")
Back to Journals » Open Access Rheumatology: Research and Reviews » Volume 13
Understanding and Managing Corticosteroid-Induced Osteoporosis
Authors Kobza AO , Herman D, Papaioannou A , Lau AN, Adachi JD
Received 2 April 2021
Accepted for publication 3 June 2021
Published 2 July 2021 Volume 2021:13 Pages 177—190
DOI https://doi.org/10.2147/OARRR.S282606
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Chuan-Ju Liu
Alexandra O Kobza,1 Deena Herman,1 Alexandra Papaioannou,2,3 Arthur N Lau,1 Jonathan D Adachi1
1Division of Rheumatology, Department of Medicine, McMaster University, Hamilton, ON, Canada; 2Division of Geriatric Medicine, Department of Medicine, McMaster University, Hamilton, ON, Canada; 3Department of Health Research Methods, Evidence & Impact, McMaster University, Hamilton, ON, Canada
Correspondence: Jonathan D Adachi 501-25 Charlton Avenue East, Hamilton, Ontario, L8N 1Y2, Canada
Tel +1 905-529-1317
Fax +1 905-521-1297
Email [email protected]
Abstract: Glucocorticoids are effective immunosuppressants used in a wide variety of diseases. Their use results in secondary osteoporosis in about 30– 50% of chronic glucocorticoid users. Glucocorticoids cause a rapid decline in bone strength within the first 3– 6 months mostly due to increased bone resorption by osteoclasts. This is followed by a more gradual loss of bone partly due to decreased osteoblastogenesis and osteoblast and osteocyte apoptosis. The loss of bone strength induced by glucocorticoids is not fully captured by bone mineral density measurements. Other tools such as the trabecular bone score and advanced imaging techniques give insight into bone quality; however, these are not used widely in clinical practice. Glucocorticoid-induced osteoporosis should be seen as a widely preventable disease. Currently, only about 15% of chronic glucocorticoid users are receiving optimal care. Glucocorticoids should be prescribed at the lowest dose and shortest duration. All patients should be counselled on lifestyle measures to maintain bone strength including nutrition and weight-bearing exercise. Pharmacological therapy should be considered for all patients at moderate to high risk of fracture as there is evidence for the prevention of bone loss and fractures with a favourable safety profile. Oral bisphosphonates are the current mainstay of therapy, whereas osteoanabolic agents may be considered for those at highest risk of fracture.
Keywords: glucocorticoid-induced osteoporosis, bone mineral density, fracture, bisphosphonate
Introduction
Synthetic glucocorticoids are prescribed in a wide range of diseases including rheumatological, pulmonary, gastrointestinal, various malignancies and following organ transplantation. The life-saving benefit that these medications provide is not without a long list of adverse effects. One of the most serious is the effect on the skeletal system where glucocorticoids can cause osteoporosis. Glucocorticoid-induced osteoporosis (GIOP) is the most common cause of secondary osteoporosis.1 Despite this well-known adverse effect, many patients are not monitored or treated appropriately.2
Osteoporosis is characterized by changes in bone architecture, leading to a decrease in bone mineral density (BMD) and an increased risk of fracture. In GIOP, the increased risk of fracture cannot be fully explained by changes in BMD. Changes in bone quality also seem to play a large role.3
This will be a review of the pathophysiology and management of glucocorticoid-induced osteoporosis.
Epidemiology
Worldwide about 1% of the adult population has been treated with long-term glucocorticoids, defined as a minimum of 3 months of consecutive use.4,5 Of this population, about 30–50% will develop GIOP. The relative risk of fracture secondary to glucocorticoid exposure is thought to be similar across age, sex and underlying disease, with about a 60% increased risk of hip fracture and a 160% increased risk of vertebral fracture.6
Unlike other causes of osteoporosis, GIOP occurs rapidly and decreases rapidly after cessation of therapy. These swift changes suggest that the risk may be partly independent of changes in BMD. It has been shown that fracture risk is higher in patients with GIOP than in women with postmenopausal osteoporosis at the same BMD.3 Reasons for this discrepancy include changes in bone quality, bone loss incurred by the underlying disease for which corticosteroids were prescribed and an increased propensity to falling due to the adverse effects of glucocorticoids on muscle strength.3,7
Glucocorticoid-induced bone loss, which mainly affects trabecular bone, is particularly rapid in the first few months of therapy initiation and then slows thereafter. Similarly, fracture risk is higher among glucocorticoid initiators (within 6 months), and then levels off in more chronic glucocorticoid users, with the highest risk being for vertebral fractures.8 A meta-analysis identified strong correlations between daily dose of glucocorticoids and fracture risk and between cumulative dose and loss of BMD.1
Fracture risk is increased even with low daily doses (2.5–7.5mg of prednisolone or its equivalent) and a direct relationship is observed between increasing dose and fracture risk.9 This was observed in a retrospective cohort study comparing glucocorticoid users with control patients selected randomly and matched by age, sex, and medical practice. With a daily dose of <2.5mg prednisolone, vertebral fracture risk was 1.55 (95% CI 1.20, 2.10) relative to control, rising to 2.59 (95% CI 2.16, 3.10) at doses of 2.5–7.5mg, and 5.18 (95% CI 4.25, 6.31) at doses ≥7.5mg prednisolone per day. For hip fracture, the relative risk was 0.99 (95% CI 0.82, 1.20), 1.77 (95% CI 1.55, 2.02), and 2.27 (95% CI 2.16, 3.10), respectively.6
There has been controversy regarding lingering effects of glucocorticoid use on fracture risk years after cessation of therapy with some studies showing no association between remote glucocorticoid use (>12 months) and fracture risk.10 However, two large meta-analyses have shown that increased risk of fracture remains following cessation of therapy independent of underlying age, gender, or disease process.1,3 This may stem from cumulative glucocorticoid dose also playing a role. A UK study showed that intermittent use of high-dose glucocorticoids (≥15mg prednisolone) with a cumulative exposure ≤1g may result in a small increase in osteoporotic fractures, whereas numerous courses of high-dose glucocorticoids amounting to a cumulative dose ≥1g have a substantially higher risk of fracture.11
Pathophysiology of Glucocorticoid Bone Loss
Glucocorticoids affect bone metabolism through a number of different pathways. They affect bone resorption, bone formation, as well as the muscle strength that protects the skeletal system.
Effects on Bone Cells
Glucocorticoids are lipophilic allowing them to pass easily through the cell membrane into the cytosol where they bind glucocorticoid receptors. This complex then translocates into the nucleus of the cell where it interacts with glucocorticoid response elements and impacts the transcription of multiple genes that affect cell function illustrated in Figure 1.12
Osteoblasts
Osteoblasts are derived from mesenchymal stem cells. They work to produce the bone matrix and eventually become embedded in this matrix as osteocytes. Glucocorticoids disrupt a number of signaling pathways in osteoblasts including bone morphogenetic protein (BMP)-Runx2, the Wnt signaling pathway, and peroxisome proliferator-activated receptor-γ2 (PPAR-γ2) all of which are important in osteoblast differentiation.13 In the Wnt signaling pathway, glucocorticoids increase the secretion of sclerostin and dickkopf-related protein 1 (Dkk-1) from osteocytes in a time- and dose-dependent manner. Both sclerostin and Dkk-1 inhibit the binding of Wnt to lipoprotein receptor-related proteins 5 and 6 (LPR 5/6). This destabilizes B-catenin leading to a cell-fate shift in pre-osteoblasts from the osteoblast lineage to the adipocyte lineage, which decreases osteoblastogenesis.14
Glucocorticoids are also implicated in osteoblast turnover in a dose-dependent manner. At physiologic doses, glucocorticoids promote osteoblast autophagy, a process of lysosomal turnover of cellular contents that is necessary to maintain osteoblast viability. At supra-physiologic doses, glucocorticoids induce apoptosis, or cell death, contributing to loss of bone strength and density.12
Osteocytes
Osteocytes function as mechanoreceptors in bone, responding to the mechanical and metabolic demands of bone and then directing the activity of osteoblasts and osteoclasts accordingly. Glucocorticoids induce osteocyte apoptosis, which is associated with loss of skeletal vascularity, angiogenesis, and a disruption of the osteocyte-canalicular circulation. This network is crucial for the formation of new bone and the repair of damaged bone. The disruption of this network is one explanation for the loss of bone strength and quality that is observed before BMD loss in glucocorticoid-treated bone.15
Osteoclasts
Sclerostin, released from osteocytes, increases circulating receptor activator of nuclear factor-κB ligand (RANKL) and decreases the amount of osteoprotegerin (OPG). The elevated RANKL-to-OPG ratio seen with glucocorticoid administration increases osteoclast differentiation and maturation16 and decreases osteoclast apoptosis,17 overall promoting bone resorption. This mechanism may explain the rapid increase in bone resorption seen in patients commencing glucocorticoid therapy. However, long-term effects of glucocorticoids on osteoclasts are unclear with some studies suggesting that glucocorticoids may disrupt the osteoclast cytoskeleton preventing ongoing bone remodelling which will in turn predispose the bone to fracture.18,19
Indirect Effects on Bone Metabolism
Glucocorticoids create a negative calcium balance in the body by increasing renal excretion of calcium and decreasing intestinal calcium reabsorption. This contributes to the secondary hyperparathyroidism seen in glucocorticoid-treated patients. There is conflicting evidence on the effects of glucocorticoids on vitamin D metabolism and parathyroid hormone (PTH) balance.20
Sex steroids are known to play a role in bone metabolism and the maintenance of bone strength. Estrogens and androgens inhibit osteoblasts from releasing local stimulating factors that promote osteoclastogenesis. Lower circulating levels of sex steroids therefore increase the production of osteoclasts, increasing bone resorption. The resulting bone loss is seen in women after menopause and in men following orchiectomy. Glucocorticoids inhibit the synthesis and secretion of sex steroids causing a similar effect and as a result contributing to GIOP.20
Insulin-like growth factor 1 (IGF-1) promotes bone formation by stimulating type 1 collagen synthesis, suppressing bone collagen degradation and suppressing osteoblast apoptosis.21 IGF-1 gene transcription is suppressed by glucocorticoids.22
Effects on Muscle Mass
Glucocorticoids induce the loss of skeletal muscle mass and muscle weakness particularly at the hip and shoulder girdle. Mechanisms for this are incompletely understood; however, changes in the muscle are appreciated as early as seven days after glucocorticoid initiation. There is evidence of increased muscle atrophy, reduced force of muscle fibers and impaired new muscle formation.23 These negative effects on muscle mass increase the risk of falls and by extension, fracture in this population.
