Back to Journals » ImmunoTargets and Therapy » Volume 14
Understanding and Managing Hyper IgE Syndromes
Authors Meric Z, Aydin M ![]() , Demir Gumus D, Yucel E, Kiykim A
, Demir Gumus D, Yucel E, Kiykim A ![]()
Received 1 September 2025
Accepted for publication 30 October 2025
Published 5 November 2025 Volume 2025:14 Pages 1233—1245
DOI https://doi.org/10.2147/ITT.S532287
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Shurin
Zeynep Meric,1,2 Muhammed Aydin,1,2 Dilan Demir Gumus,1,2 Esra Yucel,1,2 Ayca Kiykim1,2
1Istanbul University-Cerrahpasa, Cerrahpasa Medical School, Department of Pediatric Allergy and Immunology, Istanbul, Turkey; 2Istanbul University-Cerrahpasa, Cerrahpasa School of Medicine, Pediatric Allergy and Immunology Research Laboratory, Istanbul, Turkey
Correspondence: Ayca Kiykim, Istanbul University-Cerrahpasa, Cerrahpasa School of Medicine, Pediatric Allergy and Immunology, Kocamustafapasa, Fatih, Istanbul, 34098, Turkey, Tel +902124143000/67229, Email [email protected]
Abstract: Hyper-IgE syndromes represent an increasingly recognized and heterogeneous group of disorders characterized phenotypically by eczema, recurrent infections, and markedly elevated serum IgE levels. The identification of novel molecular defects in recent years has complicated definitive diagnosis, underscoring the genetic and clinical diversity of this group. In addition to immunological abnormalities, non-immunological manifestations—particularly those affecting connective tissue—contribute to significant comorbidities. The primary objectives of management are to control infections, prevent long-term complications, and improve quality of life. In this review, we summarize the clinical and laboratory features of disorders currently classified under hyper-IgE syndromes according to the most recent International Union of Immunological Societies framework and provide perspectives on their management.
Keywords: hyper IgE syndrome, eczema, Th2, atopy, STAT3
Background
In recent years, advances in clinical and molecular immunology have led to the identification of an increasing number of disorders within the spectrum of inborn errors of immunity (IEIs).1 These discoveries not only expand the spectrum of primary immunodeficiencies but also provide critical insights into fundamental immunological pathways.
Hyper-IgE syndromes (HIES) represent a heterogeneous group of rare inborn errors of immunity characterized by recurrent infections, eczema, and markedly elevated serum IgE levels. The first molecularly defined form of HIES was described with dominant-negative mutations in Signal Transducer and Activator of Transcription 3 (STAT3), initially recognized in patients with the clinical phenotype known as Job’s syndrome. This seminal finding established the role of STAT3 in immune regulation and paved the way for the subsequent discovery of additional genetic variants associated with the HIES phenotype.2
Over time, the identification of novel disease-causing mutations has clarified the involvement of STAT3 signaling pathways and related molecules in immune homeostasis, particularly in the regulation of Th2 responses.3,4 These insights have deepened our understanding of host defense mechanisms, inflammatory regulation, and the pathophysiological basis of susceptibility to infection and atopy.
In this review, we provide an overview of hyper-IgE syndromes according to the most recent International Union of Immunological Societies (IUIS) classification,1 summarizing their pathophysiology, clinical and laboratory features, and current approaches to management.
STAT3 Hyper IgE Syndrome
STAT3 Hyper IgE Syndrome (STAT3-HIES), an autosomal dominant primary immunodeficiency disorder, is characterized by elevated serum IgE levels, eczema, recurrent skin and respiratory tract infections, as well as various connective tissue and skeletal abnormalities.2 Hyper-IgE syndrome due to dominant-negative5 STAT3 mutations — formerly known as Job’s syndrome — occurs at a prevalence of less than one per million individuals.6 The JAK/STAT family comprises four Janus kinases (JAKs) and seven STAT proteins, which mediate signaling from over 50 cytokines via a phosphorylation cascade involving JAKs and STATs (Figure 1). This process begins with cytokine-receptor engagement at the cell surface and ultimately leads to nuclear translocation and transcriptional regulation.7
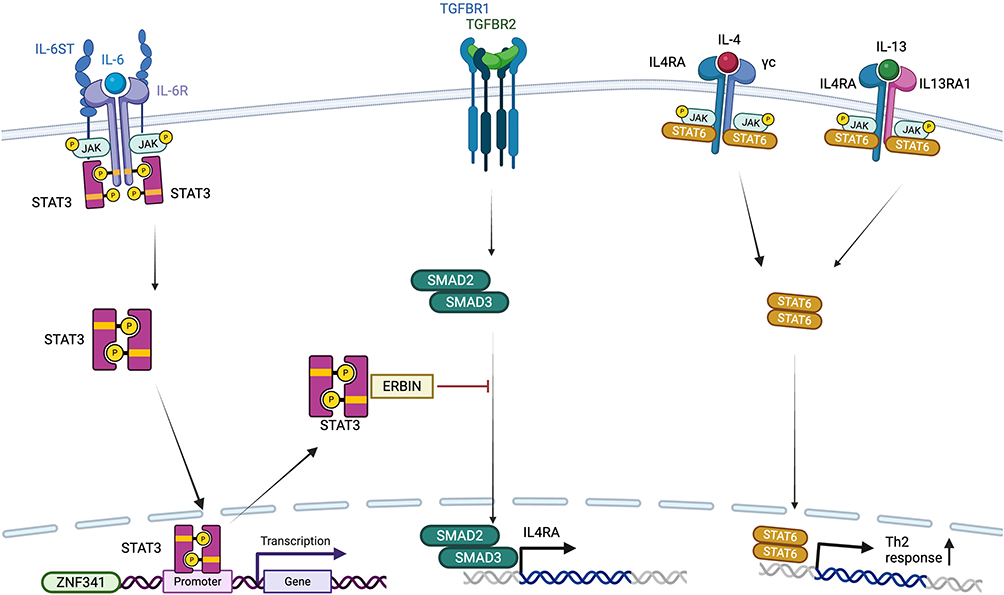 |
Figure 1 Pathways involving hyperIgE syndromes. Created in BioRender. Kiykim, A (2025) https://BioRender.com/undefined. |
Cytokine signaling through STAT3 includes IL-6, IL-10, IL-11, IL-21, IL-22 and IL-23 (4). Disruption to these pathways, combined with STAT3’s widespread expression, explains the syndrome’s multisystemic manifestations, including dermatitis, pulmonary disease, vasculopathy, and connective tissue/skeletal abnormalities8–10 (Table 1).
