Back to Journals » OncoTargets and Therapy » Volume 14
Tumor Mutation Burden and Differentially Mutated Genes Among Immune Phenotypes in Patients with Lung Adenocarcinoma
Authors Wang H, Chen S, Meng D, Wu C, Zhu J, Jiang M, Ning J, Wu S ![]() , Wu L, Li J, Chen B, Zhao S, Li W, Yu J, Fang Q, Zhu J
, Wu L, Li J, Chen B, Zhao S, Li W, Yu J, Fang Q, Zhu J ![]() , Zhao W, He Y
, Zhao W, He Y ![]() , Zhou C
, Zhou C
Received 1 December 2020
Accepted for publication 25 March 2021
Published 4 May 2021 Volume 2021:14 Pages 2953—2965
DOI https://doi.org/10.2147/OTT.S294993
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr William C. Cho
Hao Wang,1,2 Shanhao Chen,1 Die Meng,1,2 Chunyan Wu,3 Junjie Zhu,4 Minlin Jiang,1,2 Jing Ning,1,2 Shengyu Wu,1,2 Lijia Wu,5 Jingjie Li,5 Bin Chen,1 Sha Zhao,1 Wei Li,1 Jia Yu,1,2 Qiyu Fang,1,2 Jun Zhu,1,2 Wencheng Zhao,1,2 Yayi He,1 Caicun Zhou1
1Department of Medical Oncology, Shanghai Pulmonary Hospital, Shanghai, 200433, People’s Republic of China; 2Medical School, Tongji University, Shanghai, 200433, People’s Republic of China; 3Pathology Department, Shanghai Pulmonary Hospital, Shanghai, 200433, People’s Republic of China; 4Surgery Department, Shanghai Pulmonary Hospital, Shanghai, 200433, People’s Republic of China; 5Genecast Biotechnology Co., Ltd, Wuxi City, Jiangsu, 214104, People’s Republic of China
Correspondence: Yayi He; Caicun Zhou
Department of Medical Oncology, Shanghai Pulmonary Hospital, No. 507 Zhengmin Road, Shanghai, People’s Republic of China
Tel +86 13818828623
; Tel +86 13818828623
Email [email protected]; [email protected]
Introduction: Nowadays, immune checkpoint blockades (ICBs) have been extensively applied in non-small cell lung cancer (NSCLC) treatment. However, the outcome of anti-program death-1/program death ligand-1 (anti-PD-1/PD-L1) therapy is not satisfying in EGFR-mutant lung adenocarcinoma (LUAD) patients and its exact mechanisms have not been fully understood. Since tumor mutation burden (TMB) and tumor immune phenotype had been thought as potential predictors for efficacy of ICBs, we further studied the TMB and immune phenotype in LUAD patients to explore potential mechanisms for poor efficacy of ICBs in EGFR positive mutated patients and to find possible factors that could impact the tumor immune phenotype which might uncover some new therapeutic strategies or combination therapies.
Methods: We enrolled 223 LUAD patients who underwent surgery in our hospital. We evaluated TMB through targeted panel sequencing. The tumor immune phenotype, which could be divided into non-inflamed, intermediate and inflamed, was determined through immunohistochemistry using formalin-fixed paraffin-embedded samples. Enumeration data were analyzed by Chi-square test or Fisher exact test and shown as number (proportion). Logistic regression model was employed for univariate and multivariate analysis of the association between TMB levels and clinical characteristics.
Results: The median TMB level was 4.0445 mutations/Mb. Multivariate analysis showed the TMB level was significantly associated with age (P=0.026), gender (P=0.041) and EGFR mutation status (P=0.015), and in EGFR-mutant patients we found a lower proportion of patients with mutated KRAS and BRCA2. Furthermore, we found patients with or without metastatic lesions would have different immune phenotype (P=0.007). And the mutational frequencies of ALK, CDKN2A, MAP2K1, IDH2 and PTEN were significantly different among three immune phenotypes.
Conclusion: Low TMB level could be the reason for the poor efficacy of ICBs in patients having EGFR mutation. And mutational frequencies of KRAS and BRCA2 were lower in EGFR-mutant patients. Furthermore, ALK, CDKN2A, MAP2K1, IDH2 and PTEN might involve in the formation of immune phenotypes.
Keywords: lung adenocarcinoma, EGFR mutation, tumor mutation burden, immune phenotype, immune checkpoint blockade
Introduction
Nowadays, lung cancer is one of cancers with high mortality and morbidity worldwide1 and non-small cell lung cancer (NSCLC) constitutes 85% of lung cancer.2 With the progression of comprehensive genomic profiling, epidermal growth factor receptor (EGFR) and many other driver mutations have been deciphered.3 Moreover, in Chinese lung adenocarcinoma (LUAD) patients, EGFR mutation rate reached 47.5%.4 As the promising anti-tumor effect of EGFR tyrosine kinase inhibitors (TKIs) has been well demonstrated,5 EGFR-TKIs have been the first-line therapy for advanced NSCLC patients with EGFR mutation. Although target therapy can control tumor effectively, it is inevitable that patients will be resistant to the therapy eventually.6 Selection of subsequent treatment for these patients is necessary.
Recent years, immune checkpoint blockades (ICBs) related immunotherapy has developed rapidly. Some ICBs such as anti-PD-1/PD-L1 monoclonal antibodies have shown encouraging anti-tumor effect regardless of patients having pervious treatment or not.7,8 And they have been adopted as therapeutics for advanced NSCLC patients.
Thus, ICB may be an alternative therapeutics for patients who progressed after EGFR-TKIs. However, further analysis found that the outcome of anti-PD–1 therapy in patients with EGFR mutation was not satisfying.9–11 And its exact mechanisms, which are indispensable for improving the efficacy of ICB in patients after TKIs treatment and exploring new therapeutic strategies, have not been fully elucidated. Some potential biomarkers, such as tumor mutation burden (TMB)12,13 and tumor-infiltrating lymphocytes (TILs),14 have been proposed for predicting the efficacy of anti-PD-(L)1 therapy. Therefore, in this research, we collected treatment-naïve LUAD patients to evaluate the TMB landscape and mutation profile at baseline and assessed their immune phenotype according to tissue sections. Through these efforts, we tended to explore possible reasons to explain the poor outcome of ICBs in EGFR-mutant patients and found the potential factors that might influence the tumor immune phenotype.
Patients and Methods
Patients
We enrolled 223 formalin-fixed paraffin-embedded (FFPE) tissue blocks from patients who underwent surgery in Shanghai Pulmonary Hospital from Dec 2013 to Dec 2014 and reviewed their medical records. Disease staging was determined according to International Association for the Study of Lung cancer (IASLC) version 7th TNM staging system. No patients had been treated with chemotherapy, target therapy or immunotherapy before surgery. The research was approved by the Shanghai Pulmonary Hospital (NO. K18–203Y). All participants had provided their written consents and the study was conducted in accordance with the Declaration of Helsinki.