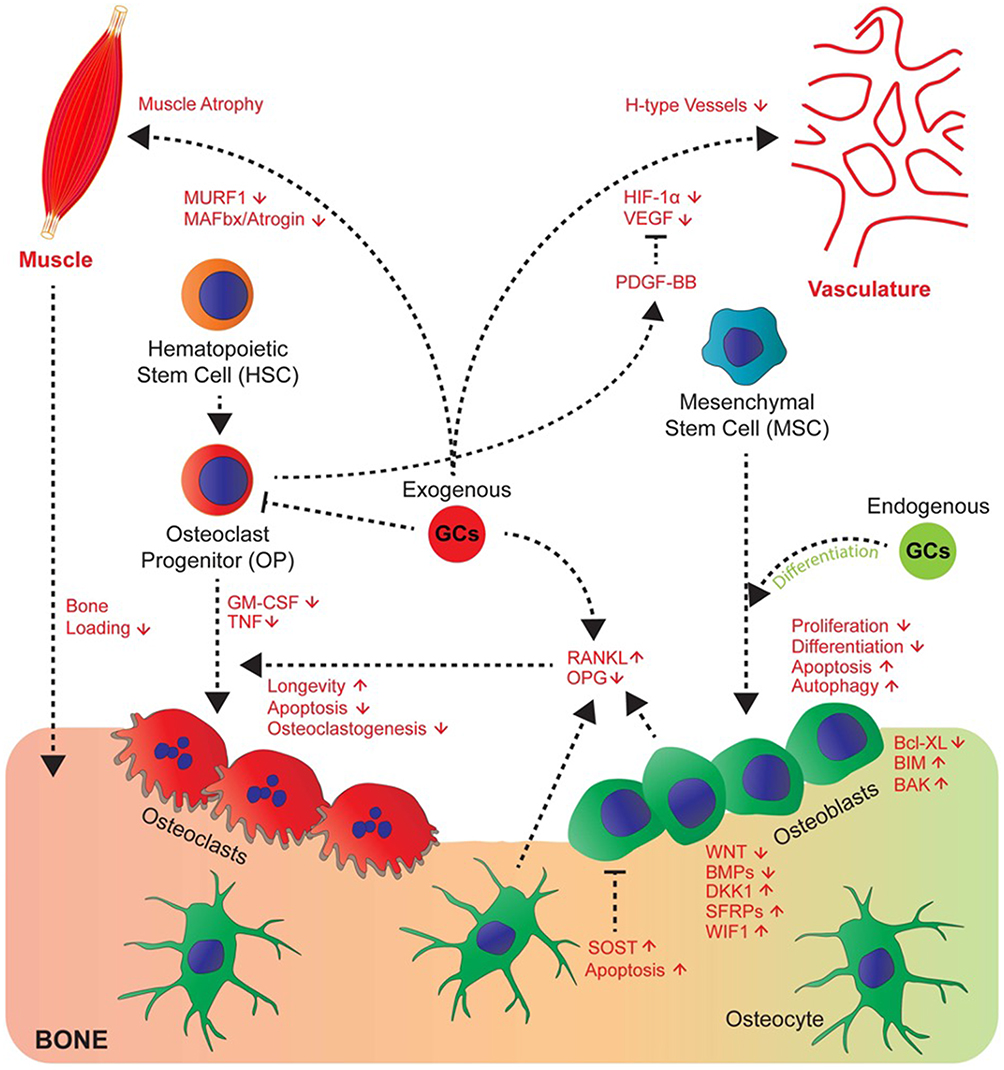 |
Figure 1 Glucocorticoids affect numerous pathways involved in bone metabolism. Endogenous glucocorticoids (green) promote the differentiation of osteoblasts. Exogenous glucocorticoids (red) inhibit the proliferation and differentiation of osteoblasts and promote their apoptosis. They increase the RANKL/OPG ratio which promotes the osteoclast lifespan and therefore bone resorption. They induce muscle atrophy and disrupt vasculature through modulation of signaling molecules such as VEGF and PDGF-BB. Abbreviations: RANKL, receptor activator of nuclear factor kappa-B ligand; OPG, osteoprotegerin; VEGF, vascular endothelial growth factor; PDGF-BB, platelet-derived growth factor-BB. Notes: Republished with permission from Ahmad M, Hachemi Y, Paxian K, Mengele F, Koenen M, Tuckermann J.AJack of All Trades: Impact of Glucocorticoids on Cellular Cross-Talk in Osteoimmunology. Front Immunol. 2019; 10:2460.24 |
Investigations
Bone Mineral Density
Bone strength depends on both quantity and quality of bone. BMD measured by dual-energy X-Ray absorptiometry (DXA) scanning is the most commonly used tool for measurement of bone strength. It allows us to quantify the amount of bone mineral at the lumbar spine, hip, and distal forearm with minimal radiation exposure. Glucocorticoids cause significantly greater early losses of trabecular bone comparing to cortical bone and these changes are seen within the first 3–6 months of treatment. The high content of trabecular bone seen at the lumbar spine puts this area at high risk of fracture early, whereas long bones, such as the radius and humerus, which are made of more cortical bone, are affected more so over time.7
However, a decline in BMD as measured by DEXA does not completely account for the increased risk of fracture seen with glucocorticoids. A large meta-analysis of 42,542 men and women followed for 176,286 person years found that those treated with corticosteroids ever compared to those with no history of corticosteroid use had a higher risk of fracture at the same BMD. The relative risk at 50 years of age was 1.98 compared to someone with no history of corticosteroid use at the same BMD and was 1.66 at 85 years of age.3
Trabecular Bone Score
Trabecular bone score (TBS) is a measurement that can be applied to DXA scans that aims to quantify the qualitative properties of trabecular bone. The score is strongly associated with trabecular number, connectivity, spacing, and bone volume/tissue volume. This makes it an attractive tool for the assessment of GIOP which has a greater effect on trabecular bone. For example, in a cross-sectional study, TBS alone and TBS with lumbar spine BMD, but not lumbar spine BMD alone, were able to discriminate between glucocorticoid-treated and glucocorticoid naïve women.25 TBS is also correlated with stiffness and mechanical behaviour of the bone. Studies have shown that TBS has better ability than BMD to discriminate between patients with fracture in a population receiving glucocorticoid therapy.26,27 TBS is inversely correlated with cumulative glucocorticoid dose and disease duration.28 The tool may also be helpful in monitoring response to treatment in GIOP. In patients with GIOP treated with alendronate and teriparatide, BMD was found to improve with both therapies, while TBS only improved with teriparatide showing it may be a more sensitive measure.29
Quantitative Ultrasound
In addition to BMD, quantitative ultrasound (QUS) is also thought to reflect structural properties of the bone such as elasticity and trabecular arrangement as ultrasound velocity depends on these factors. QUS is able to predict low BMD in patients with GIOP, but how it compares to other modalities and its ability to predict fracture risk remains unknown. Therefore, its overall utility in GIOP has not been defined.30
Advanced Imaging
Advanced bone imaging techniques provide structural information that can give insight into the quality of the bone. These 3D imaging modalities allow for a finite element analysis, which is a computer-based simulation of the stresses induced by loading of an object on the bone, the goal of which is the prediction of fractures.31
Methods for assessing macrostructure of bone include volumetric quantitative computed tomography (vQCT), high resolution quantitative computed tomography (hr-QCT), and high-resolution magnetic resonance imaging (hrMRI).31 In one study, BMD measured by QCT, but not DXA, was found to be an independent predictor of vertebral fracture in women with GIOP demonstrating the improved discriminatory capacity of this modality.32 In a cross-sectional study, women with systemic lupus erythematosus taking corticosteroids were assessed for vertebral fractures. BMD measured by DXA scan of the spine and hip was unable to discriminate between those with and without fractures, whereas hr-QCT measurements at the distal radius revealed lower levels of average BMD, cortical BMD, trabecular BMD, and trabecular thickness in those with vertebral fractures compared to those without demonstrating the higher level of detail achievable with these imaging modalities.33
Biochemical Markers
Even at low doses of corticosteroids, markers of osteoblast activity (serum propeptide of type I N-terminal procollagen [PINP], propeptide of type I C-terminal procollagen [PICP] and osteocalcin) are rapidly and significantly decreased. They then rebound to baseline with steroid withdrawal. Markers of osteoclast activity include urine and serum type I collagen N-telopeptide (NTX) and free urinary deoxypyridinoline (DPD). In one study, urine and serum NTX were unchanged throughout a 6-week course of steroid administration, whereas urinary DPD decreased significantly compared to placebo and did not return completely to baseline.34
Management
A number of therapies and interventions have been assessed for the prevention and treatment of glucocorticoid induced osteoporosis as seen in Figure 2. Management strategies are consistent for all genders and across underlying disease states for which the glucocorticoids are prescribed as glucocorticoid use is just one risk factor for osteoporosis and treatment decisions are based on absolute fracture risk.
