Back to Journals » Neuropsychiatric Disease and Treatment » Volume 11
Ultrastructural mitochondria changes in perihematomal brain and neuroprotective effects of Huperzine A after acute intracerebral hemorrhage
Authors Lu H, Jiang M, Lu L, Zheng G, Dong Q
Received 10 July 2015
Accepted for publication 20 August 2015
Published 13 October 2015 Volume 2015:11 Pages 2649—2657
DOI https://doi.org/10.2147/NDT.S92158
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Wai Kwong Tang
Haiying Lu,1,* Mei Jiang,2,* Lei Lu,3 Guo Zheng,1 Qiang Dong3
1Department of Neurology, Nanjing Children’s Hospital, Nanjing Medical University, Nanjing, 2Department of Neurology, Shanghai Pudong New Area Gongli Hospital, Shanghai, 3Department of Neurology, Huashan Hospital, Fudan University, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Aim: The purpose of the study was to observe the ultrastructural changes of neuronal mitochondria in perihematomal brain tissue and assess the therapeutic potential of Huperzine A (HA, a mitochondrial protector) following intracerebral hemorrhage (ICH).
Methods: Brain hemorrhage was induced in adult Sprague Dawley rats by injecting autologous blood into the striatum and then removing the brains 3, 6, 12, 24, or 48 hours later to analyze mitochondrial ultrastructure in a blinded manner. Parallel groups of ICH rats were treated with HA or saline immediately after ICH. Perihematomal apoptosis was determined by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), caspase-3 activation and cytochrome C translocation were tracked by immunoblots, and neurobehavioral test results were compared between the groups.
Results: Mitochondria in perihematomal neurons demonstrated dramatic changes including mitochondrial swelling, intracristal dilation, and decreased matrix density. HA treatment decreased mitochondrial injury and apoptosis, inhibited caspase-3 activation and cytochrome C translocation, and improved behavioral recovery.
Conclusion: These data show that ICH induces dramatic mitochondrial damage, and HA exhibits protective effects possibly through ameliorating mitochondrial injury and apoptosis. Collectively, these findings suggest a new direction for novel therapeutics.
Keywords: apoptosis, intracerebral hemorrhage, mitochondria, huperzine A, neuroprotection
Introduction
Intracerebral hemorrhage (ICH) is a primary event in 15% to 20% of all strokes,1 the mortality rate of which approaches 58%.2 Unfortunately, the pathophysiology of brain damage remains poorly understood, but it is known that apoptosis is the prominent form of cell death in perihematomal regions following ICH.3–6
Mitochondria directly participate in the signaling pathways that precede apoptosis by releasing cytochrome C (CytoC) and apoptosis-inducing factor (AIF),7,8 which play a central role in neuronal survival or death.9 Previous studies have reported reduced mitochondrial respiratory function10 and CytoC release4 in the perihematomal region after ICH, suggesting that mitochondrial dysfunction is associated with brain injury. However, no studies have reported morphological mitochondrial changes after ICH.
Huperzine A (HA), a lycopodium alkaloid isolated from the Chinese herb Huperzia serrata, exerts neuroprotective effects by attenuating mitochondrial dysfunction,11,12 and has been used in neurodegenerative disorders such as Alzheimer disease13 and amyotrophic lateral sclerosis.14 More importantly, it has been shown to reduce inflammation and brain injury in a rat model of cerebral ischemia.15 Therefore, it was thought useful to investigate the protective effects of HA against ICH-induced damage. In this study, we tried to identify ultrastructural changes in neuronal mitochondria and assess the therapeutic potential of HA after ICH.
Methods
Experimental design
The experiment was separated into two consecutive parts (Figure S1). First, we assessed the effect of autologous blood on the ultrastructural morphology of neuronal mitochondria. Animals with hemorrhage were sacrificed 3, 6, 12, 24, and 48 hours following ICH; control animals were killed at 12 hours. Then, the mitochondria were collected for morphology study (Figure S1). In the second part, rats were treated with HA [0.1 mg/kg, intraperitoneal (ip), used in a rat model of ischemic stroke15] or saline immediately after the surgery, once daily for 2 days (ie, D0 and D1). Mitochondrial structure, caspase-3 (Cas-3) activation, CytoC translocation, apoptosis, and behavioral scores were compared between the groups (Figure S1). To verify the optimal HA dosage, a pilot experiment was performed by applying five gradient doses and then analyzing brain edema,5 which is the most sensitive and important readout following ICH. HA doses of 0.1 mg/kg and higher (no difference among 0.1, 0.2, and 0.4 mg/kg groups) effectively reduced edema, but not lower dosages (Figure S2) did not.
Intracerebral hemorrhage
Male Sprague Dawley rats weighing 240–280 g (Shanghai SLAC Laboratory Animal Co. Ltd.) were used according to the principles of Laboratory Animal Care (NIH publication No 86-23, revised 1985). The animal study protocols were approved by the Committee on the Use and Care of Animals, Fudan University.