EGFR Mutation Status and ALK Rearrangement Examination
Genomic DNA or RNA were extracted from FFPE samples according to the manufacturer’s protocol. The extracted RNA would be reversed transcripted to cDNA for latter polymerase chain reaction (PCR) amplification and examination. EGFR mutations in exons 18-21 and EML4-ALK rearrangement were detected by AmoyDx EGFR 29 Mutations Detection Kit (Amoy, Xiamen, China) and AmoyDx EML4-ALK Fusion Gene Detection Kit (Amoy, Xiamen, China) respectively, according to the protocols. Patients with 19del, L858R, T790M, 20ins, G719X (X=A, C or S), S768I or L861Q would be classified as EGFR-mutant. Patients with positive EML4–ALK fusion were classified as ALK rearranged.
Targeted Panel Sequencing
MagPure FFPE DNA LQ kit B (Magen, Beijing, China) was used to extract DNA from FFPE samples according to the manufacturer’s instruction and the extracted DNA was stored in −20°C. 20–200ng DNA was used for fragmentation by Covaris L220 (Covaris, Massachusetts, USA). KAPA Hyper Prep Kit (Illumina platforms) (KAPA Biosystems, Massachusetts, USA) was used for library preparation and gene panel designed by NimbleGen SeqCap EZ Library (Roche, Wisconsin, USA) was applied for DNA library capturing. The sequencing was conducted through Illumina NovaSeqTM 6000 platform (Illumina, CA, USA).
TMB Evaluation
BWA aligner (v0.7.17)15 was used for data alignment after data quality control, the genome reference used was human hg19 reference. Then, the work for sorting and masking duplications in bam files were done through Picard. VarDict (v1.5.1)16 and FreeBayes (v1.2.0)17 were two callers introduced for mutations calling. Finally, all mutations were inputted into ANNOVAR (2015Jun17 −0700)18 for function annotation. After filtering the mutations with allele frequency greater than 0.002 in the Exome Aggregation Consortium (ExAC) database19 and Genome Aggregation Database (gnomAD),20 rest exonic and splicing mutations with VAF>5% and without strand bias were included into the TMB calculation. The final TMB value was normalized by the covered base.21
Immunohistochemistry (IHC)
The primary antibodies used in this study include CD8 (Dako, M7103, 1:200); FoxP3 (Biolegend, 320102, 1:100); PD-L1 (22C3)(Dako, M3653, 1:50). The immunohistochemistry staining of PD-L1 (22C3) was performed on Dako autostainerLink48 platform with Dako K8002 detection kit and amplified the signals with a mouse linker (contained in the kit) and enhanced the signals with DAB enhancer (Dako, S1961); the staining procedure was set the same to the FDA approved PD-L1 (22C3) PharmDx staining procedure. All the other 3 antibodies’ immunohistochemistry staining were performed in a humidified cube with Dako K5007 detection kit by hand, the main procedure is tissue slides were dewaxed with xylene, then rinsed with alcohol (decreasing serial concentration from 100% to 0). Antigen recovering was taken with the target retrieval solution kit (Dako, DM829) under hot high pressure for 10min. Cooling to room temperature, slides immersed with 0.3% H2O2 to reduce the background staining. Incubating the primary antibody on the slides for 1 hour at room temperature in the humiliated box. Rinsed with PBS, incubate the slides with the HRP-conjugated goat anti-Mouse/Rabbit IgG detection antibody for 30 min at room temperature. Rinsed with PBS, and visualizing the antigen with DAB, following with the standard procedures of counterstain for cell nuclear with hematoxylin and mounting of cover slides. All IHC sections were evaluated by two pathologists independently. We chose ≥1% as our cutoff for PD-L1 on tumor cells. And we defined FoxP3 ≥1% as positive.22,23 Representative IHC images were shown in Supplementary Figure 1.
Classification of Immune Phenotype
The classification was determined by viewing the IHC slides. Immunoscore I0, absence of CD8+ T cells at the tumour centre and in the invasive margin was thought as cold tumor (also called non-inflamed tumor/immune desert tumor). Immunoscore I4, high immune cell densities in both locations, was thought as hot tumor (also called inflamed tumor).24 Altered immune tumor as known as intermediate includes excluded tumor which is defined as low CD8+ T cell infiltration at the tumour centre and high at the invasive margin and immunosuppressed tumor which displays a more uniform pattern of low CD8+ T cell infiltration.
Statistics
Enumeration data were expressed as number (proportion) and were analyzed by Chi-square test or Fisher exact test. The association between TMB and clinical characteristics was assessed by univariate and multivariate analysis through logistic regression model. The statistical significance was considered as P-value less than 0.05. The statistical analysis was conducted through SPSS 22.0 (SPSS Inc., Chicago, IL, USA). All the landscape figures were plotted used the ComplexHeatmap R package and Chi–square test or Fisher exact test was used for differential analysis. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis were conducted through “clusterprofiler” R package.25
Results
Clinical Characteristics of Patients
We enrolled 223 patients totally who underwent surgery in our hospital from 2013 to 2014 (Table 1). All patients’ pathological classification was lung adenocarcinoma and had not had any treatment before. The median age of patients was 61 years old (range 28 to 83 years old). Records of smoking history were available in 168 (75.3%) patients. Among these patients, 82.7% had never smoked. Most patients were at stage I (71.5%) and one patient was microinvasive adenocarcinoma. More than half of patients (52.0%) had EGFR mutation and only 9.0% of patients had ALK rearrangement.
