Back to Journals » Neuropsychiatric Disease and Treatment » Volume 12
Uncovering the etiology of conversion disorder: insights from functional neuroimaging
Authors Ejareh dar M, Kanaan R ![]()
Received 2 September 2015
Accepted for publication 21 October 2015
Published 13 January 2016 Volume 2016:12 Pages 143—153
DOI https://doi.org/10.2147/NDT.S65880
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Roger Pinder
Maryam Ejareh dar, Richard AA Kanaan
Department of Psychiatry, University of Melbourne, Austin Health, Heidelberg, VIC, Australia
Abstract: Conversion disorder (CD) is a syndrome of neurological symptoms arising without organic cause, arguably in response to emotional stress, but the exact neural substrates of these symptoms and the underlying mechanisms remain poorly understood with the hunt for a biological basis afoot for centuries. In the past 15 years, novel insights have been gained with the advent of functional neuroimaging studies in patients suffering from CDs in both motor and nonmotor domains. This review summarizes recent functional neuroimaging studies including functional magnetic resonance imaging (fMRI), single photon emission computerized tomography (SPECT), and positron emission tomography (PET) to see whether they bring us closer to understanding the etiology of CD. Convergent functional neuroimaging findings suggest alterations in brain circuits that could point to different mechanisms for manifesting functional neurological symptoms, in contrast with feigning or healthy controls. Abnormalities in emotion processing and in emotion-motor processing suggest a diathesis, while differential reactions to certain stressors implicate a specific response to trauma. No comprehensive theory emerges from these clues, and all results remain preliminary, but functional neuroimaging has at least given grounds for hope that a model for CD may soon be found.
Keywords: conversion disorder, neuroimaging, functional neurology, hysteria, mechanisms
A Letter to the Editor has been recieved and published for this article.
Corrigendum for this paper has been published
Introduction
Patients presenting with neurological symptoms with no medical explanation are common in medical settings. These have been known under multiple names, some now historical, such as “hysteria”, some still in use, such as “functional neurological disorders, medically unexplained symptoms, and conversion disorder”. The changes in terminology reflect etiological understanding and thereby a brief history of psychiatry: from hysteria, Hippocrates’ “wandering” uterus, through witchcraft imputations in the medieval era, Freud’s psychoanalytic concept of repression through “conversion” of psychological distress into physical symptoms,1 and most recently DSM-5’s change to conversion disorder (functional neurological symptom disorder)2 with its inference to abnormalities in brain function.3 Historical review also reveals multiple approaches that did not prosper despite their plausibility.4 Such was the case with Pierre Janet who postulated that conversion symptoms were due to a selective decrease in awareness, as Ludwig later put it, caused by corticofugal inhibition.5 A broad attempt to harmonize some of the statements mentioned earlier is that conversion disorder (CD) encompasses medically unexplained, neurological symptoms that are believed to develop unintentionally in reaction to psychological and environmental factors such as trauma or daily stressors. This is an umbrella to include the Freudian concept of “unconscious conflict” under “unintentional reaction to psychological and environmental factors”.4 It is also an intent to return to a pre-Cartesian era through a more holistic mind-body vision or perhaps a way to integrate old views with modern techniques and advance toward a post-Cartesian era.
The etiology of medically unexplained symptoms, including CD, is poorly understood. Neuroimaging research is at the forefront of efforts to establish a neurobiological model for CD. Functional magnetic resonance imaging (fMRI), positron emission tomography (PET), and single photon emission computed tomography (SPECT) research is beginning to characterize dynamic pathologic changes. However, studies are often limited by subject number and typically difficult to compare due to differences in design, symptoms, and comorbidities. On the other hand, such research is only in its infancy, and holds out hope of a model for the putative brain dysfunction in CD.
There are good reasons to consider brain function a promising modality for investigation. Patients cannot or will not tell us what is going on – according to hypothesis, their relevant experiences are repressed, or dissociated – so relying on self-report is likely to be frustrating. Symptoms seem to arise quickly, in response to stress or other psychological states, and resolve spontaneously, or are cured completely with treatment, suggesting a “functional” and dynamic change rather than a structural and static one. This has led to calls to examine brain activity while a patient is experiencing putatively traumatic experiences, albeit in the artificial, designed environment of functional neuroimaging studies.6,7 In effect, this could represent an attempt to bring the holistic bio-psycho-social model under the same roof. The elements of a stress-diathesis model for CD have long been hypothesized. Traumatic life events were associated with CD even before Freud, though their relevance has been disputed.8 Equally, childhood trauma or suggestibility are venerable risk factors that have found inconsistent experimental support.9 Sustaining interest in any of these has been challenged not only by the complex nature of the investigations required, but by the sheer incomprehensibility of hysterical phenomena (just how could a divorce lead to a paralysis?) and the suspicion that something far simpler might be going on – feigning.10 Hence, there has been a welcome push for functional studies that explore mechanism, without relying on self-report, and differentiate from deliberate pretense. These could provide clues – improving our understanding of abnormal influence and connection of emotion processing areas onto brain areas responsible for sensorimotor and cognitive processes – and perhaps ultimately a pathophysiological model, for how distressing emotions are “converted” into neurological symptoms.
The general aim of functional neuroimaging studies in pathology is to identify the circuits that serve as the site of the disordered brain functions underlying abnormal cognition or behavior associated with the conditions that can facilitate the transformation of these subjective experiences into the objectively observable signs of the disorders.11,12 Several recent studies have used functional brain imaging techniques in the attempt to identify specific neural correlates associated with conversion symptoms, the review of which is given in the following section.
