Back to Journals » International Journal of Nanomedicine » Volume 17
Two-Pronged Anti-Tumor Therapy by a New Polymer-Paclitaxel Conjugate Micelle with an Anti-Multidrug Resistance Effect
Authors Du J, Zong L, Li M, Yu K, Qiao Y, Yuan Q, Pu X
Received 9 November 2021
Accepted for publication 7 March 2022
Published 22 March 2022 Volume 2022:17 Pages 1323—1341
DOI https://doi.org/10.2147/IJN.S348598
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Mian Wang
Juan Du,1 Lanlan Zong,2 Mengmeng Li,2 Keke Yu,2 Yonghui Qiao,3 Qi Yuan,2 Xiaohui Pu2
1Department of Pharmacy, The Affiliated Cancer Hospital of Zhengzhou University, Zhengzhou, Henan, 450003, People’s Republic of China; 2Institute of Pharmacy, School of Pharmacy, Henan University, Kaifeng, Henan, 475004, People’s Republic of China; 3Academy of Chinese Medical Sciences, Henan University of Chinese Medicine, Zhengzhou, Henan, 450046, People’s Republic of China
Correspondence: Juan Du; Xiaohui Pu, Tel/Fax +86-371-23880680, Email [email protected]; [email protected]
Introduction: Cancerous tumors are still a major disease that threatens human life, with tumor multidrug resistance (MDR) being one of the main reasons for the failure of chemotherapy. Thus, reversing tumor MDR has become a research focus of medical scientists.
Methods: Here, a reduction-sensitive polymer prodrug micelle, mPEG-DCA-SS-PTX (PDSP), was manufactured with a new polymer inhibitor of drug resistance as a carrier to overcome MDR and improve the anti-tumor effect of PTX.
Results: The PDSP micelles display good stability, double-responsive drug release, and excellent biocompatibility. The PDSP micelles reduced the cytotoxicity of PTX to normal HL-7702 cells and enhanced that to SMMC-7721 and MCF-7 cells in vitro. Improved sensitivity of A549/ADR to PDSP was also observed in vitro. Furthermore, in vivo experiments show reduced systemic toxicity and enhanced therapeutic efficacy of PTX to H22 subcutaneous tumor-bearing mice.
Conclusion: This work proves that the reduction-sensitive polymer prodrug micelles carried by the new polymer inhibitor can be used as an alternative delivery system to target tumors and reverse MDR for paclitaxel and other tumor-resistant drugs.
Keywords: anti-multidrug resistance, polymer prodrug micelles, paclitaxel, reduction sensitivity, liver targeting
Introduction
So far, cancer is still a leading cause of human death in the world,1–3 with multidrug resistance (MDR) being one of the main reasons for the failure of chemotherapy.4 Thus, reversing tumor MDR has become a research focus of medical scientists. At present, there are usually two ways to reverse MDR: one is to increase drug penetration into tumor cells, and the other is to inhibit the efflux of drugs. Nano-drugs can promote drug delivery to tumor tissue through the EPR effect and can increase drug penetration into cells by endocytosis. However, traditional nano-drugs cannot inhibit the drug efflux proteins on the cell membrane. Therefore, the study of nano-drugs combined with efflux protein inhibitors is gaining more and more attention.5,6 It has been reported that a reduction sensitive polycurcumin encapsulated paclitaxel micelles, pH-sensitive micelles co-loading with paclitaxel and dithiram, redox-responsive micelles co-loading with doxorubicin and indomethacin, paclitaxel-loaded d-α-tocopherol succinate modified chitosan micelles, and so on.6–9 These nano-drugs combined with efflux protein inhibitors significantly reversed tumor MDR. However, drug and small molecular inhibitors can easily leak from these nanoparticles and cause severe side effects. As reported in the literatures, external stimuli-responsive polymer conjugate micelles had attracted more and more attention due to their stability in physiological environments, such as the bloodstream and normal tissues and rapid release of drugs in specific environments. For example, reduction-sensitive mPEG-SS-PTX and HA-SS-PTX, pH-sensitive HA-dOG-PTX conjugate, and heat-responsive CPT@DOX-UCST/PPy only release about 30% of drugs within several days under physiological conditions, while they release above 50% of drugs within several hours in external stimuli environments.10–12 Therefore, polymer conjugates combining resistance inhibitors with chemotherapy drugs will be a more promising alternative strategy.
At present, the approved polymers for overcoming the MDR effect include TPGS, Pluronic, Cremophor EL, and so on.13 However, their nanomicelles have weak stability due to their large critical micelle concentration (CMC). Therefore, a new polymer, methoxy polyethylene glycol-deoxycholic acid (mPEG-DCA, as seen in Scheme 1), was synthesized to overcome MDR in our lab.14 It was confirmed that mPEG-DCA has a better anti-MDR effect than TPGS in drug-resistant cells and a subsequently better anti-tumor effect (data not shown). Therefore, this study manufactured a new nano-drug delivery system using mPEG-DCA as a carrier for antineoplastic drugs to treat tumors in a two-pronged manner.
 |
Scheme 1 The schematic diagram of the anti-tumor effect in vivo of PDSP micelles. |
Paclitaxel (PTX) is an active anti-tumor drug isolated from the bark and wood of the Pacific Taxus brevifolia.15 As an anti-cancer superstar, PTX has a therapeutic effect on various cancers, such as breast, ovarian, head and neck neoplasm, and prostate cancers.16,17 PTX is a well-known BCS class IV drug with poor solubility and permeability due to a large number of hydrophobic groups in its structure.18 Also, many studies have proven that PTX is a substrate of the membrane-bound drug efflux pump P-gp, often resulting in limited efficacy against drug-resistant tumors.13,19,20 In order to overcome these deficiencies, pharmaceutical scientists have developed a variety of PTX nanoparticles, such as nanosuspensions,21 nanoparticles,22 liposomes,23 nanoemulsions,24 micelles,25,26 nanoassemblies,27 and so on. These nanoparticles can partially avoid the side effect of commercial PTX injection (Taxel), but the overcoming tumor drug resistance is beyond their power. Therefore, a reduction sensitive polymer-paclitaxel prodrug was constructed with mPEG-DCA as an anti-MDR carrier in this study, and self-assembled into nano-micelles to improve solubility of PTX, overcome drug resistance, target tumor and even enhance antitumor (Scheme 1).
Materials and Methods
Materials
PTX was purchased from Nanjing Jingzhu Biotechnology Co., Ltd. (Nanjing, China). PTX injection (XINERJIN, 30 mg/5 mL) was bought from ShuangLu Pharm Co. Ltd. (Beijing, China). The reference substances of PTX (purity, 99.8%) and Diazepam (purity, 99.8%) were bought from Shanghai YuanYe biotechnology co., LTD. Dimethylaminopyridin (DMAP, purity, >98%), 3, 3’-Dithiodipropionic acid (DTDP, purity > 98%), succinic anhydride (SA, purity, >98%), CaH2 (purity > 98.5%), N, N’-dicyclohexyl-carbodiimide (DCC, purity, >98%), and Glutathione (GSH) were purchased from Aladdin Reagent Co., Ltd. (Shanghai, China). Succinic anhydride (purity, >99% GC), DMSO-d6, and CDCl3 were purchased from J&K Scientific Ltd. (Beijing, China). Pyrene (purity, 97%) was purchased from Shanghai Jingchun Biotechnology Co., Ltd. (Shanghai, China). Acetyl chloride was obtained from Tianjin Fuyu Fine Chemical Co., Ltd. All chemical reagents were analytical grade and used without further treatment. The mPEG-DCA was synthesized by our Lab. Dialysis tubing (MWCO 2000/350) was obtained from Shanghai Lvniao Technologies, Inc. (Shanghai, China). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl Tetrazolium bromide (MTT), RPMI 1640 medium, Dulbecco’s modified Eagle medium (DMEM), penicillin-streptomycin (Amresco, USA), 0.25% (w/v) trypsin-EDTA solution, and fetal bovine serum (FBS, Gibco, USA) were purchased from Sigma-Aldrich. H22 cells were gifted by Professor Du Gangjun at Henan University and approved by the Ethics Committee for Animal Experimentation of Henan University (License No. HUSOM-2017-236).
Synthesis of Polymer Prodrugs
Synthesis of Dithiodipropionic Anhydride (DTDPA)
Firstly, dithiodipropionic anhydride (DTDPA) was synthesized by the published methods.28 Briefly, DTDP (5 g) was dissolved in acetyl chloride (50 mL), stirred at 600 r/min, and then refluxed at 75 °C for 7 h. After the reaction, the solvent was removed by a vacuum rotary evaporator, and the product was precipitated by ether. The product was vacuum dried at 40°C to obtain DTDPA.