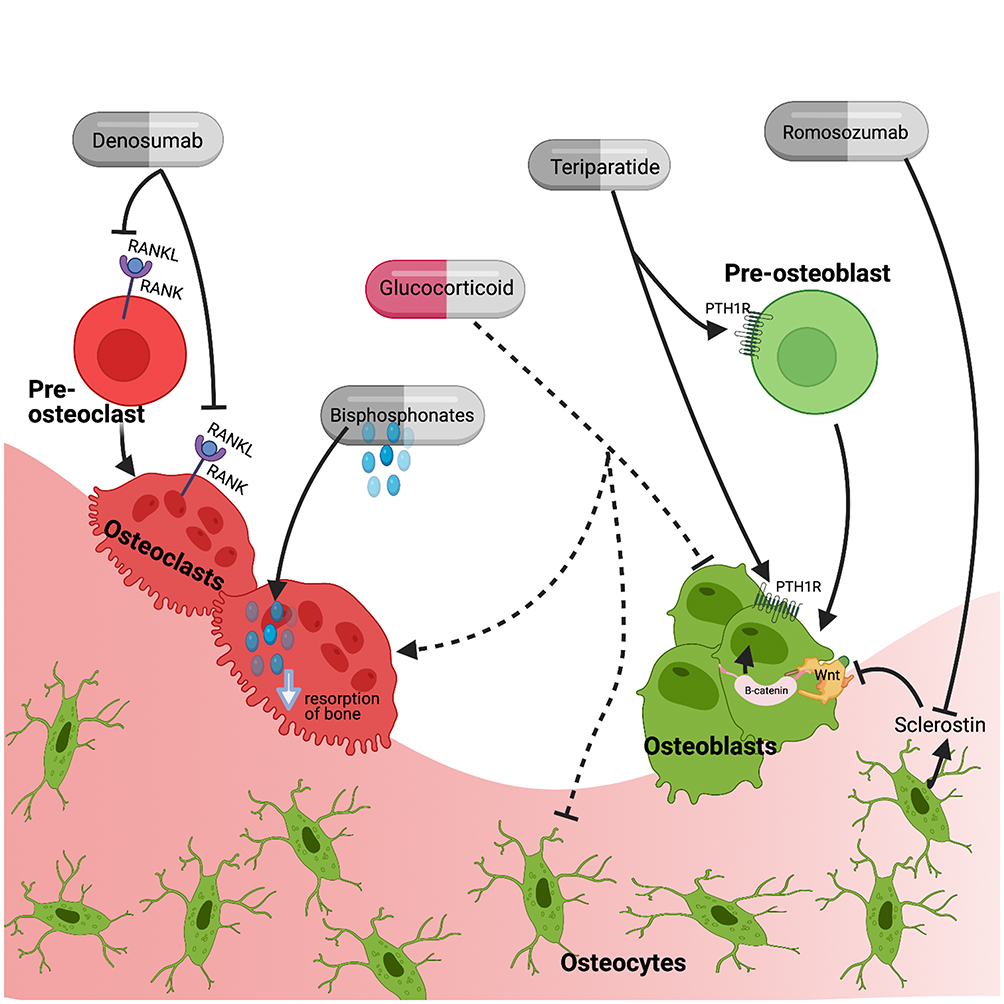 |
Figure 2 Therapeutic targets of medications used in the treatment of glucocorticoid-induced osteoporosis including anti-resorptive therapies (bisphosphonates, denosumab) and osteoanabolic agents (teriparatide and romosozumab). Abbreviations: RANK, receptor activator of nuclear factor kappa-B; RANKL, RANK ligand; PTH1R, parathyroid hormone receptor type 1. Note: Created with BioRender.com. |
Risk Stratification
There are a number of risk factors including the use of glucocorticoids that increase the risk of fracture. The decision to treat patients who are taking glucocorticoids should be based on absolute fracture risk and this can be calculated using the FRAX® tool. FRAX is a computer-based algorithm that estimates the 10-year probability of hip fracture and major osteoporotic fracture (which includes hip, clinical spine, distal forearm and humerus fractures). It can be calculated with or without BMD testing using DXA scanning. FRAX incorporates glucocorticoid use as a dichotomous risk and does not factor in daily dose or duration. It assumes an average daily dose between 2.5–7.5mg of prednisolone (or equivalent).36 Schemas have been developed to adjust this fracture risk assessment to account for lower or higher doses of glucocorticoids.37,38
The FRAX score has been incorporated into many GIOP guidelines, including the 2017 American College of Rheumatology (ACR) guidelines38 as seen in Figure 3 and the International Osteoporosis Federation and European Calcified Tissue Society joint guidelines,39 to determine which patients taking glucocorticoid should be initiated on pharmacologic therapy.