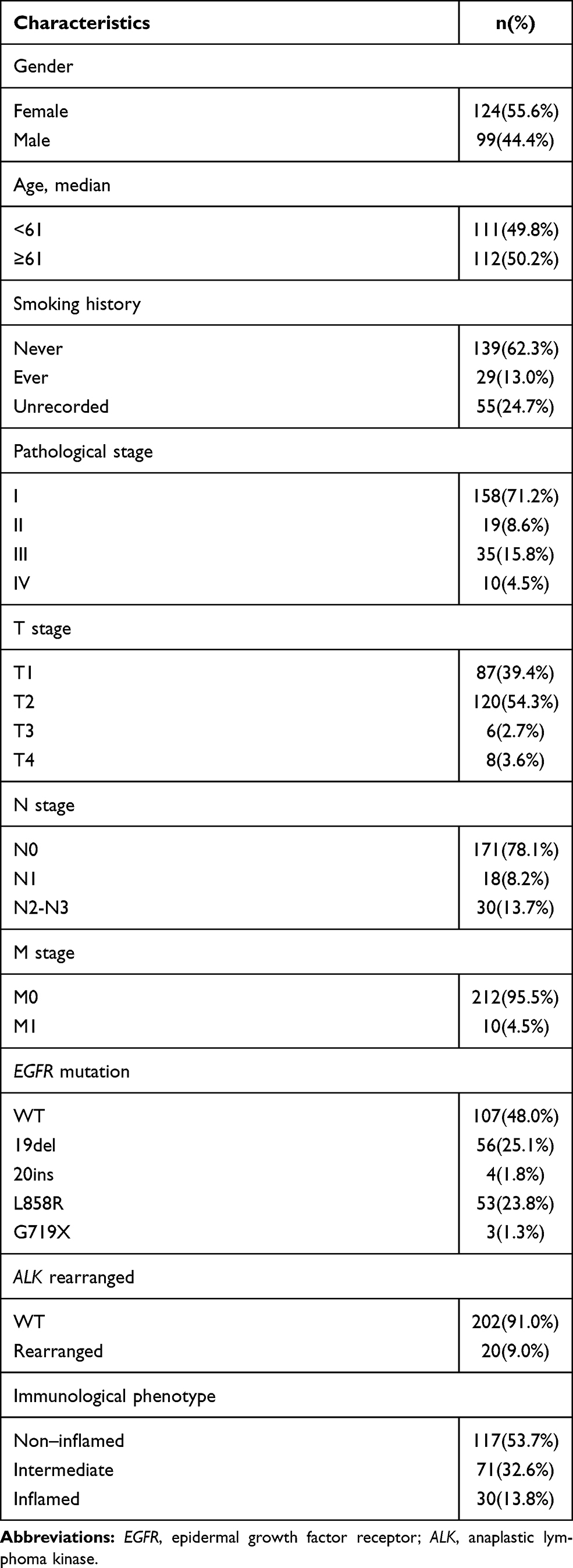 |
Table 1 Demographic and Clinical Characteristics of Patients |
TMB Analysis and Frequently Mutant Genes in RTK/RAS/RAF Signaling Pathway
We assessed the TMB level of 223 patients through targeted panel sequencing which contained 1406 genes. The median TMB level was 4.0445 mutations per megabase (range 0–47.8603, quartile range 2.0123–6.1092). The most frequent nonsynonymous mutation was missense mutation, other categories of nonsynonymous mutations, including insertions, deletions, nonsense mutations and splicing site mutations, were at relatively low frequency. And most affected genes were involved in the RTK/RAS/RAF pathway followed by the immune pathway (Figure 1A).
 |
Figure 1 The mutation profile of LUAD patients. (A) The frequency and types of mutations in genes were shown in this waterfall plots. (B) Mutations in RTK/RAS/RAF signaling pathway. |
Mutations involved in RTK/RAS/RAF signaling pathway were found in about 83.4% of LUAD patients. The most frequent genes with mutation were EGFR (48.4%) followed by KRAS (9.2%), ERBB2 (4.6%), MET (3.5%), FGFR3 (2.5%) and MAP2K1 (2.1%). Mutational frequencies of other genes in this pathway were relatively low in our cohort (Figure 1B).
Relationship Between TMB and Clinical Characteristics
We divided patients into two groups according to TMB level that had been applied in clinical trial13 (low, <10; high, ≥10). Female patients had a tendency to have lower TMB level (OR=0.222, 95% CI 0.053–0.939, P=0.041). We also got the odds ratio for younger patients was 4.266 (95% CI 1.187–15.337, P=0.026). And the odds ratio was 4.707 (95% CI 1.349–16.429, P=0.015) when comparing patients without EGFR mutation to patients with EGFR mutation. As sample size for EGFR 20ins and G719X mutation was too small, we combined them as uncommon mutation for subsequent analysis. We compared the TMB level among EGFR 19del, L858R and uncommon mutation but we did not find significant difference (P=0.611, Supplementary Table 1). As for ALK rearrangement, there was no significant correlation between ALK rearrangement and TMB level (OR=1.8×10−8, P=0.998). As few patients had ALK rearrangement in our cohort, the result should be further evaluated in larger cohorts. In our cohort, we did not find any association between TMB level and pathological stage, tumor size, metastatic status (Table 2). We also evaluated whether TMB level had an association with the expression of PD-L1 on tumor cells and no significant association was detected (OR=0.643, 95% CI 0.134–3.076, P=0.580). Furthermore, we analyzed the mutational frequencies of genes between TMB high and low groups and found some differentially mutated genes (Supplementary Table 2). GO analysis found these genes enriched in mismatch repair complex, DNA repair complex in cellular component (CC). In the biological process (BP) analysis, these genes were found to enrich in response to radiation, response to DNA damage. These genes were also enriched mainly in transcription factor binding, kinase activity and receptor binding in molecular function (MF) (Supplementary Figure 2A). KEGG analysis indicated that these genes mainly correlated with many cancers as well as canonical pathways of carcinogenesis (Supplementary Figure 2B).
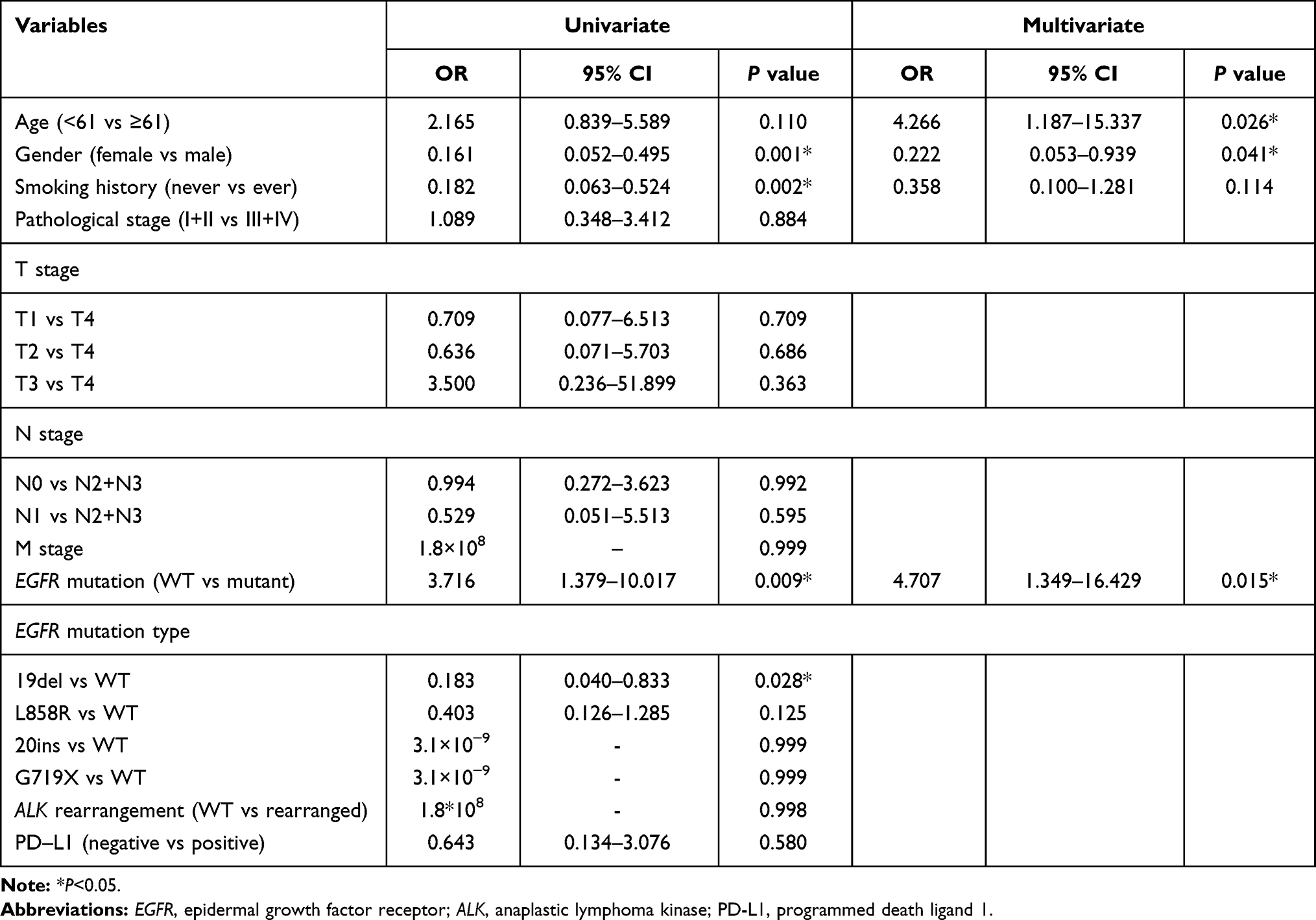 |
