Back to Journals » Therapeutics and Clinical Risk Management » Volume 17
Update on Diagnosis and Treatment of Adult Pulmonary Alveolar Proteinosis
Authors Iftikhar H, Nair GB, Kumar A ![]()
Received 14 April 2021
Accepted for publication 19 June 2021
Published 10 August 2021 Volume 2021:17 Pages 701—710
DOI https://doi.org/10.2147/TCRM.S193884
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Hira Iftikhar,1 Girish B Nair,1 Anupam Kumar2
1Division of Pulmonary and Critical Care, Beaumont Health, OUWB School of Medicine, Royal Oak, MI, USA; 2Division of Pulmonary and Critical Care, Baylor College of Medicine, Houston, TX, USA
Correspondence: Hira Iftikhar Tel +1 2485251389
Email [email protected]
Abstract: Pulmonary alveolar proteinosis (PAP) is a rare pulmonary surfactant homeostasis disorder resulting in buildup of lipo-proteinaceous material within the alveoli. PAP is classified as primary (autoimmune and hereditary), secondary, congenital and unclassifiable type based on the underlying pathogenesis. PAP has an insidious onset and can, in some cases, progress to severe respiratory failure. Diagnosis is often secured with bronchoalveolar lavage in the setting of classic imaging findings. Recent insights into genetic alterations and autoimmune mechanisms have provided newer diagnostics and treatment options. In this review, we discuss the etiopathogenesis, diagnosis and treatment options available and emerging for PAP.
Keywords: alveolar macrophage, surfactant protein, granulocyte macrophage-colony stimulating factor, whole lung lavage, lung transplant, gene therapy, alveolar proteinosis
Introduction
Pulmonary alveolar proteinosis (PAP) is a rare syndrome which was first described in 1958.1 The epidemiology of PAP remains poorly defined given the difficulty with an early and appropriate diagnosis. In a recent study, McCarthy and colleagues estimated the prevalence of PAP to be 6.87 per million in the general population, with no gender predilection.2 In another study from Japan, the prevalence was noted to be slightly lower at 6.2 per million in the general population.3
PAP is characterized by the disruption of pulmonary surfactant homeostasis and resultant accumulation of lipo-proteinaceous material within the alveoli. Pulmonary surfactant proteins are secreted by type II alveolar epithelial cells and are removed by alveolar macrophages, which are in close contact with the epithelial cells of the alveoli. Surfactant proteins maintain lung function and structural integrity of the alveolar surface as they create an air-liquid interface. The appreciation of the role of GM-CSF in the surfactant homeostasis has paved the way for a better understanding of the pathogenesis of PAP. GM-CSF plays a multifactorial role in the surfactant homeostasis. Specifically, it is key for the maturation and terminal differentiation of macrophages present in the alveoli. Furthermore, GM-CSF is crucial for the immune function of alveolar macrophages as well as myeloid cells. Studies have clearly linked the auto-antibody mediated disruption of GM-CSF signaling as an important factor in the pathogenesis of PAP.4 As a result of these findings, a lot of interest has converged on GM-CSF, both for purposes of diagnosis and as a target for treatment. Other mechanisms of pathogenesis include genetic alterations or gene mutations encoding surfactant proteins leading to ineffective surfactant. However, despite our improved understanding of pathogenesis of PAP, there are limited treatment options available for this condition.
Classification of PAP
PAP is classified according to the underlying pathogenetic mechanism (Table 1 summarizes the different types of PAP). This classification also helps determine the treatment options for PAP.
 |
Table 1 Types of Pulmonary Alveolar Proteinosis (PAP) |
Primary PAP: Autoimmune and Hereditary PAP
Primary PAP includes two separate disease entities: autoimmune PAP and hereditary PAP. Autoimmune PAP, which is the most common type of PAP accounts for 90% of cases. There is a high concentration of anti-GM-CSF immunoglobulin IgG antibodies in patients with autoimmune PAP.5,6 GM-CSF autoantibodies attack GM-CSF signaling and hamper its ability to stimulate alveolar macrophage maturation. A dysfunctional alveolar macrophage is unable to degrade the surfactant and has impaired phagocytic function. Collectively, this causes accumulation of undegraded surfactant and increases predisposition for infections. Aside from auto-antibodies, it is possible that there are other pathways that contribute to this phenomenon. It is also interesting to note that autoimmune PAP is rarely associated with other autoimmune diseases. Indeed, in a study, only seven (1.7%) of 410 patients diagnosed with PAP had a co-diagnosis of another autoimmune disease.7
Hereditary PAP accounts for only 3% of all PAP syndromes and GM-CSF antibody is negative in those patients.2 It involves the genetic mutation of the GM-CSF receptor subunits. This results in dysfunction of alveolar macrophage receptors that interact with GM-CSF, which then impairs the ability of GM-CSF to stimulate alveolar macrophages. Hereditary PAP and autoimmune PAP are, otherwise, clinically and histologically identical.
Secondary PAP
Secondary PAP accounts for 4% of all PAP syndromes.2 Here, an underlying disease causes a quantitative and/or qualitative defect of alveolar macrophages, which further results in the disruption of surfactant homeostasis and the buildup of surfactant in the alveoli. It is mainly caused by underlying hematological diseases, including myelodysplastic syndromes and malignancy. Other causes of secondary PAP include medications, chemotherapy, immune deficiencies, chronic infections, including HIV, chronic inflammatory syndromes, and dusts’ inhalation (titanium, silica, aluminum, and others).8 Hence, any clinical condition that can potentially cause quantitative or qualitative macrophage dysfunction can disrupt surfactant homeostasis and result in secondary PAP.