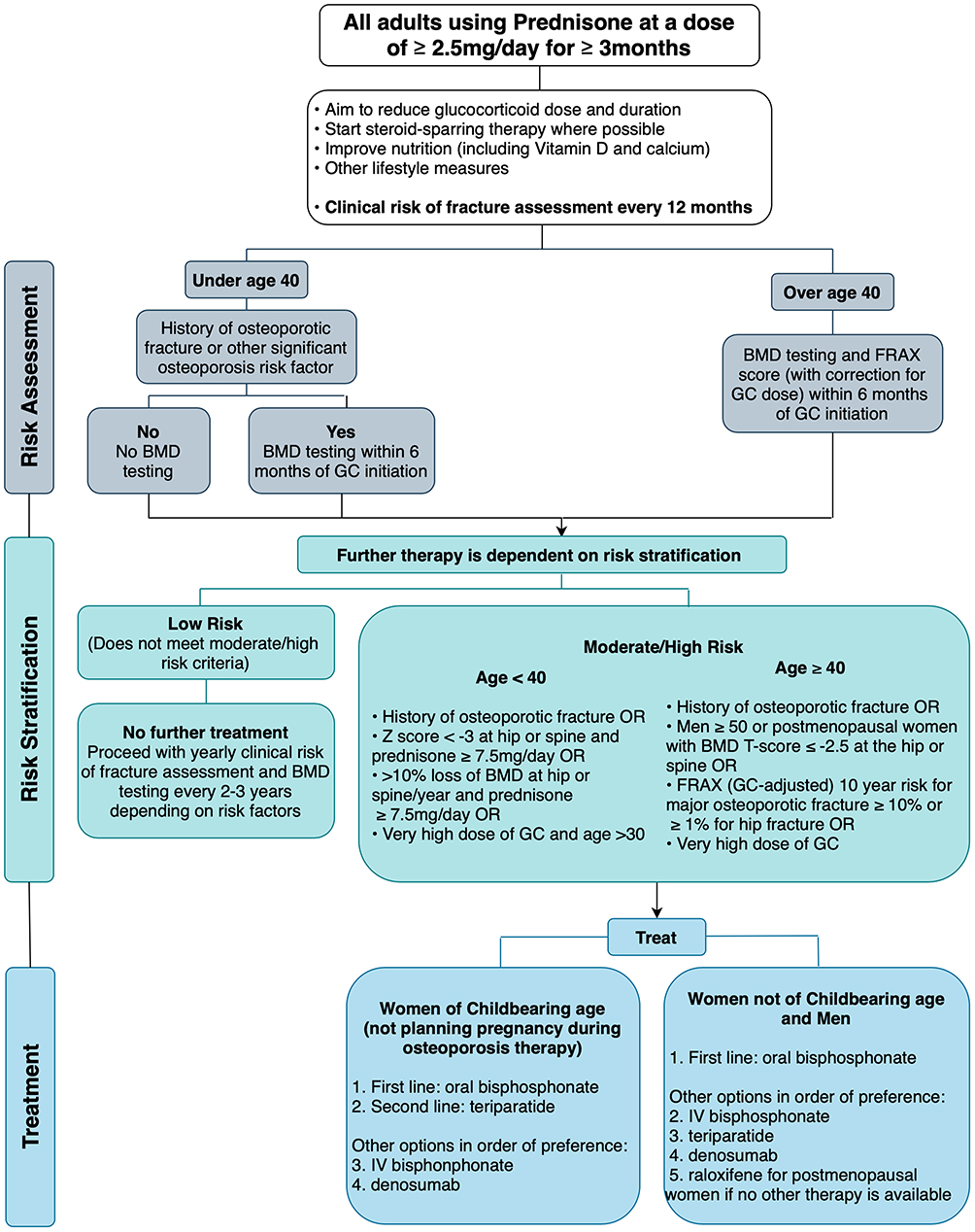 |
Figure 3 Management pathway for adults prescribed chronic glucocorticoids based on the 2017 American College of Rheumatology Guidelines.24 Recommended daily dose of vitamin D and calcium are 600–800 IU/day and 1000–2000mg/day respectively. Lifestyle measures include smoking cessation, limiting alcohol intake to 1–2 drinks per day, weight-bearing and resistance exercises, and maintaining weight in a healthy range. Very high dose of glucocorticoid is defined as ≥30mg/day and a cumulative dose of >5g in the last year. The risk of major osteoporotic fracture calculated by the FRAX tool should be increased by 1.15 and the risk of hip fracture by 1.2 if the dose of prednisone is ≥7.5mg/day. Abbreviations: BMD, bone mineral density; GC, glucocorticoid, IV, intravenous. |
Fracture Risk Assessment
Fracture risk should be assessed for all adults as soon as possible and at least within 6 months of initiation of glucocorticoid therapy and annually thereafter. This assessment includes a history of glucocorticoid use, falls, fractures, risk factors for fragility (ie, malnutrition, low body weight, endocrinopathies associated with secondary osteoporosis), family history of fracture, alcohol use and smoking, among others. A physical exam should include height and weight, testing of muscle strength, and an assessment for signs of an undiagnosed fracture. When there is suspicion of an existing fracture such as through back pain, low BMD, or documented loss of height or kyphosis, some guidelines recommend imaging through lateral imaging DXA with vertebral fracture assessment or, if that is not available, lateral X-rays of the thoracic and lumbar spine.39 Routine imaging is not recommended at this time; however, one study showed that 37% fractures in chronic glucocorticoid users may be asymptomatic.40 Fracture risk should also be estimated using the FRAX tool for adults ≥40 years old as the tool has only been validated above this age. BMD testing is indicated in adults <40 years of age with risk factors or in those ≥40 years of age. BMD testing is then advised every 1–3 years thereafter during glucocorticoid therapy. The frequency of serial testing within this 1–3 year range depends on the baseline bone density, the dose of glucocorticoid, the age and gender of the patient and the underlying disease for which the glucocorticoids were prescribed.38,39
General Measures
Glucocorticoids should be prescribed at the lowest possible dose and for the shortest duration and a steroid sparing agent should be initiated when possible. All patients prescribed long-term glucocorticoids should be assessed and counselled on strategies to improve nutrition (especially calcium and vitamin D), decrease falls risk, and other lifestyle measures including smoking cessation, avoiding excessive alcohol consumption, weight control to a recommended range and participation in regular weight-bearing exercises.38,39 Glucocorticoid-induced hyperglycemia, when present, should be treated as it is associated with increased risk of fracture secondary to altered bone turnover and decreased bone matrix competence.41
Calcium and Vitamin D
Glucocorticoids have been shown to create a negative calcium balance in the body. Therefore, many studies have assessed the efficacy of calcium supplementation as primary prevention in those using chronic low-dose glucocorticoids. Despite supplementation, a significant decline in BMD is observed, suggesting this is insufficient in preventing glucocorticoid induced bone loss.42
When simultaneous vitamin D and calcium supplementation were assessed, a meta-analysis found that the combination caused less BMD loss at the lumbar spine than calcium supplementation alone with effect size 0.60 (95% CI 0.34, 0.85).43 When vitamin D (50,000 units/week) and calcium (1000mg/day) were given to patients initiating high-dose glucocorticoids for primary prevention, there was no statistically significant difference through 36 months in terms of lumbar spine BMD.44 However, in a secondary prevention trial, in patients receiving low-dose chronic glucocorticoids, vitamin D (500 units/day) and calcium (1000mg/day) were shown to prevent loss of BMD at the lumbar spine by 0.72% (p=0.005) per year in the treatment group vs a loss of 2.0% per year in the placebo group.45
Active vitamin D analogues including calcitriol and alfacalcidol have been studied for prevention of GIOP. In a meta-analysis where the pooled effect size of active vitamin D3 metabolites was compared to the effects of placebo, calcium alone and vitamin D3 in terms of change in lumbar spine BMD. The effect was 0.35 (95% CI 0.18, 0.52) indicating the active metabolites were superior in preserving lumbar spine BMD. The active vitamin D3 analogues were also more effective in decreasing the risk of vertebral fractures.46 Bisphosphonates were, however, superior to active vitamin D3 analogues in terms of maintaining lumbar spine BMD and decreasing the risk of vertebral fractures.46,47
Bisphosphonates
Bisphosphonates are the most commonly used medication in GIOP and the first-line agent in all guidelines. This is largely due to their efficacy and low cost. Medications shown to have superior efficacy are available and will be discussed in subsequent sections. Bisphosphonates bind avidly to bone where they enter osteoclasts and cause apoptosis. Conversely, glucocorticoids increase the osteoclast lifespan, and this effect seems to predominate in the short term. Glucocorticoids also decrease osteoclastogenesis and thereby in the long term the pro-apoptotic effect of bisphosphonates may become more prominent. In addition, bisphosphonates likely protect osteoblasts from glucocorticoid-induced apoptosis and this may play a larger role in increasing BMD in GIOP.48
Bisphosphonates have been studied extensively for both the prevention and treatment of GIOP.49–51 A Cochrane systematic review of bisphosphonates in the treatment of GIOP concluded that there was a statistically significant and clinically meaningful reduction in vertebral fracture with bisphosphonates vs placebo with a relative improvement of 43% (95% CI 9, 65). Risk of non-vertebral fractures was not statistically different with a relative improvement of 21% (95% CI −33, 53). There was significant improvement in BMD at the lumbar spine and femoral neck.52
As oral bisphosphonates are poorly absorbed, it is important for patients to take this medication away from food especially those containing calcium, magnesium, or aluminum. Further, some patients experience gastrointestinal adverse effects limiting their use and efficacy.53 In an effort to improve compliance and mitigate these adverse effects, intermittent intravenous bisphosphonates have been studied in GIOP. In one study, intravenous zoledronic acid administered yearly was compared to daily oral risedronate in chronic glucocorticoid users (≥3 months) and glucocorticoid initiators (<3 months). At the 1 year follow-up mark, zoledronic acid was found to be non-inferior and possibly superior to risedronate for increase in the lumbar spine BMD in both the treatment (mean difference 1.36%; 95% CI 0.67, 2.05) and prevention (mean difference 1.96%; 95% CI 1.04, 2.88) subgroups.54
At this time, risedronate, etidronate, alendronate and IV zoledronic acid have all been approved for patients taking glucocorticoids for either prophylaxis or treatment of GIOP. Serious but rare complications of these medications are osteonecrosis of the jaw and atypical femoral fractures. Although glucocorticoids have been reported in some cases of these complications, there is no evidence to suggest that glucocorticoids are associated with increased risk of either complication in patients who are taking bisphosphonates.39,55
RANKL Inhibition: Denosumab
Glucocorticoids increase the expression of RANKL and decrease the expression of the RANKL decoy receptor OPG. Together, these actions promote the differentiation and activation of osteoclasts as well as the resorption of bone. Denosumab is a human monoclonal antibody against RANKL.