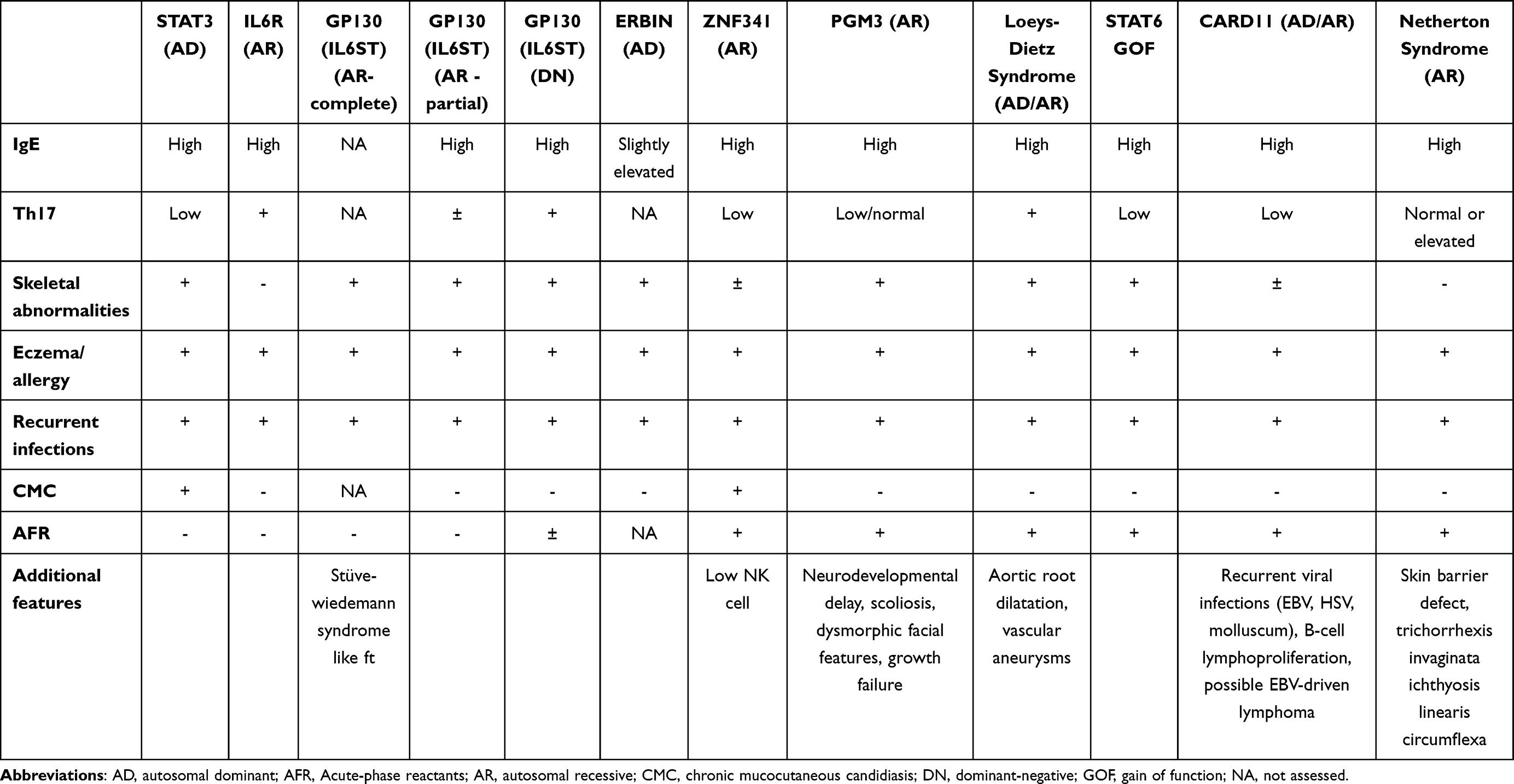 |
Table 1 Clinical and Laboratory Features of HyperIgE Syndromes |
The structural domains of the STAT3 protein implicated in disease include the Src homology 2 (SH2) domain and the DNA-binding domain (DBD). Both loss-of-function and gain-of-function (GOF) mutations can affect these regions. Dominant-negative5 STAT3 mutations result in STAT3-HIES, whereas GOF mutations are associated with large granular lymphocytic leukemia in the case of somatic mutations, and with early-onset multiorgan autoimmunity and lymphoproliferation in the case of germline mutations.8,11–13 The observation that both gain-of-function (GOF) and loss-of-function (LOF) STAT3 mutations can lead to hematopoietic malignancies reflects STAT3’s pivotal role in maintaining intracellular signaling homeostasis. GOF variants of STAT3 can suppress the STAT1-dependent Th1/IFN-γ/IL-12 axis, thereby impairing immune surveillance and promoting an immunosuppressive microenvironment characterized by elevated PD-L1, IL-10, and TGF-β expression, which collectively enhance proliferative signaling. Conversely, STAT3-LOF mutations lead to hyperactivation of STAT1 and potentially other STATs (such as STAT6) and SMADs, resulting in chronic inflammation, oxidative stress, and DNA damage that predispose hematopoietic progenitors to malignant transformation. Thus, dysregulation of STAT3—either through excessive or deficient activity—can disrupt the balance of cytokine signaling networks and contribute to hematopoietic tumorigenesis through distinct but converging mechanisms.
STAT3-DN mutations may be inherited or occur de novo, and are most commonly missense mutations or in-frame deletions. Rarely, intronic mutations have also been reported. Penetrance is generally complete, although phenotypic variability can be observed within the same family. This suggests that environmental factors, such as infection history, may influence the phenotype.14,15
Bacterial infections are common in STAT3-HIES patients and are predominantly caused by Staphylococcus aureus. These infections primarily affect the skin and lungs, but other epithelial surfaces may also be affected.16 Frequent, characteristic “cold” staphylococcal abscesses can occur anywhere on the body and may recur despite surgical or radiological drainage, necessitating prolonged courses of antibiotic therapy.17,18 This muted inflammatory response may result from impaired signaling of interleukin-6 (IL-6), a proinflammatory cytokine.19
The lungs are the second most frequently affected site of infection in STAT3-HIES. The most common causative organisms are Staphylococcus aureus, Streptococcus pneumoniae and Hemophilus influenzae.6,16 Pneumonia occurs in around 80% of patients, is often recurrent, and can lead to pleural effusion. Furthermore, it can lead to parenchymal lung disease, including bronchiectasis and pneumatocele formation. These structures can subsequently become infected with opportunistic pathogens, such as Pseudomonas aeruginosa or Aspergillus species.20
The Th17/IL-17 axis plays a crucial role in immunity against both bacteria and fungi. In STAT3-HIES patients, reduced levels of antimicrobial peptides in saliva lead to an increased susceptibility to mucosal Candida infections.21 This predisposition results in alterations to the oral microbiota and Candida overgrowth. Chronic mucocutaneous candidiasis occurs in around 70% of patients, typically manifesting as oral or genital candidiasis or onychomycosis.16
Despite having elevated serum IgE levels, STAT3-HIES patients have a lower prevalence of allergies and anaphylaxis than individuals with atopic dermatitis who have comparable IgE levels; however, this remains higher than in the general population.22 These patients exhibit impaired mast cell degranulation23 and abnormal IgE production characterized by high quantities but low allergen affinity.24
The connective tissue phenotype associated with STAT3-HIES can vary. The characteristic facial features — a prominent forehead, deeply set eyes, a broad nasal bridge and a high-arched palate — typically develop during adolescence and may not be evident in early childhood. Although permanent teeth develop normally, impaired root resorption of primary teeth prevents proper eruption of permanent teeth, consequently requiring extraction in most children.25
Vasculopathy in STAT3-HIES poses challenges in terms of both pathogenesis and clinical management. Medium-sized arterial abnormalities predominate, particularly affecting the coronary and intracranial vessels. In a prospective study, coronary artery abnormalities (including ectasia and aneurysms) were identified in 50% of patients, and small infarcts were detected in some individuals through imaging.26
The incidence of malignancy in STAT3-HIES is approximately 7%, most commonly affecting the hematopoietic system. The most frequently observed malignancy is lymphoma.16 These are typically B-cell non-Hodgkin lymphomas, though Hodgkin and T-cell lymphomas have also been reported.27
Laboratory findings in STAT3-HIES patients are typically characterized by markedly elevated serum immunoglobulin E28 levels, often exceeding 2000 IU/mL (normal adult levels are below 100 IU/mL), peripheral eosinophilia with eosinophil counts above 700/μL and reduced circulating memory T and B cells. Additionally, there is an almost complete absence of IL-17-producing Th17 cells.2
Early detection of pulmonary infections in patients with STAT3-HIES requires regular chest imaging and high clinical suspicion. Culturing skin lesions and sputum samples is useful for guiding appropriate therapy. Routine screening for scoliosis during adolescence is also recommended. Dental follow-up is necessary to ensure the timely extraction of primary teeth. Adults should be evaluated for coronary artery and cerebral aneurysms every three years. Due to the increased risk of lymphoma, regular monitoring for lymphadenopathy is also recommended.15
To prevent cutaneous and pulmonary infections, anti-staphylococcal antibiotic prophylaxis (eg cotrimoxazole) and antiseptic baths are recommended.29 For mucocutaneous Candida infections, antifungal agents such as fluconazole are recommended. If the lung parenchyma is affected, mold-active antifungals such as itraconazole should be used to prevent chronic pulmonary aspergillosis and allergic bronchopulmonary aspergillosis.28 Due to the high mortality rate associated with Aspergillus infections, prolonged use of triazoles (eg posaconazole) is recommended for pulmonary fungal infections.30
The role of hematopoietic stem cell transplantation (HSCT) in STAT3-HIES is becoming clearer. A review evaluating all reported STAT3-HIES patients who underwent HSCT demonstrated a reduction in infection frequency, improvement in skin manifestations and clinical and radiological stabilization or improvement in pulmonary function.31–33 Immunologically, decreases in IgE levels and recovery of normal IL-17-producing Th17 cells have been observed. These findings suggest that the underlying immune deficiency may be corrected and that certain aspects of the disease may improve. However, the effects on connective tissue abnormalities and vascular complications remain uncertain. For example, myocardial infarction due to coronary artery aneurysm occurred in a patient with a normalized Th17/IL-17 axis and complete donor chimerism.31,34–36
IL6ST Deficiency
IL6ST encodes gp130, the common signal-transducing subunit for the IL-6 cytokine family, including IL-6, IL-11, IL-27, leukemia inhibitory factor (LIF), oncostatin M,37 and ciliary neurotrophic factor (CNTF). Loss-of-function variants impair downstream JAK/STAT signaling, leading to defective acute-phase responses, impaired Th17 differentiation, and abnormalities in skeletal development, particularly those mediated by IL-11.5,8,38 While biallelic complete loss-of-function mutations in IL6ST cause autosomal recessive immunodeficiency, heterozygous partial loss-of-function variants have also been proposed to disrupt gp130-mediated signaling. These variants may act through dominant-negative mechanisms or haploinsufficiency, resulting in variable phenotypes. Some IL6ST variants can selectively affect specific cytokine pathways, such as IL-6 or IL-11, depending on how the mutation alters gp130 structure. This highlights the modular organization of gp130 signaling, and may help explain the diversity of clinical presentations observed in affected individuals.38,39