Congenital PAP
Congenital PAP accounts for 1.5% of PAP syndromes.2 It is a disease of the newborn but is rarely also seen in adults. It is caused by mutations that disrupt surfactant production and function. Notable mutations associated with congenital PAP are as follows: in SFTPB (SP-B), SFTPC (SP-C), ABCA3, and TTF18.
Unclassified PAP
There are few patients with PAP, who do not fit any of the criteria mentioned above. They do not have GM-CSF antibodies, any known secondary causes, mutations, or surfactant dysfunction. These patients are grouped as unclassified.
Clinical Presentation
The onset of PAP is often insidious and subtle. As the surfactant accumulates over time, the disease often has an indolent course. An acute form of PAP from excessive silica exposure has been reported but is rarely seen nowadays due to better occupational regulations.9 In a registry from Japan, only two-thirds of the patients were symptomatic at the time of diagnosis.3 Isolated dyspnea was the most common symptom, occurring in 39% of symptomatic patients, followed by dyspnea and cough (11% of the symptomatic patients) and cough only (10% of the symptomatic patients). Other infrequent symptoms included fever and weight-loss.3 The prevalence of smokers (56–80%) and occupational exposure to various inhaled dusts (23–39%) is high, especially with secondary PAP.3,10 Due to the role played by GM-CSF in immune mediated functions of alveolar macrophage function, patients with PAP are also at higher risk for systemic infections.11 Physical examination includes inspiratory crackles in about half of the patients, cyanosis is present in one-quarter of patients, and digital clubbing in a small percentage.12
Pulmonary function tests are non-specific but, in advanced diseases, may show restriction, particularly with the development of fibrosis.13 Disease severity in terms of the presence of symptoms and degree of hypoxemia correlates with reduction in DLCO. Different scoring systems, such as Severity and Prognosis Score of PAP (SPSP) and Disease Severity Score (DSS) incorporating symptoms, arterial blood gas, lung function parameters, smoking status and radiographic scores have been developed for assessing disease severity in patients with PAP.14,15 Both the scores, SPSP and DSS, are reliable to assess disease severity but are not useful for predicting disease progression or relapse.
Diagnosis of PAP
The diagnostic suspicion for PAP typically arises from the indolent nature of disease progression combined with radiographic features. Inflammatory markers including C-reactive protein (CRP), erythrocyte sedimentation rate (ESR) and lactate dehydrogenase (LDH) can be elevated. Amongst serum biomarkers, lactate dehydrogenase (LDH) has been found to be elevated in up to 80% of patients.7 However, LDH is of limited value in diagnosis due to its non-specificity. Several other biomarkers have been studied for PAP (SP-A, SP-D, KL-6, CYFRA 21–1, YKL-40 and CEA), but are not specific and are not available for routine clinical use. Hence, serological testing of biomarkers is generally not useful for PAP diagnosis.
The presence of circulating antibodies to GM-CSF is specific to auto-immune PAP and helps distinction from other types of PAP. Circulating GM-CSF antibody titers may also predict response to treatment.16 It is noteworthy, though, that GM-CSF antibody can be found in healthy individuals. However, concentration less than 10 μg/mL in serum has a good negative predictive value to rule out disease, whereas concentration greater than 19 μg/mL is specific to auto-immune PAP.17 The latex agglutination test used for the detection of GM-CSF antibodies has a diagnostic sensitivity of 100% and specificity of 98%.5 This laboratory test is performed only at highly specialized centers.
Patients diagnosed with PAP without GM-CSF autoantibodies, or a secondary cause for PAP, should be evaluated for GM-CSF concentration test and GM-CSF signaling tests for direct specific gene analyses.
Chest radiograph findings for patients with PAP can vary and are non-specific. Chest radiographs are similar in appearance to pulmonary edema with diffuse, symmetric, bilateral alveolar opacities with hilar and basal distribution. However, unlike pulmonary edema, other features of cardiac disease including cardiomegaly and pleural effusions are usually absent. CT chest, specifically high-resolution CT (HRCT) has a vital role in the diagnosis of PAP. The characteristic “crazy-paving” pattern, widely attributed to PAP, is not pathognomonic of the disease. Based on a review of 139 CT scans, the most common pattern in these patients was uniform distribution followed by lower lung zone predominance.18 Holbert and et al reported ground-glass and consolidative air-space, opacities having a homogeneous craniocaudal distribution, whereas interlobular opacities were prominent at the lung bases. Ground-glass opacities (100%) and interlobular opacities (85%) were more common; however, substantial fibrosis (7%) was reported less frequently. Serial CT scans in the same patient frequently vary over time, which suggests PAP can resolve spontaneously or can progress unremittingly, sometimes despite treatment.19 Figure 1 shows Chest CT images of patients with PAP.
