Back to Journals » Journal of Inflammation Research » Volume 15
UCHL1 Promoted Polarization of M1 Macrophages by Regulating the PI3K/AKT Signaling Pathway
Authors Huang Y, He S, Chen Y, Sheng J, Fu Y, Du X, Yang Y, Liu H, Han Z ![]() , Huang Y, Wen Q, Zhou C, Zhou X, Hu S
, Huang Y, Wen Q, Zhou C, Zhou X, Hu S ![]() , Ma L
, Ma L ![]()
Received 16 October 2021
Accepted for publication 26 January 2022
Published 4 February 2022 Volume 2022:15 Pages 735—746
DOI https://doi.org/10.2147/JIR.S343487
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Ning Quan
Yulan Huang,* Shitong He,* Yitian Chen, Junli Sheng, Yuling Fu, Xialin Du, Yalong Yang, Honglin Liu, Zhenyu Han, Yingqi Huang, Qian Wen, Chaoying Zhou, Xinying Zhou, Shengfeng Hu, Li Ma
Institute of Molecular Immunology, School of Laboratory Medicine and Biotechnology, Southern Medical University, Guangzhou, 510515, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Li Ma; Shengfeng Hu
Institute of Molecular Immunology, School of Laboratory Medicine and Biotechnology, Southern Medical University, Guangzhou, 510515, People’s Republic of China
, Email [email protected]; [email protected]
Background: As deubiquitinases (DUBs), ubiquitin C-terminal hydrolase (UCH)-L1 has been shown to play a crucial role in regulating diverse biological processes. However, its function in macrophage polarization remains unclear.
Methods: We performed in vivo and in vitro experiments to investigate the role of ubiquitin carboxyl-terminal hydrolase L1 (UCHL1), a kind of DUBs, in macrophage differentiation by using UCHL1-deficiency mice.
Results: We demonstrated that LPS stimulation induced UCHL1 expression in macrophages. The deficiency of UCHL1 expression decreased the expression of CD80 and CD86 but increased the expression of CD206. The expression of TNF-α, IL-6, iNOS, and IL-10 was downregulated, while that of Arg1, Ym1, and Fizz1 was upregulated in UCHL1 deficient macrophages. Moreover, we observed that UCHL1 promoted the degradation of p110α through autophagy, but paradoxically increased the activity of AKT, thereby promoting polarization of macrophages into pro-inflammatory states.
Conclusion: In this study, we identified UCHL1 as a positive regulator of M1 macrophage polarization. Our findings may help in developing therapeutic interventions for the treatment of inflammatory diseases and pathogenic infections.
Keywords: UCHL1, macrophages, AKT, p110α, autophagy
Background
Macrophages play a crucial role in the innate immune response, which is the first line of host defense against infection, and are also involved in the regulation of homeostasis, inflammation, and antitumor immunity.1–3 Activated macrophages are known to be classically activated or M1-type macrophages that express proinflammatory factors such as IL-6, TNF-α, IL-1β, and iNOS.4 However, macrophages can be alternatively activated (also known as M2-type macrophages), and upregulate the levels of arginase 1 (Arg1), inflammatory zone 1 (Fizz1), mannose receptor 1 (Mrc1), and chitinase 3-like-3 (YM1/CHI313).5 Several signaling pathways are involved in the balance between M1- and M2-like macrophage polarizations. Among different signaling cascades, PI3K/AKT pathway and its downstream targets play a crucial role in macrophage polarization.
AKT was discovered 25 years ago, and PI3K was identified as its upstream regulator.6 The effects of AKT on various cell types have also been widely recognized. The PI3K/AKT pathway regulates macrophage proliferation, survival, migration as well as metabolic and inflammatory signals.7 Among the PI3K/AKT signaling pathway, AKT phosphorylation has emerged as a central regulator, which is affected by the activity of PI(3) kinase, kinase PDK1, and the mammalian target of rapamycin (mTORC2).8 Phosphatases, including PHLPP1,9 PP2A,10 and CTMP,11 are known to antagonize these activities. Class IA PI3K consists of a catalytic subunit (p110) and a regulatory subunit (p85), and these form an obligatory heterodimer.12 As a major isoform of the catalytic subunit, PIK3CA (p110α) is detected in various tissues;13 mutations in this subunit are frequently seen in cancers of the colon, endometrium, breast, and prostate, as well as in the glioblastomas.14–16 Meanwhile, the degradation and resynthesis of p110α was found to depend on the proteasome.17
Post-translational modifications, particularly ubiquitination, play a crucial role in regulating diverse biological processes, including immune responses.18,19 Ubiquitination usually targets proteins in the proteasome for degradation; nonetheless, it also has several nondegradative functions.20,21 Ubiquitin chains are conjugated by three enzymes, namely, E1 ubiquitin-activating enzyme, E2 ubiquitin-conjugating enzyme, and E3 ubiquitin ligase,22 and are cleaved by deubiquitinases (DUBs).23 DUBs differentially regulate protein ubiquitination by showing specificity for protein substrates as well as ubiquitin chains; they play critical roles in both innate and adaptive immune responses,23 and are involved in several biological processes, as well as represent a class of drug targets.24,25 However, only few biological functions of DUBs, especially those in the regulation of macrophage polarization, have been investigated to date.
Ubiquitin carboxyl-terminal hydrolase L1 (UCHL1), also known as PGP9.5,26 is abundantly expressed in neurons, and functions as a deubiquitinating enzyme or ubiquitin ligase.27–29 Previous studies have shown that UCHL1 is related to human neurodegenerative disorders such as Parkinson’s disease (PD),30 Alzheimer’s disease (AD)31 and traumatic brain injury (TBI).32 However, the activity of UCHL1 and its underlying mechanisms in macrophage polarization remain unclear.