Methodology
We conducted a literature search in May 2015 using Medline and PubMed as the main search engines. The search key words were: (“conversion disorder” OR “hysteria”) AND (“functional neuroimaging” OR “functional MRI” OR “PET” OR “SPECT”). A manual search of the references included in more recent reviews was also conducted by the authors. The search was limited to studies in the English language. We found 13 fMRI studies, three SPECT, and five PET studies of CD that we will consider in the following section.
Functional magnetic resonance imaging
FMRI is a noninvasive, highly flexible, and increasingly available method for the study of brain activity among patients with neurological and psychiatric disorders that can shed light on their biological foundation, as it can reveal the site of abnormality and mechanisms of symptoms.13 We will summarize 13 recent fMRI studies of patients with CD in the subsequent section. Four of these have investigated functional sensory symptoms (paresthesia or blindness), and nine have studied motor symptoms (functional weakness or movement disorders), one of which includes specific symptom production in response to recall of stressful life events (Table 1).
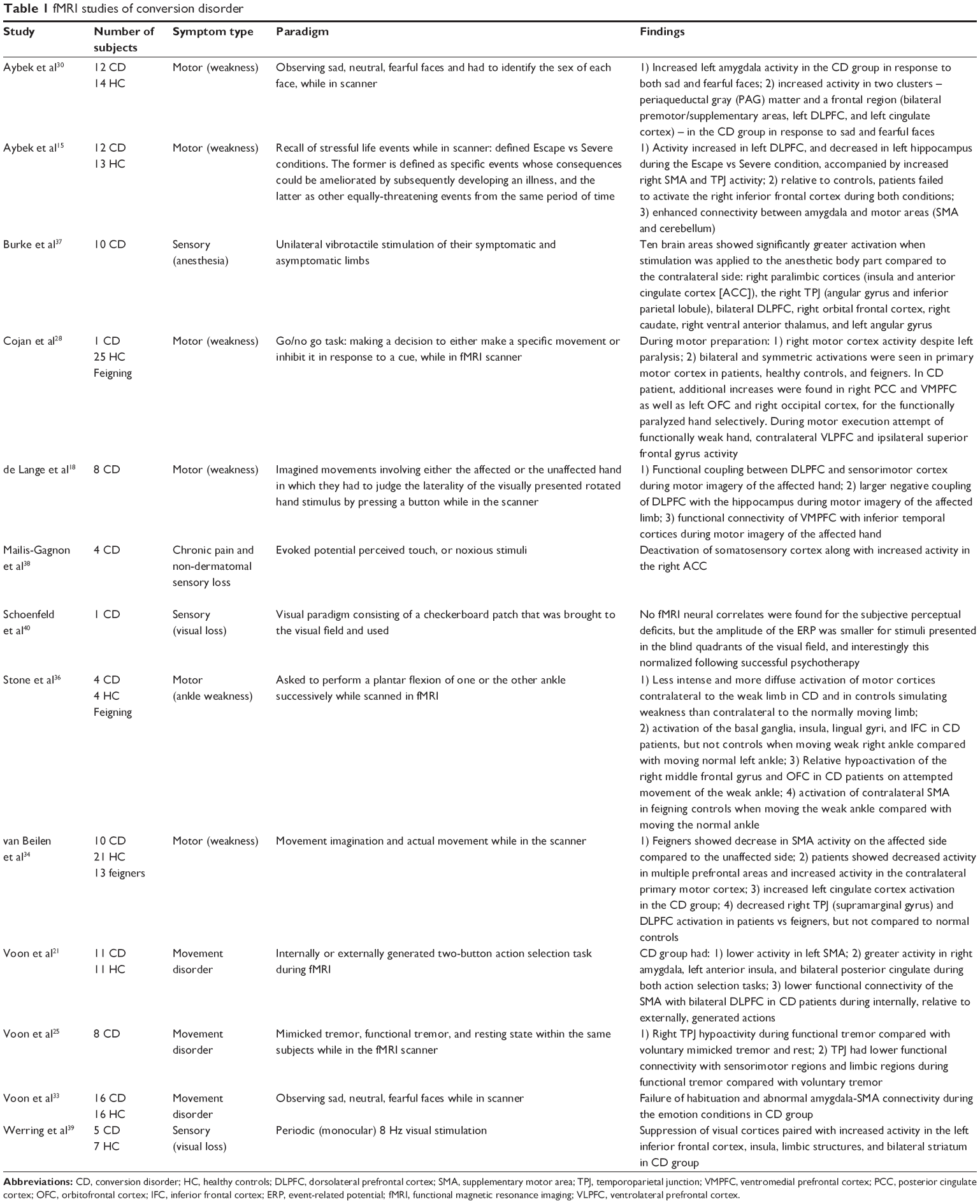 | Table 1 fMRI studies of conversion disorder |
We start with a study specifically looking at potentially etiological factors – stressful life events triggering symptom production. Following their publication of a single case study piloting this technique,14 Aybek et al investigated the recall of specific stressful life events while patients were scanned, in a case-control study of twelve patients with motor CD and 13 healthy control subjects.15 They used a comprehensive questionnaire to find stressful events that could be linked with the onset of functional symptoms, and after identifying specific events whose consequences could be ameliorated by subsequently developing an illness (so-called Escape events), they had patients recall these events while they were scanned, as well as other equally-threatening events from the same period of time (Severe events). They reported that, compared to controls, patients showed significantly increased left dorsolateral prefrontal cortex (DLPFC) and decreased left hippocampus activity during the Escape vs Severe condition. This was accompanied by increased right supplementary motor area (SMA) and temporoparietal junction (TPJ) activity. Relative to controls, patients failed to activate the right inferior frontal cortex during both conditions, and connectivity between amygdala and motor areas (SMA and cerebellum) was enhanced. This suggests a differential neural processing of Escape events in patients only. The authors formulated this under a suppression paradigm wherein conscious recall of an unwanted memory (indexed by the hippocampus) is disrupted by top-down regulation (indexed by the DLPFC) – one of the possible mechanisms involved in voluntary forgetting of unwanted memories.16,17 The activity in the SMA and TPJ were interpreted as reflecting symptoms present during recall of the Escape condition, and thus revealing an association, if not a “conversion”, of the memory suppression with symptoms. The enhanced connectivity between the amygdala and motor areas, and the inferior frontal suppression were both interpreted as possible vulnerability factors – as they were present in patients in all conditions – disordered emotion regulation, and a heightened emotion-motor connection. This study showed an attempt to go beyond mere mechanistic explanations for functional paralysis in search of potential etiological models: the journey from specific life stressors to symptoms production scrutinized in functional imaging of the brain. It remains, however, a very first step and is limited by the small number of participants, while it is strengthened by its use of a healthy control group and a very detailed interview to search for individualized life stressors.