Synthesis of mPEG-DCA-DTPA and mPEG-DCA-SA
Firstly, mPEG-DCA-DTPA and mPEG-DCA-SA was synthesized by the published methods.14 The detailed synthesis steps are as follows.
mPEG-DCA-DTPA: 3.5 mmol of DTDPA was placed into 30 mL of DMF in a three-neck flask, and stirred to dissolve entirely in a nitrogen and ice bath. 0.7 mmol of mPEG-DCA and 0.7 mmol DMAP were dissolved in 10 mL of DMF and added dropwise into the above DTDPA solution. The reaction was carried out for 24 h at 30 °C. DMF was removed by vacuum rotary evaporation. The residue was dissolved in a moderate amount of dichloromethane (DCM). DCM solution was extracted with 0.1 mol/L hydrochloric acid aqueous solution and a saturated NaCl solution twice, respectively. Dehydration of the above DCM solution was executed with the anhydrous magnesium sulfate. The solution was concentrated and mixed with anhydrous diethyl ether to obtain the precipitate, which was collected by filtration and dried under vacuum at 37 °C.
mPEG-DCA-SA: 3.5 mmol of succinic anhydride (SA), 0.7 mmol of mPEG-DCA, and 0.7 mmol DMAP were dissolved in 40 mL of DMF, and stirred in nitrogen for 2 h at room temperature. Then the reaction was continued for 24 h at 30 °C. Purification and collection of the products were the same as the above method for mPEG-DCA-DTPA.
Synthesis of mPEG-DCA-PTX Prodrugs
0.35 mmol mPEG-DCA-DTPA and 0.7 mmol DMAP were dissolved in anhydrous DCM and stirred at 30 °C for 60 min. 0.35 mmol PTX and 0.8 mmol DCC were added and reacted at 30 °C for 48 h. The reaction solution was filtered before the concentration was carried out. The concentrated solution was mixed with diethyl ether to gain a precipitate, which was collected by filtration and dried at 35 °C in a vacuum. The mPEG-DCA-SS-PTX (PDSP, shown in Scheme 2) was purified using flash chromatography with DCM: methanol (80:1) as the eluent on a silica column. The mPEG-DCA-CC-PTX (PDCP) were obtained by the same process as described above, except that mPEG-DCA-DTPA was replaced with mPEG-DCA-SA.
 |
Scheme 2 The synthesized route of PDCP and PDSP polymer prodrugs. |
Characterization of PDSP and PDCP
1H NMR spectra was recorded on an AVANCE 300 MHz spectrometer (Switzerland). Fourier transform infrared spectroscopy (FTIR) analyses were carried out with an Avatar 360 Fourier transform infrared spectrophotometer (America) using KBr pellets. The CMCs of PDSP and PDCP were also investigated by fluorescence spectroscopy with pyrene as a fluorescence probe. Briefly, PDSP and PDCP micelles were diluted to a concentration range of 5.0×10−5 mg/mL to 5.0×10−1 mg/mL. Five milliliters of diluted PDSP and PDCP micelles solutions were mixed with 0.5 mL of pyrene acetone (6.0×10−6 mol/L), respectively. The mixtures were then sonicated for 10 minutes and stored overnight in a dark place at room temperature to obliterate the acetone. The fluorescence intensity of the samples at an emission wavelength of 372 nm was recorded by a fluorescence spectrophotometer (Hitachi F-7000, Japan). The CMC was then estimated using the inflection point on the fit curve of the fluorescence intensity ratios (I333/I339) (excitation wavelengths of 333 nm and 339 nm) versus the log concentration of polymer prodrug. The CMC was inferred from the point of intersection obtained by extrapolating the intensity ratios (I333/I339) to the low- and high-concentration regions.14
Preparation of Polymer Prodrug Micelles
The PDCP and PDSP micelles were prepared by the film dispersion method.29 20 mg of PDCP was dissolved in 10 mL of CH2Cl2 through stirring. After complete dissolution, CH2Cl2 was removed to form films of the polymer prodrug by rotary evaporation, which were then dried overnight under vacuum. The films were hydrated using ultrapure water at room temperature to form a suspension. These were sonicated at 100 W using probe sonography to obtain micelles. According to the results of single-factor tests, it was found that the hydration temperature, ultrasonic power, and the counts of ultrasound were critical factors that influence the dimension and size distribution of micelles. Therefore, the central composite design (CCD)-response surface method was used to optimize the fabricated process with particle size and polydispersity index (PDI) as the responsive indexes within a minimum number of experiments.28 Three independent variables (factors) and their levels in the central composite design experiments are listed in Table 1. A total of 27 experiments and their determined dependent variables (responses) values are listed in Table 2. Design Expert 8.0.6.1 software was used to fit the experimental results to obtain an optimal polynomial mathematical model. The optimal process parameters were selected from response surface diagrams.
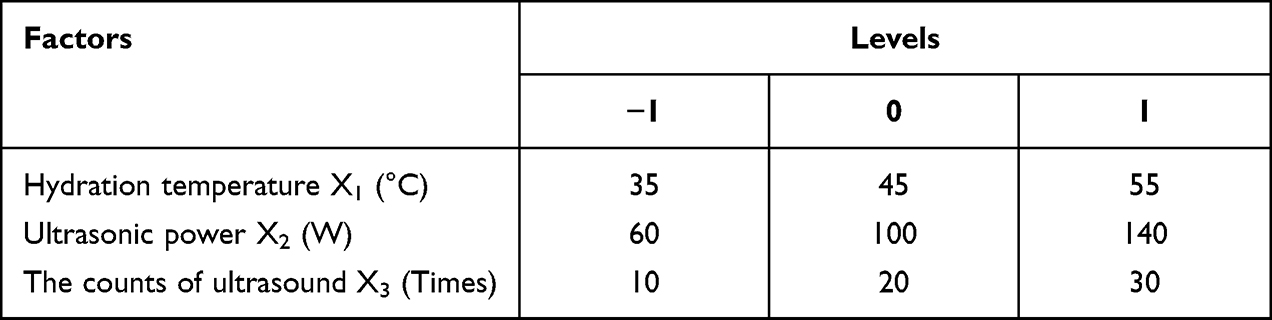 |
Table 1 The Factors and Levels of the Central Composite Design Test |
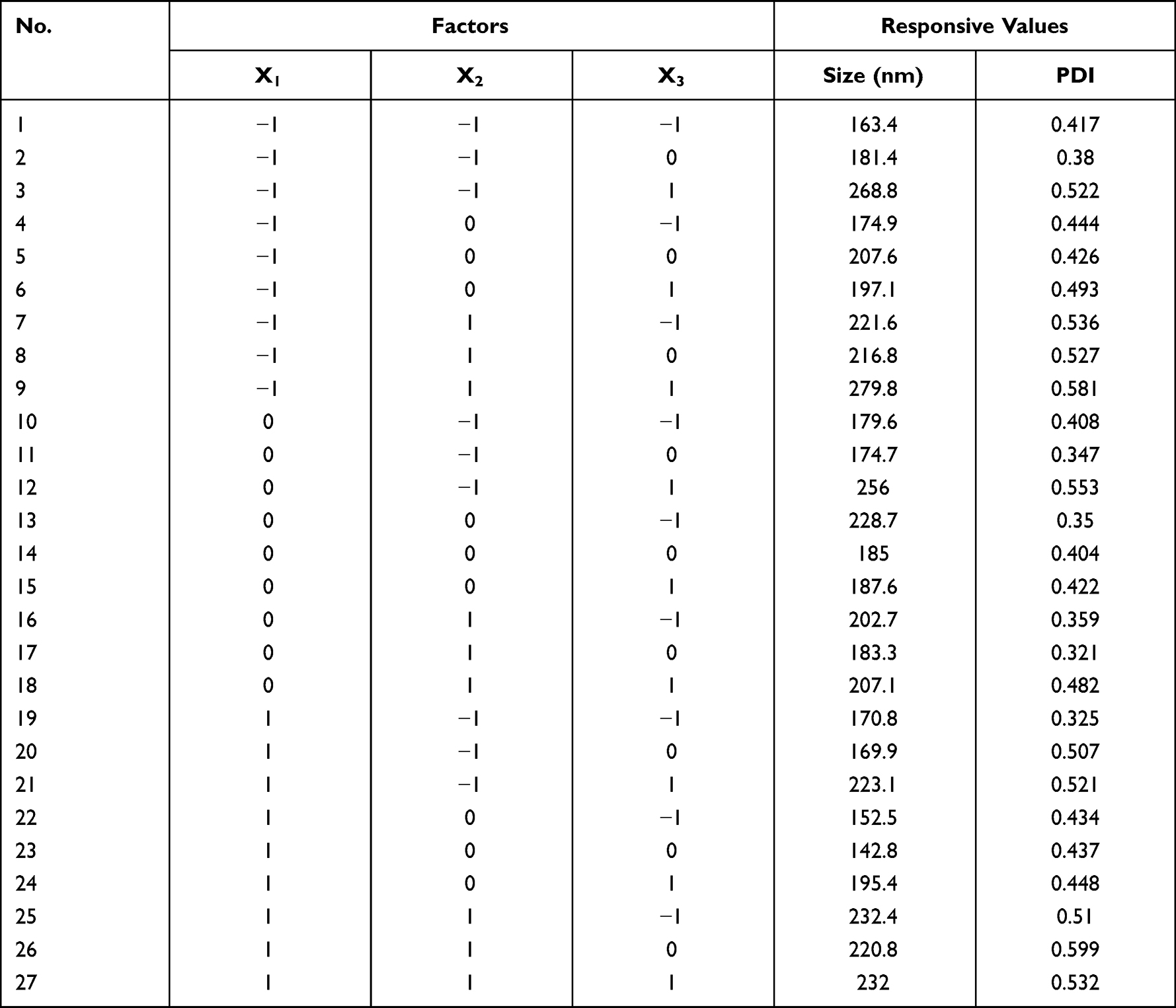 |
Table 2 The Results of the Central Composite Design Test |
The PDSP micelles and blank micelles (BM) were fabricated using these optimal process parameters, except that PDCP was replaced by PDSP or mPEG-DCA at the equivalent amount.