In this study, we observed that UCHL1 promoted the degradation of p110α through autophagy, but paradoxically increased the activity of AKT, thereby promoting the polarization of macrophages into pro-inflammatory states.
Materials and Methods
Mice
C57BL/6 mice (Wild type, WT) were provided by the Lab Animal Center of Southern Medicine University (Guangzhou, China). UCHL1-deficiency (Uchl1−/−) were from The Jackson Laboratory (USA). All of the mice were used at an age of 6–12 weeks and were randomly divided into different groups. All mice were housed in a specific pathogen-free animal facility at Southern Medical University. All experimental procedures in this study were approved by the Ethics Committee and Animal Experiment Committee of Southern Medical University (SMU-L2017022), which complies with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH publication No. 85–23, revised 1985).
Cell Culture and Reagents
Bone marrow cells were prepared from the femurs and tibias of mice and cultured in DMEM (Corning, USA) supplemented with 10% FCS (Corning, USA) and 40ng/mL murine GM-CSF (Pepro Tech, USA). The cells were collected after 7 days to be used as GM-CSF-induced bone marrow-derived macrophages (BMDMs). Female C57BL/6 mice (6–12-week old) were injected with 2 mL 3% sodium thioglycolate (Sigma, USA) solution intraperitoneally. Four days later, cells in the abdominal cavity were collected by lavaging with 15 mL DMEM without FBS and centrifugation. Cells were seeded and maintained in DMEM with 10% FBS. The adhered peritoneal macrophages were used for further experiments 24 h later. HEK293T and THP-1 cells were maintained in DMEM or RPMI-1640 (Corning, USA) medium containing 10% FCS. Cycloheximide (CHX), MG132 and bafilomycin A1 were purchased from Sigma.
Macrophage Polarization in vitro
GM-CSF-differentiated BMDMs were harvested and plated in tissue culture dishes for subsequent experiments. For M1-like activation, 5–7 × 105 BMDMs were plated in 12-well tissue culture dishes and treated with 100 ng/mL LPS (Invivogen, USA) plus 10 ng/mL IFN-γ (Peprotech, USA). For M2 polarization, cells were treated with 10 ng/mL IL-4 (Peprotech).
THP-1 cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS; Corning, USA) at 37°C atmosphere in a humidified incubator with 5% CO2 and 95% air. To stimulate differentiation, cells (5 × 105 cells/mL) were cultured with 100nM phorbol 12-myristate 13-acetate (PMA, Sigma-Aldrich, USA) for 24 h. Non-attached cells were removed by aspiration, and the adherent cells were washed with RPMI 1640 for three times. For M1-like activation, the adherent cells were treated with 100 ng/mL LPS (Invivogen, USA) plus 10 ng/mL IFN-γ (Peprotech, USA). For M2 polarization, cells were treated with 10 ng/mL IL-4 (Peprotech).
Lentivirus-Mediated UCHL1 Overexpression
HEK293 cells were transfected with phage-human or mouse UCHL1 or the empty vector along with the packaging vectors pSPAX2 and pMD2G. The medium was changed to a fresh full medium (10% FBS, 1% streptomycin-penicillin and 10 μM β-mercaptoethanol) after 8 h. Forty hours later, supernatants were GM-CSF-induced BMDMs or THP-1 cells followed by various analyses.
Enzyme-Linked Immunosorbent Assay (ELISA)
The concentration of TNF-α, IL-6, and IL-10 in the supernatants or serum was measured by ELISA kits (ExCell Bio, China) according to the manufacturer’s protocol.
Quantitative PCR (qPCR) Analysis
For quantitative PCR, total RNA was isolated with Trizol (Thermo Fisher Scientific) according to the instructions of the manufacturer. RNA (1 mg) was reverse transcribed to cDNA using the high-capacity cDNA reverse transcription kit (Applied Biosystems, USA). An Eppendorf Master Cycle Realplex 2 and the SYBR Green PCR Master Mix (Applied Biosystems) were used for quantitative real-time RT-PCR analysis. The sequences of the primers were in Supplementary Table 1.
Flow Cytometry Analysis
Cells were collected and stained with fluorescently labeled antibody for 30 minutes. The cells were then washed with PBS on ice, and the cell phenotype was analyzed by flow cytometry on a flow cytometer (BD LSR II) (BD Biosciences, USA) or Attune NxT (Thermo Fisher Scientific). Data were acquired as a fraction of labeled cells within a live-cell gate and analyzed using FlowJo software (Tree Star). All gates were set on the basis of isotype-matched control antibodies.
Plasmids Transient Transfection in HEK293 Cells
Plasmids and siRNA were transiently transfected to HEK293 cells using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufactures’ recommendation, then incubated at 37°C for 48 h. The following plasmids used in this article were manufactured by You Bio Biotechnology Co., Ltd (China): Flag-tagged p110α, and Myc-tagged UCHL1, UCHL1-mut (D30K and C90S), and then were cloned into pcDNA 3.1(+).