Clinically, IL6ST deficiency presents with a HIES-like phenotype, including severe eczema, markedly elevated serum IgE levels, and eosinophilia. Similar to autosomal dominant STAT3-HIES, affected patients frequently experience recurrent bacterial infections, particularly with Staphylococcus aureus, due to impaired mucosal and epithelial barrier immunity resulting from altered Th17 responses. Recurrent respiratory tract infections may lead to long-term pulmonary complications, including the development of bronchiectasis. The acute-phase response is often blunted in IL6ST deficiency, with low or absent C-reactive protein (CRP) levels and minimal febrile response during infections, reflecting the impact of impaired IL-6 signaling.38 In contrast to STAT3-HIES, Th17 cell numbers are relatively preserved or only modestly reduced in IL6ST deficiency, and patients typically do not develop chronic mucocutaneous candidiasis.9 This supports the hypothesis that alternative STAT3-activating cytokines can partially compensate for IL-6 signaling in driving Th17 differentiation.37 Additionally, distinctive skeletal and dental anomalies, such as delayed tooth eruption, craniosynostosis, and short stature are commonly observed, likely due to defective IL-11 signaling.38,39
Management includes prompt and targeted antimicrobial therapy, prophylactic measures for recurrent infections, and immunoglobulin replacement in patients with impaired antibody production. Early recognition and treatment of respiratory infections are essential to prevent long-term pulmonary complications such as bronchiectasis. Comprehensive skin care, including anti-inflammatory and antiseptic approaches, is important for managing eczema and preventing secondary infections. Although HSCT has been proposed as a potential treatment in IL6ST deficiency, no clinical reports have been published to date, leaving its efficacy and applicability undetermined.9,38
IL6R Deficiency
The IL6 receptor complex is composed of two different subunits, interleukin-6 receptor (IL6R) subunit, and IL6ST/GP130 subunit. The IL6R serves as the ligand-binding component of the receptor complex. It functions in association with the signal-transducing subunit, gp130, to mediate both classical and trans-signaling pathways. Loss of IL6R function disrupts several key immune processes, including fever generation, acute-phase reactant production, and Th17 cell differentiation, primarily through the JAK/STAT3 signaling axis.40–42
Clinically, similar to IL6ST deficiency, IL6R deficiency is characterized by recurrent sinopulmonary and skin infections, along with variable atopic features. Patients typically exhibit elevated serum IgE levels, peripheral eosinophilia. Mild reductions in serum IgG, IgA, and IgM levels have been reported in some cases. Notably, expected inflammatory signs, such as fever and CRP elevation are often absent, even during systemic infections.9,19,43 Despite clinical overlap with IL6ST deficiencies, IL6R deficiency exhibits distinct immunological features due to its selective impairment of IL-6 signaling, whereas IL6ST deficiency disrupts signaling of the entire IL-6 cytokine family.
Management of IL6R deficiency largely mirrors that of IL6ST deficiency and includes prompt and targeted antibiotic therapy, prophylaxis for recurrent infections, and immunoglobulin replacement in patients with impaired antibody production. Pneumococcal vaccination and close monitoring of vaccine responses are essential. Early recognition and management of bronchiectasis is critical to prevent long-term pulmonary damage. Importantly, IL-6R blocking agents such as tocilizumab should not be used in these patients, as the IL-6 receptor is already nonfunctional due to biallelic loss-of-function mutations.19 Additional immunomodulatory therapies may be considered on a case-by-case basis; however, clinical data supporting their use remain limited. Currently, there are no documented cases of HSCT in IL6R deficiency, and its potential therapeutic role remains unestablished in the absence of clinical evidence.
ZNF341 Deficiency
The ZNF341 gene encodes a zinc finger transcription factor that acts as a positive regulator of STAT3 expression. Loss-of-function mutations in ZNF341 reduce STAT3 protein levels and lead to downstream impairment of STAT3-dependent signaling pathways, notably affecting Th17 cell differentiation. As a result, the clinical phenotype closely resembles that of autosomal dominant STAT3-HIES.42,44,45
Patients typically present in early childhood with severe atopic dermatitis, recurrent respiratory and skin infections, often with Staphylococcus aureus, and markedly elevated serum IgE levels alongside peripheral eosinophilia. Bronchiectasis is a common complication due to chronic lung infections. Similar to STAT3-HIES, features such as retained primary teeth, scoliosis, and connective tissue abnormalities including joint hypermobility may also be observed. However, non-immunological manifestations tend to be milder and less frequent in ZNF341 deficiency compared to STAT3-HIES.9,44,46
Notably, recent studies have highlighted increased cellular radiosensitivity and impaired DNA repair mechanisms in individuals with ZNF341 deficiency, raising concerns about long-term malignancy risk.47 Although no definitive causal links have been established, this radiosensitivity may have implications for clinical management, particularly in the context of radiation-based imaging or preconditioning for potential transplant protocols.47
Management parallels that of STAT3-HIES and includes antimicrobial prophylaxis, particularly against staphylococcal infections, comprehensive dermatologic care, and immunoglobulin replacement therapy when indicated. Close monitoring of pulmonary status is essential to prevent long-term lung damage. Dupilumab has shown benefit in selected patients with severe atopic disease.48 To date, there are no reported cases of HSCT in patients with ZNF341 deficiency, and its role in treatment remains undefined.
PGM3 Deficiency
Phosphoglucomutase 3 (PGM3) encodes a key enzyme of the hexosamine biosynthetic pathway, catalyzing the interconversion of N-acetylglucosamine-6-phosphate to N-acetylglucosamine-1-phosphate, an essential precursor for UDP-N-acetylglucosamine (UDP-GlcNAc). UDP-GlcNAc is a critical substrate for protein glycosylation, glycosaminoglycan synthesis, and glycolipid formation.49 Deficiency of PGM3 leads to impaired glycosylation of the T-cell receptor, integrins, and cytokine receptors, resulting in defective signal transduction, immune cell adhesion, and migration. This biochemical defect disrupts multiple physiological processes, producing a multisystem disorder with both immunological and developmental features.50
The clinical spectrum of the disease is highly variable, ranging from hyper-IgE syndrome (HIES)-like phenotypes to combined immunodeficiency. Most patients present in early childhood with severe, treatment-resistant eczema and recurrent respiratory tract infections.51 These infections often progress to chronic lung disease, including bronchiectasis. Immunological findings typically include markedly elevated serum IgE, peripheral eosinophilia, and, in some cases, hypogammaglobulinemia. Additional laboratory abnormalities may include lymphopenia, neutropenia, and impaired specific antibody responses. Beyond immune dysfunction, affected individuals may display a range of non-immunologic manifestations, such as neurodevelopmental delay, intellectual disability of variable severity, scoliosis, dysmorphic facial features, and growth failure.10,52,53
Management of PGM3 deficiency is multidisciplinary. Antimicrobial prophylaxis is often required to prevent recurrent infections, with trimethoprim–sulfamethoxazole commonly used for staphylococcal coverage. Immunoglobulin replacement therapy is indicated in patients with significant antibody deficiencies.54,55 Dermatologic management is essential, with topical anti-inflammatory agents and emollients playing a key role in the control of eczema. Recently, targeted biologic agents such as dupilumab (an anti-IL-4Rα monoclonal antibody) have demonstrated efficacy in controlling severe atopic dermatitis in selected patients, highlighting the promising potential of precision biologic therapies in this context.56 Neurodevelopmental support, physical therapy, and orthopedic follow-up are essential components of long-term care.