This top-down regulation from higher order frontal regions to interfere with motor execution has been reported in other studies that have used other tasks in the fMRI scanner. de Lange et al designed a study with imagined movements involving either the affected or the unaffected hand of eight patients with functional hand weakness.18,19 Participants completed a motor imagery task, in which they had to judge the laterality of the visually presented rotated hand stimulus by pressing a button while in the scanner. They assessed the functional and effective connectivity profile of the prefrontal cortex during the task, reporting that medial prefrontal clusters were functionally connected to inferior temporal regions, but only the DLPFC was functionally connected to the motor system. They have reported multiple findings in an attempt to achieve different potential explanatory models for the development of unilateral functional hand paralysis: 1) functional coupling between DLPFC and sensorimotor cortex was observed during motor imagery of the affected hand. This specific coupling can be suggestive of a role in the selection of action plans.20 Furthermore, an increased positive coupling between DLPFC and dorsal premotor cortex was reported during imagery of the affected hand which according to the authors may reflect a compensatory mechanism, namely an increased prefrontal drive toward premotor regions supporting the imagery process of the affected hand; 2) The DLPFC had larger negative couplings with the hippocampus during motor imagery of the affected limb, which was postulated in the context of a link between (aversive) memories and functional symptoms and as a control mechanism to decouple (imagined) movements of the affected hand from aversive memories associated with those movements; 3) Ventromedial PFC (VMPFC) showed functional connectivity with inferior temporal cortices during motor imagery of the affected hand, but no functional or effective connectivity with the sensorimotor system. Given the close connections between VMPFC and the limbic areas, emotion regulation, interoceptive attention, and self-referential and autobiographical processing have been attributed to this area, which may suggest another mechanism for the functional weakness – heightened self-monitoring during motor imagery of the affected limb. While this study is offering a distinct view into a mechanistic process for conversion paralysis, it is limited by its small number of participants and lack of a control group.
Motor control and the role of the will in this process has been another focus for functional symptom models. CD patients with movement disorders were studied by Voon et al.21 They recruited eleven patients with such symptoms (tremor, dystonia, or gait abnormality) and compared them with eleven healthy volunteers while they performed either an internally or externally generated two-button action selection task during fMRI. CD patients relative to healthy subjects had lower activity in a region associated with motor preparation and inhibition, the left SMA, and greater activity in regions associated with emotional processing including the right amygdala, left anterior insula, and bilateral posterior cingulate during both action selection tasks. SMA is implicated in self-initiated action,22,23 as well as in the sense of agency, or the experience that one is in control of one’s actions.24 This study also showed lower functional connectivity of the SMA with bilateral DLPFC in CD patients during internally, relative to externally, generated actions, suggesting a potential disconnection in top-down motor control to guide internally generated action selection. Continuing the enquiry into self-agency and the possible involuntary nature of these specific movements, Voon et al designed another study with eight patients with CD and compared mimicked tremor, functional tremor, and resting state within the same subjects while in the fMRI scanner.25 They reported right TPJ hypoactivity during functional tremor when compared with voluntary mimicked tremor and rest. This region has been suggested to have an involvement in multisensory integration. Furthermore, TPJ had lower functional connectivity with sensorimotor regions and limbic regions during functional tremor when compared with voluntary tremor. While this does little to elucidate the emergence of conversion symptoms, it may clarify the subjective experience of involuntariness reported by patients. They attribute this specific distinction of internal sensory prediction and the actual sensory state to the right TPJ, a claim supported by the experimental demonstration that the opposite experience, an illusion of movement control, can be generated by stimulation of the area.26 This study also reported a lower connectivity between the TPJ and limbic regions (ventral anterior cingulate and ventral striatum) in functional tremor vs voluntary tremor that the authors propose represents reduced limbic involvement in functional movement evaluation. They postulate this is a “feed forward” abnormality, a problem in generating sensory predictions in functional tremor,27 which is compatible with the previously discussed abnormal motor preparation mechanism. However, this view is challenged by Cojan et al who examined the idea of abnormal inhibition in a task that involved making a decision to either make a specific movement or inhibit it in response to a cue, in a single patient compared with 25 controls.28 They found that motor cortical regions during movement preparation and right inferior frontal regions during movement inhibition were unchanged, which led them to conclude that both motor intention and inhibition were intact. But, like some of the studies mentioned earlier, they found functional weakness to be associated with ventrolateral prefrontal cortex activity, intriguingly a region with substantive input from areas involved in emotion processing.