A few randomized controlled trials (RCTs) have been conducted comparing denosumab to either placebo56,57 or a bisphosphonate.58–60 In general, denosumab significantly improves lumbar spine and hip BMD when compared to both placebo and a bisphosphonate over a time course of 12 and 28 months.
The largest trial was a Phase III study, including 795 patients, both men and women who had an indication for treatment of GIOP, and who were being treated with ≥7.5mg daily of glucocorticoids. In both glucocorticoid-initiators and chronic users, denosumab was superior to risedronate in increasing lumbar spine and total hip BMD at both 12 and 24 months.60 In a recent meta-analysis, denosumab was compared to control therapy in patients taking systemic glucocorticoids. Compared to bisphosphonates, denosumab significantly increased lumbar spine BMD (2.32%, 95% CI 1.73, 2.91) and hip BMD (1.52%, 95% CI 1.1, 1.94). Adverse events, serious adverse events and fractures were similar between the denosumab and bisphosphonate arms, and the studies were underpowered to detect differences in the risk of fracture.61
As a result, denosumab has been approved for use in the GIOP population. In cases where cost is not a barrier, denosumab may be used either before or after oral or IV bisphosphonates as it has superior efficacy. It is important to know that discontinuation of denosumab has been shown to result in a rapid reversal of BMD accrued during the treatment period and potentially an increased risk of multiple vertebral fractures particularly in those with underlying fractures.62,63 Therefore, denosumab discontinuation should be followed by bisphosphonate therapy for a recommended duration of 12–18 months.63
Parathyroid Hormone
Parathyroid hormone (PTH), released by the parathyroid gland, plays a critical role in calcium homeostasis in the body. When released in an intermittent and pulsatile fashion, PTH has been shown to have an anabolic effect by promoting osteoblastogenesis and differentiation.64 Teriparatide is recombinant human PTH (1–34). Given that teriparatide has proven to be an effective osteoanabolic therapy in the treatment of postmenopausal osteoporosis65 and that bone formation is impaired in the GIOP, teriparatide is a therapy with biological plausibility in the GIOP population.
A number of RCTs have been conducted in the GIOP population, where teriparatide was compared to a bisphosphonate for 18–36 months. Overall, when compared to a bisphosphonate, teriparatide had significantly greater change in BMD at the lumbar spine and total hip and a significant decrease in the incidence of vertebral fractures but no change in non-vertebral fractures.66–69 For example, in a double-blind RCT by Saag et al, 428 subjects with GIOP were randomized to teriparatide 20μg/day or alendronate 10mg/day for 36 months. Increase in BMD from baseline at 36 months for teriparatide and alendronate was 11.0% vs 5.3% at the lumbar spine, 5.2% vs 2.7% at the total hip, respectively (both p<0.001). The teriparatide group had a significant decrease in vertebral fractures 1.7% vs 7.7% (p=0.007) and no significant difference in non-vertebral fractures.69
The main limitation for widespread use of teriparatide has been the much higher cost comparing to other agents and compliance given the medications is administered as a daily subcutaneous injection. However, some argue it should be considered as first-line therapy for those at very high risk of fracture (ie, older adults starting at high doses of glucocorticoids, multiple previous fractures, very low initial bone mass) and may even be cost-effective in this group.5,70 Certainly, the availability of generic teriparatide, or other anabolic agents for GIOP, would make this a more feasible treatment plan.
Sclerostin Inhibitor: Romosozumab
Romosozumab is a humanized monoclonal antibody with high affinity and specificity for sclerostin. Sclerostin is an inhibitor of the Wnt pathway which is important for osteoblast activation, differentiation and bone formation.71 As glucocorticoids increase the secretion of sclerostin, romosozumab is a promising new therapy for GIOP. Four RCTs have been performed in primary osteoporosis where romosozumab has been compared to placebo, alendronate and teriparatide.72–75 Overall, one year of therapy with romosozumab was superior to all other standard therapies with respect to increasing BMD. At this time, studies in the GIOP population have been limited to animal models, where sclerostin antibody therapy was shown to prevent bone loss and strength.76 There is an ongoing Phase IV trial comparing romosozumab to denosumab for the treatment of osteoporosis in long-term glucocorticoid users.77 However, at this time, there is no indication for romosozumab in the treatment of GIOP.
Vertebroplasty and Kyphoplasty
In GIOP, if pain from the vertebral fracture is persistent and not-responding to pain medications, vertebroplasty and kyphoplasty may be considered. It is important to note that steroid-induced osteoporosis patients are almost twice as likely to fracture comparing to primary osteoporosis patients within one year of this procedure and therefore this approach is not favoured.78
In Summary
Two meta-analyses have been performed to compare the efficacy of osteoporosis agents in the prevention of GIOP. These studies allow for conclusions regarding fracture prevention. Amiche et al studied 27 RCTs and found that comparing to placebo, vertebral fracture rate was decreased with etidronate (RR 0.41; 95% CI 0.17, 0.90), risedronate (RR 0.30; 95% CI 0.14, 0.61), and teriparatide (RR 0.07, 95% CI 0.001, 0.48). No treatment had statistically significant reduction in non-vertebral fractures. Teriparatide showed the largest effect size in improving both vertebral and non-vertebral fractures.79 Ding et al analyzed 19 RCTs. Comparing to placebo, glucocorticoid-induced vertebral fractures were reduced with teriparatide (RR 0.11; 95% CI 0.03, 0.47), denosumab (RR 0.21, 95% CI 0.09, 0.49), and risedronate (RR 0.33, 95% CI 0.19, 0.58).80 A summary of all agents compared to placebo with regard to glucocorticoid-induced fracture risk reduction based on this meta-analysis is presented in Table 1.
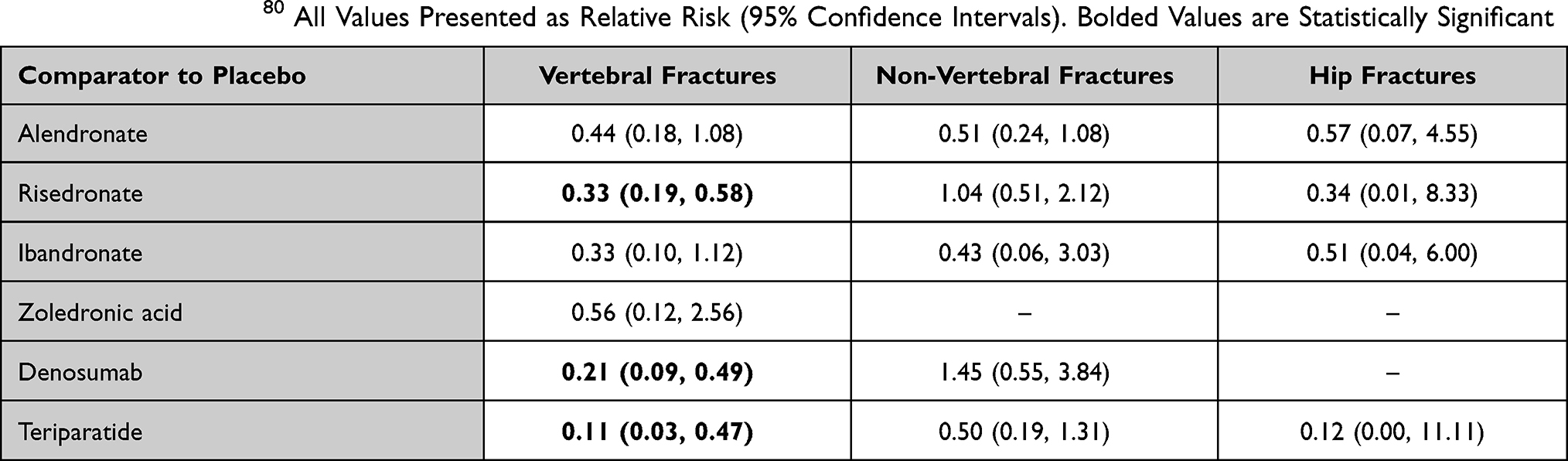 |
Table 1 Summary of Fracture Risk of Various Therapies When Compared to Placebo in Patients with GIOP Based on Recent Meta-Analysis by Ding et al.80 All Values Presented as Relative Risk (95% Confidence Intervals). Bolded Values are Statistically Significant |
Considerations in the Pediatric Population
While a complete review of the effect of chronic glucocorticoid use on bone health in the pediatric population is beyond the scope of this article, some key principles will be discussed below. As in the adult population, glucocorticoid use is an important risk factor for bone fragility in children. In addition to the mechanisms at play in the adult population, glucocorticoids have an adverse effect on the growth plate primarily through chondrocyte apoptosis and by interfering with the hypothalamic-pituitary growth hormone secretion resulting in attenuated linear growth of bone. Vertebral fractures are most common, and they tend to occur early in the treatment course and are often asymptomatic. Fortunately, some children have the ability to restore normal vertebral dimensions spontaneously following a fracture as a result of modelling bone growth. Children with less potential for vertebral body remodelling following a spinal fracture are those with poor growth, older children, and children with ongoing health concerns. Treatment to prevent further fractures remains important in all cases. IV bisphosphonates are the mainstay of therapy, but studies are underway investigating the efficacy of denosumab in this population.81