Particle Size, Zeta Potential and Particle Morphology
Particle size, size distribution (PDI), and zeta potential of polymer prodrug micelles were measured using a Nano-ZS90 ZetaSizer (Malvern Instruments, Malvern, UK). The freshly prepared micelles were placed onto a carbon-coated copper grid and dried at room temperature overnight in a vacuum oven. The dimensions and morphologies of micelles were imaged on a JEOL 2010 transmission electron microscopy (TEM, JEOL Co., Japan).
Stability
To study the stability of PDSP and PDCP micelles, storage, dilution, and plasma stability experiments were carried out.30 In the storage stability test, micelles were stored at 4 °C and room temperature for 15 days, and the sizes were determined. In the dilution stability test, micelles were diluted respectively to 10, 20, 50, 100 times with distilled water, and particle size analysis was executed half an hour later. In the plasma stability test, the micelles were incubated with rat plasma at 37 °C for 48 h in a shaker (ZWY-103D, Shanghai Zhicheng, China) at a shaking speed of 100 rpm. After that, 2 mL of the samples were withdrawn at a pre-determined time of 0, 4, 8, 16, 24, 48 h, and the sizes were analyzed. All measurements were carried out in triplicate.
In vitro Release
The GSH reduction-sensitive release test of PTX from the PDSP micelles was carried out using dialysis. One milliliter of micelles was transferred into dialysis tubes (MWCO 2000 Da) and dialyzed against 20 mL of either pH 7.4 PBS containing 0.5% Tween-80 and different concentrations of GSH (0 μM, 10 μM, 20 mM and 40 mM) or pH 5.8 PBS containing 0.5% Tween-80 and 20 mM GSH. The drug release from the PDCP micelles without a disulfide bond was also investigated as a control. The dialysis tubes were shaken in an air thermostatic oscillator at 150 rpm and 37 °C. At predefined time intervals, 1 mL of released medium was withdrawn for testing and replaced with 1 mL of fresh medium at the same temperature. The samples were filtered through 0.22 μm filters, where the concentration of PTX was determined at 227 nm with UV detection on a Waters 2695–2489 HPLC system (Waters Co., Ltd., Milford, MA, USA) and Waters Symmetry C18 column (250 mm×4.6 mm, 5 μm) whose temperature was set at 30 °C. The mobile phase consisted of acetonitrile/water (50:50, v/v) and set at a 1.0 mL/min flow rate. All experiments were performed in triplicate. Finally, time-cumulative release curves under different conditions were drawn.
Reduction-Responsibility of Micelles
To investigate reduction-response of polymer prodrug micelles, 20 mL of samples were mixed with glutathione to obtain a final GSH concentration of 10 μM, 20 mM, and 40 mM. The samples were then placed in an air thermostatic oscillator with a shaking speed of 100 rpm at 37 °C. After incubation for 24 h, the dimension of the micelles was measured by Nano-ZS90 ZetaSizer. All samples were tested three times.
Hemolysis Test
Hemolytic assays of two polymer prodrug micelles were performed using fresh rat blood.30 Firstly, the micellar solution was diluted with distilled water for a PTX concentration of 0.1, 0.3, 0.5 mg/mL. According to literature,31 the rat blood sample was prepared by the following method: 8 mL of fresh rat blood with anticoagulant was diluted with 10 mL of 0.9% sodium chloride solution to obtain the diluted blood samples. 10 mL of either micellar solution at different concentrations, distilled water (positive control), or 0.9% sodium chloride (negative control) were placed in EP tubes, respectively. Then, 0.2 mL of diluted blood samples were added into each EP tube and gently blended. All sample tubes were placed in a water thermostatic shaker with a speed of 100 rpm at 37 °C and incubated for 2 h. Finally, the samples were centrifuged at 159 g for 10 min, and the supernatant was transferred into a colorimetric plate. The absorbance of each sample was measured at 545 nm on a UV-2600 ultraviolet-visible spectrophotometer (Shimadzu Co., Ltd., Tokyo, Japan), and the relative hemolytic rates (RHR%) were calculated by the following equation.

Ds is the absorbance of the micelle samples, Dnc is the absorbance of negative control, and Dpc is the absorbance of positive control. The experiments were conducted in triplicate, and the data is shown as the mean values plus standard deviation (±SD).
In vitro Cytotoxicity Assays
Cell Culture
HL-7702 cells, SMMC-7721 cells, and MCF-7 cells were provided by Kaiji Biotech Co., Ltd. (Nanjing, China). A549/PTX cells were gifted by Professor Guang Han in Henan University and approved by the Ethics Committee for Animal Experimentation of Henan University. HL-7702 cells were cultured in DMEM medium while the other three cell lines were cultured in RPMI 1640 medium; all cell lines were then placed in a cell culture incubator with a 5% CO2 atmosphere at 37 °C. The cells were subcultured with 0.25% trypsin-EDTA (EDTA=ethylene diamine tetraacetate) upon reaching 80–90% confluence. For further experiments, cells were seeded in a 96-well plate with 1×104 cells/well. After incubation for 24 h to allow cell attachment, the cells were treated with various samples at different PTX/micelle concentrations for the following tests.
Toxicity to Normal Cells
To study the biocompatibility of the micelles and polymer, the toxicity of BM, PDCP, and PDSP micelles toward HL-7702 cells were evaluated using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.31 Briefly, the BM, PDCP, and PDSP micelles were added and co-incubated with HL-7702 cells for 24 h or 48 h at concentrations of 2, 5, 10, 20, 50, and 100 μg/mL in a 96-well plate. Untreated cells were used as control. After 24 h or 48 h of treatment, the cells were washed, and 10 µL of MTT (5 mg/mL) was added to each well. The cells were incubated for an additional 4 h at 37 °C. The medium was then discarded, and DMSO (100 µL) was added to dissolve the formazan crystals formed. The absorbance in each well was determined at a wavelength of 570 nm with a multidetector microplate reader (Hidex Chameleon V, Turku, Finland). Each drug concentration was tested in triplicate.
In vitro Cytotoxicity Assays
In this study, the efficiency of several formulations against three cancer cells (SMMC-7721, MCF-7, and A549/PTX) was studied using a free PTX solution as a reference. Briefly, three cell lines were co-incubated with PTX injection, PTX injection+BM, PDCP, PDSP, PDCP+GSH, and PDSP+GSH for 24 h or 48 h at different concentrations of PTX. For SMMC-7721 and MCF-7 cells, the concentrations of PTX were set at 0.04, 0.2, 1, 5, 25 μg/mL. PTX at concentrations of 0.4, 2, 10, 50, and 250 μg/mL was used to inhibit the growth of A549/PTX cells. After 24 h or 48 h of co-incubation, the cell viability assays were carried out by the MTT method described in the above section. Each experiment was executed in triplicate.
In vivo Antitumor Activity
Considering the proficiency and reliability of the technology of tumor cell inoculation in Kunming mice in our lab, H22 cells were selected in this study to establish a tumor-bearing mouse model for in vivo anti-tumor test. All experiments and the welfare of the animals were evaluated and approved by the Ethics Committee for Animal Experimentation of Henan University. Five to six weeks old SPF Kunming mice (20±2 g, Code No. 41003100004675) bearing an H22 tumor with a volume of 100 mm3 were randomly divided into four groups (n=10): (1) saline, (2) PTX injection, (3) PDCP micelle, and (4) PDSP micelle. No significant difference in body weight was observed between these groups. The formulations were administrated via tail vein every other day for seven injections with an equivalent PTX dose of 5 mg/kg. During the administration period, body weight and tumor size was monitored daily, and tumor volume was calculated according to the formula Volume=0.5×(width)2×(length). Mice were sacrificed at 24 h post-final injection, and the liver, kidney, heart, lungs, spleen, and tumor were collected. All tissues were washed with cold normal saline (0.9% NaCl) to remove the blood on the surface, followed by quick drying with tissue paper. After that, the tumor was weighed, and all tissues were buried in wax and sliced to perform hematoxylin and eosin (H&E) staining for pathology. The tumor weight and volume inhibition ratio (IRw and IRv) were calculated using the following formula:29

Where Dcontrol is the tumor weight or volume for the saline group, and Dtest is the tumor weight or volume for the groups treated by drugs. The images of tissue sections were carried out under a microscope after H&E staining.