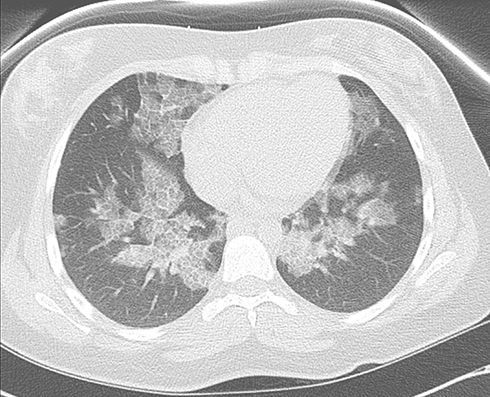 |
Figure 1 Chest CT of a patient with PAP demonstrating interlobular and intralobular septal thickening in crazy-paving pattern. |
Features not typical of PAP include focal areas of airspace disease with bulky lymphadenopathy. These features should raise the concern for alternative diagnosis, including superimposed infection and malignancy. The degree of severity on C.T. scan images is associated with disease severity and pulmonary function impairment. Ultra-low-dose CT scan can be helpful in quantifying the affected lung parenchyma, and therefore provide a sensitive and objective assessment of PAP. Furthermore, the imaging assessment has good correlation with lung function parameters.13
In most cases, bronchoalveolar lavage fluid (BALF) establishes the diagnosis with its characteristic milky and opaque appearance (lipoproteinacous material). BALF settles into a cloudy sediment layer and a translucent supernatant. In some cases, BALF might appear normal if performed in a lung zone unaffected by the disease process. BALF microscopy reveals foamy, large macrophages containing eosinophilic granules in the presence of extracellular hyaline material which is positive on Periodic Acid-Schiff (PAS) stain and negative on Alcian blue staining. Electron microscopy demonstrates lamellar bodies which are abundant in concentrically laminated structures.20 These findings are diagnostic of PAP, but they do not establish the type of PAP. Bronchoscopy, in addition, can also exclude concurrent infections, PAP mimics and secondary causes of PAP.
BAL findings in the correct clinical context and consistent CT-scan findings are often sufficient for the diagnosis. If the BAL appearance is atypical, obtaining tissue via bronchoscopy will help ascertain the diagnosis. Surgical lung biopsy is seldom required for the diagnosis of PAP.6 In a cohort of 203 PAP patients, an open lung biopsy was performed for diagnosis in 8% of cases and transbronchial biopsy in 42% of cases.3 Table 2 summarizes the diagnostic features of PAP.
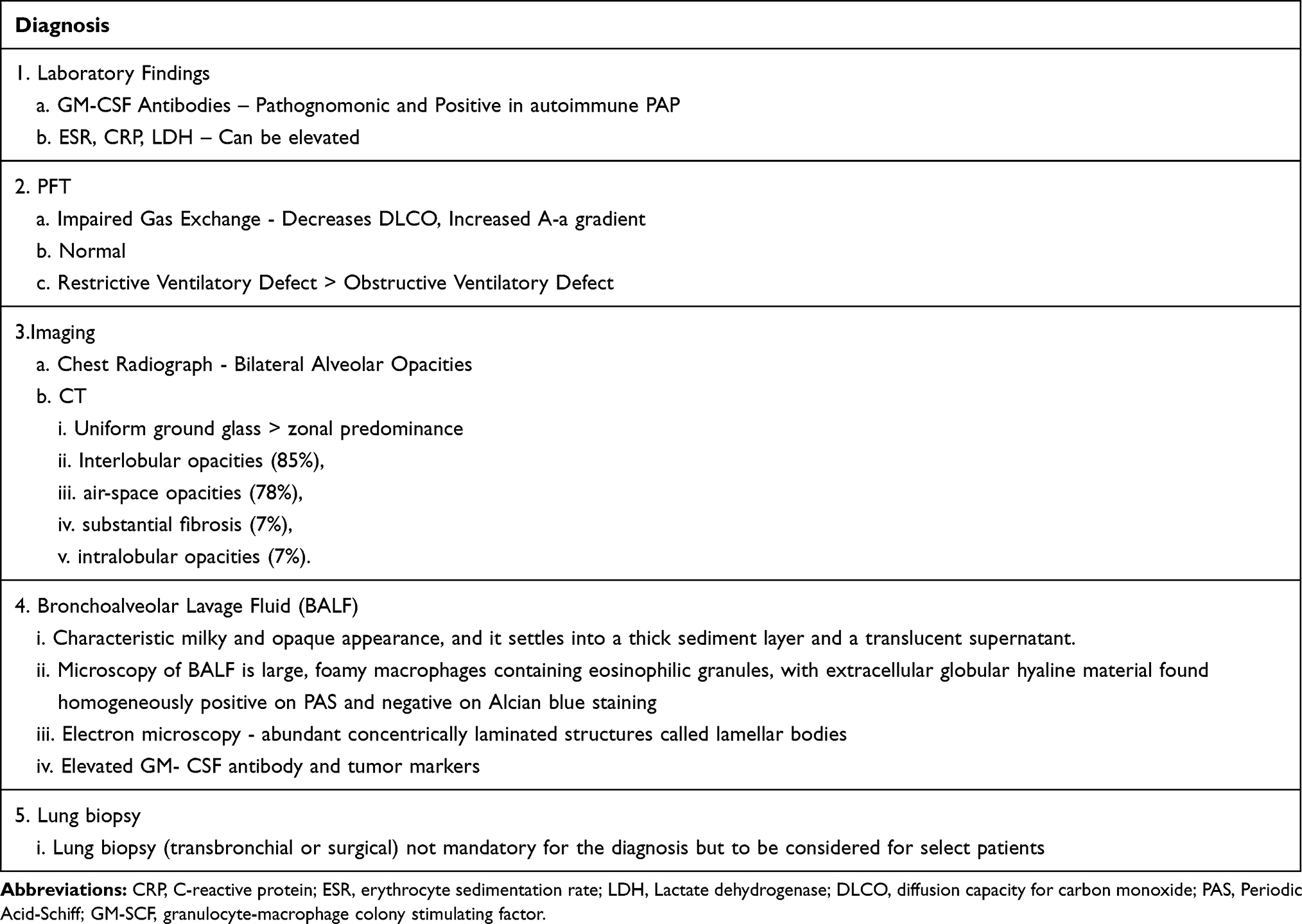 |
Table 2 Diagnosis of Pulmonary Alveolar Proteinosis (PAP) |
Treatment
Given the rare nature of the disease, there is limited evidence comparing various treatment modalities. However, Whole lung lavage (WLL) has remained at the forefront for PAP patients with worsening symptoms or lung function. Recently, several newer options targeting alveolar macrophages, reduction of GM-CSF antibodies and gene modifications have emerged. Treatment is usually directed towards the underlying pathobiology and type of PAP. In patients who fail to respond to both GM-CSF supplementation and WLL, novel therapies targeting GM-CSF antibodies or lipid metabolism have been tried. For secondary PAP, treatment of the underlying cause is of utmost importance, although often difficult to achieve. For instance, effective treatment of underlying malignancies, treatment of underlying infection or eliminating the inhalation exposure may lead to resolution of PAP in some cases.21–25 Table 3 summarizes various treatments employed for different types of PAP.