Immunoprecipitation and Immunoblot Analysis
For immunoprecipitation, cells were transfected with the indicated plasmids or were stimulated with LPS (100ng/mL) for indicated times. Whole-cell extracts were collected and incubated overnight with the appropriate antibodies, followed by incubation two hours with the protein G Plus-Agarose Immunoprecipitation reagent (GE Healthcare). Beads were washed five times with a lysis buffer (1% (vol/vol) Nonidet P-40, 1.5 M Tris-HCl (pH 8.8), 0.5 M EDTA, 5 M NaCl, protease inhibitors (Roche)), and immunoprecipitates were eluted with 2 × SDS Loading Buffer and resolved by SDS-PAGE. For deubiquitination assays, the cells were lysed with a lysis buffer, and the supernatants were denatured at 95°C for 5 min in the presence of 1% SDS. The denatured lysates were diluted with a lysis buffer to reduce the concentration of SDS below 0.1% followed by immunoprecipitation (denature-IP) with the indicated antibodies. Proteins were transferred to PVDF membranes (Merck Millipore) and further incubated with the appropriate antibodies. Immobilon Western Chemiluminescent HRP Substrate (protein sample) was used for protein detection. The relevant information about antibodies was in Supplementary Table 2.
Statistics
All experiments were performed at least thrice. When shown, multiple samples representing biological (not technical) replicates of mice were randomly sorted into each experimental group. No blinding was performed during animal experiments. Determination of statistical differences was performed with Prism 8 (GraphPad Software, Inc.) using unpaired two-tailed t-tests (to compare two groups with similar variances), or two-way ANOVA with Bonferonni’s multiple comparison test (to compare more than two groups).
Results
LPS Promoted UCHL1 Expression in Macrophages
To investigate whether UCHL1 plays a role in macrophage polarization, we initially detected UCHL1 expression in different cells stimulated with lipopolysaccharide (LPS). RNA sequencing analysis of bone marrow-derived macrophages (BMDMs) revealed that the difference in UCHL1 mRNA expression level was most significant in the UCH family when compared with the control group (Figure 1A). To confirm these results, we used qPCR analysis and Western blotting to assess UCHL1 expression in BMDMs. We found that LPS stimulation induced UCHL1 expression in BMDMs. The expression levels of UCHL1 mRNA and protein were higher in BMDMs stimulated with LPS than those in the unstimulated BMDMs (Figure 1B). These results indicate that LPS stimulation induces UCHL1 expression in macrophages.
 |
Figure 1 LPS promoted UCHL1 expression in Macrophage. (A) Heat map showing changes in expression of UCH family from RNA sequencing analyses of WT BMDMs unstimulated or stimulated with LPS (100ng/mL) for 6h. (B) UCHL1 mRNA and protein expression were assessed using qPCR analysis and Western blot in WT BMDMs stimulated with LPS (100ng/mL) for indicated time. Data shown are the mean ± SD. Data are summary of three independent experiments. |
UCHL1 Deficiency Inhibited Polarization of M1 Macrophages in vivo
The ability of UCHL1 to polarize macrophages and the underlying mechanisms remain unclear. To investigate the role of UCHL1 in macrophage polarization, we used UCHL1-deficiency (Uchl1−/−) mice. After stimulation with LPS for 48 h, the peritoneal macrophages from Uchl1−/− mice showed markedly decreased CD80 and CD86 expression, but increased CD206 levels relative to peritoneal macrophages from wild-type (WT) mice (Figure 2A). The qPCR analysis showed that the peritoneal macrophages from Uchl1−/− mice expressed reduced levels of TNF-α, IL-6, and iNOS compared to those from WT mice. However, the expression of Arg1, Ym1, and Fizz1 in peritoneal macrophages from Uchl1−/−mice was higher than that in those from WT mice (Figure 2B). The expression of TNF-α, IL-6, and NO in the serum was measured by ELISA or the Griess test. Uchl1−/− mice showed a marked decrease in the levels of TNF-α, IL-6, and NO (Figure 2C). The excessive differentiation of M1 macrophage is one of main factors leading to excessive inflammation that can cause pathological damage and death. We found that the survival rate of the Uchl1−/− mice was markedly higher than that of the WT mice after stimulation with LPS for the indicated time points (Figure 2D). Together, the above results showed that UCHL1 is a positive regulator of M1 macrophage polarization.
 |
Figure 2 UCHL1 deficiency in macrophage inhibited M1 polarization in vivo. (A–C) WT and Uchl1−/− mice were stimulated with LPS (2ug/kg) for indicated times. (A) Representative expression of CD80, CD86 and CD206 from Peritoneal macrophages after 48 hours. (B) TNF-a, IL6, iNOS, Arg1, Ym1 and Fizz1 mRNA expression from Peritoneal macrophages after 48 hours stimulation were assessed using qPCR analysis (C) TNF-α, IL-6 and IL-10 secretion in the serums was measured by ELISA after 48 hours. (D) Survival curve of WT and Uchl1−/− mice (6 weeks) stimulated with LPS (10ug/kg) for indicated times. Data shown are the mean ± SD. * P < 0.05, ** P < 0.01, *** P < 0.001. Values in (A–C) were compared using Student’s t-test. Data are summary of three independent experiments. |
UCHL1 Promoted M1 Macrophage Polarization in vitro
To determine whether UCHL1 directly affected macrophage polarization, BMDMs from WT and Uchl1−/− mice were stimulated with LPS plus IFN-γ or IL-4 to induce M1 or M2 macrophages. After stimulation for 48 h, BMDMs from Uchl1−/− mice displayed reduced CD80 and CD86 levels, but enhanced CD206 levels relative to WT BMDMs (Figure 3A). The qPCR analysis revealed that the Uchl1−/− BMDMs expressed decreased TNF-α, IL-6, iNOS, and IL-10 levels compared to WT BMDMs (Figure 3B). However, the expression of Arg1, Ym1, Fizz1, and Mrc1 in Uchl1−/− BMDMs was higher than that in WT BMDMs after stimulation with IL-4 (Supplementary Figure 1). Moreover, the expression of TNF-α, IL-6, NO, and IL-10 in the cell culture supernatant was measured. We found that Uchl1−/− BMDMs stimulated with LPS plus IFN-γ produced reduced amounts of TNF-α, IL-6, NO, and IL-10 (Figure 3C).