A double blood injection model was employed. The rats were anesthetized with pentobarbital (50 mg/kg ip) and placed in a stereotaxic frame (Kopf Instruments, Tujunga, CA, USA). Autologous blood samples were drawn from the femoral artery.16 After a burr hole was prepared, a 26-gauge needle was inserted into the striatum (location 3.0 mm lateral to the midline, 0.2 mm posterior to bregma, and 6 mm below the skull). Next, ICH was induced by a 3-minute injection of 15 μL autologous blood, and a second 5-minute injection of 30 μL 7 minutes later. The sham-injected animals received 45 μL saline over the same duration. The needle was slowly withdrawn 10 minutes after the second injection, and the incision was carefully sutured. Another ICH model induced by collagenase IV17 was also investigated and yielded similar results (data not shown).
Magnetic resonance imaging
To ensure accurate hematoma localization, T2-weighted magnetic resonance (MR) images were obtained,18 and only rats with positive hyperintense T2 signals in the appropriate area were used for the following studies (Figure 1A). A Bruker Biospec MSL-X 7/21 spectrometer (Bruker Analytik GmbH, Karlsruhe, Germany) was used. Briefly, after the rats were anesthetized with pentobarbital (50 mg/kg ip), eleven contiguous coronal T2-weighted spin echo images were acquired using a 256×256 matrix, four averages, a field of view of 3.5×3.5 cm2, a slice thickness of 1 mm, echo times of 20, 40, and 60 milliseconds, and a repetition time of 1,500 milliseconds.
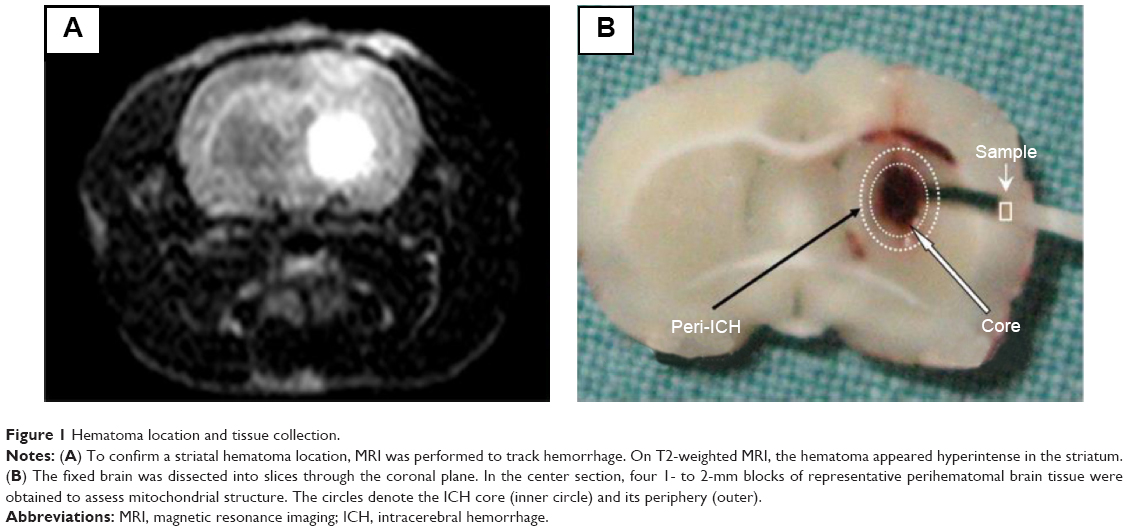 | Figure 1 Hematoma location and tissue collection. |
Electron microscopy
Electron microscopy was performed as previously described.19,20 The rat brains were perfused with normal saline solution followed by phosphate-buffered 2% glutaraldehyde and 4% paraformaldehyde. The brains were carefully removed and immersed in the same fixative before they were dissected in the coronal plane through the needle site at 0.2 mm posterior to bregma. Four 1–2 mm tissue cubes were obtained at random within the region adjacent to the hemorrhage core (Figure 1B). Each sample was then postfixed in 1% potassium ferrocyanide-reduced osmium tetroxide, dehydrated in graded acetones, and embedded in Epon 812. Thin (0.5 μm) sections were cut from the tissue blocks, stained with 0.5% toluidine blue, and then stained with lead citrate and uranyl acetate before they were photographed using a JEOL 1200EX electron microscope (JEOL, Tokyo, Japan).
Approximately 20 representative neurons per specimen, as identified by a typical nucleus and surrounding perikaryon, were randomly found and imaged for analysis. Mitochondrial injury was blindly assessing using a grading system based on the characteristic ultrastructural features of mitochondria.21
Cell death analysis
A terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) kit was used to examine the extent of cell death in the perihematomal areas.22 After an overdose of pentobarbital, the animals were perfused through the left ventricle with 100 mL 0.9% saline, followed by 100 mL 4% paraformaldehyde in 0.1 mol/L phosphate-buffered saline (PBS). Then the brains were postfixed overnight at 4°C and placed in 20% sucrose in PBS for 24 hours, followed by 30% sucrose for 24 hours. The brains were cut into 20 μm sections with a cryostat microtome (CM1800; Leica Microsystems, Wetzlar, Germany). After staining, apoptotic cells were counted on a computer screen grid from four random fields within the perihematomal region.