Hematopoietic stem cell transplantation has been attempted in a limited number of patients with severe immunological phenotypes of PGM3 deficiency, with mixed results. While immune reconstitution and decreased susceptibility to infections have been achieved in some cases, neurodevelopmental and connective tissue manifestations often persist. For example, Stray-Pedersen et al reported two patients who underwent HSCT with correction of immune defects but continued developmental delay.52 Similarly, Fusaro et al described a patient who experienced serious post-transplant complications and died eight months after HSCT despite full donor engraftment.57 In contrast, Winslow et al reported a successful early HSCT in an infant, resulting in immune recovery and normal development at one-year follow-up.58 These findings suggest that HSCT may benefit selected patients, particularly if performed early, but its role remains investigational and requires further evaluation regarding optimal timing, conditioning, and long-term outcomes.
Given its rarity, long-term natural history data remain limited. Early diagnosis through genetic testing is critical, as timely intervention can prevent irreversible organ damage. Current research is focused on defining genotype–phenotype correlations, and optimizing HSCT strategies.
ERBIN Deficiency
ERBB2-interacting protein,59 also known as ERBB2-interacting protein (ERBB2IP), is encoded by the ERBB2IP gene and plays a critical role in mediating the crosstalk between the STAT3 and TGF-β (Transforming Growth Factor Beta) signaling pathways.55,60 TGF-β is a multifunctional cytokine that possesses both immune stimulatory and inhibitory functions, influences T-lymphocyte differentiation, and promotes cell proliferation and wound healing.61,62 STAT3 negatively regulates TGF-β signaling through the ERBIN. SMAD2 and SMAD3 are intracellular mediators of TGF-β signaling; upon phosphorylation, they translocate to the nucleus to regulate gene expression related to immune modulation and cell differentiation. An ERBIN-SMAD2/3 complex forms in the cytoplasm, limiting the nuclear translocation of phosphorylated SMAD2 and SMAD3 (pSMAD2/3). In cases of STAT3 deficiency or ERBIN dysfunction, this complex is disrupted, leading to hyperactivation of TGF-β signaling due to increased nuclear pSMAD2/3.60 Abnormal regulation of TGF-β signaling has also been linked to human disorders involving connective tissue abnormalities, including Ehlers-Danlos syndrome and Loeys-Dietz syndrome.61
Both STAT3-mutant individuals and those with ERBIN deficiency exhibit increased frequencies of FOXP3⁺ regulatory T cells and elevated IL-4Rα expression. In ERBIN-deficient CD45RO+ CD4+ T cells, ex vivo analyses reveal markedly elevated production of Th2-associated cytokines (IL-4, IL-5, and IL-13), which likely contributes to the elevated IgE production, and the development of allergic diseases such as eosinophilic esophagitis and asthma.60,63 Three related individuals were identified with an AD mutation in ERBIN.60,64 These patients demonstrated multiple clinical features that closely resemble those seen in STAT3 deficiency, including recurrent infections, eczema, elevated serum IgE levels, eosinophilia, retained primary teeth, and various skeletal and vascular abnormalities.60
Recently dupilumab, an IL-4/IL-13 pathway inhibitor, has been successfully used to treat eosinophilic esophagitis and respiratory symptoms in a patient with ERBIN deficiency, highlighting the therapeutic potential of targeting this pathway in ERBIN-related allergic inflammation.61
Loeys-Dietz Syndrome
Loeys–Dietz syndrome (LDS) is a rare connective tissue disorder with a wide range of possible symptoms. Autosomal dominant forms result from heterozygous pathogenic variants in genes that regulate the TGF-β signaling pathway, including TGFBR1, TGFBR2, SMAD2, SMAD3, TGFB2 and TGFB3. Autosomal recessive forms result from mutations in the IPO8 gene.65 Approximately 75% of cases arise from de novo mutations.66 The pathogenesis is primarily based on the dysregulation of the TGF-β signaling pathway, which leads to structural weakness of the vascular wall, skeletal dysplasia and immune dysregulation.65
In LDS, the immune mechanism paradoxically involves enhanced TGF-β signaling. This aberrant signaling results in elevated IgE levels, eosinophilia, and increased type 2 cytokine responses (IL-4, IL-5 and IL-13).67 Although FOXP3+ regulatory T cells (Tregs) increase, immune tolerance is not maintained. This manifests clinically as atopy, asthma, eczema and eosinophilic gastrointestinal diseases. Furthermore, some patients have been reported to develop inflammatory bowel disease.67
The most prominent clinical manifestations of Loeys-Dietz syndrome are aortic root dilatation and dissection, as well as widespread arterial aneurysms. Arteries often have an abnormal course. As echocardiography is not always sufficient for an accurate vascular assessment, advanced imaging techniques such as magnetic resonance68 or computed tomography (CT) angiography are recommended.69 Skeletal abnormalities commonly observed in LDS include pectus deformities, scoliosis, joint hypermobility, arachnodactyly and cervical spine instability. Craniofacial anomalies typically consist of hypertelorism, a bifid uvula, a cleft palate and craniosynostosis, while cutaneous findings include thin, translucent skin; easy bruising; and dystrophic scarring.69
From an immunological standpoint, patients often present with asthma, eczema, food and environmental allergies, eosinophilic esophagitis or gastritis, and inflammatory bowel disease (IBD). Some individuals exhibit markedly elevated IgE levels and an enhanced Th2 cytokine response.67
Management and follow-up of LDS require a multidisciplinary approach. The primary objective is to prevent cardiovascular complications. Diagnosis should include baseline echocardiography, followed by regular annual evaluations. Imaging of the entire arterial system via CT or MR angiography is strongly recommended, as the risk of arterial dissection persists even when the aortic diameter is within normal limits.69 Beta-blockers and angiotensin receptor blockers, such as losartan, which modulate the TGF-β signaling pathway, can be used for therapeutic purposes.70 Surgical intervention may be indicated for smaller aortic diameters than in Marfan syndrome. Furthermore, patients should avoid contact sports, receive prophylaxis for endocarditis when necessary and undergo genetic counselling, including evaluation of at-risk family members.71
STAT6 GOF
Signal transducer and activator of transcription 6 (STAT6) GOF disease is a recently described primary atopic disorder characterized by early-onset and severe allergic inflammation.72–74 STAT6 is a transcription factor that mediates signal transduction from IL-4 and IL-13 receptors to the nucleus, orchestrating various immune functions, most notably promoting Th2 cell differentiation and IgE class switching in B cells.75,76 Upon cytokine binding to the IL-4 receptor, receptor-associated JAK kinases phosphorylate STAT6, leading to its dimerization and nuclear translocation where it regulates target gene expression.73
To date, at least 21 individuals have been reported with STAT6 GOF disease,68,77 typically presenting with severe atopic dermatitis beginning in infancy, multiple food allergies, drug allergies, asthma, eosinophilic gastrointestinal disease, recurrent anaphylaxis, and markedly elevated serum IgE levels accompanied by peripheral eosinophilia. Beyond atopic symptoms, some patients exhibited short stature, skeletal abnormalities, and recurrent infections, underscoring the multisystemic nature.72–74 Notably, a patient with STAT6 GOF disease was reported to have both early-onset atopy and follicular lymphoma, raising the possibility that STAT6 GOF mutations may confer an increased risk of malignancy. This is consistent with previous findings linking somatic STAT6 GOF mutations to lymphomagenesis, especially in follicular lymphoma; for example, in a cohort of 114 follicular lymphoma patients, somatic STAT6 GOF mutations were identified in 11% of cases.78 Patients with STAT6 GOF mutations exhibit a Th2-biased immune profile, with decreased Th1 and Th17 cell populations and, in some cases, reduced B cell counts and hypogammaglobulinemia.73
Targeted inhibition of the JAK-STAT pathway has shown notable therapeutic efficacy in STAT6 GOF–associated allergic disease. Ruxolitinib has been reported to reverse STAT6 hyperactivation, normalize Th1/Th17 responses, suppress eosinophilia, and improve atopic dermatitis. Tofacitinib led to reduction of intraepithelial eosinophils, improved esophageal endoscopic findings, and relief of dysphagia, supporting its use in eosinophilic gastrointestinal involvement.68,73 Furthermore, dupilumab, by blocking IL-4/IL-13 signaling, downregulated Th2 gene expression, decreased IL-4 receptor expression on naïve T cells, and led to marked improvement in skin inflammation and accelerated growth.68 These findings highlight the promise of targeted therapies in addressing the immune dysregulation associated with STAT6 GOF disease.