 |
Figure 3 UCHL1 deficiency in macrophage inhibited M1 polarization in vitro. BMDMs from WT and Uchl1−/− mice were stimulated with LPS (100ng/mL) plus IFN-γ (10ng/mL) for indicated times. (A) Representative expression of CD80, CD86 and CD206 from BMDMs after 48 hours. (B) TNF-a, IL-6, iNOS and IL-10 mRNA expression from BMDMs were assessed using qPCR analysis for indicated times. (C) TNF-α, IL-6, and IL-10 secretion in the supernatant were measured by ELISA after 24 hours, and concentrations of nitrate were measured by nitrate reductase assay. Data shown are the mean ± SD. * P < 0.05, ** P < 0.01, *** P < 0.001. Values in (A–C) were compared using Student’s t-test. Data are summary of three independent experiments. |
Next, we examined the role of UCHL1 in macrophage polarization by overexpressing mouse UCHL1 in BMDMs (Figure 4A and B). After stimulation with LPS plus IFN-γ, the expression of TNF-α, IL-6, and iNOS was increased in the BMDMs with UCHL1 overexpression compared to the control group (Figure 4C). However, the levels of Arg1, Ym1, Fizz1, and Mrc1 were markedly reduced after the stimulation with IL-4 (Figure 4D). Likewise, we examined the role of UCHL1 in macrophage polarization by overexpressing human UCHL1 (hUCHL1) in THP-1 induced-macrophages (Supplementary Figure 2A and B). We found that hUCHL1 overexpression markedly enhanced the expression of TNF-α, IL-6, and NO in THP-1 induced-macrophages stimulated with LPS plus IFN-γ (Supplementary Figure 2C and D). However, the levels of Arg1, Fizz1, and Mrc1 were markedly reduced after the stimulation with IL-4 (Supplementary Figure 2E). Collectively, these results suggest that UCHL1 promotes the polarization of M1 macrophages in vitro.
 |
Figure 4 Overexpression of mouse UCHL1 in macrophages promote M1 polarization in vitro. (A–D) BMDMs were infected with mouse UCHL1 lentivirus for 24 hour. (A) UCHL1 mRNA expression were assessed using qPCR. (B) UCHL1 protein expression were assessed using Western blot, and densitometry quantification of band intensity are presented in the right panel. (C) BMDMs were stimulated with LPS (100ng/mL) plus IFN-γ (10ng/mL) for indicated times. TNF-α, IL-6, iNOS, and IL-10 mRNA expression from BMDMs were assessed using qPCR analysis. (D) After stimulated with IL-4 (10 ng/mL) for 12 hours, Arg1, Ym1, Fizz1 and Mrc1 mRNA expression from BMDMs were assessed using qPCR analysis. Data shown are the mean ± SD. * P < 0.05, ** P < 0.01, *** P < 0.001. Values in (A–D) were compared using Student’s t-test. Data are summary of three independent experiments. |
UCHL1 Regulated Macrophage Polarization by Regulating the AKT Signaling Pathway
Next, we explored the UCHL1-controlled specific molecular pathway that regulates macrophage polarization. First, we investigated whether UCHL1 regulates JAK/STAT signaling. Results showed that upon stimulation with LPS plus IFN-γ or IL-4, WT and Uchl1−/− BMDMs displayed similar phosphorylation levels of STAT1 (Supplementary Figure 3A) and STAT6 (Supplementary Figure 3B). Similarly, UCHL1 deficiency did not affect p38 and EKR signaling even when stimulated with LPS plus IFN-γ (Figure 5A). However, we observed a marked reduction in AKT phosphorylation in Uchl1−/− BMDMs stimulated with LPS plus IFN-γ (Figure 5A); similar results were observed following IL-4 stimulation (Supplementary Figure 3C). We further evaluated the downstream signaling of AKT and found that phosphorylation levels of p70s6k, 4ebp1, and mTOR were reduced (Figure 5B) in Uchl1−/− BMDMs. Similarly, UCHL1 deficiency led to reduced phosphorylation levels of p70s6k, 4ebp1, and mTOR under IL-4 stimulation (Supplementary Figure 3D). Together, these results showed that UCHL1 might regulate macrophage polarization through the AKT signaling pathway.