Greater arousal or a failure to inhibit the orienting response to a familiar stimulus may be a risk factor for the development of CDs. This has been suggested in other CD types (psychogenic nonepileptic seizures [PNES]) through findings of elevated basal cortisol levels.29 To explore the possible role of abnormal arousal or threat response in a mechanism for CD, Aybek et al studied the processing of negative emotions in CD and healthy controls.30 To this end, they studied twelve patients with motor CD, and 14 age- and sex-matched healthy controls, who underwent fMRI scans while they observed sad, fearful (negative emotions), and neutral faces. The task for subjects was to identify the sex of each face, so that the emotion processing was implicit. The authors reported: 1) significantly increased left amygdala activity in the CD group in response to both sad and fearful faces. Interestingly, this distinct response in the patient group to the fearful stimuli not only did not reduce over time, as it did in the case of healthy controls, but it was further accentuated; in other words, a hyper-sensitization rather than habituation was observed; 2) significantly increased activity in two clusters – periaqueductal gray (PAG) matter and a frontal region (bilateral premotor/supplementary areas, left DLPFC, and left cingulate cortex) – was reported in the CD group when compared with healthy controls in response to sad and fearful faces. A lack of habituation in the CD group has been previously described however,31 of additional mechanistic interest is the increased activity in PAG that the authors have attributed to the “freeze response” to threat, based on the association of this area with the “fear bradycardia response”.32 Their other finding of increased frontal clusters, specifically the superior frontal gyrus that has been speculated to play a role in self-awareness and attention, may suggest another mechanism for CD. Insofar as the activation was in response to emotion, as the frontal areas involved extended to motor planning and monitoring areas such as SMA, anterior and middle cingulate gyrus, they suggest again an abnormal link from emotion to motion. A similar fMRI study was conducted by Voon et al, who recruited 16 patients with functional movement disorders, and compared them with 16 matched healthy controls33 using the same affective stimuli but with fearful, happy, and neutral faces. They also reported a failure of habituation and abnormal amygdala-SMA connectivity during the emotion conditions in patients. They hence suggested a contribution not only to symptom generation but also to why these might arise in the first place – a failure to inhibit the orienting response to a familiar stimulus – as a potential risk factor for the development of CD.
The distinction from feigning was explored by van Beilen et al who conducted an fMRI study including three groups: healthy controls, patients with unilateral functional paresis, and healthy subjects feigning unilateral sided paresis.34 They were asked to perform tasks involving movement imagination and actual movement while in the scanner. Feigners showed a similar pattern of primary and secondary motor activation to healthy controls, but a decrease in SMA activity was found on the affected side compared to the unaffected side. Patients showed decreased activity in multiple prefrontal areas and increased activity in the contralateral primary motor cortex consistent with the expected movement-related activity. They also observed increased left cingulate cortex activation in comparison to normal controls in the CD group. Furthermore, the right TPJ (supramarginal gyrus) and DLPFC showed decreased activation in patients vs feigners, but not compared to normal controls. The decrease in right TPJ has previously been reported, and is again interpreted here as a feed-forward failure,35 combining with the previously unreported DLPFC decrease to give a failure of the sense of agency and a failure of willed action, in contrast to the deliberate feigning group.
With a similar feigning contrast, Stone et al studied four patients with unilateral functional ankle weakness and healthy controls simulating weakness.36 They were all asked to perform a plantar flexion of one or the other ankle successively while scanned in fMRI. They reported: 1) less intense and more diffuse activation of motor cortices contralateral to the weak limb in patients with CD and in controls simulating weakness than contralateral to the normally moving limb; 2) activation of regions of the basal ganglia, insula, lingual gyri, and inferior frontal cortex in CD patients, but not in controls when moving weak right ankle compared with moving normal left ankle. Increased activation of inferior frontal cortex was interpreted in the context of an executive role in conscious planning or preparation of the movement, and activation of the insula, lingual gyri, superior parietal region, and precuneus as reflecting increased attention and effortful emotional processing in CD patients; 3) Relative hypoactivation of the right middle frontal gyrus and orbitofrontal cortex in CD patients on attempted movement of the weak ankle which the authors suggest as a possible a mechanism through which the movement is inhibited; 4) Activation of contralateral SMA in feigning controls when moving the weak ankle compared with moving the normal ankle, which has been suggested as an excess of movement planning activity in comparison to movement of the normal limb.
