This is due to its perturbed redox environment, often with increased MnSOD levels but insufficient levels and/or activities of peroxide-removing enzymes.64–66 Such situation has been heavily exploited in anticancer therapies, one of which includes MnP alone and/or combined with exogenously imposed oxidative stress. MnP, when administered at high concentration, exhibited an anticancer effect in its own right in a 4T1 breast cancer mouse model,71 where large inhibition of HIF-1α activity (presumably orchestrated through oxidation and subsequent inactivation of NF-kB) contributed to the suppression of angiogenesis. In the same model, the anticancer effect of MnP was further enhanced when it was given jointly with exogenous sources of H2O2 – RT, chemotherapy, or ascorbate.64 Furthermore, the massive MnP/H2O2/GSH-driven suppression of anti-apoptotic NF-κB was demonstrated in a cellular lymphoma model where lymphoma cells were exposed to MnP and dexamethasone.69,72 In addition, complexes I and III of mitochondrial respiration were S-glutathionylated, and in turn inactivated, resulting in the loss of ATP.
The excessive tumor H2O2 levels, along with up to 10-fold higher accumulation of MnPs in tumor than in normal tissue73 – as major reactants in a process of S-gluthathionylation – drive higher tumor yield of NF-κB oxidation relative to normal tissue. Consequently, apoptotic processes are favored in the tumor, whereas a suppression of inflammation occurs in normal tissue (Figure 2).66
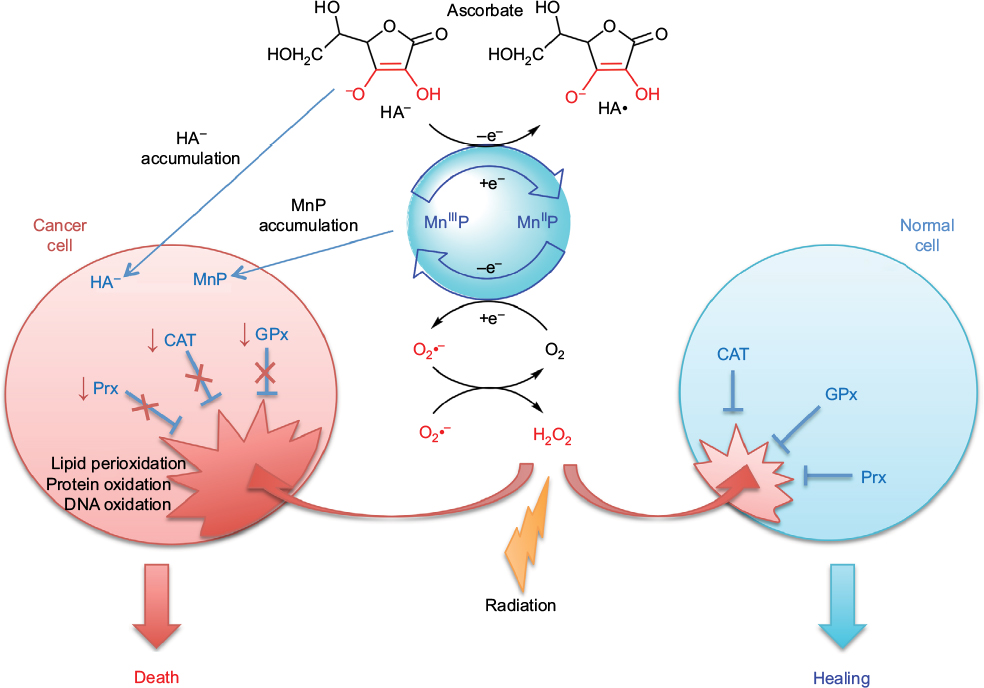 | Figure 2 Differential redox environments of cancer versus normal cells drive differential therapeutic outcomes. Notes: In tumor cells compared to normal cells, MnSOD is frequently upregulated, whereas peroxide-removing enzymes are downregulated or inactive, which gives rise to higher H2O2 levels resulting in higher oxidative stress. When anticancer therapies that further enhance tumor oxidative stress, such as redox-active drugs (MnP) in combination with radiation, and/or chemotherapy and/or ascorbate, are applied, the differential effects are observed in normal (suppression of inflammation leading to tissue healing) versus cancerous tissue (death). Adapted from Redox-Active Therapeutics. Mn porphyrin-based redox-active therapeutics. 2016:165–212. Batinić-Haberle I, Tovmasyan A, Spasojević I. © 2016, with permission of Springer.66 Abbreviations: SOD, superoxide dismutase; HA–, ascorbate; HA•, ascorbyl radical; CAT, catalase; GPx, glutathione peroxidase; Prx, peroxiredoxin; O2•–, superoxide; O2, oxygen; H2O2, hydrogen peroxide; MnIIIP and MnIIP, Mn porphyrin with manganese center in oxidized (+3) or reduced (+2) form, respectively. |
Drugs, commonly known as antioxidants/vitamins, such as ascorbate, tocopherols, and carotenoids, have been tested in clinical trials as single anticancer drugs with only marginal effect. The lack of effect is still not fully understood. Batinic-Haberle et al have contributed recently to the understanding of why that maybe so.63,73 In order to affect cancer cell metabolism, the redox active drug needs to inhibit anti-apoptotic pathways. It seems that such inhibition does not happen primarily by removing RS, as thought for a long while, but instead by using RS to oxidize signaling protein cysteines. The major function of ascorbate under physiological conditions is to protect tocopherol; it cycles with oxidized tocopherol radical thereby regenerating tocopherol. Yet at higher levels, achieved if added exogenously, ascorbate cycling with endogenous metal complexes (such as cyt P450 oxidases) will result in a large production of cytotoxic H2O2, which in turn will oxidize and inactivate thiols of critical cellular proteins (such as NF-κB and complexes I and III of mitochondrial respiration). In turn, tumor growth may be moderately suppressed. Yet, a massive anticancer effect could be achieved with increased yield of protein oxidation, when additional source(s) of H2O2 (RT and chemotherapy) and/or catalyst, optimized for ascorbate oxidation (such as MnTE-2-PyP5+ or MnTnBuOE-2PyP5+), are applied (Figure 2). 69,72–74