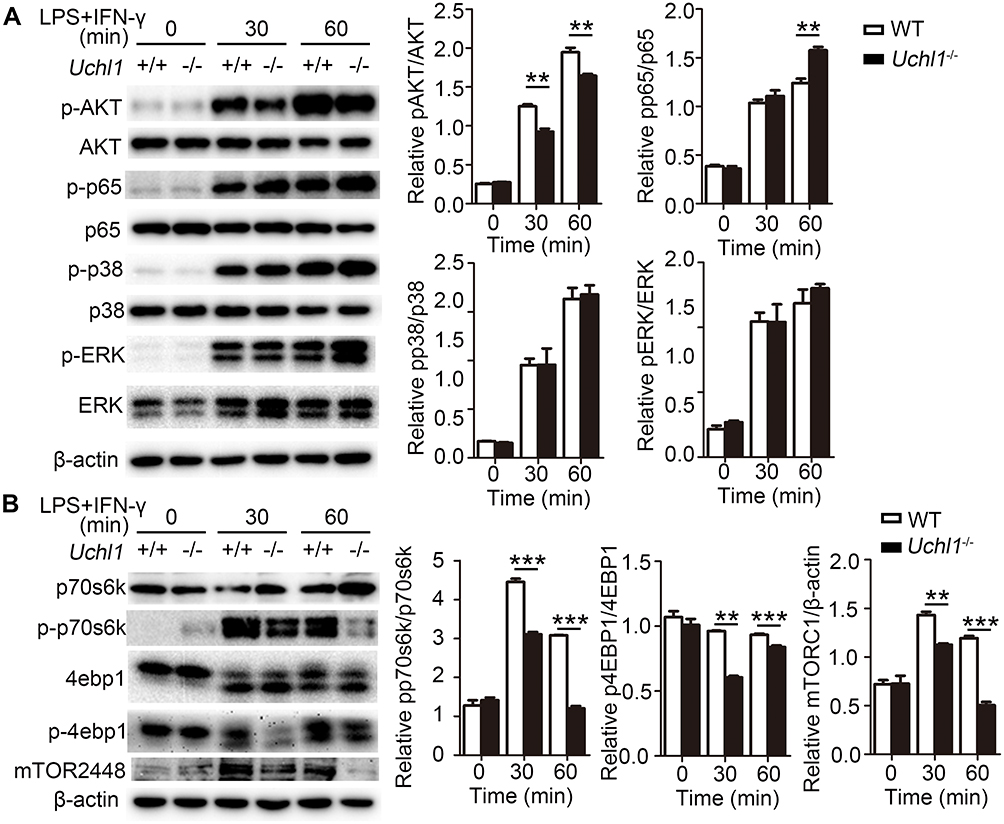 |
Figure 5 UCHL1 regulated AKT signaling pathway in BMDMs. WT and Uchl1−/− BMDMs were stimulated with LPS (100ng/mL) plus IFN-γ (10ng/mL) for indicated times. (A) Western blot analysis of the phosphorylation and total status of AKT, p65, p38, ERK. β-actin is as an internal control. And densitometry quantification of band intensity are presented in the right panel. (B) Western blot analysis of the phosphorylation and total status of p70s6k, 4ebp1 and mTOR. β-actin is as an internal control. And densitometry quantification of band intensity are presented in the right panel. Data shown are the mean ± SD. ** P < 0.01, *** P < 0.001. Values in (A and B) were compared using two-way ANOVA with Bonferroni’s test. Data are representative of three independent experiments with similar results. |
UCHL1 Promoted the Degradation of p110α Through Autophagy
To further investigate how UCHL1 regulates the AKT signaling pathway, WT and Uchl1−/− BMDMs were stimulated with LPS plus IFN-γ. The levels of AKT phosphorylation regulators, including p85, p110, PDK1, and PTEN, were analyzed; the results showed that only p110α was upregulated in the Uchl1−/− BMDMs (Figure 6A). Meanwhile, WT and Uchl1−/− BMDMs stimulated with IL-4 also displayed the same results, ie, only p110α was found to be upregulated in these Uchl1−/− BMDMs (Supplementary Figure 4A). To determine whether UCHL1 affected p110α expression, we incubated BMDMs with cycloheximide (CHX) before the stimulation. CHX blocks protein synthesis, making it easy to see how quickly the protein degrades. We found that the expression of p110α in WT BMDMs decreased over time, whereas UCHL1 deficiency suppressed this reduction (Figure 6B). Previous studies have shown that p110α deficiency promoted the differentiation of M1 macrophages but inhibited that of M2 macrophages. Consistent with these results, we found that HS-173, a p110α inhibitor, promoted the expression of TNF-α, IL-6, INOS, and IL-10 (Supplementary Figure 4B) in BMDMs after the stimulation with LPS plus IFN-γ; however, it inhibited the expression of Fizz1, YM-1, and Arg-1 in BMDMs after the stimulation with IL-4 (Supplementary Figure 4C). These results showed that UCHL1 might regulate macrophage polarization by promoting the degradation of p110α.