 |
Table 3 Treatment for Pulmonary Alveolar Proteinosis (PAP) |
Whole Lung Lavage
Whole Lung Lavage (WLL) is currently the first-line therapy for PAP. Although it is the most widely used treatment for PAP, there are no specific guidelines for WLL procedure itself or standardized indications for patients who might benefit from WLL. The main indication is the limitation of daily activities due to dyspnea and disease progression. In the literature, it has been suggested that patients with hypoxemia with partial pressure of oxygen (PaO2) less than 70 mmHg on room air or poor gas exchange indicated by increased alveolar-arterial (A-a) oxygen gradient greater than 40 mmHg are more likely to have disease progression and thus may benefit from WLL.26,27 Other indications for WLL are respiratory symptoms, declining oxygenation, worsening disease on imaging, and declining lung function, including gas exchange or forced vital capacity.28 Most of the patients undergo WLL in the first year of diagnosis.7
Over the years, the procedure of WLL has been modified but is not yet standardized. WLL is an invasive procedure performed via double-lumen endobronchial intubation in order to physically remove the lipoproteinaceous material from the alveoli. WLL requires single lung ventilation. In patients who cannot tolerate single lung ventilation, extra corporeal membrane oxygenation (ECMO) support may be required for the procedure. Most centers perform WLL in separate sessions for each lung and treat the most severely affected lung, as per imaging, first. If imaging is equivocal, obtaining a perfusion scan may help determine a better-perfused lung, and the lung that can tolerate single lung ventilation better.29 The lavage of the second lung is usually safer as the disease improves after the treatment of the diseased lung with first WLL. Multiple aliquots of warm saline are infused with a total of 5–40 L per lung to emulsify and remove the PAP sediment. Chest percussion device used during the lavage procedure may augment the lavage output. In a global survey over different centers with 1110 WLL procedures, the most commonly reported complications were fever (18%) followed by hypoxemia (14%), wheezing (6%), pneumonia (5%), fluid leakage (4%), pleural effusion (3.1%) and pneumothorax (0.8%).28
WLL has been associated with improvement in dyspnea, PaO2, [A-a] gradient, forced expiratory volume (FEV1), vital capacity, and diffusion capacity of carbon monoxide (DLCO), six-minute walk test.30,31 Forced vital capacity (FVC) usually improves immediately after the first WLL and continues to improve thereafter. DLCO, however, is slow to improve and shows incomplete recovery in most studies.
The timing of subsequent WLL treatments is variable and usually dictated by patient’s symptoms and disease severity based on pulmonary function and radiographs. There is considerable variability in patient response to WLL and time in between different treatments. For instance, in one of the studies around 70% of patients remained without PAP recurrence at seven years.31 Another study reported a median duration before requiring a repeat therapeutic whole lung lavage at approximately 15 months.7 A global survey of 368 patients with PAP reported that the mean number of procedures per patient was 2.5 (Standard deviation of 1.5) in a five year period.28
In conclusion, whole lung remains the gold-standard treatment for PAP. While there is a lack of data in the form of large scale randomized controlled trials comparing WLL to other modalities, it is considered a safe procedure in an experienced setting, with potential for providing long-term benefits in patients with PAP. Experienced centers for WLL remain relatively limited.