Western blot
Fresh brain tissue around the hematoma was removed, and protein extraction of both the cytosolic and mitochondrial fractions was performed using a multiple centrifugation method.23 Briefly, the homogenized tissue was centrifuged at 750× g for 10 minutes at 4°C and then at 10,000× g for 20 minutes. The pellets were used to obtain the mitochondrial fraction. Then the supernatant was further centrifuged at 100,000× g for 60 minutes and was used for the cytosolic analysis. After the protein concentrations were determined, equal amounts of the samples were loaded, and the proteins were separated by 10%–20% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. The primary antibodies were as follows: 1:1,000 anti-cleaved Cas-3 (9664, Cell Signaling Technology, Danvers, MA, USA); 1:1,000 anti-CytoC (556433; BD, Franklin Lakes, NJ, USA); and 1:5,000 anti-COX (A21348; Thermo Fisher Scientific, Waltham, MA, USA). After incubation with secondary antibodies, protein bands were detected and captured, and the intensities were quantified with the Quantity One version 16.0 software (Bio-Rad Laboratories Inc., Hercules, CA, USA).
Behavioral tests
Both behavioral tests were performed in a blinded fashion.24 For the forelimb placing test, the rats were held by their torsos, allowing the forelimbs to hang free, and forelimb movement was induced by brushing the vibrissae on the edge of a countertop. The intact limb (ipsilateral to ICH) would immediately be placed on the countertop in response to the stimuli, but the paralyzed forelimb (contralateral to ICH) might fail to move. The number of positive reactions was counted out of ten. Asymmetric forelimb use was recorded by videotaping rats placed in a transparent cylinder for 5 minutes. We calculated the number of times each animal used the paralyzed (C), normal (I), or both forelimbs (B) to contact the wall during a full rear. The limb-use asymmetry score was calculated as (I–C)/(I+C+B).
Statistical analysis
Data are presented as mean ± SD. Multiple comparisons of mitochondrial injury scores among different groups and the optical density (OD) values of immunoblots were obtained through one-way analyses of variance (ANOVA) followed by Bonferroni post hoc tests. Differences in apoptosis and mitochondrial scores between the vehicle and HA-treated groups were compared by independent sample t-tests. Repeated measures ANOVA followed by the Tukey–Kramer test was used to compare behavioral test results. All statistical analyses were performed using SPSS 16.0 (SPSS Inc., Chicago, IL, USA). Differences were considered statistically significant at P<0.05.
Results
All rats survived ICH and had an appropriate T2 hyperintense signal on MR imaging (Figure 1A). The center of the hematoma was located 0.2 mm posterior to the bregma, where the needle for blood injection was inserted. Physiological parameters such as arterial blood pressure, heart rate, temperature, and arterial blood gases were not significantly different between the groups (data not shown). Electron microscopy did not show any fixative artifacts.
Normal mitochondria structure
Neurons in the sham group had normal nuclei and cytoplasmic contents without signs of edema, nuclear membrane breaks, or clumped tigroid chromatin (Figure 2A). Mitochondria also appeared to have normal morphology, demonstrating no evidence of swelling, outer membrane breaks, or intracristal dilation (Figure 2B).
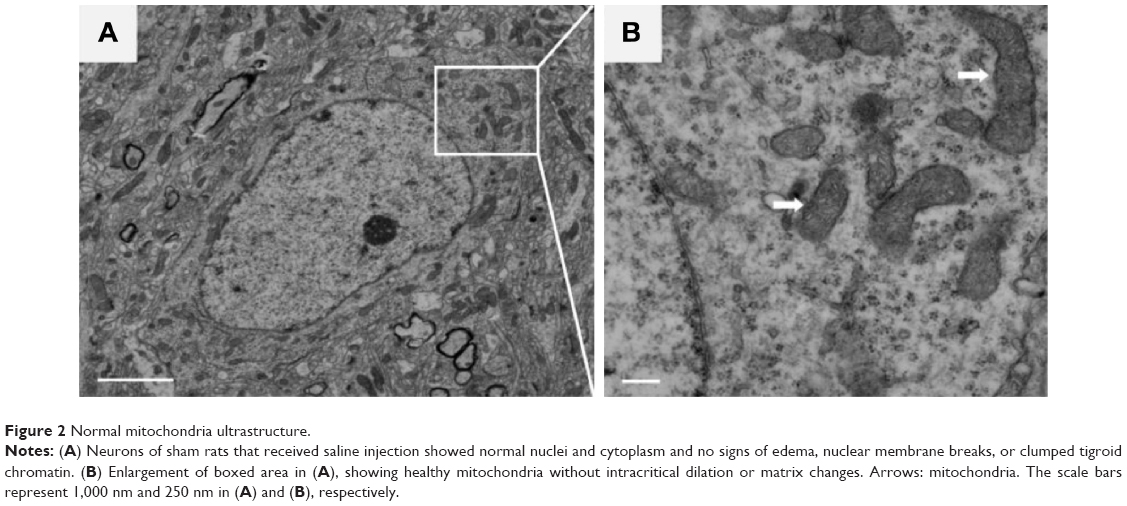 | Figure 2 Normal mitochondria ultrastructure. |
ICH led to mitochondrial changes
Mitochondrial injury was assessed using a scoring system based on known ultrastructural characteristics.21 The mitochondrial morphology changes in ICH rats were time dependent. At 3 hours after blood injection, few neuronal mitochondria around the hematoma were abnormal (Figure 3A). However, we observed significant morphological transformation at 12 hours, including mitochondrial swelling, intracristal dilation, and decreased electron density of the matrix (Figure 3B). These findings are similar to previously reported results.19–21 Most severely damaged mitochondria were prominent at 24 hours, with some exhibiting broken surrounding membranes (Figure 3C, arrow) and endoplasmic reticulum (ER) swelling (Figure 3C, stars). Mitochondrial ultrastructural damages were also dramatic at 48 hours (Figure 3D). The time-dependent characteristics of injury are summarized in Figure 3E.