 |
Figure 6 UCHL1 promoted the degradation of p110α through autophagy-dependent degradation pathways. (A) Western blot analysis of the phosphorylation or total status of p110, p85, PDK1 and PTEN in WT or Uchl1−/− BMDMs after stimulated with LPS (100ng/mL) plus IFN-γ (10ng/mL) for indicated time. β-actin is as an internal control. And densitometry quantification of band intensity are presented in the right panel. (B) Western blot analysis of p110 and β-actin in WT and Uchl1−/− BMDMs stimulated with LPS (100ng/mL) plus IFN-γ (10ng/mL) for indicated time in the presence of cycloheximide (CHX, 100 μg/mL). And densitometry quantification of band intensity are presented in the below panel. (C) Extracts of BMDMs unstimulated or stimulated with LPS (100 ng/mL) for 6 hours were subjected to immunoprecipitation with anti-IgG or anti-p110α beads and immunoblot analysis with anti-p110α and anti-UCHL1. Input, immunoblot analysis of whole-cell lysates without immunoprecipitation (throughout). (D) 293T cells were transfected with vectors for Myc-UCHL1 and Flag-p110α, then immunoprecipitation (IP) with anti-Flag beads and immunoblot analysis with anti-Flag and anti-Myc. Input, immunoblot analysis of whole-cell lysates without immunoprecipitation (throughout). (E) Extracts of WT and KO BMDMs treated with Mg132 after stimulated with LPS (100 ng/mL) for 6 hours were subjected to immunoprecipitation with anti-p110α beads and immunoblot analysis with anti-p110α and anti-Ubiquitin (Ub). Input, immunoblot analysis of whole-cell lysates without immunoprecipitation (throughout). (F) Western blot analysis of p110 and β-actin in WT and KO BMDMs stimulated with LPS (100ng/mL) plus IFN-γ (10ng/mL) for indicated time in the presence of DMSO or MG132 (20 μg/mL) or bafilomycin-A1 (BafA1, 50 nM). And densitometry quantification of band intensity are presented in the below panel. Data shown are the mean ± SD. ** P < 0.01, *** P < 0.001. Values in (A and B) were compared using two-way ANOVA with Bonferroni’s test, and using Student's t-test in (F). Data are representative of three independent experiments with similar results. |
Next, we determined how UCHL1 regulates the stability of p110α protein. Immunoprecipitation and Western blot analyses revealed that UCHL1 did not interact with p110α in BMDMs (Figure 6C). Consistent with this result, UCHL1 did not interact with p110α in 293T cells transfected with Myc-UCHL1 and Flag-p110α (Figure 6D). Furthermore, we analyzed the ubiquitination of p110α and found that UCHL1 depletion remarkably decreased the poly-ubiquitination of p110α (Figure 6E). Previous studies have shown that although UCHL1 is a deubiquitinating enzyme, it possesses both ubiquitination and deubiquitination activities. To investigate whether the expression of p110α depended on UCHL1 ubiquitination and deubiquitination enzyme activities, 293T cells were transfected with plasmids encoding Flag-p110α and increasing amounts of Myc-UCHL1 (WT), Myc-UCHL1 (K30D, functional defects in ubiquitination and deubiquitination enzyme activities), or Myc-UCHL1 (C90S, functional defects in deubiquitination enzyme activity). However, functional defects in UCHL1 ubiquitination and deubiquitination enzyme activities had no effect on p110α (Supplementary Figure 4D–F). The degradation of p110α in UCHL1-deficiency cells was not affected by the proteasome inhibitor MG132, but was completely blocked by the autophagy inhibitor bafilomycin-A1 (BafA1) (Figure 6F). Collectively, these results suggest that UCHL1 promotes the degradation of p110α through autophagy.
Discussion
DUBs play a crucial role in controlling the ubiquitination and regulating macrophage polarization. Several studies in related fields have clearly demonstrated that DUBs, such as OTUD5, USP14, USP19, ITCH, and Mysm1, triggered macrophage polarization.33–37 In previous studies, UCHL1 was detected as “brain-specific protein”.38 Most research pertaining to UCHL1 has been carried out in the context of human neurodegenerative disorders such as Parkinson’s disease (PD),30 Alzheimer’s disease (AD)31 and traumatic brain injury (TBI).32 However, in this study, we showed for the first time that UCHL1 plays a crucial role in macrophage polarization.
Several studies have focused on the regulatory functions of UCHL1 in immune cells. Previous studies have shown that UCHL1 leads to the activation of AKT, as evidenced by the upregulation of phosphorylated AKT.39–43 Likewise, our results showed that UCHL1 deficiency decreased the phosphorylation level of AKT in macrophages. In response to the microenvironment, activation of the NF-κB signaling pathway is mainly required for M1-orientated immune processes in macrophages. A previous in vitro study showed that UCHL1 mediated the degradation of the NF-κB essential modulator, resulting in the suppression of p65 phosphorylation.44 Similarly, we found that UCHL1 knockdown effectively increased the p65 phosphorylation levels. However, this was inconsistent with the fact that UCHL1 mainly promoted polarization of M1 macrophages. The functions of UCHL1 in the regulation of macrophage polarization may be varied and contradictory, and further studies are required to address this. A previous study showed that silencing UCHL1 upregulated the phosphorylation level of ERK to promote gastric cancer metastasis.42 However, in this study, we did not observe any effect of UCHL1 on ERK phosphorylation in macrophages. UCHL1 may have different regulatory mechanisms for different cell types.
As a deubiquitinase, UCHL1 regulates the AKT signaling pathway, depending on its enzymatic activity. A previous study showed that UCHL1 boosted the AKT signaling pathway by reducing the antagonistic phosphatase PHLPP1, which required its deubiquitinase activity.45 In addition, other studies have also found that UCHL1 interacts with AKT and removes K48 polyubiquitin chains, whereas it also promotes K63 ubiquitination of AKT.40,46 Meanwhile, UCHL1 has been reported to disrupt a complex between the DDB1-CUL4 ubiquitin ligase complex and raptor, which counteracts DDB1-CUL4-mediated raptor ubiquitination.43 However, we found that UCHL1 upregulated AKT phosphorylation by promoting ubiquitin-mediated degradation of p110α, which did not require its ubiquitinase or deubiquitinase activity but was rather dependent on autophagy. However, we were unable to confirm any direct binding between UCHL1 and p110α. The mechanism by which UCHL1 regulates p110α degradation through autophagy is also unclear. Further studies are required to delineate the specific regulatory mechanisms.
In conclusion, we identified UCHL1 as a positive regulator of macrophage polarization, promoting the M1 phenotype and upregulating inflammatory cytokines and decreasing anti-inflammatory cytokines through the p110α-mediated AKT signaling pathway. Our study highlights the significance of UCHL1 in the regulation of innate immunity. Our findings suggest that UCHL1 may act as a potential drug target for developing therapeutic interventions for the treatment of inflammatory diseases.
Conclusions
In this study, we identified UCHL1 as a positive regulator of M1 macrophage polarization. Our findings may help in developing therapeutic interventions for the treatment of inflammatory diseases and pathogenic infections.