GM-CSF Supplementation
Two routes of recombinant GM-CSF treatment (molgramostim and sargramostim), inhaled and subcutaneous, have been trialed in patients with autoimmune PAP. The use of recombinant GM-CSF is based on the premise that patients with neutralizing antibodies to GM-CSF have reduced bioavailability of GM-CSF. At present, GM-CSF supplementation therapy is generally considered for patients who cannot undergo WLL or have autoimmune PAP refractory to WLL. The primary single-patient experience demonstrated that the use of subcutaneous recombinant human GM-CSF led to a substantial improvement of oxygenation generated hypothesis for larger studies.32 Subsequently, in an open-label study on 25 patients with PAP, subcutaneous GM-CSF administration was well tolerated and resulted in clinical improvement in terms of symptoms and radiographic appearance.33
GM-CSF can be reconstituted in saline and administered via nebulizer (Inhaled GM-CSF). For inhaled GM-CSF (sargramostim), a smaller prospective trial initially demonstrated good tolerance and a modest improvement in dyspnea and six-minute walk distance.34 More recently, a multicenter, double-blind trial involving 64 patients with mild to moderate PAP, inhaled GM-CSF (sargramostim), compared with placebo, was associated with a statistically significant, but modest improvement in the alveolar-arterial oxygen gradient but with no clinically important changes in outcomes.35 In another six-month Phase II randomized study involving 36 autoimmune PAP patients, inhaled GM-CSF (molgramostim) for six months had no impact on the alveolar-arterial (A-a) oxygen gradient in patients who had mild to moderate pulmonary alveolar proteinosis.36 Health-related quality of life measures did improve after three months of treatment, and there was a marginal improvement in terms of total lung capacity and diffusion capacity. In the recently concluded largest randomized trial of 138 patients, a continuous regimen of 300 mcg of molgramostim, in comparison with an intermittent regimen and placebo, demonstrated statistically significant improvement in pulmonary gas transfer, clinical parameters (including biomarkers and ground glass on CT scan) and functional health status compared to placebo, with similar rates of adverse events.37
In the pooled analysis of 10 observational studies by Sheng et al, including 115 patients with autoimmune PAP, GM-CSF demonstrated response rate of 80%, with relapse rate of 22% and with improvement in oxygenation indices.38 The pooled response rate of GM-CSF therapy was not inferior to WLL therapy in terms of disease severity score, PFT and 6-min walking test.38 Inhaled GM-CSF therapy showed a higher response rate, greater improvement in A-a gradient and PaO2 than subcutaneous GM-CSF treatment, suggesting inhaled GM-CSF therapy could potentially have more benefits in autoimmune PAP patients. Another large randomized control trial is underway at this time to look at inhaled GM-CSF molgramostim in autoimmune PAP.
Thus, GM-CSF is a feasible option as a primary treatment for those who cannot tolerate WLL or as an adjuvant therapy for those with persistent disease. Cumulative evidence from studies so far demonstrates variable and modest improvements in both subjective and physiologic parameters. Inhaled GM-CSF, when used daily in patients with moderate to severe autoimmune, PAP, has an edge over subcutaneously administered GM-CSF. Definitive evidence regarding the use of recombinant GM-CSF as primary therapy is lacking.
Rituximab
Rituximab is a monoclonal antibody targeting CD20 of B cells with proven efficacy for various hematological and autoimmune disorders. It works by depletion of B cells and can decrease production of the (GM‐CSF) autoantibodies. By targeting CD20 of B cells and reducing antibodies, the autoantibody levels can be reduced in autoimmune PAP. In a retrospective analysis of patients with autoimmune PAP, 13 patients who were treated with rituximab did not show any improvement six months after treatment, but four patients (30%) presented a significant decrease in A-a difference in oxygen after one year (which might not be related to rituximab therapy but to a spontaneous improvement).39 No serious adverse events were noticed in patients treated with rituximab. In contrast, in an open-label, phase II study of 10 patients with autoimmune PAP, rituximab was infused intravenously for two doses about two weeks apart. Compared to pre-treatment values, arterial oxygen, A-aO2 difference and imaging significantly improved following rituximab treatment, and anti-GM-CSF IgG level decreased in the BALF.40 Thus, available evidence is not sufficient for rituximab to be considered as first-line therapy but in selected patients with refractory PAP, as long as an underlying systemic infection has been ruled out, a trial of rituximab may be reasonable.
Plasmapheresis
GM-CSF antibodies play a pathogenic role in autoimmune PAP, and plasmapheresis can potentially reduce circulating antibodies and consequently restore the surfactant hemostasis. However, the evidence for plasmapheresis in patients with PAP is limited to case reports. These case reports have shown a variable and modest benefit at best - some showing lower level of systemic anti‐GM‐CSF antibodies, improvement in chest radiograph, and arterial oxygen concentration increment from 50 to 70 mmHg,16,41 while others showing no significant clinical change despite a reduction in autoantibody levels.42
Lung Transplantation
Lung transplantation is a possibility in patients who have minimal to no response to the above measures and have progressive clinical deterioration. This is mainly in patients with progressive pulmonary fibrosis. There have been case reports of a recurrence of PAP after double lung transplantation.43–45 Hence lung transplantation should be reserved for selected candidates and recipients should be monitored for recurrence.
Glucocorticoids
Autoimmune diseases are often treated with corticosteroids and therefore have been used in the treatment of PAP. However, systemic corticosteroids have not been shown to be beneficial in PAP patients. In a retrospective analysis of 31 patients from different centers, PAP deteriorated during corticosteroid therapy in 74% of patients and an accelerated rate of worsening noticed in patients treated with high-dose steroids.46 There was associated increased mortality, which was attributed to new infections in these patients. While the exact reason for corticosteroids causing harm is unclear, presently its use is not recommended for PAP.
Other Agents of Potential Use
In a mouse PAP model, the lipid content of both alveolar macrophages and type 2 epithelial cells, two primary cells involved in surfactant homeostasis, was high compared to wild-type animals.47 These mice, when treated with statins, had a diminished BAL turbidity, decreased cholesterol level, and improvement of PAP because of the efflux of cholesterol from alveolar macrophages.48 In PAP, surfactant composition has been noted to have an altered cholesterol/phospholipid ratio, with markedly higher amount of cholesterol and less amount of phospholipid. A recent publication also reported the use of pioglitazone, a thiazolidinedione, to ameliorate PAP.49 Whether the limited evidence of these agents will evolve into a meaningful impact on large-scale trials remains to be seen.