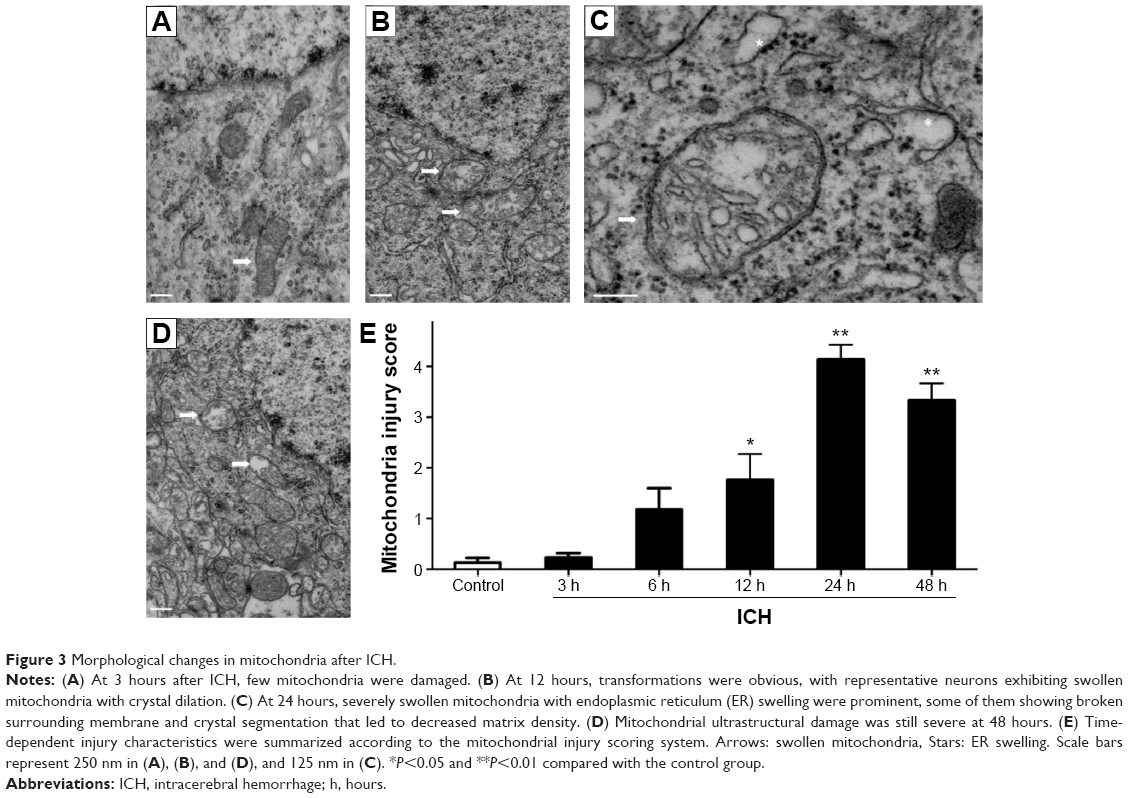 | Figure 3 Morphological changes in mitochondria after ICH. |
HA attenuated mitochondrial damage and apoptosis
Accumulating evidence suggests that HA exerts neuroprotective functions by attenuating mitochondrial injury.13 Compared to the vehicle-treated group, the HA-treated group showed significantly reduced mitochondrial damage score (Figure 4A, P<0.05).
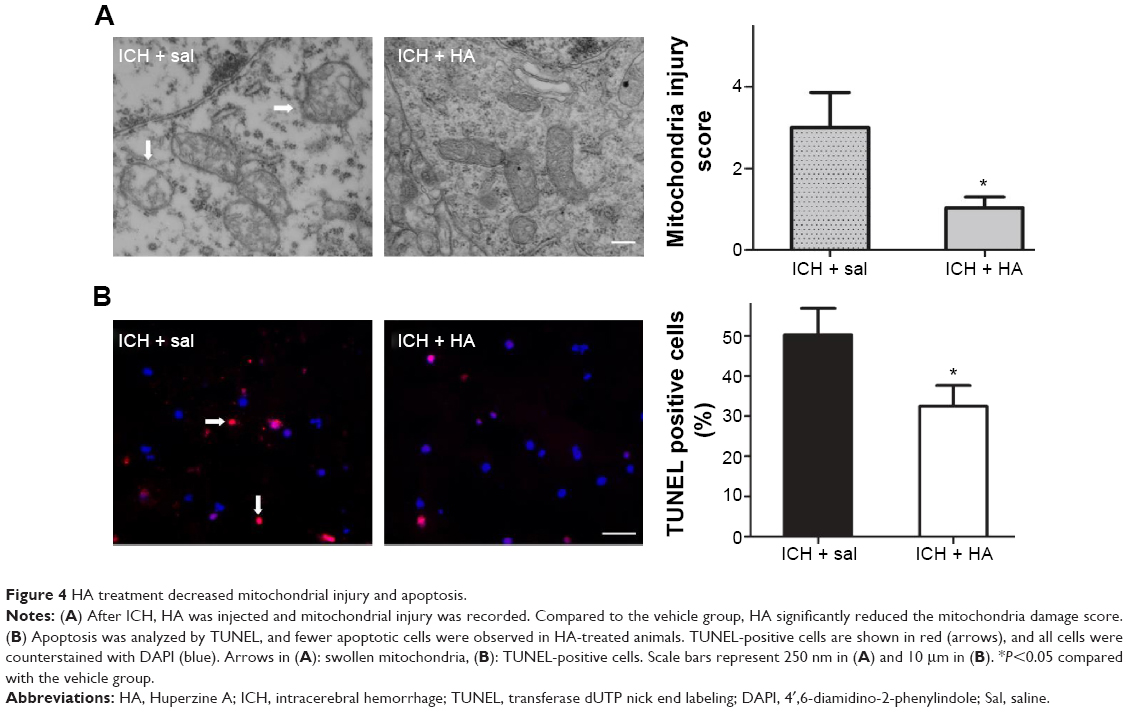 | Figure 4 HA treatment decreased mitochondrial injury and apoptosis. |
Programmed cell death is the main form of ICH-induced cell death,3–6 and TUNEL was used to detect apoptosis within the perihematomal tissue. Compared to saline injection, HA significantly decreased the number of apoptotic cells (Figure 4B, P<0.05).