A number of studies focused on sensory CD. They are relatively consistent in their finding of reduced activity in primary sensory areas, and increased activity elsewhere. In one study, ten subjects underwent unilateral vibrotactile stimulation of their symptomatic and asymptomatic limbs.37 Ten brain areas showed significantly greater activation when stimulation was applied to the anesthetic body part compared to the contralateral side: right paralimbic cortices (insula and anterior cingulate cortex [ACC]), the right TPJ (angular gyrus and inferior parietal lobule), bilateral DLPFC, right orbital frontal cortex, right caudate, right ventral anterior thalamus, and left angular gyrus. Mailis-Gagnon et al reported on four patients with chronic pain and non-dermatomal sensory loss, who had deactivation of somatosensory cortex along with increased activity in the right ACC during evoked potential perceived touch, or noxious stimuli compared to unperceived stimuli.38 In a sensory fMRI study of five patients with functional visual loss and seven normal volunteers, Werring et al evaluated brain activation induced by periodic (monocular) 8 Hz visual stimulation.39 Patients showed suppression of visual cortices paired with increased activity in the left inferior frontal cortex, insula, limbic structures, and bilateral striatum.39
Schoenfeld et al used fMRI and event-related potentials (ERP) to investigate a patient with conversional blindness, before and after successful psychotherapy treatment.40 This is an interesting report in that the patient had blindness only in two quadrants of the visual field, which allowed using the remainder of the visual field as a control. They used a visual paradigm consisting of a checkerboard patch that was brought to the visual field in a specific manner. They could not find any fMRI neural correlates for the subjective perceptual deficits, perhaps due to the low power of fMRI in a single case. In contrast, the ERP differentiated the two: the amplitude of the ERP component had smaller amplitudes for stimuli presented in the blind quadrants of the visual field, and interestingly this normalized following successful psychotherapy.
Single photon emission computerized tomography
SPECT is a neuroimaging technique used primarily to assess cerebral blood flow by measuring the distribution of radioactive tracer uptake and retention in different areas of the brain.41 We will describe three SPECT studies of CD in the following section (Table 2). In one of the first functional neuroimaging studies of CD, Tiihonen et al reported a case study of a single patient with a left-sided sensory deficit.42 During electrical sensory stimulation of the left side, SPECT revealed hypoperfusion of the right parietal cortex (somatosensory cortex) and simultaneous hyperperfusion of the right frontal lobe areas. Another report of an early study of five cases of motor CD who underwent SPECT found reductions in regional cerebral blood in the dominant (left) temporal lobe in all cases as well as right temporal lobe in one case.43 Vuilleumier et al assessed brain responsiveness to passive sensory stimulation in seven patients suffering from unilateral sensorimotor loss in a SPECT study44 measuring blood flow during tuning fork stimulation of the affected and unaffected limbs during the active phase of the CD and following its resolution. The results showed hypoactivation of the thalamus and basal ganglia contralateral to the affected limb, which normalized with symptom resolution, suggesting that the functional symptom is associated with altered somatosensory processing. They believe this is of particular interest as basal ganglia, especially the caudate nucleus, might be particularly well situated to modulate motor processes based on emotional and situational cues from the limbic system. This was taken to support a model of functional disruption of cortico-striato-thalamo-cortical loops underlying symptoms.45
 | Table 2 SPECT studies of conversion disorder |
Positron emission tomography
PET, like SPECT, is a molecular imaging technique for mapping tracer uptake in target tissues in vivo.46 We will describe three PET studies of CD in the following section (Table 3). In an early study, Marshall et al used H215O-PET to investigate brain perfusion in a 43-year-old female suffering from functional paralysis of the left leg.47 When the patient was asked to move her left leg, no neural activation of the primary motor cortex but enhanced activation of the right orbitofrontal cortex and right anterior cingulate cortex was observed. This can be formulated in the context of top-down regulation: increased cerebral blood flow in frontal cortical areas could be the correlate of active inhibitory mechanisms directed toward primary sensorimotor areas which are translated into limb paralysis dictated by the frontal cortex. Spence et al studied three patients with unilateral functional motor weakness moving a joystick while undergoing PET and compared with four healthy individuals feigning weakness and six controls who did normal movements in the same task.48 They found decreased left DLPFC activity in patients when attempting to move their affected limb, regardless of which limb was affected. The selective dysfunction of left DLPFC in patients with functional motor symptoms and its specific activation by the internal generation (“choice”) of action was interpreted as the involvement of “higher” components of volition in these patients. This again supports the previously mentioned studies that pointed to motor execution and planning abnormalities as a possible underlying mechanism for symptom production in CD.
 | Table 3 PET studies of conversion disorder |
In yet a different CD symptom type, Schrag et al studied six patients with psychogenic dystonia and compared them to five patients with genetically determined dystonia and six healthy controls, all of whom underwent PET scans at rest, during fixed posturing of the right leg and during paced ankle movements.49 They found that on averaging regional cerebral blood flow across all tasks, the organic dystonia group showed abnormal increases in the primary motor cortex and thalamus compared with controls, with decreases in the cerebellum. In contrast, the psychogenic dystonia group showed an opposing pattern, with abnormally increased blood flow in the cerebellum and basal ganglia, with decreases in the primary motor cortex. Furthermore, during movement, compared with rest, there was abnormal activation in the right DLPFC that was common to both organic and psychogenic dystonia groups compared with control subjects. This again is consistent with previous proposals about the subcortical connections mediating psychogenic symptoms; in this case, the basal ganglia, cerebellum, and thalamus mediate fronto-subcortical loops that relate to motor intention or attention, with further modulation possible by reciprocal connections with the amygdala and orbitofrontal cortex. It has been proposed that the basal ganglia facilitate a form of focused motor attention subserving the automatic binding of cortical motor areas in a coherent sequence of motor activity and thought.50 Psychogenic dystonia may reflect dysfunction of these fronto-subcortical circuits.
Arthuis et al studied 16 patients with intractable seizures who underwent 18FDG-PET, and were later found to have PNES with co-morbid epilepsy, and compared these with 16 healthy controls.51 A voxel by voxel intergroup analysis was performed to look for significant differences in interictal (resting state) cerebral metabolism as well as metabolic connectivity using voxel-wise interregional correlation analysis. Patients with PNES exhibited significant hypometabolism within the right inferior parietal and central region and within the bilateral anterior cingulate cortex, and a significant increase in metabolic correlation, in comparison to healthy participants, between the right inferior parietal/central region and the bilateral cerebellum, and between the bilateral anterior cingulate cortex and the left parahippocampal gyrus. The hypometabolism of the anterior cingulate region stands in contrast to the hyperactivation reported in other studies, including those in PNES,34 but interpretation is complicated by the comorbid epilepsy.