Gene Therapy
Gene therapy can play an important part in hereditary PAP. Induced pluripotent stem cells (iPSC) represent a possible source for in vitro generation of macrophages.50 Protocols to obtain functional macrophages that recapitulate the pathognomonic defects in GM-CSF signaling and macrophage function have been established.51 These gene-corrected macrophages can become a possible autologous cell source for therapeutic strategy in hereditary PAP.52 Hematopoietic stem cell transplantation with subsequent clinical improvement has been reported in PAP patients with GM-CSF receptor deficiency.53 In another approach in children with hereditary PAP due to CSF2RA or CSF2RB mutations, Suzuki and colleagues report Csf2rb-gene-corrected pulmonary macrophage transplantation to have a significant clinical improvement with only one administration, and was well tolerated.54
Supportive Care
Patients with PAP are at high risk for infectious complications given the macrophage dysfunction. Patients should be counseled for immunizations, including annual influenza and, where appropriate, pneumococcal and COVID-19 vaccine. Avoidance of any potential inhalational insult, both occupational or recreational in nature, should be emphasized. Cigarette smoking has been found not only to be a possible trigger but it also increases the number of sessions of WLL required by patients.55 Smoking avoidance should be advised in these patients. Long-term oxygen therapy is sometimes necessary in patients with poor gas exchange and decreased PaO2. Pulmonary rehabilitation should also be considered on a case-by-case basis. Since PAP is a chronic disease, there is a possible burden associated with treatment, in addition to the treatment itself. Patients with PAP, like all other chronic diseases, often require chronic disease management in order to control their symptoms and avoid complications associated with the disease.
Conclusion
Early diagnosis and targeted treatment remain the cornerstone of the management of PAP. WLL is the most widely used and accepted treatment for primary PAP. In the last few years, our knowledge of PAP pathogenesis has improved with the identification of the role of GM-CSF signaling; as a result, inhaled GM-CSF has emerged as a potential therapy. In addition, approaches like statin therapy and pulmonary alveolar macrophage transplantation are other promising areas that need further research. There is a scarcity of randomized control trials in patients with PAP, given the rarity of the disease, making data interpretation difficult. Cohort enrichment efforts at specialized PAP centers following the footsteps of other rare disease states, such as cystic fibrosis, could potentially serve as a beacon for further clinical trials. In addition, patient advocacy-based foundation plays an important role in the management of rare diseases like PAP.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval for the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
Dr Girish B Nair reports Speaker’s Bureau from Boehringer Ingelheim, outside the submitted work. None of the authors report any other potential conflicts of interest with the materials presented.
References
1. Rosen SH, Castleman B, Liebow AA, Enzinger FM, Hunt RTN. Pulmonary alveolar proteinosis. N Engl J Med. 1958;258(23):1123–1142. doi:10.1056/nejm195806052582301
2. McCarthy C, Avetisyan R, Carey BC, Chalk C, Trapnell BC. Prevalence and healthcare burden of pulmonary alveolar proteinosis. Orphanet J Rare Dis. 2018;13(1):129. doi:10.1186/s13023-018-0846-y
3. Inoue Y, Trapnell BC, Tazawa R, et al. Characteristics of a large cohort of patients with autoimmune pulmonary alveolar proteinosis in Japan. Am J Respir Crit Care Med. 2008;177(7):752–762. doi:10.1164/rccm.200708-1271OC
4. Tanaka N, Watanabe J, Kitamura T, Yamada Y, Kanegasaki S, Nakata K. Lungs of patients with idiopathic pulmonary alveolar proteinosis express a factor which neutralizes granulocyte-macrophage colony stimulating factor. FEBS Lett. 1999;442(2–3):246–250. doi:10.1016/s0014-5793(98)01668-8
5. Kitamura T, Uchida K, Tanaka N, et al. Serological diagnosis of idiopathic pulmonary alveolar proteinosis. Comparative study research support, non-U.S. gov’t. Am J Respir Crit Care Med. 2000;162(2 Pt 1):658–662. doi:10.1164/ajrccm.162.2.9910032
6. Delaval P, Brinchault G, Corre R, Jouneau S, Meunier C, Briens E. [Pulmonary alveolar phospholipoproteinosis]. Rev Pneumol Clin. 2005;61(3):186–192. Lipoprotéinose alvéolaire pulmonaire. French. doi:10.1016/s0761-8417(05)84811-4
7. Seymour JF, Presneill JJ. Pulmonary alveolar proteinosis: progress in the first 44 years. Research support, non-U.S. gov’t review. Am J Respir Crit Care Med. 2002;166(2):215–235. doi:10.1164/rccm.2109105
8. Borie R, Danel C, Debray MP, et al. Pulmonary alveolar proteinosis. Eur Respir Rev. 2011;20(120):98–107. doi:10.1183/09059180.00001311
9. Xipell JM, Ham KN, Price CG, Thomas DP. Acute silicoproteinosis. Thorax. 1977;32(1):104–111. doi:10.1136/thx.32.1.104
10. Briens E, Delaval P, Mairesse MP, et al. [Pulmonary alveolar proteinosis]. Rev Mal Respir. 2002;19(2Pt1):166–182. Lipoprotéinose alvéolaire pulmonaire. French.