Conclusion
This meeting covered large themes of the hypoxia field and highlighted the complexity of the oxygen sensing pathway. It opened discussion about perspectives to better understand its role in physiology and disease occurrence and to specifically target it for therapies.
Acknowledgments
The meeting “Hypoxia Nantes 2106” (https://hypoxia.univ-nantes.fr) was financially supported by Alliance Bio Expertise, Baker Ruskinn, the Région Pays de la Loire, project “ERRATA” and “EryCan”; Laboratory of Excellence GR-Ex, reference ANR-11-LABX-0051; Nantes University; and Dove Press. We thank the organizing committee for their invaluable help: Franck Verrecchia, Christophe Merceron, Richard Pace, Jérôme Guicheux, Elizabeth Bernardo, Stéphanie Olivo, and Sabine Goude.
Disclosure
IBH is a consultant with BioMimetix JVLLC and holds equities in BioMimetix JVLLC. IBH and Duke University have patent rights and have licensed technologies to BioMimetix JVLLC. The authors report no other conflicts of interest in this work.
References
Maxwell PH, Wiesener MS, Chang GW et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399(6733):271–275. | ||
Stebbins CE, Kaelin WG Jr, Pavletich NP. Structure of the VHL-ElonginC-ElonginB complex: implications for VHL tumor suppressor function. Science. 1999;284(5413):455–4561. | ||
Berra E, Benizri E, Ginouvès A, Volmat V, Roux D, Pouysségur J. HIF prolylhydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J. 2003;22(16):4082–4090. | ||
Gardie B, Percy MJ, Hoogewijs D, et al. The role of PHD2 mutations in the pathogenesis of erythrocytosis. Hypoxia (Auckl). 2014;2:71–90. | ||
Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE. 2005;(306):re12. | ||
Mole DR, Blancher C, Copley RR, et al. Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcripts. J Biol Chem. 2009;284(25):16767–16775. | ||
Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3(10):721–32. | ||
Pouysségur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006;441(7092):437–443. | ||
Bishop T, Ratcliffe PJ. Signaling hypoxia by hypoxia-inducible factor protein hydroxylases: a historical overview and future perspectives. Hypoxia (Auckl). 2014; 5;2:197–213. | ||
Loenarz C, Coleman ML, Boleininger A, et al. The hypoxia-inducible transcription factor pathway regulates oxygen sensing in the simplest animal, Trichoplax adhaerens. EMBO Rep. 2011;12(1):63–70. | ||
Appelhoff RJ, Tian YM, Raval RR, et al. Differential function of the prolyl hydroxylases PHD1, PHD2 and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279(37):38458–38465. | ||
Chowdhury R, Leung IK, Tian YM, et al. Structural basis for oxygen degradation domain selectivity of the HIF prolyl hydroxylases. Nat Commun. 2016;7:12673. | ||
Chan MC, Atasoylu O, Hodson E, et al. Potent and selective triazole-based inhibitors of the hypoxia-inducible factor prolyl-hydroxylases with activity in the murine brain. PLoS One. 2015;10(7):e0132004. | ||
Chan MC, Holt-Martyn JP, Schofield CJ, Ratcliffe PJ. Pharmacological targeting of the HIF hydroxylases – a new field in medicine development. Mol Aspects Med. 2016;47–48:54–75. | ||
Franke K, Gassmann M, Wielockx B. Erythrocytosis: the HIF pathway in control. Blood. 2013;122(7):1122–1128. | ||