In vivo Tissue Distribution
To evaluate the biodistribution of two polymer prodrug micelles in this study, H22 tumor-bearing mice with a tumor volume of 500 mm3 were randomly divided into four groups (n=24) (saline, PDCP micelles, PDSP micelles, and PTX injection). With saline as control, drug formulations were administrated intravenously at a dose of 10 mg/kg PTX via the caudal vein. All animal experiments were evaluated and approved by the Ethics Committee for Animal Experimentation of Henan University (License No. HUSOM2021-078). Six mice in each group were sacrificed at 2, 6, 12, and 24 h after administration. The liver, kidney, heart, lung, spleen, and tumor were collected, respectively. All tissue samples were stored at −20 °C until analysis. To determine PTX in tissues, samples were prepared using the modified method reported by Cao.13 Briefly, the 0.1 g of tissue sample was weighed accurately and homogenized using a DY89-1 electric glass tissue homogenizer (Ningbo Scientz Biotechnology Co., Ltd. China) after adding 300 μL of physiologic saline including 10% (v/v) methanol. 100 μL of tissue homogenate were put in an EP tube, and 100 μL of methanol and 100 μL of internal standard solution (Diazepam, 2.65 μg/mL) were added into, vortexed for 3 min. Then the samples were shaken in an ultrasonic instrument for 1 min and centrifuged at 12,000 rpm for 10 min. The supernatant was transferred to a new EP tube and bent-dried with nitrogen at 60 °C. The residue was dissolved with 100 µL methanol and centrifuged at 12,000 rpm for 10 min. The concentration of PTX in samples was determined by HPLC as described above, using acetonitrile/water (50:50, v/v) as the mobile phase. The area under the concentration-time curve (AUC) in all the tissues was calculated using the trapezoid method. We calculated the tumor-targeting rate (TTR) of three formulations according to the following formula:

Results and Discussion
Synthesis and Characterization of MDCP and MDSP
The 1H-NMR and IR spectra confirmed the formation of the PDSP and PDCP prodrugs (Figure S1–3) (Supplementary Materials). As shown in Figure S1, the reaction between mPEG-DCA and DTDPA is verified by forming an ester bond. The peak of the new forming ester bond between DTDPA and mPEG-DCA appears at δ 2.65 ppm. Additionally, the signals of methylene (-CH2-CH2-S-S-CH2-CH2-) in DTDPA were at δ 2.96 ppm, δ 2.81 ppm, and δ 2.65 ppm. All results indicate that mPEG-DCA-DTPA is successfully synthesized. Figure S2 shows the characteristic peaks of PTX with aromatic protons at δ 7.0~8.0 ppm, acetyl and methyl protons at δ 1.0~2.5 ppm. The most reactive position for structure modification of PTX is the C-2’-OH. Therefore, the activated mPEG-DCA-DTPA is prone to react with the C-2’-OH to form an ester bond. As shown in Figure S2, the comparison of the 1H NMR spectra of PTX and PDSP indicates that the peak of C-20-H shifts from δ 4.79 ppm to δ 5.45 ppm. All characteristic peaks of PTX and mPEG-DCA-DTPA observed from Figure S2 indicate the successful synthesis of PDSP. Similar peak changes can also be found in PDCP, indicating that PDCP was successfully synthesized.
The FT-IR diagrams of mPEG-DCA, PTX, mPEG-DCA-SA, mPEG-DCA-DTPA, PDCP, and PDSP are shown in Figure S3. In the spectra of PDSP and PDCP, the asymmetric and symmetric stretching vibration peaks of VC-H in DCA’s structure were shown at 2945.3 cm−l and 2887.4 cm−1, while the stretching vibration peaks of VC=O in the carboxyl and ester were shown at 1730.2 cm−1. These peaks were similar to those in the spectra of mPEG-DCA-SA and mPEG-DCA-DTPA. The characteristic absorption peaks of PTX also appeared in the spectra of the two polymer prodrugs, particularly the ketone carbonyl VC=O at 1737.9 cm−1; the carbonyl peak of the amide group at 1651.1 cm−1, and the monosubstituted benzene ring at 707.9 cm−1. Therefore, the results suggest that PTX was successfully bonded with mPEG-DCA-SA or mPEG-DCA-DTPA. The results also showed that the CMCs of PDSP and PDCP were 19.1 μg/mL and 9.3 μg/mL, which was smaller than the CMCs of polymers CSA-ss-DOCA, TPGS, and Pluronic reported in the kinds of literature.32–34 Small CMC will be beneficial to the formation of micelles.26,28
The Results of Process Optimization
To prepare uniform and stable polymer prodrug micelles, the preparation process was optimized with the central composite design, which is a statistical tool to present the quantitative relationship between independent variables and responses values.35 In this study, the different levels of three experimental factors (seen in Table 1) were employed in the central composite design experiments. The particle size (Y1) and PDI (Y2) were used as the responses (dependent variables). The experimental results were fitted to multiple linear equations, quadratic polynomials, and cubic polynomials (seen in Eq. S1~ S6) of the effects of hydration temperature (X1), ultrasonic power (X2), and the counts of ultrasound (X3) on the responses (Supplementary Materials) using the Design Expert 8.0.6.1 software. The fitting degree of the equation is good if its correlation coefficient R value is close to 1. The cubic polynomial model was found to be optimal to describe the correlation between the change of variables and the subsequent responses (seen in Eq. S5 and S6).
The three-dimensional response surface was drawn in line with the fitted cubic polynomial equation (Figure S4). As the predicted values come closer to the actual ones, the points on the scatterplot fall closer to the line. If they are not, another model must be checked to improve the fit. If the points are all very close to the line, the model is expected to offer good predictability.36 Figure S4 shows good prediction capability of the selected models for both Y1 and Y2. According to the optimal region on the surface, the best process was obtained as follows: hydration temperature at 43 °C, ultrasonic power at 90 W, and ultrasonic frequency at 14 times. Under the optimized conditions, the experimental validation of the proposed model was conducted using three batches of the sample. The polymer prodrug micelles used in the rest of the study were manufactured using this optimization process.
Characterization of Particle Size, Zeta Potential, and Morphology
To confirm the form of the polymer prodrug micelles, the mean particle sizes, PDIs, zeta potentials, and morphology of PDSP and PDCP micelles were studied. Figure 1 shows that the average particle size of PDSP and PDCP were both about 200 nm with a generally uniform size distribution, as indicated by their low PDIs in Table S1. Compared to microparticles, PTX nanoparticles can increase solubility, control PTX release, minimize adverse effects and dosing frequency, decreased dose fluctuations in plasma level.13,21,37 Zeta potential of the micelles was approximately −9 to −15 mV, as shown in Table S1, indicating that these nanoparticles might have good stability because of the strong homogeneous charge repulsion between particles.
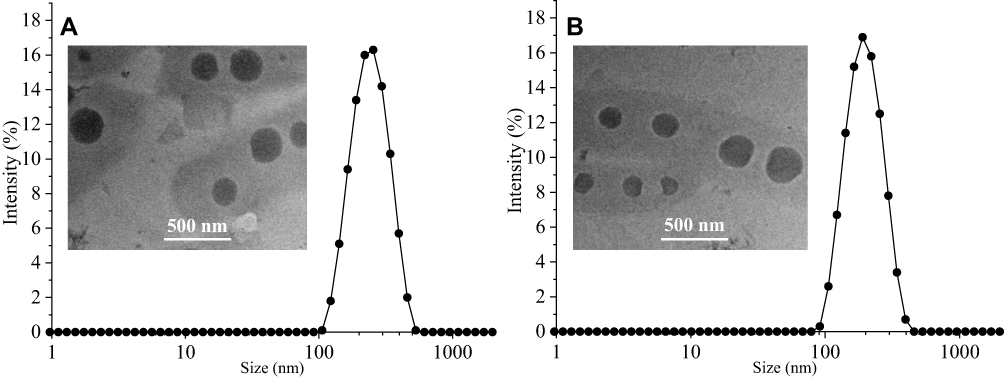 |
Figure 1 The particle size and TEM photos of PDCP (A) and PDSP (B) micelles. |
TEM images (Figure 1A and B) demonstrate that the PDSP and PDCP micelles possess spherical morphology at smaller measured sizes compared to the size data obtained by dynamic light scattering, which was attributed to the fact that these micelles were dried and their polyethylene glycol outer layer shrunk after losing water in the TEM experiments.