11. Rosen LB, Rocha Pereira N, Figueiredo C, et al. Nocardia-induced granulocyte macrophage colony-stimulating factor is neutralized by autoantibodies in disseminated/extrapulmonary nocardiosis. Clin Infect Dis. 2015;60(7):1017–1025. doi:10.1093/cid/ciu968
12. Trapnell BC, Whitsett JA, Nakata K. Pulmonary alveolar proteinosis. N Engl J Med. 2003;349(26):2527–2539. doi:10.1056/NEJMra023226
13. Sui X, Du Q, Xu KF, et al. Quantitative assessment of Pulmonary Alveolar Proteinosis (PAP) with ultra-dose CT and correlation with Pulmonary Function Tests (PFTs). PLoS One. 2017;12(3):e0172958. doi:10.1371/journal.pone.0172958
14. Bai J, Xu J, Yang W, et al. A new scale to assess the severity and prognosis of pulmonary alveolar proteinosis. Can Respir J. 2016;2016:3412836. doi:10.1155/2016/3412836
15. Inoue Y, Nakata K, Arai T, et al. Epidemiological and clinical features of idiopathic pulmonary alveolar proteinosis in Japan. Research support, non-U.S. gov’t. Respirology. 2006;11(Suppl):S55–60. doi:10.1111/j.1440-1843.2006.00810.x
16. Bonfield TL, Kavuru MS, Thomassen MJ. Anti-GM-CSF titer predicts response to GM-CSF therapy in pulmonary alveolar proteinosis. Clin Immunol. 2002;105(3):342–350. doi:10.1006/clim.2002.5301
17. Sakagami T, Beck D, Uchida K, et al. Patient-derived granulocyte/macrophage colony-stimulating factor autoantibodies reproduce pulmonary alveolar proteinosis in nonhuman primates. Am J Respir Crit Care Med. 2010;182(1):49–61. doi:10.1164/rccm.201001-0008OC
18. Holbert JM, Costello P, Li W, Hoffman RM, Rogers RM. CT features of pulmonary alveolar proteinosis. AJR Am J Roentgenol. 2001;176(5):1287–1294. doi:10.2214/ajr.176.5.1761287
19. Godwin JD, Müller NL, Takasugi JE. Pulmonary alveolar proteinosis: CT findings. Radiology. 1988;169(3):609–613. doi:10.1148/radiology.169.3.3186983
20. Costello JF, Moriarty DC, Branthwaite MA, Turner-Warwick M, Corrin B. Diagnosis and management of alveolar proteinosis: the rôle of electron microscopy. Thorax. 1975;30(2):121–132. doi:10.1136/thx.30.2.121
21. Cordonnier C, Fleury-Feith J, Escudier E, Atassi K, Bernaudin JF. Secondary alveolar proteinosis is a reversible cause of respiratory failure in leukemic patients. Am J Respir Crit Care Med. 1994;149(3 Pt 1):788–794. doi:10.1164/ajrccm.149.3.8118651
22. Chaulagain CP, Pilichowska M, Brinckerhoff L, Tabba M, Erban JK. Secondary pulmonary alveolar proteinosis in hematologic malignancies. Hematol Oncol Stem Cell Ther. 2014;7(4):127–135. doi:10.1016/j.hemonc.2014.09.003
23. Ishii H. Knowledge of secondary pulmonary alveolar proteinosis complicated with myelodysplastic syndrome. Intern Med. 2020;59(8):1019–1020. doi:10.2169/internalmedicine.4193-19
24. Ladeb S, Fleury-Feith J, Escudier E, Tran Van Nhieu J, Bernaudin JF, Cordonnier C. Secondary alveolar proteinosis in cancer patients. Support Care Cancer. 1996;4(6):420–426. doi:10.1007/bf01880639
25. Chung JH, Pipavath SJ, Myerson DH, Godwin D. Secondary pulmonary alveolar proteinosis: a confusing and potentially serious complication of hematologic malignancy. J Thorac Imaging. 2009;24(2):115–118. doi:10.1097/RTI.0b013e3181930ed6
26. Cooper JD, Duffin J, Glynn MF, et al. Combination of membrane oxygenator support and pulmonary lavage for acute respiratory failure. J Thorac Cardiovasc Surg. 1976;71(2):304–308. doi:10.1016/S0022-5223(19)40247-X
27. Shah PL, Hansell D, Lawson PR, Reid KB, Morgan C. Pulmonary alveolar proteinosis: clinical aspects and current concepts on pathogenesis. Thorax. 2000;55(1):67–77. doi:10.1136/thorax.55.1.67
28. Campo I, Luisetti M, Griese M, et al. Whole lung lavage therapy for pulmonary alveolar proteinosis: a global survey of current practices and procedures. Orphanet J Rare Dis. 2016;11(1):115. doi:10.1186/s13023-016-0497-9
29. Awab A, Khan MS, Youness HA. Whole lung lavage-technical details, challenges and management of complications. J Thorac Dis. 2017;9(6):1697–1706. doi:10.21037/jtd.2017.04.10
30. Rogers RM, Levin DC, Gray BA, Moseley LW
31. Beccaria M, Luisetti M, Rodi G, et al. Long-term durable benefit after whole lung lavage in pulmonary alveolar proteinosis. Eur Respir J. 2004;23(4):526–531. doi:10.1183/09031936.04.00102704
32. Seymour JF, Dunn AR, Vincent JM, Presneill JJ, Pain MC. Efficacy of granulocyte-macrophage colony-stimulating factor in acquired alveolar proteinosis. N Engl J Med. 1996;335(25):1924–1925. doi:10.1056/nejm199612193352513
33. Venkateshiah SB, Yan TD, Bonfield TL, et al. An open-label trial of granulocyte macrophage colony stimulating factor therapy for moderate symptomatic pulmonary alveolar proteinosis. Chest. 2006;130(1):227–237. doi:10.1378/chest.130.1.227