CARD11 Deficiency
CARD11 (also known as CARMA1) is a protein belonging to the membrane-associated guanylate kinase (MAGUK) family. It cooperates with B-cell lymphoma/leukaemia 10 (BCL10) and mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1) to form the “CBM complex”.79
Biallelic null mutations in CARD11 result in severe T- and B-cell immunodeficiency in humans and mouse models.80 Somatic gain-of-function (GOF) mutations in CARD11 are frequently observed in non-Hodgkin B-cell lymphomas, whereas germline GOF mutations in humans cause B-cell expansion with NF-κB activation and T-cell hypo responsiveness (BENTA) syndrome.81–83 Conversely, hypomorphic or dominant-negative mutations lead to partial rather than complete loss of function, resulting in variable immunodeficiency phenotypes often accompanied by a strong predisposition towards atopic disease.79,84,85
In CARD11 deficiency, T cell proliferation and balanced T helper differentiation are impaired due to defects in multiple T cell receptor (TCR) signaling pathways that depend on CARD11. Reduced TCR signal strength can lead to a Th2 bias under certain conditions.86 Defects in both NF-κB and mTORC1 impair the IL-2 feedback loop and the metabolic reprogramming required for T cell proliferation, thereby limiting expansion. Decreased mTORC1 activity suppresses Th1 differentiation and favors a dominant Th2 response.87 Similarly, NF-κB blockade preferentially disrupts the Th1 response, predisposing individuals to Th2-driven allergic inflammation.88
Gain-of-function and loss-of-function mutations in CARD11 both reduce circulating memory B cells and impair immunoglobulin production.89 This suggests that CARD11 plays a vital role in modulating the BCR signaling necessary for normal B cell maturation and differentiation. Under physiological conditions, IgE signaling drives B cells towards short-lived plasmablasts, which are then subject to apoptosis. However, the loss of BCR signaling molecules can lead to the accumulation of long-lived IgE+ memory/plasma cells.90 Reduced CARD11 signaling in humans may therefore contribute to sustained IgE production and the development of allergic phenotypes.85
Clinically, patients present with recurrent infections, atopic dermatitis, allergic manifestations and various immunodeficiency-related findings. Central features of the clinical phenotype include impaired T cell proliferation, reduced IFN-γ production, and Th2 skewing.87 In addition, defects in B cell differentiation contribute to impaired immunoglobulin responses.90 Cutaneous viral infections, such as molluscum contagiosum and HSV-1, are frequently observed, most likely due to impaired immune surveillance by CD8+ T cells. The same mechanism may also account for tumor development in certain cases. Patients with BENTA syndrome, in particular, often present with recurrent viral infections (eg EBV) and molluscum contagiosum, and carry an increased risk of developing lymphoma or leukemia.82
Currently, there is no standard curative therapy for CARD11 deficiency, and patient management is supportive. Nevertheless, glutamine supplementation has emerged as a promising strategy that may alleviate disease severity by restoring T cell glutamine uptake and mTORC1 signaling. In the context of CARD11 deficiency, glutamine may partially restore T cell function via the glutamine transporter Alanine-Serine-Cysteine Transporter 2 (ASCT2), which acts as a “glutamine gate”.91 This approach has the potential to enhance immune responses and attenuate the atopic phenotype.92
Netherton Syndrome
Comél-Netherton syndrome (NS) is a rare autosomal recessive genetic disorder caused by mutations in the SPINK5 gene (serine protease inhibitor Kazal type 5).93 It occurs at a rate of approximately 1 in 200,000 births, with an estimated prevalence of 1–9 per 1,000,000 people. SPINK5 is located on chromosome 5q32, consists of 33 exons and spans approximately 61 kilobases (kb) in total. It encodes a serine protease inhibitor protein called LEKTI (lymphoepithelial Kazal-type-related inhibitor). LEKTI is expressed in the granular layer of the epidermis and in the inner root sheaths of hair follicles.94 It plays a role in maintaining skin barrier integrity, the activation of T and B lymphocytes, complement activation, cytokine secretion and the recruitment of inflammatory cells.93
The majority of reported SPINK5 mutations result in premature stop codons, leading to the loss of LEKTI expression.93 Thus far, approximately 80 distinct genetic mutations in the SPINK5 gene have been identified. These include 19 missense mutations, 25 splice site mutations, 20 small deletions, 12 small insertions, two indels, one large deletion and one compound heterozygous mutation.95
NS is characterized clinically by congenital ichthyosiform erythroderma, trichorrhexis invaginata (“bamboo hair”) and atopic manifestations such as food allergies, asthma and rhino-conjunctivitis. These are often accompanied by elevated IgE levels and eosinophilia.94 Patients are especially at risk of life-threatening complications due to excessive transepidermal water loss during the first year of life, including hypernatremic dehydration, hypothermia, growth retardation, and sepsis.93 In most patients, the erythema present at birth evolves into ichthyosis linearis circumflexa, which is characterized by distinctive “double-edged” scaling.93 This condition presents as pruritic, polycyclic, erythematous plaques with circinate or serpiginous scaling and double edges. Another distinctive feature is “bamboo hair” (trichorrhexis invaginata), which is characterized by invagination of the hair shaft, producing a “ball-in-cup” appearance.93
Information regarding treatment options and outcomes in Netherton syndrome is limited, and the therapeutic regimens used to date have primarily been supportive. First-line treatment options include topical corticosteroids.96 Topical calcineurin inhibitors and narrowband ultraviolet B (NB-UVB) phototherapy have also been reported in patients with NS. Many patients require systemic agents. Retinoids have been described in various studies. New systemic treatment approaches have been proposed, such as immunoglobulins and biologic agents that target specific inflammatory pathways.97
Dupilumab, which targets the IL-4 and IL-13 pathways, is the most frequently used biologic agent. Most studies have reported significant improvements in skin lesions and pruritus. Secukinumab and ixekizumab target the IL-17/IL-23 axis, which has recently been shown to be overexpressed in patients with NS. TNF-α blockers (eg infliximab and adalimumab), omalizumab (anti-IgE) and ustekinumab (an IL-12/23 inhibitor) have been used in a limited number of cases.97 A Phase 1 trial published in 2019 demonstrated the safety and feasibility of gene therapy using autologous keratinocytes transduced with a lentiviral vector encoding the SPINK5 gene. Although still in its early stages, this method is being considered as a potential therapeutic option.98
Conclusion
Hyper-IgE syndromes comprise a heterogeneous group of disorders characterized by eczema, recurrent infections, and markedly elevated serum IgE levels. The appearance of non-immunological manifestations later in life often complicates the distinction of these syndromes from severe atopic disease in infancy. HIES should be suspected particularly when symptoms begin in the neonatal period, respond poorly to standard therapies, arise in atypical locations, or are accompanied by recurrent infections. A detailed family history is essential, and when clinical suspicion remains, immunological assessment followed by molecular genetic testing should be pursued.