Discussion
As mentioned earlier, an examination of past attempts at understanding CD will reveal an almost historical review of psychiatry. It is a condition of particular interest as it sits right on the fence between the brain and the mind, challenging those who would seek a simple classification. From the Hippocratic notion of wandering womb, which designated an organic seat for the symptoms, via the Cartesian dualism of separating the mind and the body altogether, back to the search for an organic cause by Pierre Briquet who proposed a “neurocerebral” origin for the condition,2 we have come a long and tortuous way. A brief window for an organic perspective for CD was opened prior to Freud’s psychoanalytic theory, but that view was clouded by imputations of malingering,52 and Freud served as a step toward the separation of the mind and body again, albeit reluctantly.53 After a pause imposed by psychoanalytic domination this effort was followed later, in tune with the evolution of new cognitive psychological models proposed for psychiatric disorders as well as the emergence of new research methodologies such as functional neuroimaging, by a somewhat closer look into the functioning of the brain.
It is, however, important to consider the many limitations of this type of study of CD. There are familiar problems of low subject numbers, and symptom and paradigm heterogeneity. This makes the task of demonstrating a unique explanatory model an arduous one, and raises the question of whether a unique neuroanatomical basis for CD is even plausible. Like all research, imaging experiments also impose conditions that do not conform to the real world.12 This is particularly problematic for CD, as the “real world” of a clinical presentation may disappear under experimental conditions: patients with CD who are weak in one leg are not weak all the time – they are not weak when they are asleep, or distracted, or when they move the leg as “tricked” by a neurologist seeking Hoover’s sign – so their symptom may simply not be present when scanned.54 Equally, it is hard to conclude that the contrasting condition of deliberate feigning, though often artfully experimentally contrived, is anything like that of a patient who has been carefully, fearfully, and, perhaps with time, semi-automatically perfecting it. This is one reason why the few studies not testing symptoms,15,30,33,51 arguably,18,19 seem more informative, particularly as avenues for exploring etiology, as opposed to mechanisms of symptom production.
There are nevertheless many clues to etiology from the studies reviewed. Firstly, in terms of feigning, while it is not possible to give a unifying account of the difference, all studies concurred that there is a difference from feigning– which, despite the caveats above, is hard to find via other means, except perhaps electrophysiology.55 This is crucial for the status of the disorder.
Secondly, in support of the previously mentioned statement, there are elements of a plausible mechanism for at least some symptoms. This is equally crucial, since the inference to feigning draws much of its strength from the difficulty in understanding how these symptoms could otherwise be,56 even if they do not define CD as an entity.57 Inhibition of normal networks (motor, sensory, or special sensory) as a common pathway to functional symptoms39,48 has been reported in studies that found an increased activity in prefrontal areas (or possibly the cingulate cortex58) associated with reduced activity in sensorimotor areas.47,59 A deficit in motor planning has been suggested, with the repeated implication of the SMA.15,18,21 This has been replicated in nonimaging studies.55,60 Abnormal self-monitoring has also been proposed,19 with the TPJ, DLPFC, and VMPFC implicated,18,19 and the latter’s dense connections with limbic areas suggesting a link with interoceptive attention and self-referential and autobiographical processing61 during symptom production.
Thirdly, abnormalities in emotion regulation and connectivity have been identified, in non-symptom studies, potentially pointing to a diathesis or vulnerability. Two studies found an abnormal emotion-motor connectivity,15,33 and two a failure of normal habituation.30,33 Though the subjects may have been symptomatic at the time of scanning, the paradigms were not directly symptom-invoking or provoking, which suggests the abnormalities may be present independently of symptoms. If so, these would suggest both a hyper-reactivity to normal emotional stimuli and an established pathway linking that reactivity to motor output, and therefore perhaps symptoms. The pre-morbid presence of such factors would obviously require explanation in itself, however.
Fourthly and finally, specific traumatic life events have been shown to be differentially processed and linked to symptoms in CD, and this has been interpreted in a way familiar to any student of Freud:14,15 the recall of events judged to be of etiological relevance were associated with brain activation analogous to memory suppression and with brain activation in the now-familiar SMA and TPJ, plausible sites of symptom production.
It cannot yet be claimed that a comprehensive theory emerges from this, and, of course, it would be absurd to ignore other investigative modalities currently yielding exciting insights (notably, but not exclusively, electrophysiology), but functional neuroimaging has given etiological hints to the specific diatheses, stressors, and mechanisms for enacting this most confounding condition. The results are in all cases preliminary, with confirmation typically relying on activation overlap rather than on paradigm replication, but given the tantalizing glimpses of answers to an age-old puzzle, the call for more studies must be as strident as it was predictable. Further studies could improve methodology by increasing the number of participants, recruiting more arms into the same studies (control groups consisting of patients with same neurological symptoms as well as healthy controls), as well as longitudinal cohort designs that would follow up patients with CD following symptoms resolution.
Disclosure
The authors report no conflicts of interest in this work.