Table 2 Association Between TMB and Clinical Characteristics |
TMB Landscape at Different EGFR Mutation Status
As we found EGFR mutation status was associated with the TMB level of LUAD patients, we further compared if the mutant genes were different between patients with and without EGFR mutation. Thus, we divided patients into two groups according to their EGFR mutation status and found 116 (52.0%) patients had EGFR mutation. We analyzed mutations in both groups separately (Figure 2) and compared mutational frequencies of different genes between two groups. And differentially mutant genes were KRAS (3.45% vs 20.56%, P=0.0002, Supplementary Figure 3A) and BRCA2 (3.45% vs 12.15%, P=0.0283, Supplementary Figure 3B). As there were also EGFR-mutant patients having high TMB, we further evaluated differentially mutated genes between high TMB+EGFR-mutant and low TMB+EGFR-mutant patients. We found that the mutational frequencies of BRCA2 (33.33% vs 1.82%, P=0.0129), RB1 (33.33% vs 1.82%, P=0.0129), CDKN2A (33.33% vs 2.73%, P=0.0209), CYP2D6 (33.33% vs 2.73%, P=0.0209) and TP53 (83.33% vs 39.09%, P=0.0230) were lower in low TMB+EGFR-mutant patients (Supplementary Figure 4).
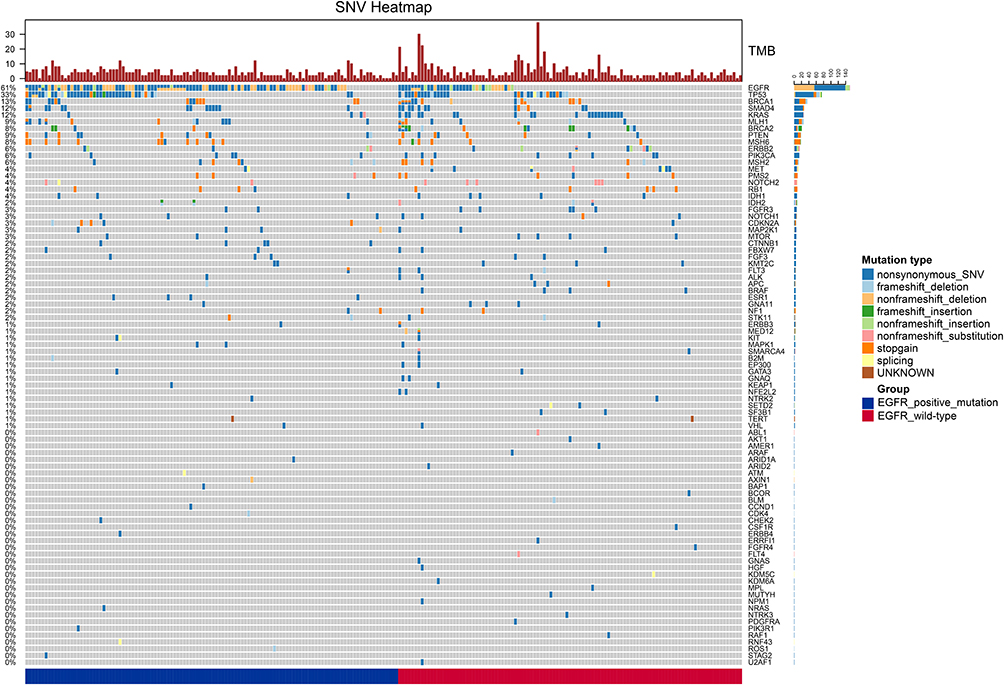 |
Figure 2 Profile of mutated genes in EFGR-mutant and wild-type patients. |
Relationship Between Immunological Phenotype and Clinical Characteristics
We were also curious about whether immune phenotype was different among patients with different EGFR mutation status and TMB level and tended to find which mutant gene might influence the tumor immune microenvironment. After reviewing tumor tissue sections, 218 (97.8%) specimens were available and according to IHC data we divided tumors into non-inflamed (117, 53.7%), intermediate (71, 32.6%) and inflamed (30, 13.8%). After further analysis, we found proportions of three immune phenotypes were similar among patients with different TMB level and EGFR mutation status (Table 3). Similar result was also observed when we compare the immune phenotypes among EGFR 19del, L858R and uncommon mutation (P=0.812, Supplementary Table 3). Meanwhile, we did not find that ALK rearrangement correlate to the immune phenotype (P=0.714). However, we found proportions of immune phenotypes were of statistical difference among patients at different M stage (P=0.007). We also analyzed the correlation between EGFR mutation and regulatory T cell (Treg) infiltration but no significant difference was found (P=0.066, Supplementary Table 4). Then, we compared the mutant genes among three groups (Figure 3). Mutational frequencies of ALK in inflamed, intermediate and non-inflamed patients were 6.67%, 1.41% and 0%, respectively (P=0.0200, Supplementary Figure 5A). Mutational frequency of CDKN2A was the highest in intermediate immune phenotype followed by inflamed phenotype (3.33% vs 7.04% vs 0%, P=0.0163, Supplementary Figure 5B). Mutational frequencies of IDH2 were also significantly different among three phenotypes (6.67% vs 2.82% vs 0%, P=0.0396, Supplementary Figure 5C). We also found that the proportions of patients with MAP2K1 mutation (6.67% vs 4.23% vs 0%, P=0.0390, Supplementary Figure 5D) and PTEN mutation (20% vs 8.45% vs 5.13%, P=0.0306, Supplementary Figure 5E) were different among three phenotypes.