The primary goals of management are to control infections, prevent long-term complications such as chronic lung disease, and improve quality of life. In cases where the underlying molecular mechanism is defined, targeted therapies may offer benefit for patients who do not respond adequately to conventional treatment. Although hematopoietic stem cell transplantation has been attempted in a limited number of cases with encouraging outcomes, non-immunological manifestations are often not corrected by this approach. In this regard, emerging gene therapy strategies may represent a promising future avenue of treatment.
Disclosure
The authors declare that they have no competing interests.
References
1. Poli MC, Aksentijevich I, Bousfiha AA, et al. Human inborn errors of immunity: 2024 update on the classification from the International Union of Immunological Societies Expert Committee. J Human Immun. 2025;1(1):e20250003. doi:10.70962/jhi.20250003
2. Hsu AP, Davis J, Puck JM, Holland SM, Freeman AF. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. [GeneReviews® Internet]. STAT3 Hyper IgE Syndrome. STAT3 Hyper IgE Syndrome. Seattle (WA), Seattle (WA): University of Washington; 2010. PMID: 20301786.
3. Gharehzadehshirazi A, Amini A, Rezaei N. Hyper IgE syndromes: a clinical approach. Clin Immunol. 2022;237:108988. doi:10.1016/j.clim.2022.108988
4. Ogulur I, Mitamura Y, Yazici D, et al. Type 2 immunity in allergic diseases. Cell Mol Immunol. 2025;1–32.
5. Gardner S, Jin Y, Fyfe PK, et al. Structural insights into IL-11-mediated signalling and human IL6ST variant-associated immunodeficiency. Nat Commun. 2024;15(1):2071. doi:10.1038/s41467-024-46235-6
6. Gernez Y, Freeman AF, Holland SM, et al. Autosomal dominant hyper-IgE syndrome in the USIDNET registry. J Allergy Clin Immunol. 2018;6(3):996–1001. doi:10.1016/j.jaip.2017.06.041
7. Villarino AV, Kanno Y, Ferdinand JR, O’Shea JJ. Mechanisms of Jak/STAT signaling in immunity and disease. J Immunol. 2015;194(1):21–27. doi:10.4049/jimmunol.1401867
8. O’Shea JJ, Holland SM, Staudt LM. JAKs and STATs in immunity, immunodeficiency, and cancer. N Engl J Med. 2013;368(2):161–170. doi:10.1056/NEJMra1202117
9. Chen Y-H, Spencer S, Laurence A, Thaventhiran JE, Uhlig HH. Inborn errors of IL-6 family cytokine responses. Curr Opin Immunol. 2021;72:135–145. doi:10.1016/j.coi.2021.04.007
10. Sassi A, Lazaroski S, Wu G, et al. Hypomorphic homozygous mutations in phosphoglucomutase 3 (PGM3) impair immunity and increase serum IgE levels. J Allergy Clin Immunol. 2014;133(5):1410–9.e13. doi:10.1016/j.jaci.2014.02.025
11. Milner JD, Vogel TP, Forbes L, et al. Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood J Am Soc Hematol. 2015;125(4):591–599.
12. Rajala HL, Porkka K, Maciejewski JP, Loughran TP Jr, Mustjoki S. Uncovering the pathogenesis of large granular lymphocytic leukemia—novel STAT3 and STAT5b mutations. Ann Med. 2014;46(3):114–122. doi:10.3109/07853890.2014.882105
13. Jägle S, Heeg M, Grün S, et al. Distinct molecular response patterns of activating STAT3 mutations associate with penetrance of lymphoproliferation and autoimmunity. Clin Immunol. 2020;210:108316. doi:10.1016/j.clim.2019.108316
14. Heimall J, Davis J, Shaw PA, et al. Paucity of genotype–phenotype correlations in STAT3 mutation positive Hyper IgE Syndrome (HIES). Clin Immunol. 2011;139(1):75–84. doi:10.1016/j.clim.2011.01.001
15. Wolach O, Kuijpers T, Ben-Ari J, et al. Variable clinical expressivity of STAT3 mutation in hyperimmunoglobulin E syndrome: genetic and clinical studies of six patients. J Clin Immunol. 2014;34(2):163–170. doi:10.1007/s10875-014-9988-4
16. Chandesris M-O, Melki I, Natividad A, et al. Autosomal dominant STAT3 deficiency and hyper-IgE syndrome: molecular, cellular, and clinical features from a French national survey. Medicine. 2012;91(4):e1–e19. doi:10.1097/MD.0b013e31825f95b9
17. Redor A, Danion F, Parize P, et al. Devastating gynecological infections in women with STAT3 deficiency. Clinl Infect Dis. 2020;71(7):e186–e90. doi:10.1093/cid/ciaa020
18. Parisi X, Bergerson J, Urban A, Darnell D, Stratton P, Freeman AF. Obstetric and gynecological care in patients with STAT3-deficient hyper IgE syndrome. J Clin Immunol. 2020;40(7):1048–1050. doi:10.1007/s10875-020-00827-1
19. Spencer S, Köstel BS, Egner W, et al. Loss of the interleukin-6 receptor causes immunodeficiency, atopy, and abnormal inflammatory responses. J Exp Med. 2019;216(9):1986–1998. doi:10.1084/jem.20190344
20. Freeman AF, Olivier KN. Hyper IgE syndromes and the lung. Clinics Chest Med. 2016;37(3):557. doi:10.1016/j.ccm.2016.04.016
21. Conti H, Baker O, Freeman A, et al. New mechanism of oral immunity to mucosal candidiasis in hyper-IgE syndrome. Mucosal Immunol. 2011;4(4):448–455. doi:10.1038/mi.2011.5
22. Siegel AM, Stone KD, Cruse G, et al. Diminished allergic disease in patients with STAT3 mutations reveals a role for STAT3 signaling in mast cell degranulation. J Allergy Clin Immunol. 2013;132(6):1388–96.e3. doi:10.1016/j.jaci.2013.08.045
23. Hox V, O’Connell MP, Lyons JJ, et al. Diminution of signal transducer and activator of transcription 3 signaling inhibits vascular permeability and anaphylaxis. J Allergy Clin Immunol. 2016;138(1):187–199. doi:10.1016/j.jaci.2015.11.024
24. Boos A, Hagl B, Schlesinger A, et al. Atopic dermatitis, STAT 3‐and DOCK 8‐hyper‐IgE syndromes differ in IgE‐based sensitization pattern. Allergy. 2014;69(7):943–953. doi:10.1111/all.12416
25. Meixner I, Hagl B, Kröner CI, et al. Retained primary teeth in STAT3 hyper-IgE syndrome: early intervention in childhood is essential. Orphanet J Rare Dis. 2020;15(1):244. doi:10.1186/s13023-020-01516-3
26. Chandesris M-O, Azarine A, Ong K-T, et al. Frequent and widespread vascular abnormalities in human signal transducer and activator of transcription 3 deficiency. Circulation. 2012;5(1):25–34. doi:10.1161/CIRCGENETICS.111.961235
27. Dziadzio M, Chee R, McNamara C, Burns S, Seneviratne S, Brimbacher B. Lymphoma complicating STAT3 hyper-IgE syndrome. In: Clinical and Experimental Immunology. NJ USA: Wiley-Blackwell; 2013.