Care Gap in the Screening, Prevention and Treatment of GIOP
Despite the existence of comprehensive guidelines, suboptimal prevention and treatment of GIOP persists. Studies show a general increase in BMD testing and osteoporosis management in GC patients from the late 90s (when the first American College of Rheumatology guidelines were published) to 2010s. However, primary prevention for GIOP remains suboptimal in the general population with ~25% of chronic glucocorticoid users receiving BMD testing or osteoporosis therapy.2,82
It is helpful to conceptualize the care gap in terms of provider, patient, and system barriers. Comparing to the general population, primary care physicians and even more so rheumatologists show improved adherence to GIOP guidelines.83,84 For example, in a Canadian rheumatology practice, following prednisone prescription, 79% of patients were taking vitamin D, 86% were taking calcium, 25% had a DXA scan ordered, and 50% were prescribed osteoporosis therapy. Overall, there was suboptimal adherence to guidelines, with only 15% of patients following all recommendations according to the Osteoporosis Canada 2010 practice guidelines.84
Regardless of prescriber specialty, certain cohorts of patients are overlooked in terms of GIOP prevention, including pre-menopausal women, non-white patients, men and rural patients.82,84 Concern regarding pregnancy while on bisphosphonates is a barrier to treatment of pre-menopausal women.85
Furthermore, the care gap is exacerbated by ambivalence regarding whose responsibility it is to manage GIOP: the specialist who prescribed the GCs or the family physician. Finally, doctors cite insufficient time in clinical encounters to address GIOP in light of weightier health issues being managed.86 Proposed strategies to improve knowledge translation include patient-accessible handouts in waiting rooms and pharmacist reminders to physicians triggered by prescription of glucocorticoids.86
Conclusion
Glucocorticoids are effective immunosuppressants with widespread use. They exhibit a predictable effect on bone characterized by rapid bone resorption within the first 3–6 months, followed by a more gradual decreased bone formation. Some bone strength is regained with cessation of glucocorticoid therapy; however, there are lasting effects on bone integrity associated with cumulative glucocorticoid dose. As a result, glucocorticoid dose and duration should be limited for all patients. All patients should be counselled on lifestyle measures to maintain bone strength and mass. All patients receiving chronic glucocorticoids should be assessed for absolute fracture risk and treatment should be considered for those at increased risk of fracture. Oral bisphosphonates are the current mainstay of treatment; however, denosumab does provide greater increases in BMD and osteoanabolic agents such as teriparatide may be considered for those at highest risk of fracture. As more osteoanabolic agents become available and cost decreases, this may become a feasible first-line option for those at highest risk.
Disclosure
A Kobza and D Herman have no conflicts of interest or competing interests to report. A Papaioannou sits on the advisory board and has received grants from Amgen outside the submitted work. A Lau has received personal fees from Amgen and Eli Lilly as well as grants from Amgen and Eli Lilly, all of which are outside the submitted work. J Adachi reports grants and personal fees from Amgen outside the submitted work, grants from Radius and has been an advisor to Gilead and is part of their speaker’s bureau, outside the submitted work. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
1. Van Staa TP, Leufkens HGM, Cooper C. The epidemiology of corticosteroid-induced osteoporosis: a meta-analysis. Osteoporos Int. 2002;13(10):777–787. doi:10.1007/s001980200108
2. Majumdar SR, Lix LM, Morin SN, Yogendran M, Metge CJ, Leslie WD. The disconnect between better quality of glucocorticoid-induced osteoporosis preventive care and better outcomes: a population-based cohort study. J Rheumatol. 2013;40(10):1736–1741. doi:10.3899/jrheum.130041
3. Kanis JA, Johansson H, Oden A, et al. A meta-analysis of prior corticosteroid use and fracture risk. J Bone Miner Res. 2004;19(6):893–899. doi:10.1359/JBMR.040134
4. Overman RA, Yeh J, Deal CL. Prevalence of oral glucocorticoid usage in the United States: a general population perspective. Arthritis Care Res (Hoboken). 2013;65(2):294–298. doi:10.1002/acr.21796
5. Adami G, Saag KG. Glucocorticoid-induced osteoporosis: 2019 concise clinical review. Osteoporos Int. 2019;30(6):1145–1156. doi:10.1007/s00198-019-04906-x
6. Van Staa TP, Leufkens HGM, Abenhaim L, Zhang B, Cooper C. Use of oral corticosteroids and risk of fractures. J Bone Miner Res. 2000;15(6):993–1000. doi:10.1359/jbmr.2000.15.6.993
7. Lane NE, Lukert B. The science and therapy of glucocorticoid-induced bone loss. Endocrinol Metab Clin North Am. 1998;27(2):465–483. doi:10.1016/S0889-8529(05)70017-7
8. Amiche MA, Albaum JM, Tadrous M, et al. Fracture risk in oral glucocorticoid users: a Bayesian meta-regression leveraging control arms of osteoporosis clinical trials. Osteoporos Int. 2016;27(5):1709–1718. doi:10.1007/s00198-015-3455-9
9. Van Staa TP, Leufkens HGM, Abenhaim L, Zhang B, Cooper C. Oral corticosteroids and fracture risk: relationship to daily and cumulative doses. Rheumatology. 2000;39(12):1383–1389. doi:10.1093/rheumatology/39.12.1383
10. Majumdar SR, Morin SN, Lix LM, Leslie WD. Influence of recency and duration of glucocorticoid use on bone mineral density and risk of fractures: population-based cohort study. Osteoporos Int. 2013;24(9):2493–2498. doi:10.1007/s00198-013-2352-3
11. De Vries F, Bracke M, Leufkens HGM, Lammers JWJ, Cooper C, Van Staa TP. Fracture risk with intermittent high-dose oral glucocorticoid therapy. Arthritis Rheum. 2007;56(1):208–214. doi:10.1002/art.22294
12. Wang L, Heckmann BL, Yang X, Long H. Osteoblast autophagy in glucocorticoid-induced osteoporosis. J Cell Physiol. 2019;234(4):3207–3215. doi:10.1002/jcp.27335
13. Hayashi K, Yamaguchi T, Yano S, et al. BMP/Wnt antagonists are upregulated by dexamethasone in osteoblasts and reversed by alendronate and PTH: potential therapeutic targets for glucocorticoid-induced osteoporosis. Biochem Biophys Res Commun. 2009;379(2):261–266. doi:10.1016/j.bbrc.2008.12.035
14. Song L, Liu M, Ono N, Bringhurst FR, Kronenberg HM, Guo J. Loss of wnt/β-catenin signaling causes cell fate shift of preosteoblasts from osteoblasts to adipocytes. J Bone Miner Res. 2012;27(11):2344–2358. doi:10.1002/jbmr.1694
15. Weinstein RS. Glucocorticoids, osteocytes, and skeletal fragility: the role of bone vascularity. Bone. 2010;46(3):564–570. doi:10.1016/j.bone.2009.06.030
16. Wijenayaka AR, Kogawa M, Lim HP, Bonewald LF, Findlay DM, Atkins GJ. Sclerostin stimulates osteocyte support of osteoclast activity by a RANKL-dependent pathway. PLoS One. 2011;6(10):e25900. doi:10.1371/journal.pone.0025900
17. Jia D, O’brien CA, Stewart SA, Manolagas SC, Weinstein RS. Glucocorticoids act directly on osteoclasts to increase their life span and reduce bone density. Endocrinology. 2006;147(12):5592–5599. doi:10.1210/en.2006-0459