Stability of Micelles
Figure 2A shows the dilution stability of the PDSP and PDCP micelles. The particle size of PDSP and PDCP micelles did not change significantly when diluted up to 100 times, suggesting that PDSP and PDCP micelles could have good stability when diluted by the blood in body.26 Figure 2B shows the storage stability of PDSP and PDCP micelles at room temperature and 4 °C. The particle size of all micelles hardly changed at room temperature and 4 °C within 12 days. These results clearly show that both micelles had good storage stability over the measured period. Figure 2C shows the plasma stability of the polymer prodrug micelles. The particle sizes of PDSP and PDCP micelle did not change significantly for 48 h after being mixed with rat plasma, suggesting that they could remain stable when administered intravenously.29
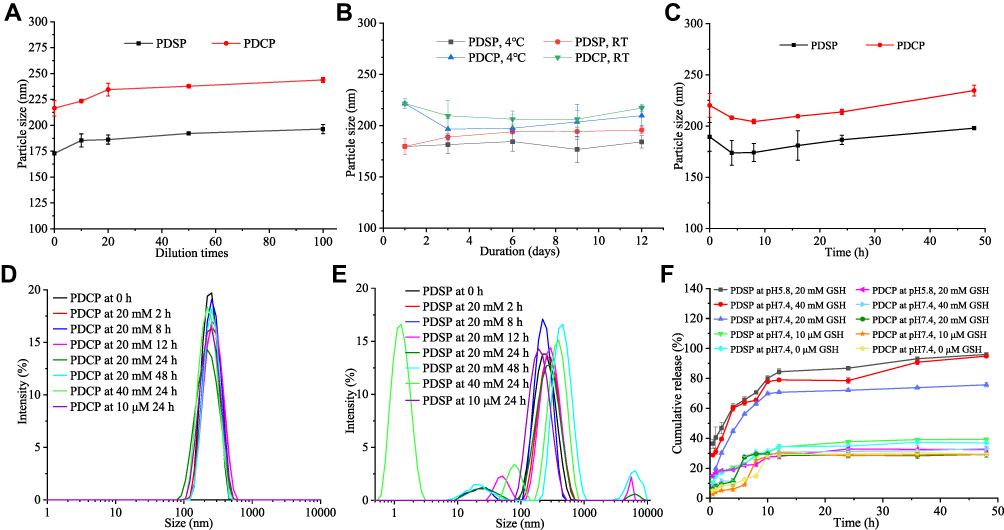 |
Figure 2 The results of in vitro stability and reduction-sensitivity test. (A) Dilution stability. (B) Storage stability. (C) Plasma stability. (D) Reduction-sensitive release in the environment with different levels of glutathione (GSH). (E) Reduction-sensitivity of mPEG-DCA-CC-PTX (PDCP) micelles. (F) Reduction-sensitivity of mPEG-DCA-SS-PTX (PDSP) micelles. |
In vitro Release
In some studies, it has been confirmed that the NP prodrugs developed with disulfide-bonds could be sensitive to reductants, thereby releasing drugs on-demand.38,39 In this study, we determined the reduction-responsive drug release in GSH-containing medium at different concentrations and pH values to predict the stability in blood circulation and the efficiency of disulfide-bond-bridged prodrug micelles response to the microenvironment of tumor cells. As shown in Figure 2D, the disulfide-bond-bridged prodrug micelles exhibited GSH-triggered drug release. It can be seen that about 75% and 95% of PTX were released from PDSP micelles within 48h when incubated in PBS (pH 7.4) and 20 mM and 40 mM of GSH. However, only about 30% of PTX was generated within 48 h in 10 μM and 0 μM GSH solution (pH 7.4), which are equivalent concentrations of GSH in blood and extracellular matrix (about 2~20 μM).40 These results imply that the PDSP prodrug had good stability in blood and a keen response to a high concentration of GSH in tumor cells. Thus, the prodrug could specifically deliver a drug into tumor cells.26 In contrast, less than 30% of PTX was released from PDCP micelles after incubation in all mediums with different concentrations of GSH and pH value, which indicated that PDCP micelles were not sensitive even when co-incubated with large concentrations of GSH. Additionally, Figure 2D shows that over 95% of PTX was released from PDSP prodrugs when incubated in PBS (pH 5.8) with 20 mM of GSH, implying that PDSP micelles should be sensitive to an acidic environment in tumor cells. Consequently, reduction-sensitive prodrugs can selectively release drugs based on the amount of external GSH and the weak acid environment.
Reduction-Response of Micelles
The size changes of PDSP and PDCP prodrug micelles after incubation with different concentrations of GSH could further reflect their sensibility to a reductive environment. In this section, the micelle size’s response to the reductive environment was investigated, and the results are shown in Figure 2E and F. As shown in Figure 2E, the average size of PDCP micelles had not changed after incubation for 24 h at many different concentrations of GSH, and hardly changed even at high GSH concentrations (40 mM GSH for 24 h) or for longer periods (20 mM GSH for 48 h). This indicates that PDCP was not sensitive to the reductive environment in tumor cells. However, the particle size of PDSP micelles increased significantly with time when incubated in 20 mM GSH for 48 h or 40 mM GSH for 24 h, as shown in Figure 2F. These results suggest that PDSP micelle had good reduction sensitivity, which was consistent with the in vitro release test results.
Biocompatibility in vitro
The hemolysis test could be used to estimate the toxicity of nanoparticles to red blood cells. When the hemolysis ratios are lower than the maximum hemolysis rate (5%) for the medical device requested by the FDA,41,42 the nanoparticles are considered good hemocompatibility. In this study, hemolysis was not observed after the two polymer prodrug micelles were incubated with rat blood for 2 h (as shown in Figure 3A). The hemolysis rates calculated by the absorbance were less than 5% even for the highest concentration of PDSP and PDCP micelles (0.5 mg/mL as shown in Figure 3B), which indicates that the two polymer prodrug micelles did not contribute to RBC membrane related toxicity. Based on these results, it seems that all of the PTX micelles had good blood compatibility.
 |
Figure 3 In vitro biosafety tests. (A) Hemolysis (i: Distilled water, ii: Saline, iii-v: mPEG-DCA-SS-PTX (PDSP) at 0.1, 0.3, 0.5 mg/mL, vi-viii: mPEG-DCA-CC-PTX (PDCP) at 0.1, 0.3, 0.5 mg/mL). (B) Hemolysis rate. (C) The cytotoxicity of blank micelle (BM) to HL-7702 cells. (D) The cytotoxicity of PDSP, PDCP, Paclitaxel (PTX) injection and PTX injection+BM to HL-7702 cells at 24 h. (E) The cytotoxicity of PDSP, PDCP, PTX injection and PTX injection+BM to HL-7702 cells at 48 h. Data were presented as mean ±SD. *P<0.05, **P<0.01. |
To study the biosafety of polymer prodrug micelles, the in vitro cytotoxicity tests of BM, PTX, PTX+BM, PDCP, and PDSP micelles to normal HL-7702 cells was carried out by an MTT assay. Figure 3C shows the effect of BM on the proliferation of HL-7702 cells, where its inhibition rate was less than 10%. This implies that mPEG-DCA had very good biosafety. As shown in Figure 3D, the viability of cells treated by the two drug-loaded micelles was more than 75% at a concentration of 25 μg/mL after incubation for 24 h, and the cell viability was still over 65% even incubated for 48 h. This indicates that the PDCP and PDSP micelles were safe. The cytotoxicity of PDCP and PDSP micelles to HL-7702 cells was not significantly different. However, the PTX injection and PTX injection+BM showed strong cytotoxicity for HL-7702 cells, where cell viability was lower than 40% when incubated at a 25 μg/mL concentration for 48 h (seen in Figure 3E). This suggests that the PTX injection had high cytotoxicity to normal cells, primarily attributed to excipients such as Cremophor EL.21,26 However, the cytotoxicity of the PTX injection and PTX injection+BM to HL-7702 cells were almost equivalent, which further confirmed that mPEG-DCA was biocompatible. Therefore, PDCP and PDSP micelles could eliminate toxicities induced by the solubilizer in the PTX injection and are highly biocompatible.
In vitro Cytotoxicity and Anti-Drug Resistance of Tumor Cells