28. Denning DW, Cadranel J, Beigelman-Aubry C, et al. Chronic pulmonary aspergillosis: rationale and clinical guidelines for diagnosis and management. Eur Respir J. 2015;47(1):45–68. doi:10.1183/13993003.00583-2015
29. Woellner C, Gertz EM, Schäffer AA, et al. Mutations in STAT3 and diagnostic guidelines for hyper-IgE syndrome. J Allergy Clin Immunol. 2010;125(2):424–32.e8. doi:10.1016/j.jaci.2009.10.059
30. Vinh DC, Sugui JA, Hsu AP, Freeman AF, Holland SM. Invasive fungal disease in autosomal-dominant hyper-IgE syndrome. J Allergy Clin Immunol. 2010;125(6):1389–1390. doi:10.1016/j.jaci.2010.01.047
31. Goussetis E, Peristeri I, Kitra V, et al. Successful long-term immunologic reconstitution by allogeneic hematopoietic stem cell transplantation cures patients with autosomal dominant hyper-IgE syndrome. J Allergy Clin Immunol. 2010;126(2):392–394. doi:10.1016/j.jaci.2010.05.005
32. Patel N, Gallagher J, Torgerson T, Gilman A. Successful haploidentical donor hematopoietic stem cell transplant and restoration of STAT3 function in an adolescent with autosomal dominant hyper-IgE syndrome. J Clin Immunol. 2015;35(5):479–485. doi:10.1007/s10875-015-0167-z
33. Yanagimachi M, Ohya T, Yokosuka T, et al. The potential and limits of hematopoietic stem cell transplantation for the treatment of autosomal dominant hyper-IgE syndrome. J Clin Immunol. 2016;36(5):511–516. doi:10.1007/s10875-016-0278-1
34. Nester TA, Wagnon AH, Reilly WF, Spitzer G, Kjeldsberg CR, Hill HR. Effects of allogeneic peripheral stem cell transplantation in a patient with job syndrome of hyperimmunoglobulinemia E and recurrent infections. Am J Med. 1998;105(2):162–164. doi:10.1016/S0002-9343(98)00200-9
35. Gennery A, Flood T, Abinun M, Cant A. Bone marrow transplantation does not correct the hyper IgE syndrome. Bone Marrow Transplant. 2000;25(12):1303–1305. doi:10.1038/sj.bmt.1702446
36. Harrison SC, Tsilifis C, Slatter MA, et al. Hematopoietic stem cell transplantation resolves the immune deficit associated with STAT3-dominant-negative hyper-IgE syndrome. J Clin Immunol. 2021;41(5):934–943. doi:10.1007/s10875-021-00971-2
37. Annunziato F, Cosmi L, Liotta F, Maggi E, Romagnani S. Defining the human T helper 17 cell phenotype. Trends Immunol. 2012;33(10):505–512. doi:10.1016/j.it.2012.05.004
38. Schwerd T, Twigg SR, Aschenbrenner D, et al. A biallelic mutation in IL6ST encoding the GP130 co-receptor causes immunodeficiency and craniosynostosis. J Exp Med. 2017;214(9):2547–2562. doi:10.1084/jem.20161810
39. Shahin T, Aschenbrenner D, Cagdas D, et al. Selective loss of function variants in IL6ST cause Hyper-IgE syndrome with distinct impairments of T-cell phenotype and function. Haematologica. 2018;104(3):609. doi:10.3324/haematol.2018.194233
40. Taga T, Kishimoto T. Gp130 and the interleukin-6 family of cytokines. Ann Rev Immunol. 1997;15(1):797–819. doi:10.1146/annurev.immunol.15.1.797
41. Nahum A, Sharfe N, Broides A, et al. Defining the biological responses of IL-6 by the study of a novel IL-6 receptor chain immunodeficiency. J Allergy Clin Immunol. 2020;145(3):1011–5.e6. doi:10.1016/j.jaci.2019.11.015
42. Minegishi Y. Hyper-IgE syndrome, 2021 update. Allergol Int. 2021;70(4):407–414. doi:10.1016/j.alit.2021.07.007
43. Esparza-Gordillo J, Schaarschmidt H, Liang L, et al. A functional IL-6 receptor (IL6R) variant is a risk factor for persistent atopic dermatitis. J Allergy Clin Immunol. 2013;132(2):371–377. doi:10.1016/j.jaci.2013.01.057
44. Frey-Jakobs S, Hartberger JM, Fliegauf M, et al. ZNF341 controls STAT3 expression and thereby immunocompetence. Sci Immunol. 2018;3(24):eaat4941. doi:10.1126/sciimmunol.aat4941
45. Béziat V, Li J, J-X L, et al. A recessive form of hyper-IgE syndrome by disruption of ZNF341-dependent STAT3 transcription and activity. Sci Immunol. 2018;3(24):eaat4956. doi:10.1126/sciimmunol.aat4956
46. Béziat V, Fieschi C, Momenilandi M, et al. Inherited human ZNF341 deficiency. Curr Opin Immunol. 2023;82:102326. doi:10.1016/j.coi.2023.102326
47. Cekic S, Huriyet H, Hortoglu M, et al. Increased radiosensitivity and impaired DNA repair in patients with STAT3-LOF and ZNF341 deficiency, potentially contributing to malignant transformations. Clin Exp Immunol. 2022;209(1):83–89. doi:10.1093/cei/uxac041
48. Dick JK, Boull C, Pozos TC, Maguiness SM. Improvement in atopic dermatitis and recurrent infection with dupilumab in children with distinct genetic types of hyper‐ige syndrome: a case series and literature review. Pediatr Dermatol. 2025;42(2):376–382. doi:10.1111/pde.15780
49. Greig KT, Antonchuk J, Metcalf D, et al. Agm1/Pgm3-mediated sugar nucleotide synthesis is essential for hematopoiesis and development. Mol Cell Biol. 2007;27(16):5849–5859. doi:10.1128/MCB.00802-07
50. Yang L, Zerbato B, Pessina A, et al. PGM3 insufficiency: a glycosylation disorder causing a notable T cell defect. Front Immunol. 2024;15:1500381. doi:10.3389/fimmu.2024.1500381
51. Zhang Y, Yu X, Ichikawa M, et al. Autosomal recessive PGM3 mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. J Allergy Clin Immunol. 2014;133(5):1400. doi:10.1016/j.jaci.2014.02.013
52. Stray-Pedersen A, Backe PH, Sorte HS, et al. PGM3 mutations cause a congenital disorder of glycosylation with severe immunodeficiency and skeletal dysplasia. Am J Hum Genet. 2014;95(1):96–107. doi:10.1016/j.ajhg.2014.05.007
53. Lundin KE, Hamasy A, Backe PH, et al. Susceptibility to infections, without concomitant hyper-IgE, reported in 1976, is caused by hypomorphic mutation in the phosphoglucomutase 3 (PGM3) gene. Clin Immunol. 2015;161(2):366–372. doi:10.1016/j.clim.2015.10.002
54. Park B, Liu GY. Staphylococcus aureus and hyper-IgE syndrome. Int J Mol Sci. 2020;21(23):9152. doi:10.3390/ijms21239152
55. Salehi M, Neshati Z, Ahanchian H, et al. Hyper IgE syndromes: understanding, management, and future perspectives: a narrative review. Health Sci Rep. 2025;8(3):e70497. doi:10.1002/hsr2.70497
56. Kao AS, Deirawan H, Poowuttikul P, Daveluy S. Hyper IgE syndrome‐related disease treated with dupilumab: a case report. Clin Case Rep. 2023;11(9):e7614. doi:10.1002/ccr3.7614
57. Fusaro M, Vincent A, Castelle M, et al. Two novel homozygous mutations in phosphoglucomutase 3 leading to severe combined immunodeficiency, skeletal dysplasia, and malformations. J Clin Immunol. 2021;41(5):958–966. doi:10.1007/s10875-021-00985-w
58. Winslow A, Jalazo ER, Evans A, Winstead M, Moran T. A De Novo cause of PGM3 deficiency treated with hematopoietic stem cell transplantation. J Clin Immunol. 2022;42(3):691–694. doi:10.1007/s10875-021-01196-z
59. Barzaghi F, Hernandez LCA, Neven B, et al. Long-term follow-up of IPEX syndrome patients after different therapeutic strategies: an international multicenter retrospective study. J Allergy Clin Immunol. 2018;141(3):1036–49.e5. doi:10.1016/j.jaci.2017.10.041
60. Lyons J, Liu Y, Ma C, et al. ERBIN deficiency links STAT3 and TGF-β pathway defects with atopy in humans. J Exp Med. 2017;214(3):669–680. doi:10.1084/jem.20161435