Hodson EJ, Nicholls LG, Turner PJ, et al. Regulation of ventilatory sensitivity and carotid body proliferation in hypoxia by the PHD2/HIF-2 pathway. J Physiol. 2016;594(5):1179–1195. | ||
Rosenberger C, Mandriota SJ, Jurgensen JS, et al. Expression of hypoxia-inducible factor1a and -2a in hypoxic and ischemic rat kidneys. A Soc Nephrol. 2002;13:1721–1732. | ||
Zhou T, Chien M-S, Kaleem S, Matsunami H. Single cell transcriptome analysis of mouse carotid body glomus cells. J Physiol. 2016;594:4225–4251. | ||
Schödel J, Oikonomopoulos S, Ragoussis J, Pugh CW, Ratcliffe PJ, Mole DR. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood. 2012;117(23):e207–e217. | ||
Schödel J, Mole DR, Ratcliffe PJ. Pan-genomic binding of hypoxia-inducible transcription factors. Biol Chem. 2013;394(4):507–517. | ||
Choudhry H, Schodel J, Oikonomopoulos S, et al. Extensive regulation of the non-coding transcriptome by hypoxia: role of HIF in releasing paused RNApol2. EMBO Rep. 2014;15(1):70–76. | ||
Salama R, Masson N, Simpson P, et al. Heterogeneous effects of direct hypoxia pathway activation in kidney cancer. PLoS One. 2015;10(8):e0134645. | ||
Platt JL, Salama R, Smythies J, et al. Capture-C reveals preformed chromatin interactions between HIF-binding sites and distant promoters. EMBO Rep. 2016;17(10):1410–1421. | ||
Choudhry H, Harris AL, McIntyre A. The tumour hypoxia induced non-coding transcriptome. Mol Aspects Med. 2016;47–48:35–53. | ||
Choudhry H, Albukhari A, Morotti M, et al. Tumor hypoxia induces nuclear paraspeckle formation through HIF-2α dependent transcriptional activation of NEAT1 leading to cancer cell survival. Oncogene. 2015;34(34):4546. | ||
Voellenkle C, Garcia-Manteiga JM, Pedrotti S, et al. Implication of long noncoding RNAs in the endothelial cell response to hypoxia revealed by RNA-sequencing. Sci Rep. 2016;6:24141. | ||
Sena JA, Wang L, Heasley LE, Hu CJ. Hypoxia regulates alternative splicing of HIF and non-HIF target genes. Mol Cancer Res. 2014;12(9): 1233–1243. | ||
Jakubauskiene E, Vilys L, Makino Y, Poellinger L, Kanopka A. Increased serine-arginine (SR) protein phosphorylation changes pre-mRNA splicing in hypoxia. J Biol Chem. 2015;290(29):18079–18089. | ||
Memon D, Dawson K, Smowton CSF, Xing W, Dive C, Miller CJ. Hypoxia-driven splicing into noncoding isoforms regulates the DNA damage response. Npj Genom Med. 2016;1:16020. | ||
Yao Y, Shang J, Song W, Deng Q, Liu H, Zhou Y. Global profiling of the gene expression and alternative splicing events during hypoxia-regulated chondrogenic differentiation in human cartilage endplate-derived stem cells. Genomics. 2016;107(5):170–177. | ||
Sharon D, Tilgner H, Grubert F, Snyder M. A single-molecule long-read survey of the human transcriptome. Nat Biotechnol. 2013;31(11):1009–1014. | ||
Oikonomopoulos S, Wang YC, Djambazian H, Badescu D, Ragoussis J. 2016. Benchmarking of the Oxford Nanopore MinION sequencing for quantitative and qualitative assessment of cDNA populations. Sci Rep. 2016;6:31602. | ||
Rahtu-Korpela L, Karsikas S, Hörkkö S, et al. HIF prolyl 4-hydroxylase-2 inhibition improves glucose and lipid metabolism and protects against obesity and metabolic dysfunction. Diabetes. 2014;63(10):3324–3333. | ||
Rahtu-Korpela L, Määttä J, Dimova EY, et al. Hypoxia-inducible factor prolyl 4-hydroxylase-2 inhibition protects against development of atherosclerosis. Arterioscler Thromb Vasc Biol. 2016;36(4):608–617. | ||
Matsuura H, Ichiki T, Inoue E, et al. Prolyl hydroxylase domain protein 2 plays a critical role in diet-induced obesity and glucose intolerance. Circulation. 2013;127(21):2078–2087. | ||
Michailidou Z, Morton NM, Moreno Navarrete JM, et al. Adipocyte pseudohypoxia suppresses lipolysis and facilitates benign adipose tissue expansion. Diabetes. 2015;64(3):733–745. | ||