The results of cytotoxicity for the PTX injection, PTX injection+BM, PDCP, PDCP+GSH, PDSP, and PDSP+GSH to SMMC-7721, MCF-7, A549/PTX cells are shown in Figure 4. As shown in Figure 4, the killing effect of PTX on the three kinds of tumor cells was weaker compared with other formulations, and its IC50 was similar to the results reported in literatures.26,43,44 As shown in Figure 4A and B, the survival rate of SMMC-7721 cells decreased with an increase of PTX concentration after 24 h or 48 h incubation. Moreover, the IC50 of six formulations against SMMC-7721 cells after incubation for 48 h was significantly lower than that for 24 h (Table S2), suggesting that the toxicity of each formulation to SMMC-7721 cells increased with an increase in incubation time. These results also suggest that the anti-tumor activity of each formulation was not only time-dependent but also concentration-dependent. More importantly, it was found that the cell survival rate (51% and 42%) in the PTX injection+BM group was lower than that of PTX injection (59% and 53%) at 24 h and 48 h, and that the corresponding IC50 was 1.88 and 0.83 times lower than that of the PTX injection, respectively. This suggests that BM may have activity in killing tumor cells. At 24 h and 48 h, the IC50 values of the PDCP+GSH group were slightly lower but not significantly different than that of the PDCP group, which indicated that even high concentrations of GSH could not influence the anti-tumor activity of PDCP because of its non-responsiveness to GSH. However, the IC50 of the PDSP group was 1.94, 1.79 times greater than that of the PDSP+GSH group, and significantly lower than those of the other four groups, indicating that a high concentration of GSH in cells could enhance the anti-tumor activity of PTX in PDSP due to its reduction-sensitivity. It can be seen from Figure 4C and D, the cytotoxicity of all formulations to MCF-7 cells was similar to those SMMC-7721 cells. It also confirms that the reduced sensitivity of PDSP could enhance the anti-tumor activity of PTX. These results suggest that PDSP could deliver PTX to tumor cells and release PTX in response to the strong reductive environment of the tumor, thus improving the anti-tumor activity of PTX.27,45
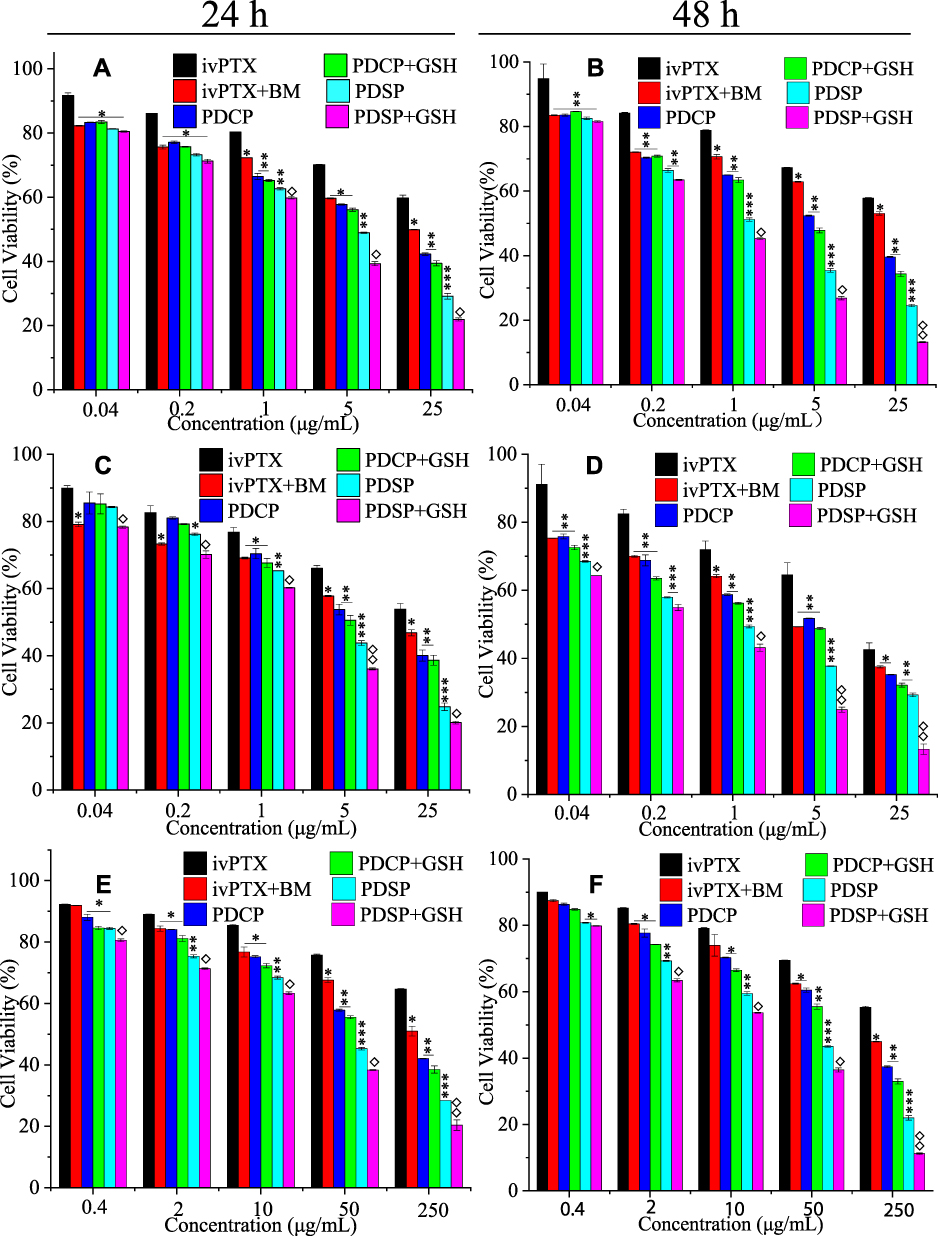 |
Figure 4 The cytotoxicity of six PTX formulations to SMCC-7721 cells (A and B), MCF-7 cells (C and D), and A549/PTX cells (E and F) at 24 h and 48 h. Data were presented as mean ±SD. The asterisk indicates the statistical significance between other formulations to Paclitaxel (PTX) injection, the rhombus indicates the statistical significance between mPEG-DCA-SS-PTX (PDSP) and PDSP+ glutathione (GSH). *P<0.05, **P<0.01, ***P<0.001, and ◊P<0.05, ◊◊P<0.01. BM, PDCP represents blank micelle and mPEG-DCA-CC-PTX, respectively. |
In order to study the effect of various formulations on anti-tumor drug resistance, their cytotoxicity to the drug-resistant cell line A549/PTX cells was investigated. From Figure 4E and F, it can be seen that the inhibitory efficiency of each formulation on the growth of A549/PTX cells increased as PTX concentration and incubation time also increased, indicating that all formulations inhibited the proliferation of A549/PTX cells in a concentration-dependent and time-dependent manner. At 24 h and 48 h, the inhibitory effect of PTX injection+BM on the growth of A549/PTX cells was 7.34 and 2.42 times lower than those of the PTX injection, which this difference was more significant than those of sensitive cell lines SMMC-7721 and MCF-7. These results suggest that BM can not only kill tumor cells but also reverse tumor drug resistance. This anti-tumor activity of BM would cooperate with PTX to kill the tumor in a two-pronged way for PDSP micelles. For PDCP+GSH and PDCP, the difference of their IC50 against A549/PTX cells was similar to those for drug-sensitive cell lines SMMC-7721 and MCF-7, which indicates that high concentrations of GSH could not improve the activity of PDCP to drug-resistant cells. This result was attributed to the fact that PDCP did not respond to GSH to release drugs and mPEG-DCA in tumor cells, so mPEG-DCA did not reverse the drug resistance of tumor cells. On the other hand, the IC50 of PDSP+GSH against A549/PTX was 1.08 and 1.16 times lower than those of PDSP, indicating that the high level of GSH increased the anti-tumor activity of PDSP due to its reduction sensitivity. In addition, we can also find that the IC50 ratios of PDSP to PDCP against A549/PTX cells were larger than those of drug-sensitive cell lines SMMC-7721 and MCF-7, suggesting that PDSP increases the inhibitory effect of PTX on A549/PTX cells more dramatically. The above result indicates that PDSP can significantly reverse the drug resistance of the tumor.
In vivo Anti-Tumor Activity and Toxicity Evaluation
Figure 5A lists the dimension, shape of the tumor, and the trends of change in tumor volume in various groups after intravenous administration over 14 days. Tumors treated with saline grew rapidly and reached 2465 mm3 on the 14th day. However, the tumors in mice administered with PTX injection, PDCP micelles, and PDSP micelles only grew to approximately 1427, 896, and 352 mm3, respectively. This indicates that the tumor growth was inhibited by these drug-loaded formulations at different levels. Compared with the PTX injection, the PDSP micelles could significantly inhibit tumor growth (P<0.01), which may be attributed to its reduction-sensitive drug release. Moreover, the reduction insensitive PDCP micelles displayed much weaker inhibitory effects on tumor growth compared with the PDSP micelles (P<0.01). The tumor inhibition rates were calculated by comparing the average tumor volume and mass in the mice treated by the three PTX formulations compared to those treated by saline (Figures 5B and S5). Overall formulations, PDSP micelles displayed the highest tumor inhibition rates, which suggests that the PDSP micelles have better antitumor activity compared with the other two formulations.
 |
Figure 5 The results of in vivo anti-tumor activity of the different Paclitaxel (PTX) formulations (such as PTX injection, mPEG-DCA-CC-PTX (PDCP) and mPEG-DCA-SS-PTX (PDSP)) to H22 tumor in mice. (A) The change in tumor volume and the tumor shape (n=10), **P<0.01, ***P<0.001. (B) The inhibition rate of tumor volume, *P<0.05, ***P<0.001. (C) The change in the bodyweight of mice for 14 days. (D) The curve of mice survival for 14 days. |
The changing trend in the bodyweight of mice is shown in Figure 5C, from which it can be seen that there was no significant change in body weight of all animals within 14 days of administration. For the groups treated with PDSP and PDCP prodrug micelles, the low fluctuation of body weight might be attributed to the fact that the two polymer prodrug micelles did not induce observed systemic toxicity. The bodyweight of mice in the PTX injection group had no significant change, which was consistent with the results mentioned in previous literature.38,46 Therefore, the systemic toxicity of each formulation could not be accurately reflected by the change of body weight, such that further studies on biosafety by other methods are necessary. Figure 5D showed the survival rates of tumor-bearing mice in various test groups. The survival rates of mice injected with the PDSP micelles groups reached 100%, but that of the PTX injection group was only 60%, which is being interpreted as the following: In PTX injection, a 50:50 mixture of Cremophor EL and ethanol has been used as a solvent to overcome its poor aqueous solubility.47 As is well known, Cremophor EL has shown many side effects (including hypersensitivity, nephrotoxicity, and neurotoxicity48,49), which seriously limit its clinical application; However, PDSP micelles completely avoids the use of biologically unacceptable solvents as Cremophor EL. Therefore, the PDSP micelles may have better biosafety compared with the PTX injection.
The images of primary organ slices stained by hematoxylin-eosin (H&E) are shown in Figure 6. No obvious abnormalities were observed in the slices of the heart, liver, and kidney for the PDSP and PDCP groups. However, the PTX injection-induced severe pathological changes in the heart, liver, and kidney with distinct cell degeneration, inflammatory cell infiltration, and hemorrhage. These results suggest that PDSP and PDCP had good biocompatibility and that the PTX injection had obvious side effects.23,31 In histological images of tumor slices, all the groups except for the saline group showed different levels of tumor cell necrosis, which was characterized by distinct debris. Among these treatment groups, tumor tissues treated by PDSP micelles exhibited the highest level of necrosis and the fewest number of proliferating cells, indicating strong inhibition of malignant proliferation. In summary, our results suggest that the PDSP micelles exhibit enhanced antitumor activity and less systemic toxicity.