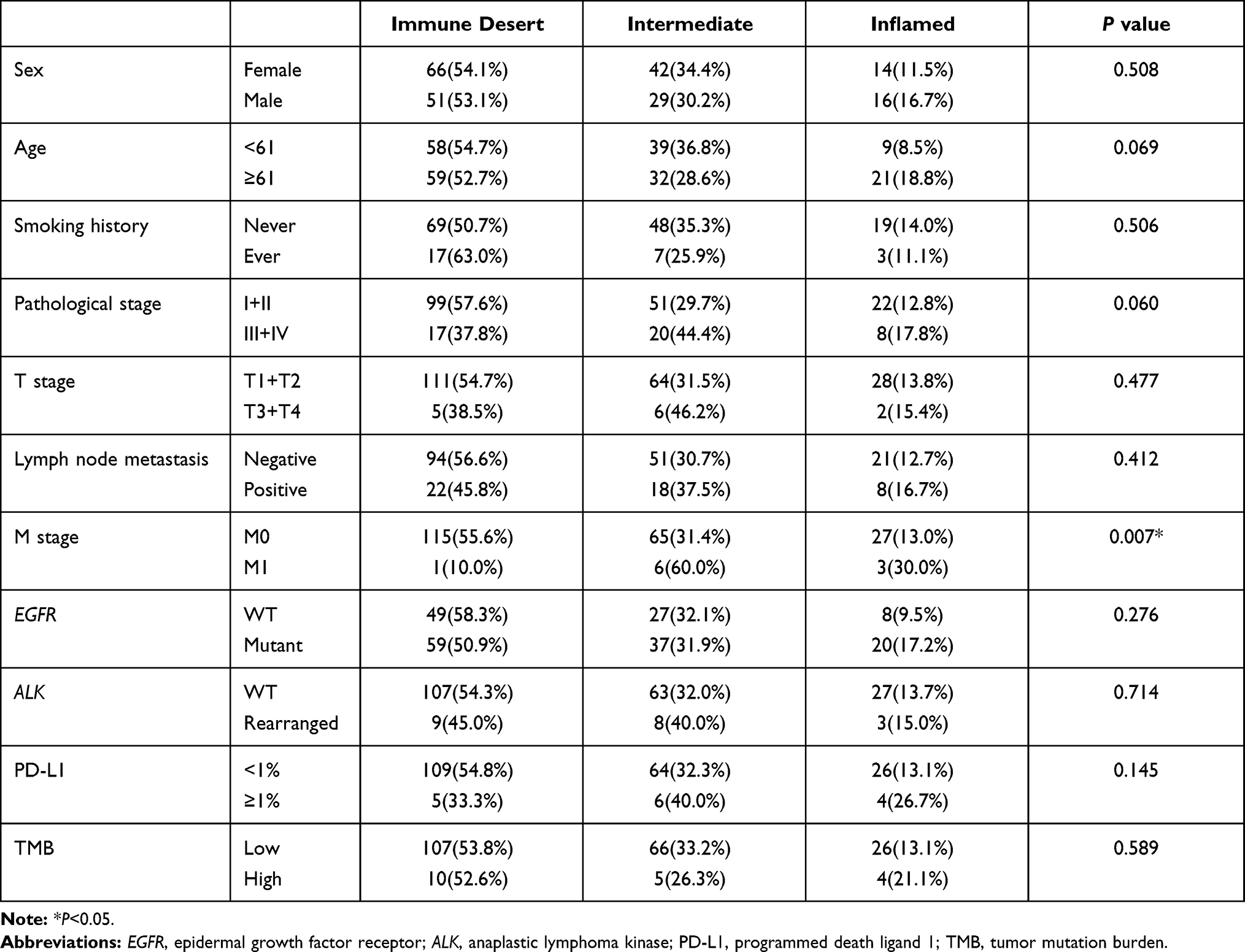 |
Table 3 Immune Phenotype and Clinical Characteristics |
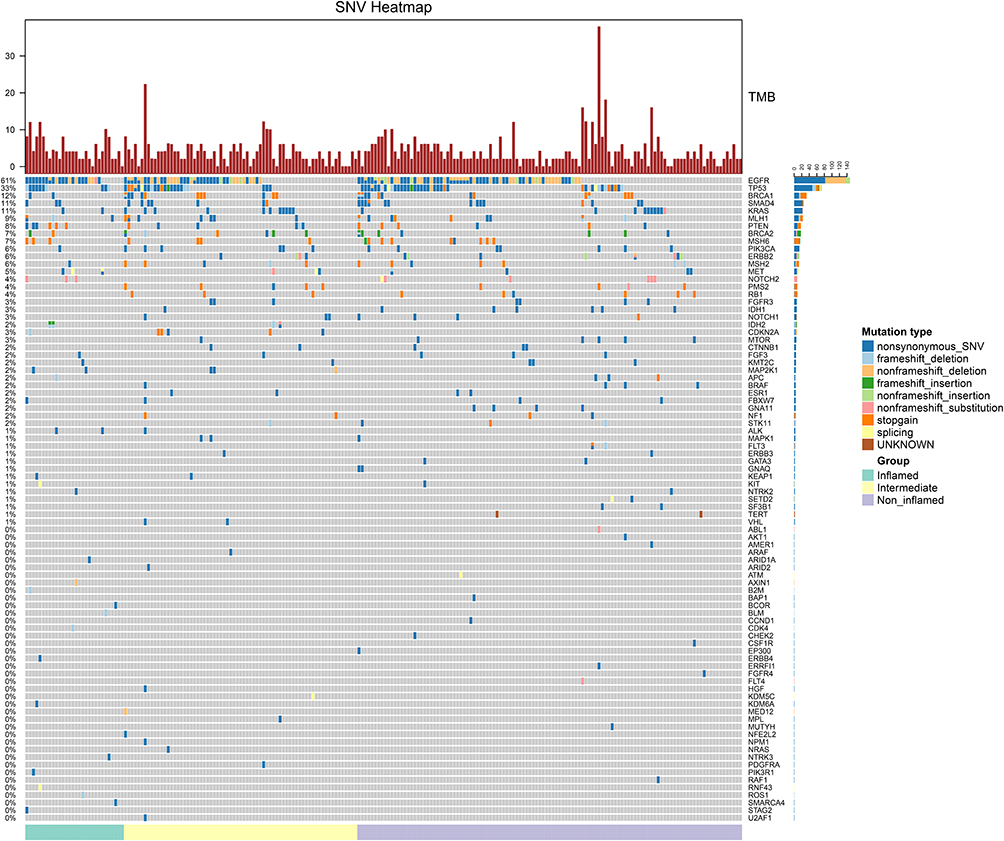 |
Figure 3 Mutated genes in inflamed, intermediate and non-inflamed patients. |
Discussion
Although ICBs had shown their promising anti-tumor effect, EGFR mutant patients did not get clinical benefit as expected.10,11 A recent study using tumor tissue showed EGFR mutant patients tended to have lower PD-L1 expression.26 This result was incompatible with the finding in cell lines27 which might be caused by a more complex environment the tumor cells actually lived in. Therefore, altered PD-L1 expression might not fully explain the poor efficacy of ICBs in EGFR mutant patients.