18. Teitelbaum SL. Glucocorticoids and the osteoclast. Clin Exp Rheumatol. 2015;33(4 Suppl 92):S37–S39.
19. Kim H-J. New understanding of glucocorticoid action in bone cells. BMB Rep. 2010;43(8):524–529. doi:10.5483/BMBRep.2010.43.8.524
20. Patschan D, Loddenkemper K, Buttgereit F. Molecular mechanisms of glucocorticoid-induced osteoporosis. Bone. 2001;29(6):498–505. doi:10.1016/S8756-3282(01)00610-X
21. Canalis E, Centrella M, Burch W, McCarthy TL. Insulin-like growth factor I mediates selective anabolic effects of parathyroid hormone in bone cultures. J Clin Invest. 1989;83(1):60–65. doi:10.1172/JCI113885
22. Delany AM, Durant D, Canalis E. Glucocorticoid suppression of IGF I transcription in osteoblasts. Mol Endocrinol. 2001;15(10):1781–1789. doi:10.1210/mend.15.10.0704
23. Sato AY, Richardson D, Cregor M, et al. Glucocorticoids induce bone and muscle atrophy by tissue-specific mechanisms upstream of E3 ubiquitin ligases. Endocrinology. 2017;158(3):664–677. doi:10.1210/en.2016-1779
24. Ahmad M, Hachemi Y, Paxian K, Mengele F, Koenen M, Tuckermann J. A Jack of all trades: impact of glucocorticoids on cellular cross-talk in osteoimmunology. Front Immunol. 2019;10:2460. doi:10.3389/fimmu.2019.02460
25. Paggiosi MA, Peel NFA, Eastell R. The impact of glucocorticoid therapy on trabecular bone score in older women. Osteoporos Int. 2015;26(6):1773–1780. doi:10.1007/s00198-015-3078-1
26. Leib ES, Winzenrieth R. Bone status in glucocorticoid-treated men and women. Osteoporos Int. 2016;27(1):39–48. doi:10.1007/s00198-015-3211-1
27. Florez H, Hernández-Rodríguez J, Muxi A, et al. Trabecular bone score improves fracture risk assessment in glucocorticoid-induced osteoporosis. Rheumatology. 2020;59(7):1574–1580. doi:10.1093/rheumatology/kez464
28. Sandru F, Carsote M, Dumitrascu MC, Albu SE, Valea A. Glucocorticoids and trabecular bone score. J Med Life. 2020;13(4):449–453. doi:10.25122/jml-2019-0131
29. Saag KG, Agnusdei D, Hans D, et al. Trabecular bone score in patients with chronic glucocorticoid therapy–induced osteoporosis treated with alendronate or teriparatide. Arthritis Rheumatol. 2016;68(9):2122–2128. doi:10.1002/art.39726
30. Maricic M, Gluck O. Densitometry in glucocorticoid-induced osteoporosis. J Clin Densitom. 2004;7(4):359–363. doi:10.1385/JCD:7:4:359
31. Kalpakcioglu BB, Engelke K, Genant HK. Advanced imaging assessment of bone fragility in glucocorticoid-induced osteoporosis. Bone. 2011;48(6):1221–1231. doi:10.1016/j.bone.2011.02.005
32. Lian K-C, Lang TF, Keyak JH, et al. Differences in hip quantitative computed tomography (QCT) measurements of bone mineral density and bone strength between glucocorticoid-treated and glucocorticoid-naïve postmenopausal women. Osteoporos Int. 2005;16(6):642–650. doi:10.1007/s00198-004-1736-9
33. Li EK, Zhu TY, Tam L-S, et al. Bone microarchitecture assessment by high-resolution peripheral quantitative computed tomography in patients with systemic lupus erythematosus taking corticosteroids. J Rheumatol. 2010;37(7):1473LP–1479. doi:10.3899/jrheum.091231
34. Ton FN, Gunawardene SC, Lee H, Neer RM. Effects of low-dose prednisone on bone metabolism. J Bone Miner Res. 2005;20(3):464–470. doi:10.1359/JBMR.041125
35. Chiodini I, Merlotti D, Falchetti A, Gennari L. Treatment options for glucocorticoid-induced osteoporosis. Expert Opin Pharmacother. 2020;21(6):721–732. doi:10.1080/14656566.2020.1721467
36. Kanis JA, Johnell O, Oden A, Johansson H, McCloskey E. FRAX and the assessment of fracture probability in men and women from the UK. Osteoporos Int. 2008;19(4):385–397. doi:10.1007/s00198-007-0543-5
37. Kanis JA, Johansson H, Oden A, McCloskey EV. Guidance for the adjustment of FRAX according to the dose of glucocorticoids. Osteoporos Int. 2011;22(3):809–816. doi:10.1007/s00198-010-1524-7
38. Buckley L, Guyatt G, Fink HA, et al. 2017 American College of Rheumatology Guideline for the Prevention and Treatment of Glucocorticoid-Induced Osteoporosis. Arthritis Rheumatol. 2017;69(8):1521–1537. doi:10.1002/art.40137
39. Lekamwasam S, Adachi JD, Agnusdei D, et al. A framework for the development of guidelines for the management of glucocorticoid-induced osteoporosis. Osteoporos Int. 2012;23(9):2257–2276. doi:10.1007/s00198-012-1958-1
40. Angeli A, Guglielmi G, Dovio A, et al. High prevalence of asymptomatic vertebral fractures in post-menopausal women receiving chronic glucocorticoid therapy: a cross-sectional outpatient study. Bone. 2006;39(2):253–259. doi:10.1016/j.bone.2006.02.005
41. Starup-Linde J, Vestergaard P. MANAGEMENT OF ENDOCRINE DISEASE: diabetes and osteoporosis: cause for concern? Eur J Endocrinol. 2015;173(3):R93–R99. doi:10.1530/EJE-15-0155
42. Sambrook P, Birmingham J, Kelly P, et al. Prevention of corticosteroid osteoporosis–a comparison of calcium, calcitriol, and calcitonin. N Engl J Med. 1993;328(24):1747–1752. doi:10.1056/NEJM199306173282404
43. Amin S, LaValley MP, Simms RW, Felson DT. The role of vitamin D in corticosteroid-induced osteoporosis: a meta- analytic approach. Arthritis Rheum. 1999;42(8):1740–1751. doi:10.1002/1529-0131(199908)42:8<1740::AID-ANR25>3.0.CO;2-E
44. Adachi JD, Bensen WG, Bianchi F, et al. Vitamin D and calcium in the prevention of corticosteroid induced osteoporosis: a 3 year followup. J Rheumatol. 1996;23(6):995–1000.
45. Buckley LM, Leib ES, Cartularo KS, Vacek PM, Cooper SM. Calcium and vitamin D3 supplementation prevents bone loss in the spine secondary to low-dose corticosteroids in patients with rheumatoid arthritis: a randomized, double-blind, placebo-controlled trial. Ann Intern Med. 1996;125(12):961–968. doi:10.7326/0003-4819-125-12-199612150-00004
46. De Nijs RNJ, Jacobs JWG, Algra A, Lems WF, Bijlsma JWJ. Prevention and treatment of glucocorticoid-induced osteoporosis with active vitamin D 3 analogues: a review with meta-analysis of randomized controlled trials including organ transplantation studies. Osteoporos Int. 2004;15(8):589–602. doi:10.1007/s00198-004-1614-5
47. de Nijs RNJ, Jacobs JWG, Lems WF, et al. Alendronate or alfacalcidol in glucocorticoid-induced osteoporosis. N Engl J Med. 2006;355(7):675–684. doi:10.1056/NEJMoa053569
48. Weinstein RS, Chen J-R, Powers CC, et al. Promotion of osteoclast survival and antagonism of bisphosphonate-induced osteoclast apoptosis by glucocorticoids. J Clin Invest. 2002;109(8):1041–1048. doi:10.1172/JCI14538
49. Adachi JD, Bensen WG, Brown J, et al. Intermittent etidronate therapy to prevent corticosteroid-induced osteoporosis. N Engl J Med. 1997;337(6):382–388. doi:10.1056/NEJM199708073370603
50. Cohen S, Levy RM, Keller M, et al. Risedronate therapy prevents corticosteroid-induced bone loss: a twelve- month, multicenter, randomized, double-blind, placebo-controlled, parallel- group study. Arthritis Rheum. 1999;42(11):2309–2318. doi:10.1002/1529-0131(199911)42:11<2309::AID-ANR8>3.0.CO;2-K
51. Saag KG, Emkey R, Schnitzer TJ, et al. Alendronate for the prevention and treatment of glucocorticoid-induced osteoporosis. N Engl J Med. 1998;339(5):292–299. doi:10.1056/NEJM199807303390502
52. Allen CS, Yeung JHS, Vandermeer B, Homik J. Bisphosphonates for steroid-induced osteoporosis. Cochrane Database Syst Rev. 2016;10. doi:10.1002/14651858.CD001347.pub2
53. Siris ES, Fan C-PS, Yang X, Sajjan S, Sen SS, Modi A. Association between gastrointestinal events and compliance with osteoporosis therapy. Bone Rep. 2015;4:5–10. doi:10.1016/j.bonr.2015.10.006