HA inhibited Cas-3 activation and CytoC translocation
To further analyze the impact of HA on mitochondria-mediated apoptosis, Western blot was performed to test Cas-3 activation and CytoC. Compared to saline, HA significantly inhibited Cas-3 cleavage and CytoC cytoplasmic translocation (Figure 5A and B, P<0.05).
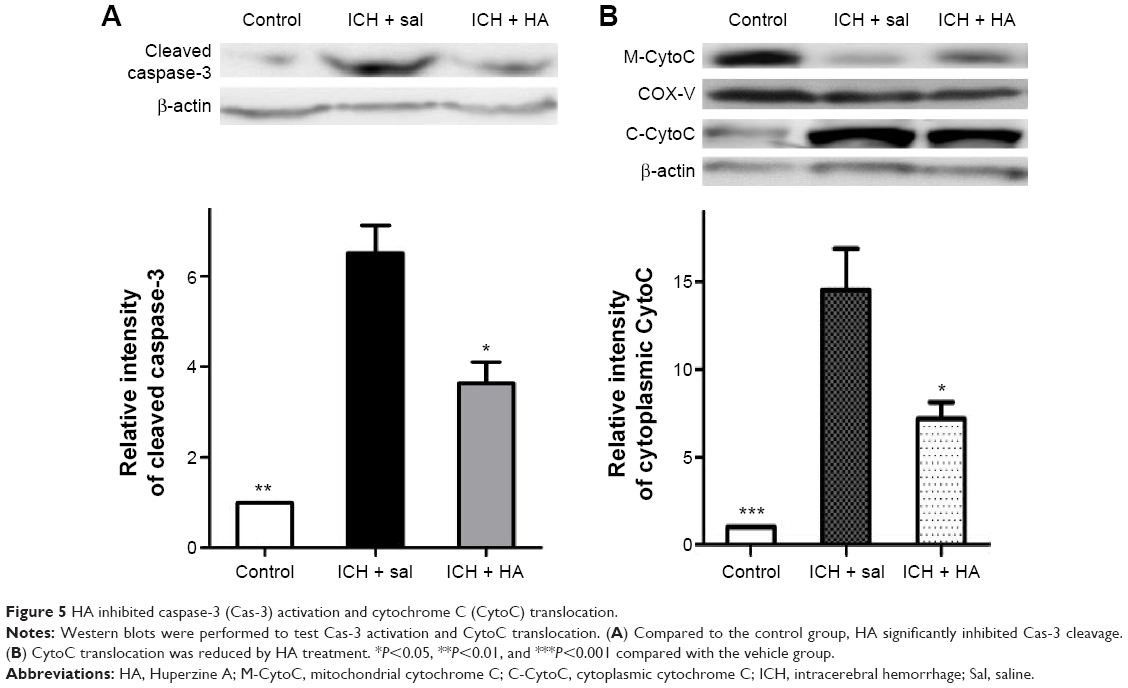 | Figure 5 HA inhibited caspase-3 (Cas-3) activation and cytochrome C (CytoC) translocation. |
HA improved locomotion recovery
All animals resumed normal weight gain after ICH surgery, and there were no differences in body weight between the groups. Two behavioral tests were performed at different time points. One day after ICH, there was no significant difference between the groups. However, after D2, rats that received HA showed significantly improved locomotor recovery compared to control (Figure 6, P<0.05).