TMB has been considered as another potentially independent predictive factor for the efficacy of anti-PD-1 therapy12,13,28,29 and FDA has approved TMB >10 mutations per megabase as an indicator for using pembrolizumab for unresected or metastatic solid tumor.30 We compared the TMB level of different EGFR mutant status. EGFR wild-type patients would show a higher TMB level (OR= 4.707, 95% CI 1.349–16.429). Thus, low TMB level might be a potential cause for the ineffectiveness of anti-PD-1 therapy in EGFR mutant patients. Our result was consistent with other studies in different study populations and ethnic background.31,32 Current research also pointed out that some clinical parameters such as male, smoking, lung squamous cell carcinoma, metastatic status, tumor size and gene mutation status like TP53 and CDKN2A mutation would also affect TMB level.33,34 Similar, we also found that mutational frequencies of some genes, such as TP53, genes involved in DNA damage repair and so on, were higher in patients having high TMB level. But we did not find tumor size was independently correlated to TMB level after multivariate analysis. This might be due to different population and the cutoff value of tumor size. Research has found a lower TMB in EGFR 19del mutation than that in L858R mutation,31,35 but in our study, we did not find different TMB level among EGFR 19del, L858R and uncommon mutations (Supplementary Table 1). Relatively small sample size and different ethnic background might cause the inconsistent result.
We further analyzed the different mutations between EGFR wild-type and mutant patients to explore the potential reason to explain why TMB level was low in EGFR mutant patients. We found BRCA2 and KRAS mutation frequency were lower in EGFR mutant patients. BRCA2 is an important compartment in homologous recombination repair (HRR), a method predominantly to repair replication-associated DNA double-strand breaks, through which it can protect the accuracy of genome during DNA replication.36 Recently, pulmonary sarcomatoid carcinoma with BRCA2 mutation having higher TMB has been reported.37 Furthermore, Wang et al identified co-mutations in DNA damage response (DDR) pathways such as HRR and mismatch repair (MMR) or HRR and base excision repair (BER) were correlated to increased TMB and better response to ICBs.38 Thus, we hypothesized that the low frequency of BRCA2 mutation caused the low TMB level in EGFR mutant patients. This was further demonstrated when we narrowed population into EGFR mutant patients in our cohort. As some patients would have EGFR and BRAC2 mutation simultaneously, it should be investigated whether patients with co-mutation would get benefit from ICBs. KRAS mutation, another vital driver mutation, has been demonstrated being associated with higher TMB than KRAS wild-type,39,40 which was also demonstrated in our cohort. Liu et al found expression of some DDR pathway-related genes in KRAS-mutant tumor was significantly decreased.40 It was a partial mechanism of elevated TMB in KRAS mutant patients. But the exact mechanisms are still not fully understood. It is necessary to delineate how KRAS mutation affects the DDR pathways in LUAD which can lead us to find out novel indicators of high TMB and biomarkers for predicting the efficacy of ICBs. As driver mutations were mutually exclusive with each other, patients with EGFR mutation rarely had KRAS and other driver mutations. Thus, we presumed that KRAS mutation might drive the carcinogenesis of partial EGFR wild-type patients in our cohort and make the TMB increased in these patients while the low proportion of KRAS mutation in EGFR-mutant patients caused low TMB in these patients.
As TMB alone may not be a perfect predictor and cancer cells have complex interactions with immune cells,30 tumor immune microenvironment should also be considered as an impact factor for immune therapy. Studies have suggested that tumor immune microenvironment could impact the outcome of ICBs therapy and TILs could be a biomarker for predicting the efficacy of ICBs.14,41 As currently immune phenotype could be divided into non-inflamed, intermediate and inflamed according to IHC, we also compared the immune phenotype among different EGFR mutations in our cohort. However, no difference was found neither between EGFR-mutant and wild-type nor among different EGFR mutation subtypes in our study. In others’ studies lack of infiltration of CD8+T cell and lower co-expression level of PD-L1+/CD8+ in EGFR-mutant patients have been demonstrated by both IHC and bioinformatic analysis.9,26,42,43 Moreover, higher proportion of patients with PD-L1+/CD8+ was also found in uncommon EGFR mutant patients than that in EGFR 19del and L858R.44 Thus, affecting tumor immune microenvironment might be one of the mechanisms of decreasing the efficacy of ICBs in EGFR-mutant patients. But in our data, we did not find the differences. This discrepancy might be caused by our relatively small sample size.
TMB could reflect the neoantigen burden of the tumor indirectly and neoantigen partially reflected the tumor immunogenicity which also played a significant role in determining tumor immune microenvironment. A study also demonstrated TMB level was positively correlated with local immune cytolytic activity in many cancers including LUAD.45 Thus, we also wondered if immune phenotypes were different between high and low TMB level in our cohort. However, our current study found the immune phenotypes in the high TMB group were similar to that in the low TMB group. As higher mutational frequency of B2M was found in high TMB group, deficient antigen presentation might be the reason for preventing the inflamed immune phenotype forming in high TMB group in our cohort. It also demonstrated that TMB was not the sole factor that affects T cell activation, which was suggested in other studies as well.46,47 Further studies are needed for exploring more factors which could influence the immune phenotype determination.
In our study, we further analyzed differentially mutant genes among three immune phenotypes. ALK expression was mainly restricted in immune-privilege location during normal development, fused and overexpressed ALK would activate humoral and cellular immune reaction against the tumor.48 And in our cohort, we also found ALK mutational frequency was higher in inflamed phenotype. This consequence seemed to be contradictory with the poor clinical efficacy of ICBs in ALK rearranged patients.49 This discrepancy might be explained by the complex role of ALK in immunity. On the one hand, fused or mutant ALK might activate the immune system and induce inflammatory. On the other hand, fused ALK could induce expression of immunosuppressive factors and suppressed cell surface expression of HLA I molecule.48 Different fusion patterns of ALK were also found having different impacts on tumor immune microenvironment.50 Further studies are indispensable to detailed analyzing various influence on tumor immunity of different mutant or fused ALK. IDH is an important family of enzymes, including IDH1 and IDH2, in cellular metabolism. IDH1 R132H mutation in glioma would cause decreased CD8+T cell infiltration and reduced expression of chemokines which recruit cytotoxic T lymphocyte.51 However, in our analysis, we found the proportion of mutant IDH2 was higher in inflamed patients. This inconsistent result might be explained by different tumor type and variants. More studies are necessary to explore the significance of IDH mutation in LUAD and its role in modulating tumor immunity. PTEN, a vital tumor suppressor, was also found having relationship with tumor immune microenvironment but the finding was inconsistent. In melanoma and glioma loss of function mutation in PTEN was correlated with low T cell infiltration and an unfavorable immune microenvironment.52,53 But in microsatellite unstable colorectal cancer, frameshift mutation in PTEN was correlated with higher density of infiltrating lymphocytes.54 Our finding was consistent with the latter one, PTEN mutation tended to be more frequent in patients with inflamed phenotype. Further clinical studies are required to investigate the outcome of ICBs in PTEN–mutant LUAD patients to explore its exact role in lung cancer immune therapy. Recent studies have found MEK inhibitor could upregulate MHC I expression on tumor cells and reverse the immunosuppressive microenvironment to inflamed phenotype.55–57 But in our study patients with inflamed phenotype showed a higher proportion of MAP2K1 (also known as MEK1) mutation. The possible reason might be that different variants in MAP2K1 would have different roles in the formation of tumor microenvironment. More studies are necessary for analyzing the impact of specific MAP2K1 mutation on tumor immunity in LUAD to improve immune therapy and combination therapy. CDKN2A had been well studied as a cell cycle inhibitor and its inactivation would cause CDK activation and cell cycle progression.58 Uncontrolled cell division might cause the lack of nutrition and necrosis of tumor cells, which could promote antigen presentation and anti-tumor immunity. Uncontrolled cell cycle also could promote the accumulation of errors of the genome during cell division which might facilitate the formation of neoantigens. Meanwhile, the errors might also affect other genes involved in immune pathways and potentially negatively modulate the ability of a tumor to activate immunity and the immune reaction against the tumor. Thus, its exact impact on tumor immune microenvironment still needs to be further elucidated.