34. Tazawa R, Trapnell BC, Inoue Y, et al. Inhaled granulocyte/macrophage-colony stimulating factor as therapy for pulmonary alveolar proteinosis. Am J Respir Crit Care Med. 2010;181(12):1345–1354. doi:10.1164/rccm.200906-0978OC
35. Tazawa R, Ueda T, Abe M, et al. Inhaled GM-CSF for pulmonary alveolar proteinosis. N Engl J Med. 2019;381(10):923–932. doi:10.1056/NEJMoa1816216
36. Tian X, Yang Y, Chen L, et al. Inhaled granulocyte-macrophage colony stimulating factor for mild-to-moderate autoimmune pulmonary alveolar proteinosis - a six month phase II randomized study with 24 months of follow-up. Orphanet J Rare Dis. 2020;15(1):174. doi:10.1186/s13023-020-01450-4
37. Trapnell BC, Inoue Y, Bonella F, et al. Inhaled molgramostim therapy in autoimmune pulmonary alveolar proteinosis. N Engl J Med. 2020;383(17):1635–1644. doi:10.1056/NEJMoa1913590
38. Sheng G, Chen P, Wei Y, Chu J, Cao X, Zhang HL. Better approach for autoimmune pulmonary alveolar proteinosis treatment: inhaled or subcutaneous granulocyte-macrophage colony-stimulating factor: a meta-analyses. Respir Res. 2018;19(1):163. doi:10.1186/s12931-018-0862-4
39. Soyez B, Borie R, Menard C, et al. Rituximab for auto-immune alveolar proteinosis, a real life cohort study. Respir Res. 2018;19(1):74. doi:10.1186/s12931-018-0780-5
40. Kavuru MS, Malur A, Marshall I, et al. An open-label trial of rituximab therapy in pulmonary alveolar proteinosis. Eur Respir J. 2011;38(6):1361–1367. doi:10.1183/09031936.00197710
41. Garber B, Albores J, Wang T, Neville TH. A plasmapheresis protocol for refractory pulmonary alveolar proteinosis. Lung. 2015;193(2):209–211. doi:10.1007/s00408-014-9678-2
42. Luisetti M, Rodi G, Perotti C, et al. Plasmapheresis for treatment of pulmonary alveolar proteinosis. Eur Respir J. 2009;33(5):1220–1222. doi:10.1183/09031936.00097508
43. Parker LA, Novotny DB. Recurrent alveolar proteinosis following double lung transplantation. Chest. 1997;111(5):1457–1458. doi:10.1378/chest.111.5.1457
44. Gal AA, Bryan JA, Kanter KR, Lawrence EC. Cytopathology of pulmonary alveolar proteinosis complicating lung transplantation. J Heart Lung Transplant. 2004;23(1):135–138. doi:10.1016/s1053-2498(03)00032-9
45. Yousem SA. Alveolar lipoproteinosis in lung allograft recipients. Hum Pathol. 1997;28(12):1383–1386. doi:10.1016/s0046-8177(97)90228-8
46. Akasaka K, Tanaka T, Kitamura N, et al. Outcome of corticosteroid administration in autoimmune pulmonary alveolar proteinosis: a retrospective cohort study. BMC Pulm Med. 2015;15(1):88. doi:10.1186/s12890-015-0085-0
47. Sallese A, Suzuki T, McCarthy C, et al. Targeting cholesterol homeostasis in lung diseases. Sci Rep. 2017;7(1):10211. doi:10.1038/s41598-017-10879-w
48. McCarthy C, Lee E, Bridges JP, et al. Statin as a novel pharmacotherapy of pulmonary alveolar proteinosis. Case reports research support, N.I.H., extramural. Nat Commun. 2018;9(1):3127. doi:10.1038/s41467-018-05491-z
49. Dupin C, Hurtado M, Cazes A, et al. Pioglitazone in pulmonary alveolar proteinosis: promising first clinical experience. Respir Med Res. 2020;78:100756. doi:10.1016/j.resmer.2020.100756
50. Suzuki T, Mayhew C, Sallese A, et al. Use of induced pluripotent stem cells to recapitulate pulmonary alveolar proteinosis pathogenesis. Am J Respir Crit Care Med. 2014;189(2):183–193. doi:10.1164/rccm.201306-1039OC
51. Mucci A, Kunkiel J, Suzuki T, et al. Murine iPSC-derived macrophages as a tool for disease modeling of hereditary pulmonary alveolar proteinosis due to Csf2rb deficiency. Stem Cell Rep. 2016;7(2):292–305. doi:10.1016/j.stemcr.2016.06.011
52. Lachmann N, Happle C, Ackermann M, et al. Gene correction of human induced pluripotent stem cells repairs the cellular phenotype in pulmonary alveolar proteinosis. Am J Respir Crit Care Med. 2014;189(2):167–182. doi:10.1164/rccm.201306-1012OC
53. Fremond ML, Hadchouel A, Schweitzer C, et al. Successful haematopoietic stem cell transplantation in a case of pulmonary alveolar proteinosis due to GM-CSF receptor deficiency. Clinical conference research support, non-U.S. gov’t. Thorax. 2018;73(6):590–592. doi:10.1136/thoraxjnl-2017-211076
54. Suzuki T, Arumugam P, Sakagami T, et al. Pulmonary macrophage transplantation therapy. Research support, N.I.H., extramural research support, non-U.S. gov’t. Nature. 2014;514(7523):450–454. doi:10.1038/nature13807
55. Suzuki T, Trapnell BC. Pulmonary alveolar proteinosis syndrome. Clin Chest Med. 2016;37(3):431–440. doi:10.1016/j.ccm.2016.04.006
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.