Bakris GL, Yu K-P, Leong R, et al. Effects of a novel anemia treatment, FG-4592 –an oral hypoxia-inducible prolyl hydroxylase inhibitor (HIF-PHI) on blood pressure and cholesterol in patients with chronic kidney disease J Clin Hypertension. 2013;14:487–489. | ||
Olson E, Demopoulos L, Haws TF, et al. Short-term treatment with a novel HIF-prolyl hydroxylase inhibitor (GSK1278863) failed to improve measures of performance in subjects with claudication-limited peripheral artery disease. Vasc Med. 2014;19(6):473–482. | ||
Marsch E, Demandt JA, Theelen TL, et al. Deficiency of the oxygen sensor prolyl hydroxylase 1 attenuates hypercholesterolaemia, atherosclerosis, and hyperglycaemia. Eur Heart J. 2016;37(39):2993–2997. | ||
Myllyharju J, Schipani E. Extracellular matrix genes as hypoxia-inducible targets. Cell Tissue Res. 2010;339(1):19–29. | ||
Myllyharju J. Prolyl 4-hydroxylases, key enzymes in the synthesis of collagens and regulation of the response to hypoxia, and their roles as treatment targets. Ann Med. 2008;40(6):402–417. | ||
Mäki JM. Lysyl oxidases in mammalian development and certain pathological conditions. Histol Histopathol. 2009;24(5):651–660. | ||
Cox TR, Gartland A, Erler JT. Lysyl oxidase, a targetable secreted molecule involved in cancer metastasis. Cancer Res. 2016;76(2):188–192. | ||
Semenza GL. The hypoxic tumor microenvironment: a driving force for breast cancer progression. Biochim Biophys Acta. 2016;1863(3):382–391. | ||
Aro E, Khatri R, Gerard-O’Riley R, Mangiavini L, Myllyharju J, Schipani E. Hypoxia-inducible factor-1 (HIF-1) but not HIF-2 is essential for hypoxic induction of collagen prolyl 4-hydroxylases in primary newborn mouse epiphyseal growth plate chondrocytes. J Biol Chem. 2012;287(44):37134–37144. | ||
Holster T, Pakkanen O, Soininen R, et al. Loss of assembly of the main basement membrane collagen, type IV, but not fibril-forming collagens and embryonic death in collagen prolyl 4-hydroxylase I null mice. J Biol Chem. 2007;282(4):2512–2519. | ||
Aro E, Salo AM, Khatri R, et al. Severe extracellular matrix abnormalities and chondrodysplasia in mice lacking collagen prolyl 4-hydroxylase isoenzyme II in combination with a reduced amount of isoenzyme I. J Biol Chem. 2015;290(27):16964–16978. | ||
Mäki JM, Räsänen J, Tikkanen H, et al. Inactivation of the lysyl oxidase gene Lox leads to aortic aneurysms, cardiovascular dysfunction, and perinatal death in mice. Circulation. 2002;106(19):2503–2509. | ||
Mäki JM, Sormunen R, Lippo S, et al. Lysyl oxidase is essential for normal development and function of the respiratory system and for the integrity of elastic and collagen fibers in various tissues. Am J Pathol. 2005;167(4):927–936. | ||
Kutchuk L, Laitala A, Soueid-Bomgarten S, et al. Muscle composition is regulated by a Lox-TGFβ feedback loop. Development. 2015;142(5):983–993. | ||
Gelderblom H, Hogendoorn PCW, Dijkstra SD, et al. The clinical approach towards chondrosarcoma. Oncologist. 2008;13:320–329. | ||
Speetjens FM, de Jong Y, Gelderblom H, Bovee JV. Molecular oncogenesis of chondrosarcoma: impact for targeted treatment. Curr Opin Oncol. 2016;28:314–322. | ||
van Oosterwijk JG, Herpers B, Meijer D, et al. Restoration of chemosensitivity for doxorubicin and cisplatin in chondrosarcoma in vitro: BCL-2 family members cause chemoresistance. Ann Oncol. 2011;23:1617–1626. | ||
Bovee JV, Hogendoorn PCW, Wunder JS, Alman BA. Cartilage tumours and bone development: molecular pathology and possible therapeutic targets. Nat Rev Cancer 2010;10:481–488. | ||
Gibson JS, Milner PI, White R, Fairfax TP, Wilkins RJ. Oxygen and reactive oxygen species in articular cartilage: modulators of ionic homeostasis. Pflugers Arch. 2008;45:563–573. | ||