Our study also had some limitations. Firstly, the sample size was not large enough in our study. Secondly, we used targeted panel sequencing to assess the TMB. Thus, mutations in genes which are not contained in the panel could not be identified and analyzed. Thirdly, we did not use multi-site samples to evaluate the TMB so that the potential influence of heterogeneity of tumor TMB could not be taken into consideration in this study.
Conclusions
In this research, lower TMB was found in EGFR mutant patients, which indicated that low TMB could be a potential reason for the unsatisfied efficacy of ICBs in EGFR mutant patients. Although we did not find a statistical difference of immune phenotype between EGFR mutant and wild-type patients, changes in immune phenotype should still be considered as a potential critical factor impacting on the efficacy of ICBs in EGFR mutant patients. It should be further investigated in larger cohort retrospectively and prospectively. We also identified ALK, IDH2, CDKN2A, MAP2K1 and PTEN have different mutational frequencies among three immune phenotypes. This indicated that the pathways modulated by these genes could affect the tumor immune microenvironment. Further studies are indispensable for understanding their roles in building tumor immune microenvironment.
Abbreviations
BER, base excision repair; BP, biological process; CC, cellular component; DDR, DNA damage response; FFPE, formalin-fixed paraffin embedded; HRR, homologous recombination repair; ICB, immune checkpoint blockade; IHC, immunohistochemistry; GO, gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; LUAD, lung adenocarcinoma; MF, molecular function; MMR, mismatch repair; NSCLC, non-small cell lung cancer; OS, overall survival; PCR, polymerase chain reaction; PD-1/PD-L1, program death-1/program death ligand-1; PFS, progression-free survival; SCLC, small cell lung cancer; TILs, tumor-infiltrating lymphocytes; TKIs, tyrosine kinase inhibitors; TMB, tumor mutation burden; Treg, regulatory T cell.
Data Sharing Statement
The data was available at corresponding author with reasonable request.
Ethics Approval and Informed Consent
The research was approved by the Shanghai Pulmonary Hospital (NO. K18-203Y). All participants had provided their written consents and the study was conducted in accordance with the Declaration of Helsinki.
Consent for Publication
All authors are in agreement with the content of the manuscript and agree the submission.
Acknowledgments
Hao Wang and Shanhao Chen are joint first authors.
Author Contributions
All authors contributed to data analysis, drafting or revising the article, have agreed on the journal to which the article will be submitted, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Funding
This study was supported in part by a grant of young talents in Shanghai, National Natural Science Foundation of China (81802255), Young Talents in Shanghai (2019QNBJ), “Dream Tutor” Outstanding Young Talents Program (fkyq1901), Clinical Research Project of Shanghai Pulmonary Hospital (fk18005), Key Discipline in 2019 (oncology), Project of Shanghai Municipal Science and Technology Commission (Project of Municipal Science and Technology Commission), Scientific research project of Shanghai Pulmonary Hospital (fkcx1903), Shanghai Municipal Commission of Health and Family Planning (2017YQ050), Innovation Training Project of SITP of Tongji University, and key projects of leading talent (19411950300). Youth project of hospital management research fund of Shanghai Hospital Association (Q1902037).
Disclosure
There are no conflicts of interest.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7–34. doi:10.3322/caac.21551
2. Youlden DR, Cramb SM, Baade PD. The international epidemiology of lung cancer: geographical distribution and secular trends. J Thorac Oncol. 2008;3(8):819–831. doi:10.1097/JTO.0b013e31818020eb
3. Mayekar MK, Bivona TG. Current landscape of targeted therapy in lung cancer. Clin Pharmacol Ther. 2017;102(5):757–764. doi:10.1002/cpt.810
4. Yatabe Y, Kerr KM, Utomo A, et al. EGFR mutation testing practices within the Asia Pacific region: results of a multicenter diagnostic survey. J Thorac Oncol. 2015;10(3):438–445. doi:10.1097/JTO.0000000000000422
5. Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362(25):2380–2388. doi:10.1056/NEJMoa0909530
6. Steuer CE, Ramalingam SS. Targeting EGFR in lung cancer: lessons learned and future perspectives. Mol Aspects Med. 2015;45:67–73. doi:10.1016/j.mam.2015.05.004
7. Garon EB, Rizvi NA, Hui R, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372(21):2018–2028. doi:10.1056/NEJMoa1501824
8. Herbst RS, Baas P, Kim DW, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet (London, England). 2016;387(10027):1540–1550. doi:10.1016/S0140-6736(15)01281-7
9. Gainor JF, Shaw AT, Sequist LV, et al. EGFR mutations and ALK rearrangements are associated with low response rates to PD-1 pathway blockade in non-small cell lung cancer: a retrospective analysis. Clin Cancer Res. 2016;22(18):4585–4593. doi:10.1158/1078-0432.CCR-15-3101
10. Lee CK, Man J, Lord S, et al. Checkpoint inhibitors in metastatic EGFR-mutated non-small cell lung cancer-a meta-analysis. J Thorac Oncol. 2017;12(2):403–407. doi:10.1016/j.jtho.2016.10.007
11. Bylicki O, Paleiron N, Margery J, et al. Targeting the PD-1/PD-L1 immune checkpoint in EGFR-mutated or ALK-translocated non-small-cell lung cancer. Target Oncol. 2017;12(5):563–569. doi:10.1007/s11523-017-0510-9
12. Rizvi H, Sanchez-Vega F, La K, et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol. 2018;36(7):633–641. doi:10.1200/JCO.2017.75.3384
13. Hellmann MD, Ciuleanu TE, Pluzanski A, et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. 2018;378(22):2093–2104. doi:10.1056/NEJMoa1801946
14. Yi M, Jiao D, Xu H, et al. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol Cancer. 2018;17(1):129. doi:10.1186/s12943-018-0864-3
15. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv: Genomics. 2013.