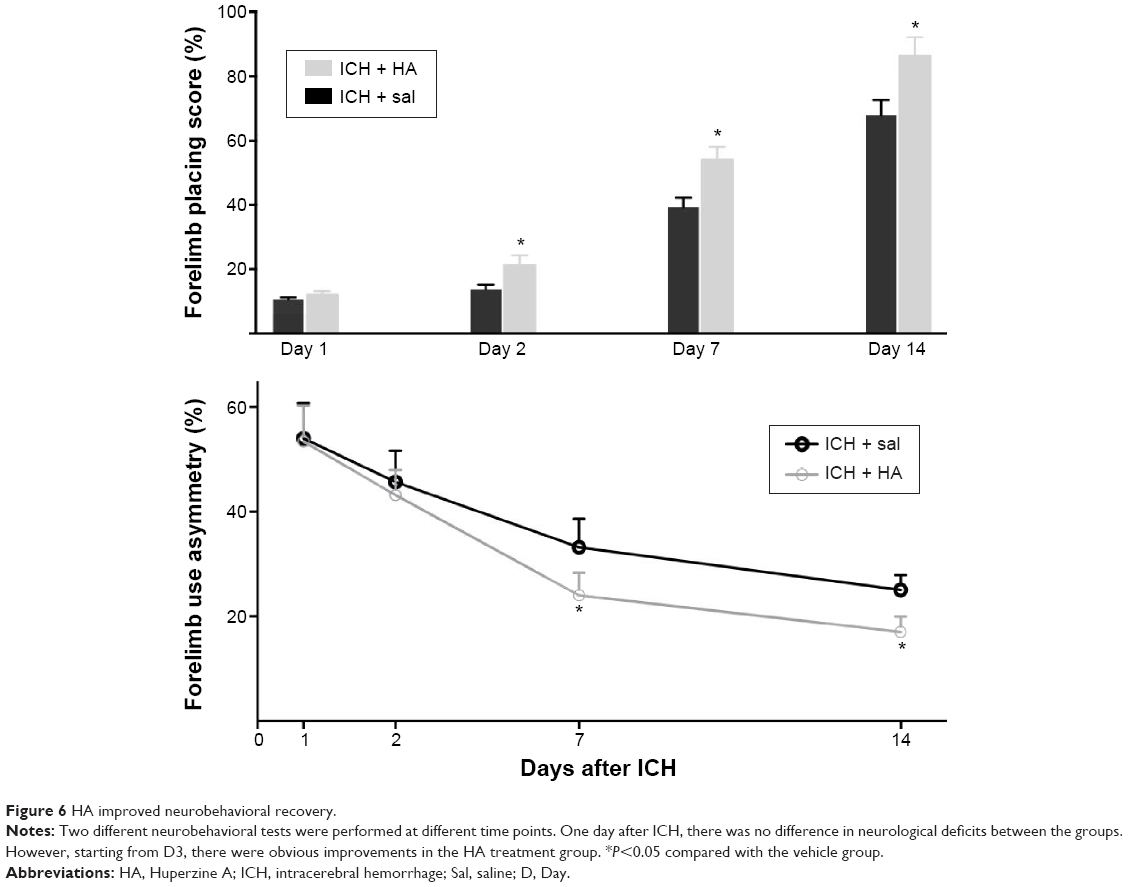 | Figure 6 HA improved neurobehavioral recovery. |
Discussion
Mitochondrial swelling, first described ~80 years ago, is one of the most universal ultrastructural changes after cerebral ischemia.25 Since then, several histopathological studies have described mitochondrial changes following ischemia.19,25,26 However, there is a paucity of modern ultrastructural investigations examining mitochondrial morphology after ICH, though reduced mitochondrial respiratory function27 and CytoC release4 have been reported in perihemorrhage areas. To the best of our knowledge, this is the first investigation designed to observe ultrastructural mitochondrial changes after ICH. Our study was designed to build upon previous findings; the present results show that ICH altered the mitochondrial structure, which could be the reason for or result of functional changes. Combining our and other findings, a fair conclusion is that mitochondrial damage might be playing a role in ICH pathogenesis.
Mitochondria initiate common forms of programmed cell death by releasing proteins from the intermembrane and intracristal spaces. These include CytoC, which induces apoptosis through activating caspases, and AIF, which is linked with caspase-independent apoptosis.7,8 Permeabilization of the outer membrane of mitochondria results in the release of caspase-activating molecules and caspase-independent death effectors and/or mitochondrial metabolic failure, which lead to mitochondrial swelling and neuronal death.7 We found that ultrastructural changes occurred very soon after ICH and would be a leading cause for apoptosis, although we did not find any direct evidence to support the hypothesis that neuronal death was triggered by mitochondrial damage.
A previous study reported that reduced State 3 respiration was evident 6 hours following hemorrhage.27 We found obvious mitochondrial ultrastructure changes taking place at a similar time point, suggesting that mitochondrial swelling is associated with cellular respiratory dysfunction and leads to apoptosis. To further prove this, we treated the ICH rat model with HA, which attenuates apoptosis by inhibiting mitochondria-dependent Cas-3 activation.12 Surprisingly, in addition to reducing apoptosis, HA treatment enhanced neurobehavioral recovery, possibly by attenuating mitochondrial injury. More importantly, HA inhibited Cas-3 cleavage and cytoplasmic translocation of CytoC, suggesting that a mitochondrial-dependent pathway of apoptosis was prevented by HA treatment after ICH. As the efficacy and safety of HA has been tested by many clinical trials for Alzheimer disease,3 it would be equally important and interesting to observe its effects of HA in ICH patients after additional preclinical studies are performed to explore the benefits, molecular mechanisms, and safety of HA in various stroke models.
Conclusion
Mitochondrial morphology damage is apparent in perihematomal areas after ICH, and mitochondrial dysfunction may contribute to secondary injury. HA is protective against neurological damage, possibly via its ability to ameliorate mitochondrial injury. These findings suggest a new direction for developing novel ICH therapies.