References
Allison DB, Roberts MS. On constructing the disorder of hysteria. J Med Philos. 1994;19(3):239–259. | ||
Crommelinck M. Neurophysiology of conversion disorders: a historical perspective. Neurophysiol Clin. 2014;44(4):315–321. | ||
Kanaan RA, Armstrong D, Wessely SC. The function of ‘functional’: a mixed methods investigation. J Neuro, Neurosurg Psychiatry. 2012(3):248. | ||
Ali S, Jabeen S, Pate RJ, et al. Conversion disorder- mind versus Body: a review. Innov Clin Neurosci. 2015;12(5–6):27–33. | ||
Ludwig AM. Hysteria. A neurobiological theory. Arch Gen Psychiatry. 1972;27(6):771–777. | ||
Kozlowska K. Healing the disembodied mind: contemporary models of conversion disorder. Harv Rev Psychiatry. 2005;13(1):1–13. | ||
Aybek S, Kanaan RA, David AS. The neuropsychiatry of conversion disorder. Curr Opin Psychiatry. 2008;21(3):275–280. | ||
Stone J, Edwards MJ. How “psychogenic” are psychogenic movement disorders? Mov Disord. 2011;26(10):1787–1788. | ||
Nicholson TRJ, Stone J, Kanaan RAA. Conversion disorder: a problematic diagnosis. J Neurol, Neurosur Psychiatry. 2011(11):1267. | ||
Kanaan RA. Hysteria and the varieties of deception. AJOB Neuroscience. 2010;1(1):55. | ||
Fielding JM, Marwede D. The anatomy of the image: toward an applied onto-psychiatry. Philos Psychiatr Psychol. 2011(4):287. | ||
Kanaan RAA, McGuire PK. Conceptual challenges in the neuroimaging of psychiatric disorders. Philos Psychiatr Psychol. 2011;18(4):323–332. | ||
Weiller C, May A, Sach M, Buhmann C, Rijntjes M. Role of functional imaging in neurological disorders. J Magn Reson Imaging. 2006;23(6):840–850. | ||
Kanaan RA, Craig TK, Wessely SC, David AS. Imaging repressed memories in motor conversion disorder. Psychosom Med. 2007;69(2):202–205. | ||
Aybek S, Nicholson TR, Zelaya F, et al. Neural correlates of recall of life events in conversion disorder. JAMA psychiatry. 2014;71(1):52–60. | ||
Benoit RG, Anderson MC. Opposing mechanisms support the voluntary forgetting of unwanted memories. Neuron. 2012;76(2):450–460. | ||
Anderson MC, Ochsner KN, Kuhl B, et al. Neural systems underlying the suppression of unwanted memories. Science. 2004;303(5655):232–235. | ||
de Lange FP, Toni I, Roelofs K. Altered connectivity between prefro ntal and sensorimotor cortex in conversion paralysis. Neuropsychologia. 2010;48(6):1782–1788. | ||
de Lange FP, Roelofs K, Toni I. Increased self-monitoring during imagined movements in conversion paralysis. Neuropsychologia. 2007;45(9):2051–2058. | ||
Rowe J, Friston K, Frackowiak R, Passingham R. Attention to action: specific modulation of corticocortical interactions in humans. Neuroimage. 2002;17(2):988–998. | ||
Voon V, Brezing C, Gallea C, Hallett M. Aberrant supplementary motor complex and limbic activity during motor preparation in motor conversion disorder. Mov Disord. 2011;26(13):2396–2403. | ||
Nachev P, Kennard C, Husain M. Functional role of the supplementary and pre-supplementary motor areas. Nat Rev Neurosci. 2008;9(11):856–869. | ||
Passingham RE, Bengtsson SL, Lau HC. Medial frontal cortex: from self-generated action to reflection on one’s own performance. Trends Cogn Sci. 2010;14(1):16–21. | ||
Moore JW, Ruge D, Wenke D, Rothwell J, Haggard P. Disrupting the experience of control in the human brain: pre-supplementary motor area contributes to the sense of agency. Proc Biol Sci. 2010;277(1693):2503–2509. | ||
Voon V, Gallea C, Hattori N, Bruno M, Ekanayake V, Hallett M. The involuntary nature of conversion disorder. Neurology. 2010;74(3):223–228. | ||
Desmurget M, Reilly KT, Richard N, Szathmari A, Mottolese C, Sirigu A. Movement intention after parietal cortex stimulation in humans. Science. 2009;324(5928):811–813. | ||
Edwards MJ, Adams RA, Brown H, Pareés I, Friston KJ. A Bayesian account of ‘hysteria’. Brain. 2012;135(11):3495–3512. | ||
Cojan Y, Waber L, Carruzzo A, Vuilleumier P. Motor inhibition in hysterical conversion paralysis. Neuroimage. 2009;47(3):1026–1037. | ||
Bakvis P, Spinhoven P, Giltay EJ, et al. Basal hypercortisolism and trauma in patients with psychogenic nonepileptic seizures. Epilepsia. 2010;51(5):752–759. | ||
Aybek S, Nicholson TR, O’Daly O, Zelaya F, Kanaan RA, David AS. Emotion-motion interactions in conversion disorder: an FMRI study. PLoS One. 2015;10(4):e0123273. | ||
Horvath T, Friedman J, Meares R. Attention in hysteria: a study of Janet’s hypothesis by means of habituation and arousal measures. The American journal of psychiatry. 1980;137(2):217–220. | ||
Hermans EJ, Henckens MJ, Roelofs K, Fernandez G. Fear bradycardia and activation of the human periaqueductal grey. Neuroimage. 2013;66:278–287. | ||