Kubo T, Sugita T, Shimose S, Matsuo T, Arihiro K, Ochi M. Expression of hypoxia-inducible factor-1alpha and its relationship to tumour angiogenesis and cell proliferation in cartilage tumours. J Bone Joint Surg Br. 2008;90:364–370. | ||
Boeuf S, Bovee JVMG, Lehner B, Hogendoorn PCW, Richter W. Correlation of hypoxic signalling to histological grade and outcome in cartilage tumours. Histopathology. 2010;56:641–651. | ||
Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2hydroxyglutarate. Nature. 2010;465:966. | ||
Zhao S, Lin Y, Xu W, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science. 2009;324:261–265. | ||
Chowdhury R, Yeoh KK, Tian YM, et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011;12:463–469. | ||
Koivunen P, Lee S, Duncan CG, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutaratelinked to EGLN activation. Nature. 2012;483(7390):484–488. | ||
Xu, W, Yang H, Liu Y, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. | ||
Pansuriya TC, van ER, d’Adamo P, et al. Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat Genet. 2011;43:1256–1261. | ||
Batinic-Haberle I, Tovmasyan A, Roberts ER, et al. SOD therapeutics: latest insights into their structure-activity relationships and impact on the cellular redox-based signaling pathways. Antioxid Redox Signal. 2014;20(15):2372–2415. | ||
Batinic-Haberle I, Tovmasyan A, Spasojevic I. An educational overview of the chemistry, biochemistry and therapeutic aspects of Mn porphyrins – from superoxide dismutation to H2O2-driven pathways. Redox Biol. 2015;5(0):43–65. | ||
Batinić-Haberle I, Tovmasyan A, Spasojević I. Mn porphyrin-based redox-active therapeutics. In: Batinić-Haberle I, Rebouças JS, Spasojević I, editors. Redox-Active Therapeutics. Cham: Springer International Publishing; 2016;165–212. | ||
Oliver KM, Garvey JF, Ng CT, et al. Hypoxia activates NF-kappaB-dependent gene expression through the canonical signaling pathway. Antioxid Redox Signal. 2009;11(9):2057–2564. | ||
Zhou J, Schmid T, Brune B. Tumor necrosis factor-alpha causes accumulation of a ubiquitinated form of hypoxia inducible factor-1α through a nuclear factor-κB dependent pathway. Mol Biol Cell. 2003;14(6):2216–2225. | ||
Jaramillo MC, Briehl MM, Crapo JD, Batinic-Haberle I, Tome ME. Manganese porphyrin, MnTE-2-PyP5+, acts as a pro-oxidant to potentiate glucocorticoid-induced apoptosis in lymphoma cells. Free Radic Biol Med. 2012;52(8):1272–1284. | ||
Tse HM, Milton MJ, Piganelli JD. Mechanistic analysis of the immunomodulatory effects of a catalytic antioxidant on antigen-presenting cells: implication for their use in targeting oxidation-reduction reactions in innate immunity. Free Radic Biol Med. 2004;36(2):233–247. | ||
Rabbani ZN, Spasojevic I, Zhang X, et al. Antiangiogenic action of redox-modulating Mn(III) meso-tetrakis(N-ethylpyridinium-2-yl)porphyrin, MnTE-2-PyP(5+), via suppression of oxidative stress in a mouse model of breast tumor. Free Radic Biol Med. 2009;47(7):992–1004. | ||
Jaramillo MC, Briehl MM, Batinic-Haberle I, Tome ME. Manganese (III) meso-tetrakis N-ethylpyridinium-2-yl porphyrin acts as a pro-oxidant to inhibit electron transport chain proteins, modulate bioenergetics, and enhance the response to chemotherapy in lymphoma cells. Free Radic Biol Med. 2015;83(0):89–100. | ||
Tovmasyan A, Sampaio RS, Boss MK, et al. Anticancer therapeutic potential of Mn porphyrin/ascorbate system. Free Radic Biol Med. 2015;89:1231–1247. | ||
Tovmasyan, Go, Y-M, Jones D, Spasojevic I, Batinic-Haberle I. Redox proteomics of 4T1 breast cancer cell after treatment with MnTE-2-PyP5+/ascorbate system. Free Radic Biol Med. 2016;100(S1):S93. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.