61. Droghini HR, Abonia JP, Collins MH, et al. Targeted IL-4Rα blockade ameliorates refractory allergic eosinophilic inflammation in a patient with dysregulated TGF-β signaling due to ERBIN deficiency. J Allergy Clin Immunol. 2022;10(7):1903–1906. doi:10.1016/j.jaip.2022.01.012
62. Morikawa M, Derynck R, Miyazono K. TGF-β and the TGF-β family: context-dependent roles in cell and tissue physiology. Cold Spring Harbor Perspect Biol. 2016;8(5):a021873. doi:10.1101/cshperspect.a021873
63. Milner JD. ERBIN and phosphoglucomutase 3 deficiency. Curr Opin Immunol. 2023;84:102353. doi:10.1016/j.coi.2023.102353
64. Redmond MT, Scherzer R, Prince BT. Novel genetic discoveries in primary immunodeficiency disorders. Clin Rev Allergy Immunol. 2022;63(1):55–74. doi:10.1007/s12016-021-08881-2
65. Loeys BL, Chen J, Neptune ER, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nature Genet. 2005;37(3):275–281. doi:10.1038/ng1511
66. Loeys BL, Schwarze U, Holm T, et al. Aneurysm syndromes caused by mutations in the TGF-β receptor. N Engl J Med. 2006;355(8):788–798. doi:10.1056/NEJMoa055695
67. Frischmeyer-Guerrerio PA, Guerrerio AL, Oswald G, et al. TGFβ receptor mutations impose a strong predisposition for human allergic disease. Sci Trans Med. 2013;5(195):195ra94–ra94. doi:10.1126/scitranslmed.3006448
68. Sharma M, Suratannon N, Leung D, et al. Human germline gain-of-function in STAT6: from severe allergic disease to lymphoma and beyond. Trends Immunol. 2024;45(2):138–153. doi:10.1016/j.it.2023.12.003
69. MacCarrick G, Black JH, Bowdin S, et al. Loeys–Dietz syndrome: a primer for diagnosis and management. Genet Med. 2014;16(8):576–587. doi:10.1038/gim.2014.11
70. Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312(5770):117–121. doi:10.1126/science.1124287
71. Teixidó-Tura G, Franken R, Galuppo V, et al. Heterogeneity of aortic disease severity in patients with Loeys–Dietz syndrome. Heart. 2016;102(8):626–632. doi:10.1136/heartjnl-2015-308535
72. Takeuchi I, Yanagi K, Takada S, et al. STAT6 gain-of-function variant exacerbates multiple allergic symptoms. J Allergy Clin Immunol. 2023;151(5):1402–9.e6. doi:10.1016/j.jaci.2022.12.802
73. Baris S, Benamar M, Chen Q, et al. Severe allergic dysregulation due to a gain of function mutation in the transcription factor STAT6. J Allergy Clin Immunol. 2023;152(1):182–94.e7. doi:10.1016/j.jaci.2023.01.023
74. Suratannon N, Ittiwut C, Dik WA, et al. A germline STAT6 gain-of-function variant is associated with early-onset allergies. J Allergy Clin Immunol. 2023;151(2):565–71.e9. doi:10.1016/j.jaci.2022.09.028
75. Takeda K, Tanaka T, Shi W, et al. Essential role of Stat6 in IL-4 signalling. Nature. 1996;380(6575):627–630. doi:10.1038/380627a0
76. Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for the development of Th2 cells. Immunity. 1996;4(3):313–319. doi:10.1016/S1074-7613(00)80439-2
77. AlYafie R, Velayutham D, Van Panhuys N, Jithesh PV. The genetics of hyper IgE syndromes. Front Immunol. 2025;16:1516068. doi:10.3389/fimmu.2025.1516068
78. Minskaia E, Maimaris J, Jenkins P, et al. Autosomal dominant STAT6 gain of function causes severe atopy associated with lymphoma. J Clin Immunol. 2023;43(7):1611–1622. doi:10.1007/s10875-023-01530-7
79. Ma CA, Stinson JR, Zhang Y, et al. Germline hypomorphic CARD11 mutations in severe atopic disease. Nature Genet. 2017;49(8):1192–1201. doi:10.1038/ng.3898
80. Juilland M, Thome M. Role of the CARMA1/BCL10/MALT1 complex in lymphoid malignancies. Current Opinion Hematol. 2016;23(4):402–409. doi:10.1097/MOH.0000000000000257
81. Turvey SE, Durandy A, Fischer A, et al. The CARD11-BCL10-MALT1 (CBM) signalosome complex: stepping into the limelight of human primary immunodeficiency. J Allergy Clin Immunol. 2014;134(2):276–284. doi:10.1016/j.jaci.2014.06.015
82. Arjunaraja S, Angelus P, Su HC, Snow AL. Impaired control of epstein–barr virus infection in B-cell expansion with NF-κB and T-cell anergy disease. Front Immunol. 2018;9:198. doi:10.3389/fimmu.2018.00198
83. Snow AL, Xiao W, Stinson JR, et al. Congenital B cell lymphocytosis explained by novel germline CARD11 mutations. J Exp Med. 2012;209(12):2247–2261. doi:10.1084/jem.20120831
84. Dadi H, Jones TA, Merico D, et al. Combined immunodeficiency and atopy caused by a dominant negative mutation in caspase activation and recruitment domain family member 11 (CARD11). J Allergy Clin Immunol. 2018;141(5):1818–30.e2. doi:10.1016/j.jaci.2017.06.047
85. Jun JE, Wilson LE, Vinuesa CG, et al. Identifying the MAGUK protein Carma-1 as a central regulator of humoral immune responses and atopy by genome-wide mouse mutagenesis. Immunity. 2003;18(6):751–762. doi:10.1016/S1074-7613(03)00141-9
86. Yamane H, Paul WE. Early signaling events that underlie fate decisions of naive CD 4+ T cells toward distinct T‐helper cell subsets. Immunol Rev. 2013;252(1):12–23. doi:10.1111/imr.12032
87. Pollizzi KN, Powell JD. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat Rev Immunol. 2014;14(7):435–446. doi:10.1038/nri3701
88. Aronica MA, Mora AL, Mitchell DB, et al. Preferential role for NF-κB/Rel signaling in the type 1 but not type 2 T cell-dependent immune response in vivo. J Immunol. 1999;163(9):5116–5124. doi:10.4049/jimmunol.163.9.5116
89. Arjunaraja S, Snow AL. Gain-of-function mutations and immunodeficiency: at a loss for proper tuning of lymphocyte signaling. Curr Opin Allergy Clin Immunol. 2015;15(6):533–538. doi:10.1097/ACI.0000000000000217
90. Haniuda K, Fukao S, Kodama T, Hasegawa H, Kitamura D. Autonomous membrane IgE signaling prevents IgE-memory formation. Nat Immunol. 2016;17(9):1109–1117. doi:10.1038/ni.3508
91. Nakaya M, Xiao Y, Zhou X, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity. 2014;40(5):692–705. doi:10.1016/j.immuni.2014.04.007
92. Hamilton KS, Phong B, Corey C, et al. T cell receptor–dependent activation of mTOR signaling in T cells is mediated by Carma1 and MALT1, but not Bcl10. Sci Signaling. 2014;7(329):ra55–ra. doi:10.1126/scisignal.2005169
93. Hovnanian A. Netherton syndrome: skin inflammation and allergy by loss of protease inhibition. Cell Tissue Res. 2013;351(2):289–300. doi:10.1007/s00441-013-1558-1
94. Bitoun E, Chavanas S, Irvine AD, et al. Netherton syndrome: disease expression and spectrum of SPINK5 mutations in 21 families. J Invest Dermatol. 2002;118(2):352–361. doi:10.1046/j.1523-1747.2002.01603.x
95. Sarri CA, Roussaki-Schulze A, Vasilopoulos Y, et al. Netherton syndrome: a genotype-phenotype review. Mol Diagn Ther. 2017;21(2):137–152. doi:10.1007/s40291-016-0243-y
96. Petrova E, Hovnanian A. Advances in understanding of Netherton syndrome and therapeutic implications. Expert Opin Orphan Drugs. 2020;8(11):455–487. doi:10.1080/21678707.2020.1857724
97. Nouwen AE, Schappin R, Nguyen NT, et al. Outcomes of systemic treatment in children and adults with Netherton syndrome: a systematic review. Front Immunol. 2022;13:864449.
98. Di W-L, Lwin SM, Petrova A, et al. Generation and clinical application of gene-modified autologous epidermal sheets in Netherton syndrome: lessons learned from a phase 1 trial. Human Gene Ther. 2019;30(9):1067–1078. doi:10.1089/hum.2019.049
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.