 |
Figure 6 The images of the heart, liver, kidney, and tumor slices stained with H&E dyes after treatment by the different Paclitaxel (PTX) formulations, such as PTX injection, mPEG-DCA-CC-PTX (PDCP) and mPEG-DCA-SS-PTX (PDSP). Black arrows indicate tissue injury in this area, such as cell degeneration, necrosis, inflammatory cell infiltration and intra-tissue bleeding. |
Biodistribution Test
As shown in Figure 7, the biodistribution of PTX was significantly influenced by the PTX micelle formulations. Firstly, the PTX injection displayed high concentrations in various tissues at 2 h after administration and was quickly eliminated from all the tissues thereafter, which indicated that the PTX injection is rapidly distributed and eliminated in vivo. The concentration of free PTX from all formulations was very high at 2 h in the kidney, which may be attributed to the fact that the kidney is one of the main organs for the excretion of drugs. PTX concentration determined from the two micelle groups at 2 h showed the similar to that from PTX injection group in various normal tissues besides the liver and kidney and was high in the kidney within 6 h, which was similar to that reported by Zhang et al.23 These results may be attributed to the abrupt release and leakage of a small amount of PTX from micelles in the physiological environment, as indicated by the results of in vitro release test in pH7.2 medium. We do not know what the long-term effects of this may be, but our results warrant close examination of potential side effects in future preclinical studies. From the PDSP micelles group, most PTX can be observed in the liver within 24 h, possibly due to the liver-targeting property of deoxycholic acid in the polymer prodrug, which could mean that PDSP would have a better effect on an in-situ tumor of the liver.50–53 The accumulation of PTX from PDSP and PDCP micelles in tumor tissues at 24 h was significantly higher than that of PTX injection (5.66 and 2.72 times, P<0.001 and P<0.05). Moreover, the TTR of PDSP and PDCP micelles in tumor tissues within 24 h was higher than that of PTX injection (seen from Figure S6, 1.62 and 1.33 times). This phenomenon may be attributed to the EPR effect,45,54–56 which was favorable for suppressing tumor cells. As shown in Figure 5, the level of free PTX detected in tumors treated with PDSP micelles was higher than that of PDCP micelles within 6 h~24 h, which may be due to the specific and rapid release of PTX from PDSP micelles induced by a high level of GSH in the tumor cells, and the inhibitory effect of mPEG-DCA on drug efflux. These results explain why tumors’ dimension in mice treated with PDSP micelles was significantly smaller than those treated with PDCP micelles.
 |
Figure 7 Body distribution of Paclitaxel (PTX) after i.v. administration of mPEG-DCA-CC-PTX (PDCP), mPEG-DCA-SS-PTX (PDSP) and PTX injection to mice at 2 h, 6 h, 12 h, 24 h (n=6), *P<0.05, ***P<0.001. |
Conclusions
In this study, a new reduction-sensitive polymer prodrug was successfully synthesized and self-assembled into nano-micelles by a thin-film dispersion-sonication method to reverse the tumor’s MDR. The polymer prodrug micelles had a particle size of about 200 nm, spherical morphology, and good physical stability. These micelles did not have aggregation and protein adsorption in plasma. PDSP micelles have strong reduction and acidic sensitivity. The micelles would have good biocompatibility for intravenous administration, as indicated by the in vitro biosafety test results. In vitro cell test, mPEG-DCA could improve the anti-tumor activity of PTX, and especially enhance the sensitivity of drug-resistant cell lines to PTX. Moreover, PDSP micelles had a more substantial killing effect on tumor cells than the PTX injection and PDCP micelles, especially for drug-resistant cells. It is preliminarily proved that the PDSP could also combine with PEG-DCA and PTX to kill tumors in a two-pronged way and overcome tumor MDR. In vivo test, PDSP micelles showed strong anti-tumor activity, good biosafety, and effectively targeted tumors in tumor-bearing mice by releasing PTX in tumor tissues. To sum up, the PDSP micelles in this study can target PTX into tumor cells, and exert synergistic anti-tumor effects of chemotherapeutic drugs and MDR polymer inhibitors. This is expected to become an alternative delivery system for chemotherapeutic drugs such as PTX and other substrates of P-gp. For this two-pronged drug delivery system, the pharmacokinetics, the specific mechanism of anti-MDR and anti-tumor, and potential long-term side effects of intact micelles in vivo may need to be further studied in the future.
Data Sharing Statement
The data in our manuscript and supporting information are available from Xiaohui Pu ([email protected]) or Lanlan Zong ([email protected]).
Acknowledgments
We are grateful for financial support from the National Natural Science Foundation of China (No. U1904155), the Henan provincial key research development, and a special project for promotion (No. 202102310483).
Disclosure
All authors have no conflicts of interest for this work to disclose.
References
1. Smith RA, Andrews KS, Brooks D, et al. Cancer screening in the United States, 2019: a review of current American Cancer Society guidelines and current issues in cancer screening. CA Cancer J Clin. 2019;69(3):184–210. doi:10.3322/caac.21557
2. Zheng P, Liu Y, Chen J, Xu W, Li G, Ding J. Targeted pH-responsive polyion complex micelle for controlled intracellular drug delivery. Chin Chem Letters. 2020;31(5):1178–1182. doi:10.1016/j.cclet.2019.12.001
3. Wang Y, Wang X, Zhang J, et al. Gambogic acid-encapsulated polymeric micelles improved therapeutic effects on pancreatic cancer. Chin Chem Letters. 2019;30(4):885–888. doi:10.1016/j.cclet.2019.02.018
4. Paul S, Moye-Rowley WS. Multidrug resistance in fungi: regulation of transporter-encoding gene expression. Front Physiol. 2014;5(143). doi:10.3389/fphys.2014.00143
5. Wu J, Lu Y, Lee A, et al. Reversal of multidrug resistance by transferrin-conjugated liposomes co-encapsulating doxorubicin and verapamil. J Pharm Pharm Sci. 2007;10(3):350–357.
6. Huo Q, Zhu J, Niu Y, et al. pH-triggered surface charge-switchable polymer micelles for the co-delivery of paclitaxel/disulfiram and overcoming multidrug resistance in cancer. Int J Nanomedicine. 2017;12:8631–8647. doi:10.2147/ijn.s144452
7. Wang J, Wang F, Li F, et al. A multifunctional poly(curcumin) nanomedicine for dual-modal targeted delivery, intracellular responsive release, dual-drug treatment and imaging of multidrug resistant cancer cells. J Mater Chem B. 2016;4(17):2954–2962. doi:10.1039/C5TB02450A
8. Zhou Y, Wang S, Ying X, et al. Doxorubicin-loaded redox-responsive micelles based on dextran and indomethacin for resistant breast cancer. Int J Nanomedicine. 2017;12:6153–6168. doi:10.2147/IJN.S141229
9. Zhang X, He F, Xiang K, et al. CD44-targeted facile enzymatic activatable chitosan nanoparticles for efficient antitumor therapy and reversal of multidrug resistance. Biomacromolecules. 2018;19(3):883–895. doi:10.1021/acs.biomac.7b01676
10. Yin S, Huai J, Chen X, et al. Intracellular delivery and antitumor effects of a redox-responsive polymeric paclitaxel conjugate based on hyaluronic acid. Acta Biomater. 2015;26:274–285. doi:10.1016/j.actbio.2015.08.029
11. Zhong Y, Goltsche K, Cheng L, et al. Hyaluronic acid-shelled acid-activatable paclitaxel prodrug micelles effectively target and treat CD44-overexpressing human breast tumor xenografts in vivo. Biomaterials. 2016;84:250–261. doi:10.1016/j.biomaterials.2016.01.049
12. Zhang X, Wang S, Cheng G, Yu P, Chang J, Chen: X. Cascade drug-release strategy for enhanced anticancer therapy. Matter. 2021;4(1):26–53. doi:10.1016/j.matt.2020.10.002
13. Cao Y, Wei Z, Li M, et al. Formulation, pharmacokinetic evaluation and cytotoxicity of an enhanced-penetration paclitaxel nanosuspension. Curr Cancer Drug Targets. 2019;19(4):338–347. doi:10.2174/1568009618666180629150927
14. Zong L, Wang H, Hou X, et al. A novel GSH-triggered polymeric nanomicelles for reversing MDR and enhancing antitumor efficiency of hydroxycamptothecin. Int J Pharm. 2021;600:120528. doi:10.1016/j.ijpharm.2021.120528
15. Huizing MT, Misser VHS, Pieters RC, et al. Taxanes: a new class of antitumor agents. Cancer Invest. 1995;13(4):381–404. doi:10.3109/07357909509031919
16. Kou L, Sun R, Xiao S, et al. OCTN2-targeted nanoparticles for oral delivery of paclitaxel: differential impact of the polyethylene glycol linker size on drug delivery in vitro, in situ, and in vivo. Drug Deliv. 2020;27(1):170–179. doi:10.1080/10717544.2019.1710623
17. Li H, Li J, He X, et al. Histology and antitumor activity study of PTX-loaded micelle, a fluorescent drug delivery system prepared by PEG-TPP. Chin Chem Letters. 2019;30(5):1083–1088. doi:10.1016/j.cclet.2019.01.003
18. Zhang B, Xue A, Zhang C, Yu J, Chen W, Sun: D. Bile salt liposomes for enhanced lymphatic transport and oral bioavailability of paclitaxel. Pharmazie. 2016;71(6):320–326.
19. Jang SH, Wientjes MG, Au: JLS. Kinetics of P-glycoprotein-mediated efflux of paclitaxel. J Pharmacol Exp Ther. 2001;298(3):1236–1242.
20. Gallo JM, Li S, Guo P, Reed K, Ma: J. The effect of P-glycoprotein on paclitaxel brain and brain tumor distribution in mice. Cancer Res. 2003;63(16):5114–5117.