Acknowledgment
This work was supported by grants from the Nanjing Science and Technology Development Program (201402021 to HYL) and National Natural Science Foundation of China (81070935 to QD).
Disclosure
The authors report no conflicts of interest in this work.
References
Qureshi AI, Tuhrim S, Broderick JP, Batjer HH, Hondo H, Hanley DF. Medical progress: spontaneous intracerebral hemorrhage. N Engl J Med. 2001;344(19):1450–1460. | ||
Sutherland GR, Auer RN. Primary intracerebral hemorrhage. J Clin Neurosci. 2006;13(5):511–517. | ||
Delgado P, Cuadrado E, Rosell A, et al. Apoptosis involvement after human spontaneous intracerebral hemorrhage: fas system and caspase-3 in plasma and brain tissue. Stroke. 2007;38(2):544–544. | ||
Felberg RA, Grotta JC, Shirzadi AL, et al. Cell death in experimental intracerebral hemorrhage: the “black hole” model of hemorrhagic damage. Ann Neurol. 2002;52(3):S22–S22. | ||
Gong C, Boulis N, Qian J, Turner DE, Hoff JT, Keep RF. Intracerebral hemorrhage-induced neuronal death. Neurosurgery. 2001; 48(4):875–882. | ||
Qureshi AI, Suri MF, Ostrow PT, et al. Apoptosis as a form of cell death in intracerebral hemorrhage. Neurosurgery. 2003;52(5):1041–1047. | ||
Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305(5684):626–629. | ||
Shiozaki EN, Shi YG. Caspases, IAPs and Smac/DIABLO: mechanisms from structural biology. Trends Biochem Sci. 2004;29(9):486–494. | ||
Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiol Rev. 2000;80(1):315–360. | ||
Kim-Han JS, Kopp SJ, Dugan LL, Diringer MN. Perihematomal mitochondrial dysfunction after intracerebral hemorrhage. Stroke. 2006; 37(10):2457–2462. | ||
Xiao XQ, Zhang HY, Tang XC. Huperzine A attenuates amyloid beta-peptide fragment 25–35-induced apoptosis in rat cortical neurons via inhibiting reactive oxygen species formation and caspase-3 activation. J Neurosci Res. 2002;67(1):30–36. | ||
Zhou J, Tang XC. Huperzine A attenuates apoptosis and mitochondria-dependent caspase-3 in rat cortical neurons. FEBS Lett. 2002; 526(1–3):21–25. | ||
Zhang HY, Tang XC. Neuroprotective effects of huperzine A: new therapeutic targets for neurodegenerative disease. Trends Pharmacol Sci. 2006;27(12):619–625. | ||
Hemendinger RA, Armstrong EJ 3rd, Persinski R, et al. Huperzine A provides neuroprotection against several cell death inducers using in vitro model systems of motor neuron cell death. Neurotox Res. 2008;13(1):49–61. | ||
Wang ZF, Wang J, Zhang HY, Tang XC. Huperzine A exhibits anti-inflammatory and neuroprotective effects in a rat model of transient focal cerebral ischemia. J Neurochem. 2008;106(4):1594–1603. | ||