Data Sharing Statement
Source data are provided in this paper. The datasets generated and analyzed during the current study are available from the corresponding authors on reasonable requests.
Ethics Approval
All experimental procedures in this study were approved by the Ethics Committee and Animal Experiment Committee of Southern Medical University (SMU-L2017022), which complies with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH publication No. 85–23, revised 1985).
Consent for Publication
All participants in this study gave the signed consent for the publication of identifiable details to be published in the Journal of Inflammation Research.
Acknowledgments
We thank all members of the Prof. Li Ma’s laboratory for helpful discussions and input.
Author Contributions
YLH and SFH designed research; YLH, STH, YTC, JLS, YLF, XLD, YLY, HLL, ZYH, BZ and YQH conducted research; YLH, STH, QW, XYZ, CYZ, SFH, and LM analyzed data; YLH, SFH, and LM wrote the paper. SFH and LM provided essential reagents, or provided essential materials; SFH and LM as the corresponding author conducted the experiment. All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval for the version to be published, and agree to be accountable for all aspects of the work.
Funding
This work was supported National Natural Science Foundation of China (82072242, 81772150, 81901614, and 82070906), Guangdong Basic and Applied Basic Research Foundation (2019A1515011103, and 2021A1515010933).
Disclosure
The authors declare no competing financial/non-financial interests.
References
1. Phan AT, Goldrath AW, Glass CK. Metabolic and epigenetic coordination of T cell and macrophage immunity. Immunity. 2017;46(5):714–729. doi:10.1016/j.immuni.2017.04.016
2. Ginhoux F, Schultze JL, Murray PJ, Ochando J, Biswas SK. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat Immunol. 2016;17(1):34–40. doi:10.1038/ni.3324
3. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496(7446):445–455. doi:10.1038/nature12034
4. Murray PJ. Macrophage polarization. Annu Rev Physiol. 2017;79(1):541–566. doi:10.1146/annurev-physiol-022516-034339
5. Kimura T, Nada S, Takegahara N, et al. Polarization of M2 macrophages requires Lamtor1 that integrates cytokine and amino-acid signals. Nat Commun. 2016;7(1):13130. doi:10.1038/ncomms13130
6. Franke TF, Yang SI, Chan TO, et al. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81(5):727–736. doi:10.1016/0092-8674(95)90534-0
7. Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med. 2005;9(1):59–71. doi:10.1111/j.1582-4934.2005.tb00337.x
8. Franke TF. PI3K/Akt: getting it right matters. Oncogene. 2008;27(50):6473–6488. doi:10.1038/onc.2008.313
9. Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell. 2005;18(1):13–24. doi:10.1016/j.molcel.2005.03.008
10. Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122(2):261–273. doi:10.1016/j.cell.2005.05.012
11. Maira SM, Galetic I, Brazil DP, et al. Carboxyl-terminal modulator protein (CTMP), a negative regulator of PKB/Akt and v-Akt at the plasma membrane. Science. 2001;294(5541):374–380. doi:10.1126/science.1062030
12. Geering B, Cutillas PR, Nock G, Gharbi SI, Vanhaesebroeck B. Class IA phosphoinositide 3-kinases are obligate p85-p110 heterodimers. Proc Natl Acad Sci USA. 2007;104:7809–7814.
13. Kok K, Geering B, Vanhaesebroeck B. Regulation of phosphoinositide 3-kinase expression in health and disease. Trends Biochem Sci. 2009;34(3):115–127. doi:10.1016/j.tibs.2009.01.003
14. Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9(8):550–562. doi:10.1038/nrc2664
15. Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27(41):5497–5510. doi:10.1038/onc.2008.245
16. Samuels Y, Wang Z, Bardelli A, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi:10.1126/science.1096502
17. Yuan TL, Wulf G, Burga L, Cantley LC. Cell-to-cell variability in PI3K protein level regulates PI3K-AKT pathway activity in cell populations. Curr Biol. 2011;21(3):173–183. doi:10.1016/j.cub.2010.12.047
18. Hu H, Sun SC. Ubiquitin signaling in immune responses. Cell Res. 2016;26(4):457–483. doi:10.1038/cr.2016.40
19. Fu Y, Wang P, Zhao J, et al. USP12 promotes CD4(+) T cell responses through deubiquitinating and stabilizing BCL10. Cell Death Differ. 2021;28(10):2857–2870. doi:10.1038/s41418-021-00787-y
20. Jarome TJ, Devulapalli RK. The ubiquitin-proteasome system and memory: moving beyond protein degradation. Neuroscientist. 2018;24(6):639–651. doi:10.1177/1073858418762317
21. Varshavsky A. The ubiquitin system, autophagy, and regulated protein degradation. Annu Rev Biochem. 2017;86(1):123–128. doi:10.1146/annurev-biochem-061516-044859
22. Kulathu Y, Komander D. Atypical ubiquitylation - the unexplored world of polyubiquitin beyond Lys48 and Lys63 linkages. Nat Rev Mol Cell Biol. 2012;13(8):508–523. doi:10.1038/nrm3394
23. Sun SC. Deubiquitylation and regulation of the immune response. Nat Rev Immunol. 2008;8(7):501–511. doi:10.1038/nri2337
24. Wertz IE, Wang X. From discovery to bedside: targeting the ubiquitin system. Cell Chem Biol. 2019;26(2):156–177. doi:10.1016/j.chembiol.2018.10.022
25. Bedford L, Lowe J, Dick LR, Mayer RJ, Brownell JE. Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets. Nat Rev Drug Discov. 2011;10(1):29–46. doi:10.1038/nrd3321
26. Doran JF, Jackson P, Kynoch PA, Thompson RJ. Isolation of PGP 9.5, a new human neurone-specific protein detected by high-resolution two-dimensional electrophoresis. J Neurochem. 1983;40(6):1542–1547. doi:10.1111/j.1471-4159.1983.tb08124.x
27. Liu Y, Fallon L, Lashuel HA, Liu Z, Lansbury PT
28. Larsen CN, Krantz BA, Wilkinson KD. Substrate specificity of deubiquitinating enzymes: ubiquitin C-terminal hydrolases. Biochemistry. 1998;37:3358–3368.