54. Reid DM, Devogelaer JP, Saag K, et al. Zoledronic acid and risedronate in the prevention and treatment of glucocorticoid-induced osteoporosis (HORIZON): a multicentre, double-blind, double-dummy, randomised controlled trial. Lancet. 2009;373(9671):1253–1263. doi:10.1016/S0140-6736(09)60250-6
55. Schilcher J, Michaëlsson K, Aspenberg P. Bisphosphonate use and atypical fractures of the femoral shaft. N Engl J Med. 2011;364(18):1728–1737. doi:10.1056/NEJMoa1010650
56. Dore RK, Cohen SB, Lane NE, et al. Effects of denosumab on bone mineral density and bone turnover in patients with rheumatoid arthritis receiving concurrent glucocorticoids or bisphosphonates. Ann Rheum Dis. 2010;69(5):872–875. doi:10.1136/ard.2009.112920
57. Ishiguro S, Ito K, Nakagawa S, Hataji O, Sudo A. The clinical benefits of denosumab for prophylaxis of steroid-induced osteoporosis in patients with pulmonary disease. Arch Osteoporos. 2017;12(1):44. doi:10.1007/s11657-017-0336-1
58. Mok CC, Ho LY, Ma KM. Switching of oral bisphosphonates to denosumab in chronic glucocorticoid users: a 12-month randomized controlled trial. Bone. 2015;75:222–228. doi:10.1016/j.bone.2015.03.002
59. Iseri K, Iyoda M, Watanabe M, et al. The effects of denosumab and alendronate on glucocorticoid-induced osteoporosis in patients with glomerular disease: a randomized, controlled trial. PLoS One. 2018;13(3):e0193846–e0193846. doi:10.1371/journal.pone.0193846
60. Saag KG, Pannacciulli N, Geusens P, et al. Denosumab versus risedronate in glucocorticoid-induced osteoporosis: final results of a twenty-four–month randomized, double-blind, double-dummy trial. Arthritis Rheumatol. 2019;71(7):1174–1184. doi:10.1002/art.40874
61. Yanbeiy ZA, Hansen KE. Denosumab in the treatment of glucocorticoid-induced osteoporosis: a systematic review and meta-analysis. Drug Des Devel Ther. 2019;13:2843. doi:10.2147/DDDT.S148654
62. Anagnostis P, Paschou SA, Mintziori G, et al. Drug holidays from bisphosphonates and denosumab in postmenopausal osteoporosis: EMAS position statement. Maturitas. 2017;101:23–30. doi:10.1016/j.maturitas.2017.04.008
63. Tsourdi E, Langdahl B, Cohen-Solal M, et al. Discontinuation of Denosumab therapy for osteoporosis: a systematic review and position statement by ECTS. Bone. 2017;105:11–17. doi:10.1016/j.bone.2017.08.003
64. Dempster DW, Cosman F, Parisien M, Shen V, Lindsay R. Anabolic actions of parathyroid hormone on bone*. Endocr Rev. 1993;14(6):690–709. doi:10.1210/edrv-14-6-690
65. Neer RM, Arnaud CD, Zanchetta JR, et al. Effect of parathyroid hormone (1-34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001;344(19):1434–1441. doi:10.1056/NEJM200105103441904
66. Devogelaer JP, Adler RA, Recknor C, et al. Baseline glucocorticoid dose and bone mineral density response with teriparatide or alendronate therapy in patients with glucocorticoid-induced osteoporosis. J Rheumatol. 2010;37(1):141–148. doi:10.3899/jrheum.090411
67. Glüer CC, Marin F, Ringe JD, et al. Comparative effects of teriparatide and risedronate in glucocorticoid- induced osteoporosis in men: 18-month results of the EuroGIOPs trial. J Bone Miner Res. 2013;28(6):1355–1368. doi:10.1002/jbmr.1870
68. Lane NE, Sanchez S, Modin GW, Genant HK, Pierini E, Arnaud CD. Parathyroid hormone treatment can reverse corticosteroid-induced osteoporosis: results of a randomized controlled clinical trial. J Clin Invest. 1998;102(8):1627–1633. doi:10.1172/JCI3914
69. Saag KG, Zanchetta JR, Devogelaer JP, et al. Effects of teriparatide versus alendronate for treating glucocorticoid-induced osteoporosis: thirty-six-month results of a randomized, double-blind, controlled trial. Arthritis Rheum. 2009;60(11):3346–3355. doi:10.1002/art.24879
70. Lespessailles E, Chapurlat R. High fracture risk patients with glucocorticoid-induced osteoporosis should get an anabolic treatment first. Osteoporos Int. 2020;31(10):1829–1834. doi:10.1007/s00198-020-05568-w
71. Kobza AO, Papaioannou A, Lau AN, Adachi JD. Romosozumab in the treatment of osteoporosis. Immunotherapy. 2020;12(13):965–981. doi:10.2217/imt-2020-0158
72. Cosman F, Crittenden DB, Adachi JD, et al. Romosozumab treatment in postmenopausal women with osteoporosis. N Engl J Med. 2016;375(16):1532–1543. doi:10.1056/NEJMoa1607948
73. Saag KG, Petersen J, Brandi ML, et al. Romosozumab or alendronate for fracture prevention in women with osteoporosis. N Engl J Med. 2017;377(15):1417–1427. doi:10.1056/NEJMoa1708322
74. Langdahl BL, Libanati C, Crittenden DB, et al. Romosozumab (sclerostin monoclonal antibody) versus teriparatide in postmenopausal women with osteoporosis transitioning from oral bisphosphonate therapy: a randomised, open-label, Phase 3 trial. Lancet (London, England). 2017;390(10102):1585–1594. doi:10.1016/S0140-6736(17)31613-6
75. Lewiecki EM, Blicharski T, Goemaere S, et al. A Phase III randomized placebo-controlled trial to evaluate efficacy and safety of romosozumab in men with osteoporosis. J Clin Endocrinol Metab. 2018;103(9):3183–3193. doi:10.1210/jc.2017-02163
76. Yao W, Dai W, Jiang L, et al. Sclerostin-antibody treatment of glucocorticoid-induced osteoporosis maintained bone mass and strength. Osteoporos Int. 2016;27(1):283–294. doi:10.1007/s00198-015-3308-6
77. NIH U.S. National Library of Medicine. ClinicalTrials.gov. Available from: https://clinicaltrials.gov/ct2/results?cond=Osteoporosis&term=romosozumab&cntry=&state=&city=&dist=.
78. Syed MI, Patel NA, Jan S, Shaikh A, Grunden B, Morar K. Symptomatic refractures after vertebroplasty in patients with steroid-induced osteoporosis. Am J Neuroradiol. 2006;27(9):1938 LP- 1943.
79. Amiche MA, Albaum JM, Tadrous M, et al. Efficacy of osteoporosis pharmacotherapies in preventing fracture among oral glucocorticoid users: a network meta-analysis. Osteoporos Int. 2016;27(6):1989–1998. doi:10.1007/s00198-015-3476-4
80. Ding L, Hu J, Wang D, et al. Efficacy and safety of first- and second-line drugs to prevent glucocorticoid-induced fractures: network meta-analysis. J Clin Endocrinol Metab. 2020;105(3):600–613. doi:10.1210/clinem/dgz023
81. Ward LM. Glucocorticoid-induced osteoporosis: why kids are different. Front Endocrinol (Lausanne). 2020;11:576. doi:10.3389/fendo.2020.00576
82. Albaum JM, Lévesque LE, Gershon AS, Liu G, Cadarette SM. Glucocorticoid-induced osteoporosis management among seniors, by year, sex, and indication, 1996–2012. Osteoporos Int. 2015;26(12):2845–2852. doi:10.1007/s00198-015-3200-4
83. Buckley LM, Marquez M, Feezor R, Ruffin DM, Benson LL. Prevention of corticosteroid-induced osteoporosis: results of a patient survey. Arthritis Rheum. 1999;42(8):1736–1739. doi:10.1002/1529-0131(199908)42:8<1736::AID-ANR24>3.0.CO;2-E
84. Koller G, Katz S, Charrois TL, Ye C. Glucocorticoid-induced osteoporosis preventive care in rheumatology patients. Arch Osteoporos. 2019;14(1):16. doi:10.1007/s11657-019-0570-9
85. Duyvendak M, Naunton M, Van Roon EN, Bruyn GAW, Brouwers JRBJ. Systematic review of trends in prophylaxis of corticosteroid-induced osteoporosis: the need for standard audit guidelines. Osteoporos Int. 2008;19(10):1379–1394. doi:10.1007/s00198-008-0598-y
86. Guzman-Clark JRS, Fang MA, Sehl ME, Traylor L, Hahn TJ. Barriers in the management of glucocorticoid-induced osteoporosis. Arthritis Care Res. 2007;57(1):140–146. doi:10.1002/art.22462
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.