Voon V, Brezing C, Gallea C, et al. Emotional stimuli and motor conversion disorder. Brain. 2010;133(Pt 5):1526–1536. | ||
van Beilen M, de Jong BM, Gieteling EW, Renken R, Leenders KL. Abnormal parietal function in conversion paresis. PLoS One. 2011;6(10):e25918. | ||
Voon V. Functional neurological disorders: imaging. Neurophysiol Clin/Clin Neurophysiol. 2014;(4):339. | ||
Stone J, Zeman A, Simonotto E, et al. FMRI in patients with motor conversion symptoms and controls with simulated weakness. Psychosomatic medicine. 2007;69(9):961–969. | ||
Burke MJ, Ghaffar O, Staines WR, Downar J, Feinstein A. Functional neuroimaging of conversion disorder: the role of ancillary activation. Neuroimage Clin. 2014;6:333–339. | ||
Mailis-Gagnon A, Giannoylis I, Downar J, et al. Altered central somatosensory processing in chronic pain patients with “hysterical” anesthesia. Neurology. 2003;60(9):1501–1507. | ||
Werring DJ, Weston L, Bullmore ET, Plant GT, Ron MA. Functional magnetic resonance imaging of the cerebral response to visual stimulation in medically unexplained visual loss. Psychol Med. 2004;34(4):583–589. | ||
Schoenfeld MA, Hassa T, Hopf JM, Eulitz C, Schmidt R. Neural correlates of hysterical blindness. Cereb Cortex. 2011;21(10):2394–2398. | ||
Cragar DE, Berry DTR, Fakhoury TA, Cibula JE, Schmitt FA. A review of diagnostic techniques in the differential diagnosis of epileptic and nonepileptic seizures. Neuropsychol Rev. 2002;12(1):31–64. | ||
Tiihonen J, Kuikka J, Viinamäki H, Lehtonen J, Partanen J. Altered cerebral blood flow during hysterical paresthesia. Biol Psychiatry. 1995;37(2):134–135. | ||
Yaźići KM, Kostakoglu L. Cerebral blood flow changes in patients with conversion disorder. Psychiatry Res. 1998;83(3):163–168. | ||
Vuilleumier P, Chicherio C, Assal F, Schwartz S, Slosman D, Landis T. Functional neuroanatomical correlates of hysterical sensorimotor loss. Brain. 2001;124(Pt 6):1077–1090. | ||
Carson AJ, Brown R, David AS, et al. Functional (conversion) neurological symptoms: research since the millennium. J Neurol Neurosurg Psychiatry. 2012;83(8):842–850. | ||
Singhal T. Positron emission tomography applications in clinical neurology. Semin Neurol. 2012;32(4):421–431. | ||
Marshall JC, Halligan PW, Fink GR, Wade DT, Frackowiak RS. The functional anatomy of a hysterical paralysis. Cognition. 1997;64(1):B1–B8. | ||
Spence SA, Crimlisk HL, Cope H, Ron MA, Grasby PM. Discrete neurophysiological correlates in prefrontal cortex during hysterical and feigned disorder of movement. Lancet. 2000;355(9211):1243–1244. | ||
Schrag AE, Mehta AR, Bhatia KP, et al. The functional neuroimaging correlates of psychogenic versus organic dystonia. Brain. 2013;136(Pt 3):770–781. | ||
Brown P, Marsden CD. What do the basal ganglia do? Lancet. 1998;351(9118):1801–1804. | ||
Arthuis M, Micoulaud-Franchi JA, Bartolomei F, McGonigal A, Guedj E. Resting cortical PET metabolic changes in psychogenic non-epileptic seizures (PNES). J Neurol Neurosur Psychiatry. 2015;86(10):1106–1112. | ||
Kanaan RA, Wessely SC. The origins of factitious disorder. Hist Human Sci. 2010;23(2):68–85. | ||
Kanaan RAA. Freud’s Hysteria and its Legacy. In: Hallett M, Stone J, Carson A, editors. Functional Neurological Disorders. Amsterdam: Elsevier. In press 2015. | ||
Kanaan RAA, McGuire PK. Challenges for psychiatric neuroimaging: the case of conversion disorder. Philos Psychiatr and Psychol. 2011;18(4):345–348. | ||
Blakemore RL, Hyland BI, Hammond-Tooke GD, Anson JG. Deficit in late-stage contingent negative variation provides evidence for disrupted movement preparation in patients with conversion paresis. Biolol Psychol. 2015;109:73–75. | ||
Kanaan RA. Hysteria and the Varieties of Deception. Am J Bioethics –Neurosci. 2010;1(1):55–64. | ||
Spence SA. Hysterical paralyses as disorders of action. Cogn Neuropsychiatry. 1999;4(3):203–226. | ||
van Beilen M, Vogt BA, Leenders KL. Increased activation in cingulate cortex in conversion disorder: what does it mean? J Neurol Sci. 2010;289(1–2):155–158. | ||
Halligan PW, Athwal BS, Oakley DA, Frackowiak RS. Imaging hypnotic paralysis: implications for conversion hysteria. Lancet. 2000;355(9208):986–987. | ||
Blakemore RL, Hyland BI, Hammond-Tooke GD, Anson JG. Distinct modulation of event-related potentials during motor preparation in patients with motor conversion disorder. PLoS One. 2013;8(4):e62539. | ||
Gilbert SJ, Spengler S, Simons JS, et al. Functional specialization within rostral prefrontal cortex (area 10): a meta-analysis. J Cogn Neurosci. 2006;18(6):932–948. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.