21. Li M, Chen Y, Yin L, et al. Formulation and stability evaluation of structure-altered paclitaxel nanosuspensions stabilized by a biocompatible amino acid copolymer. Sci Adv Mater. 2017;9(10):1713–1723. doi:10.1166/sam.2017.3229
22. Yu M, Su D, Yang Y, et al. D-T7 peptide-modified PEGylated bilirubin nanoparticles loaded with cediranib and paclitaxel for antiangiogenesis and chemotherapy of glioma. ACS Appl Mater Interfaces. 2019;11(1):176–186. doi:10.1021/acsami.8b16219
23. Zhang J, Du Q, Song X, et al. Evaluation of the tumor-targeting efficiency and intratumor heterogeneity of anticancer drugs using quantitative mass spectrometry imaging. Theranostics. 2020;10(6):2621–2630. doi:10.7150/thno.41763
24. Ding D, Sun B, Cui W, et al. Integration of phospholipid-drug complex into self-nanoemulsifying drug delivery system to facilitate oral delivery of paclitaxel. Asian J Pharmaceut Sci. 2019;14(5):552–558. doi:10.1016/j.ajps.2018.10.003
25. Liu Y, Sun J, Cao W, et al. Dual targeting folate-conjugated hyaluronic acid polymeric micelles for paclitaxel delivery. Int J Pharm. 2011;421(1):160–169. doi:10.1016/j.ijpharm.2011.09.006
26. Chang S, Wang Y, Zhang T, et al. Redox-responsive disulfide bond-bridged mpeg-pbla prodrug micelles for enhanced paclitaxel biosafety and antitumor efficacy. Front Oncol. 2019;9(823). doi:10.3389/fonc.2019.00823
27. Luo C, Sun B, Wang C, et al. Self-facilitated ROS-responsive nanoassembly of heterotypic dimer for synergistic chemo-photodynamic therapy. J Controlled Release. 2019;302:79–89. doi:10.1016/j.jconrel.2019.04.001
28. Zong L, Wang Y, Qiao P, et al. Reduction-sensitive poly(ethylene glycol)–polypeptide conjugate micelles for highly efficient intracellular delivery and enhanced antitumor efficacy of hydroxycamptothecin. Nanotechnology. 2020;31(16):165102. doi:10.1088/1361-6528/ab6749
29. Pu X, Zhao L, Li J, et al. A polymeric micelle with an endosomal pH-sensitivity for intracellular delivery and enhanced antitumor efficacy of hydroxycamptothecin. Acta Biomater. 2019;88:357–369. doi:10.1016/j.actbio.2019.02.039
30. Qiao Y, Wei Z, Qin T, et al. Combined nanosuspensions from two natural active ingredients for cancer therapy with reduced side effects. Chin Chem Letters. 2021;32(9):2877–2881. doi:10.1016/j.cclet.2021.03.049
31. Luo C, Sun J, Sun B, et al. Facile fabrication of tumor redox-sensitive nanoassemblies of small-molecule oleate prodrug as potent chemotherapeutic nanomedicine. Small. 2016;12(46):6353–6362. doi:10.1002/smll.201601597
32. Liu H, Wu S, Yu J, et al. Reduction-sensitive micelles self-assembled from amphiphilic chondroitin sulfate A-deoxycholic acid conjugate for triggered release of doxorubicin. Mater Sci Eng C Mater Biol Appl. 2017;75:55–63. doi:10.1016/j.msec.2017.02.030
33. Collnot EM, Baldes C, Wempe MF, et al. Mechanism of inhibition of P-glycoprotein mediated efflux by vitamin E TPGS: influence on ATPase activity and membrane fluidity. Mol Pharm. 2007;4(3):465–474. doi:10.1021/mp060121r
34. Gao Y, Li LB, Zhai: G. Preparation and characterization of Pluronic/TPGS mixed micelles for solubilization of camptothecin. Colloids Surf B Biointerfaces. 2008;64(2):194–199. doi:10.1016/j.colsurfb.2008.01.021
35. Tung NT, Tran CS, Pham TM, et al. Development of solidified self-microemulsifying drug delivery systems containing l-tetrahydropalmatine: design of experiment approach and bioavailability comparison. Int J Pharm. 2018;537(1–2):9–21. doi:10.1016/j.ijpharm.2017.12.027
36. Taheri M, Bagheri M, Moazeni-Pourasil RS, Ghassempour: A. Response surface methodology based on central composite design accompanied by multivariate curve resolution to model gradient hydrophilic interaction liquid chromatography: prediction of separation for five major opium alkaloids. J Sep Sci. 2017;40(18):3602–3611. doi:10.1002/jssc.201700416
37. Alavi M, Nokhodchi: A. Micro- and nanoformulations of paclitaxel based on micelles, liposomes, cubosomes, and lipid nanoparticles: recent advances and challenges. Drug Discov Today. 2021. doi:10.1016/j.drudis.2021.10.007
38. Sun B, Luo C, Yu H, et al. Disulfide bond-driven oxidation- and reduction-responsive prodrug nanoassemblies for cancer therapy. Nano Lett. 2018;18(6):3643–3650. doi:10.1021/acs.nanolett.8b00737
39. Yu W, Shevtsov M, Chen X, Gao: H. Advances in aggregatable nanoparticles for tumor-targeted drug delivery. Chin Chem Letters. 2020;31(6):1366–1374. doi:10.1016/j.cclet.2020.02.036
40. Chi Y, Yin X, Sun K, et al. Redox-sensitive and hyaluronic acid functionalized liposomes for cytoplasmic drug delivery to osteosarcoma in animal models. J Control Release. 2017;261:113–125. doi:10.1016/j.jconrel.2017.06.027
41. Liu MC, Liu B, Sun XY, et al. Core/Shell structured Fe(3)O(4)@TiO(2)-DNM nanospheres as multifunctional anticancer platform: chemotherapy and photodynamic therapy research. J Nanosci Nanotechnol. 2018;18(7):4445–4456. doi:10.1166/jnn.2018.15338
42. Yang X, Cai X, Yu A, Xi Y, Zhai: G. Redox-sensitive self-assembled nanoparticles based on alpha-tocopherol succinate-modified heparin for intracellular delivery of paclitaxel. J Colloid Interface Sci. 2017;496:311–326. doi:10.1016/j.jcis.2017.02.033
43. Zhang J, Zhao X, Chen Q, et al. Systematic evaluation of multifunctional paclitaxel-loaded polymeric mixed micelles as a potential anticancer remedy to overcome multidrug resistance. Acta Biomater. 2017;50:381–395. doi:10.1016/j.actbio.2016.12.021
44. Gu Y, Zhong Y, Meng F, Cheng R, Deng C, Zhong: Z. Acetal-linked paclitaxel prodrug micellar nanoparticles as a versatile and potent platform for cancer therapy. Biomacromolecules. 2013;14(8):2772–2780. doi:10.1021/bm400615n
45. Yang B, Wang K, Zhang D, et al. Polydopamine-modified ROS-responsive prodrug nanoplatform with enhanced stability for precise treatment of breast cancer. RSC Adv. 2019;9(16):9260–9269. doi:10.1039/c9ra01230c
46. Sun B, Luo C, Zhang X, et al. Probing the impact of sulfur/selenium/carbon linkages on prodrug nanoassemblies for cancer therapy. Nat Commun. 2019;10(1):3211. doi:10.1038/s41467-019-11193-x
47. Fjallskog ML, Frii L, Bergh: J, Is Cremophor EL. Solvent for paclitaxel, cytotoxic? Lancet. 1993;342(8875):873. doi:10.1016/0140-6736(93)92735-C
48. Rowinsky EK, Donehower: RC. Paclitaxel (taxol). N Engl J Med. 1995;332(15):1004–1014. doi:10.1056/NEJM199504133321507
49. Malingrг© MM, Terwogt JM, Beijnen JH, et al. Phase I and pharmacokinetic study of oral paclitaxel. J clin oncol. 2000;18(12):2468. doi:10.1200/JCO.2000.18.12.2468
50. Chen ZP, Zhu JB, Chen HX, et al. Synthesis of a novel polymer bile salts-(polyethylene glycol)2000-bile salts and its application to the liver-selective targeting of liposomal DDB. Drug Dev Ind Pharm. 2010;36(6):657–665. doi:10.3109/03639040903410342
51. Chen ZP, Xiao L, Liu W, et al. Novel materials which possess the ability to target liver cells. Expert Opin Drug Deliv. 2012;9(6):649–656. doi:10.1517/17425247.2012.679261
52. Putz G, Schmider W, Nitschke R, Kurz G, Blum: HE. Synthesis of phospholipid-conjugated bile salts and interaction of bile salt-coated liposomes with cultured hepatocytes. J Lipid Res. 2005;46(11):2325–2338. doi:10.1194/jlr.M500144-JLR200
53. Parks DJ, Blanchard SG, Bledsoe RK, et al. Bile acids: natural ligands for an orphan nuclear receptor. Science. 1999;284(5418):1365–1368. doi:10.1126/science.284.5418.1365
54. Li Z, Zhu J, Wang Y, et al. In situ apolipoprotein E-enriched Corona guides dihydroartemisinin-decorating nanoparticles towards LDLr-mediated tumor-homing chemotherapy. Asian J Pharm Sci. 2020;15(4):482–491. doi:10.1016/j.ajps.2019.05.002
55. Wang Y, Song J, Chow SF, Chow AH, Zheng: Y. Particle size tailoring of ursolic acid nanosuspensions for improved anticancer activity by controlled antisolvent precipitation. Int J Pharm. 2015;494(1):479–489. doi:10.1016/j.ijpharm.2015.08.052
56. Hashizume H, Baluk P, Morikawa S, et al. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000;156(4):1363–1380. doi:10.1016/s0002-9440(10)65006-7
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.