29. Wilkinson KD, Lee KM, Deshpande S, Duerksen-Hughes P, Boss JM, Pohl J. The neuron-specific protein PGP 9.5 is a ubiquitin carboxyl-terminal hydrolase. Science. 1989;246(4930):670–673. doi:10.1126/science.2530630
30. Lunati A, Lesage S, Brice A. The genetic landscape of Parkinson’s disease. Rev Neurol. 2018;174(9):628–643. doi:10.1016/j.neurol.2018.08.004
31. Tramutola A, Di Domenico F, Barone E, Perluigi M, Butterfield DA. It is all about (U)biquitin: role of altered ubiquitin-proteasome system and UCHL1 in Alzheimer disease. Oxid Med Cell Longev. 2016;2016:2756068. doi:10.1155/2016/2756068
32. Wang KK, Yang Z, Zhu T, et al. An update on diagnostic and prognostic biomarkers for traumatic brain injury. Expert Rev Mol Diagn. 2018;18(2):165–180. doi:10.1080/14737159.2018.1428089
33. Zhang Y, Fan Y, Jing X, et al. OTUD5-mediated deubiquitination of YAP in macrophage promotes M2 phenotype polarization and favors triple-negative breast cancer progression. Cancer Lett. 2021;504:104–115. doi:10.1016/j.canlet.2021.02.003
34. Xu F, Ma Y, Huang W, et al. Typically inhibiting USP14 promotes autophagy in M1-like macrophages and alleviates CLP-induced sepsis. Cell Death Dis. 2020;11(8):666. doi:10.1038/s41419-020-02898-9
35. Liu T, Wang L, Liang P, et al. USP19 suppresses inflammation and promotes M2-like macrophage polarization by manipulating NLRP3 function via autophagy. Cell Mol Immunol. 2020;18(10):2431–2442.
36. Lin X, Zhang H, Boyce BF, Xing L. Ubiquitination of interleukin-1α is associated with increased pro-inflammatory polarization of murine macrophages deficient in the E3 ligase ITCH. J Biol Chem. 2020;295(33):11764–11775. doi:10.1074/jbc.RA120.014298
37. Zhao X, Huang XH, Dong XH, et al. Deubiquitinase Mysm1 regulates macrophage survival and polarization. Mol Biol Rep. 2018;45(6):2393–2401. doi:10.1007/s11033-018-4405-3
38. Day IN, Thompson RJ. UCHL1 (PGP 9.5): neuronal biomarker and ubiquitin system protein. Prog Neurobiol. 2010;90(3):327–362. doi:10.1016/j.pneurobio.2009.10.020
39. Hussain S, Bedekovics T, Ali A, et al. A cysteine near the C-terminus of UCH-L1 is dispensable for catalytic activity but is required to promote AKT phosphorylation, eIF4F assembly, and malignant B-cell survival. Cell Death Discov. 2019;5(1):152. doi:10.1038/s41420-019-0231-1
40. Luo Y, He J, Yang C, et al. UCH-L1 promotes invasion of breast cancer cells through activating Akt signaling pathway. J Cell Biochem. 2018;119(1):691–700. doi:10.1002/jcb.26232
41. Jin Y, Zhou H, Gong L, Xia J, Yu G, Chen Y. Prognostic potential and oncogenic effects of UCH-L1 expression in hilar cholangiocarcinoma. Int J Clin Exp Pathol. 2017;10(11):10802–10811.
42. Gu YY, Yang M, Zhao M, et al. The de-ubiquitinase UCHL1 promotes gastric cancer metastasis via the Akt and Erk1/2 pathways. Tumour Biol. 2015;36(11):8379–8387. doi:10.1007/s13277-015-3566-0
43. Hussain S, Feldman AL, Das C, Ziesmer SC, Ansell SM, Galardy PJ. Ubiquitin hydrolase UCH-L1 destabilizes mTOR complex 1 by antagonizing DDB1-CUL4-mediated ubiquitination of raptor. Mol Cell Biol. 2013;33(6):1188–1197. doi:10.1128/MCB.01389-12
44. Karim R, Tummers B, Meyers C, et al. Human papillomavirus (HPV) upregulates the cellular deubiquitinase UCHL1 to suppress the keratinocyte’s innate immune response. PLoS Pathog. 2013;9(5):e1003384. doi:10.1371/journal.ppat.1003384
45. Hussain S, Foreman O, Perkins SL, et al. The de-ubiquitinase UCH-L1 is an oncogene that drives the development of lymphoma in vivo by deregulating PHLPP1 and Akt signaling. Leukemia. 2010;24(9):1641–1655. doi:10.1038/leu.2010.138
46. Mitra S, Sammani S, Wang T, et al. Role of growth arrest and DNA damage-inducible α in Akt phosphorylation and ubiquitination after mechanical stress-induced vascular injury. Am J Respir Crit Care Med. 2011;184(9):1030–1040. doi:10.1164/rccm.201103-0447OC
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.