DelBigio MR, Yan HJ, Buist R, Peeling J. Experimental intracerebral hemorrhage in rats – magnetic resonance imaging and histopathological correlates. Stroke. 1996;27(12):2312–2319. | ||
Mayne M, Ni W, Yan HJ, et al. Antisense oligodeoxynucleotide inhibition of tumor necrosis factor-alpha expression is neuroprotective after intracerebral hemorrhage. Stroke. 2001;32(1):240–247. | ||
Power C, Henry S, Del Bigio MR, et al. Intracerebral hemorrhage induces macrophage activation and matrix metalloproteinages. Ann Neurol. 2003;53(6):731–742. | ||
Solenski NJ, diPierro CG, Trimmer PA, Kwan AL, Helms GA. Ultrastructural changes of neuronal mitochondria after transient and permanent cerebral ischemia. Stroke. 2002;33(3):816–824. | ||
Sun MG, Williams J, Munoz-Pinedo C, et al. Correlated three-dimensional light and electron microscopy reveals transformation of mitochondria during apoptosis. Nat Cell Biol. 2007;9(9):1057–U1021. | ||
Crouser ED, Julian MW, Blaho DV, Pfeiffer DR. Endotoxin-induced mitochondrial damage correlates with impaired respiratory activity. Crit Care Med. 2002;30(2):276–284. | ||
Rodrigues CM, Sola S, Nan Z, et al. Tauroursodeoxycholic acid reduces apoptosis and protects against neurological injury after acute hemorrhagic stroke in rats. Proc Natl Acad Sci U S A. 2003;100(10):6087–6092. | ||
Endo H, Kamada H, Nito C, Nishi T, Chan PH. Mitochondrial translocation of p53 mediates release of cytochrome c and hippocampal CA1 neuronal death after transient global cerebral ischemia in rats. J Neurosci. 2006;26(30):7974–7983. | ||
Hua Y, Schallert T, Keep RF, Wu J, Hoff JT, Xi G. Behavioral tests after intracerebral hemorrhage in the rat. Stroke. 2002;33(10):2478–2484. | ||
Solenski NJ, diPierro CG, Trimmer PA, Helm GA. Ultrastructural changes of cortical neuronal mitochondria following transient cerebral ischemia: evidence of reperfusion injury. Ann Neurol. 1998;44(3):493–493. | ||
Li J, Ma X, Yu W, et al. Reperfusion promotes mitochondrial dysfunction following focal cerebral ischemia in rats. PLoS One. 2012; 7(9):e46498. | ||
Kim-Han JS, Kopp SJ, Dugan LL, Diringer MN. Perihematomal mitochondrial dysfunction after intracerebral hemorrhage. Stroke. 2006;37(12):3057–3057. |
Supplementary materials
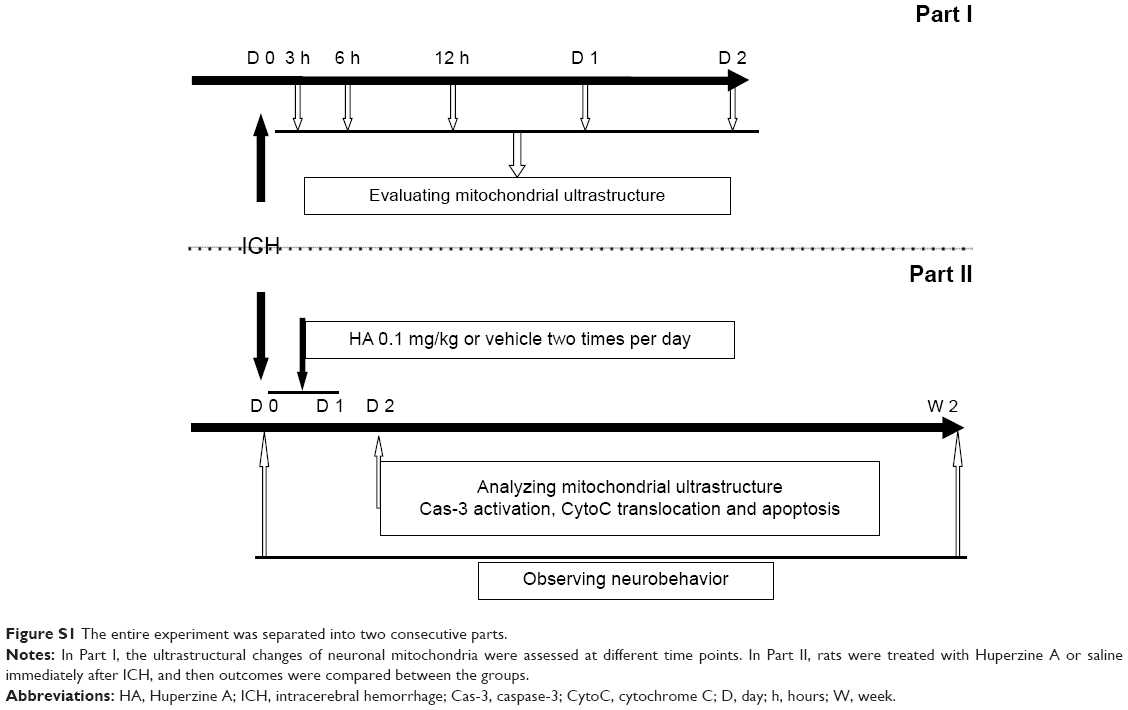 | Figure S1 The entire experiment was separated into two consecutive parts. |
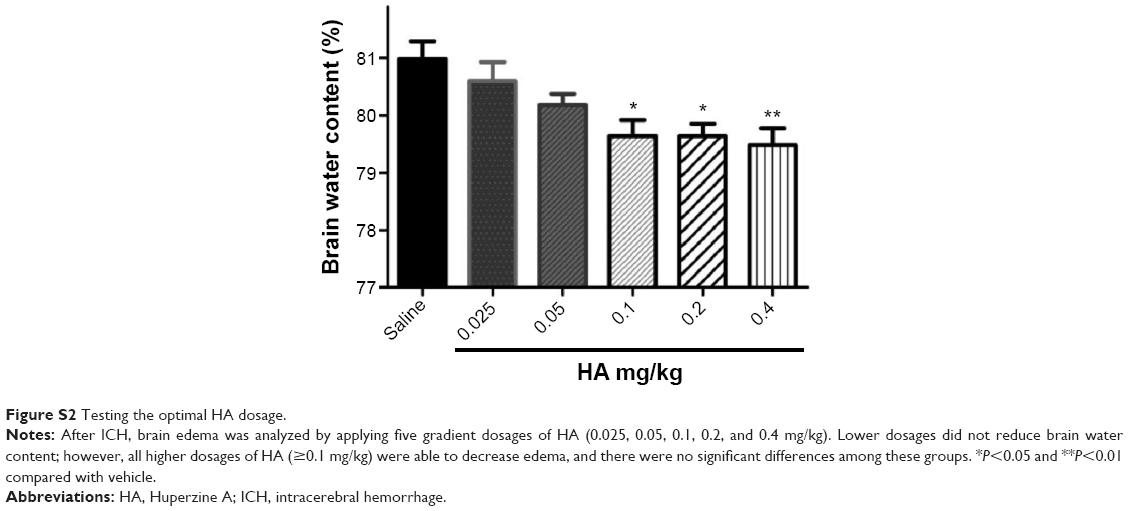 | Figure S2 Testing the optimal HA dosage. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.