16. Lai Z, Markovets A, Ahdesmaki M, et al. VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res. 2016;44(11):e108. doi:10.1093/nar/gkw227
17. Garrison E, Marth GT. Haplotype-based variant detection from short-read sequencing. arXiv: Genomics. 2012.
18. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi:10.1093/nar/gkq603
19. Karczewski KJ, Weisburd B, Thomas B, et al. The ExAC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res. 2017;45(D1):D840–d845. doi:10.1093/nar/gkw971
20. Karczewski KJ, Francioli L. The genome aggregation database (gnomAD). MacArthur Lab. 2017.
21. Chalmers ZR, Connelly CF, Fabrizio D, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9(1):34. doi:10.1186/s13073-017-0424-2
22. Usó M, Jantus-Lewintre E, Bremnes RM, et al. Analysis of the immune microenvironment in resected non-small cell lung cancer: the prognostic value of different T lymphocyte markers. Oncotarget. 2016;7(33):52849–52861. doi:10.18632/oncotarget.10811
23. Adams SF, Levine DA, Cadungog MG, et al. Intraepithelial T cells and tumor proliferation: impact on the benefit from surgical cytoreduction in advanced serous ovarian cancer. Cancer. 2009;115(13):2891–2902. doi:10.1002/cncr.24317
24. Galon J, Mlecnik B, Bindea G, et al. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J Pathol. 2014;232(2):199–209. doi:10.1002/path.4287
25. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16(5):284–287. doi:10.1089/omi.2011.0118
26. Dong ZY, Zhang JT, Liu SY, et al. EGFR mutation correlates with uninflamed phenotype and weak immunogenicity, causing impaired response to PD-1 blockade in non-small cell lung cancer. Oncoimmunology. 2017;6(11):e1356145. doi:10.1080/2162402X.2017.1356145
27. Chen N, Fang W, Zhan J, et al. Upregulation of PD-L1 by EGFR activation mediates the immune escape in EGFR-driven NSCLC: implication for optional immune targeted therapy for NSCLC patients with EGFR mutation. J Thorac Oncol. 2015;10(6):910–923. doi:10.1097/JTO.0000000000000500
28. Hellmann MD, Callahan MK, Awad MM, et al. Tumor mutational burden and efficacy of nivolumab monotherapy and in combination with ipilimumab in small–cell lung cancer. Cancer Cell. 2018;33(5):853–861.e854. doi:10.1016/j.ccell.2018.04.001
29. Marabelle A, Fakih M, Lopez J, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, Phase 2 KEYNOTE-158 study. Lancet Oncology. 2020;21(10):1353–1365. doi:10.1016/S1470-2045(20)30445-9
30. Sung WWY, Chow JCH, Cho WCS. Tumor mutational burden as a tissue-agnostic biomarker for cancer immunotherapy. Expert Rev Clin Pharmacol. 2020;1–3.
31. Offin M, Rizvi H, Tenet M, et al. Tumor mutation burden and efficacy of EGFR-tyrosine kinase inhibitors in patients with EGFR-mutant lung cancers. Clin Cancer Res. 2019;25(3):1063–1069. doi:10.1158/1078-0432.CCR-18-1102
32. Jiao XD, He X, Qin BD, et al. The prognostic value of tumor mutation burden in EGFR-mutant advanced lung adenocarcinoma, an analysis based on cBioPortal data base. J Thorac Dis. 2019;11(11):4507–4515. doi:10.21037/jtd.2019.11.04
33. Ozaki Y, Muto S, Takagi H, et al. Tumor mutation burden and immunological, genomic, and clinicopathological factors as biomarkers for checkpoint inhibitor treatment of patients with non-small-cell lung cancer. Cancer Immunol Immunother. 2020;69(1):127–134. doi:10.1007/s00262-019-02446-1
34. Alborelli I, Leonards K, Rothschild SI, et al. Tumor mutational burden assessed by targeted NGS predicts clinical benefit from immune checkpoint inhibitors in non-small cell lung cancer. J Pathol. 2020;250(1):19–29. doi:10.1002/path.5344
35. Hastings K, Yu HA, Wei W, et al. EGFR mutation subtypes and response to immune checkpoint blockade treatment in non-small-cell lung cancer. Ann Oncol. 2019;30(8):1311–1320. doi:10.1093/annonc/mdz141
36. Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat Rev Cancer. 2011;12(1):68–78. doi:10.1038/nrc3181
37. Liang X, Li Q, Xu B, et al. Mutation landscape and tumor mutation burden analysis of Chinese patients with pulmonary sarcomatoid carcinomas. Int J Clin Oncol. 2019;24(9):1061–1068. doi:10.1007/s10147-019-01454-6
38. Wang Z, Zhao J, Wang G, et al. Comutations in DNA damage response pathways serve as potential biomarkers for immune checkpoint blockade. Cancer Res. 2018;78(22):6486–6496. doi:10.1158/0008-5472.CAN-18-1814
39. Yang S, Yu X, Fan Y, Shi X, Jin Y. Clinicopathologic characteristics and survival outcome in patients with advanced lung adenocarcinoma and KRAS mutation. J Cancer. 2018;9(16):2930–2937. doi:10.7150/jca.24425
40. Liu C, Zheng S, Jin R, et al. The superior efficacy of anti-PD-1/PD-L1 immunotherapy in KRAS-mutant non–small cell lung cancer that correlates with an inflammatory phenotype and increased immunogenicity. Cancer Lett. 2020;470:95–105. doi:10.1016/j.canlet.2019.10.027
41. Riaz N, Havel JJ, Makarov V, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell. 2017;171(4):934–949.e916. doi:10.1016/j.cell.2017.09.028
42. Liu SY, Dong ZY, Wu SP, et al. Clinical relevance of PD-L1 expression and CD8+ T cells infiltration in patients with EGFR-mutated and ALK-rearranged lung cancer. Lung Cancer. 2018;125:86–92. doi:10.1016/j.lungcan.2018.09.010
43. Jin R, Liu C, Zheng S, et al. Molecular heterogeneity of anti-PD-1/PD-L1 immunotherapy efficacy is correlated with tumor immune microenvironment in East Asian patients with non-small cell lung cancer. Cancer Biol Med. 2020;17(3):768–781.
44. Chen K, Cheng G, Zhang F, et al. PD-L1 expression and T cells infiltration in patients with uncommon EGFR-mutant non-small cell lung cancer and the response to immunotherapy. Lung Cancer. 2020;142:98–105. doi:10.1016/j.lungcan.2020.02.010
45. Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160(1–2):48–61. doi:10.1016/j.cell.2014.12.033
46. Chan TA, Yarchoan M, Jaffee E, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol. 2019;30(1):44–56. doi:10.1093/annonc/mdy495
47. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–74. doi:10.1126/science.aaa4971
48. Wang L, Lui VWY. Emerging roles of ALK in immunity and insights for immunotherapy. Cancers. 2020;12(2).
49. Oya Y, Kuroda H, Nakada T, Takahashi Y, Sakakura N, Hida T. Efficacy of immune checkpoint inhibitor monotherapy for advanced non-small-cell lung cancer with ALK rearrangement. Int J Mol Sci. 2020;21(7):2623. doi:10.3390/ijms21072623
50. Yang CY, Liao WY, Ho CC, et al. Association of programmed death-ligand 1 expression with fusion variants and clinical outcomes in patients with anaplastic lymphoma kinase-positive lung adenocarcinoma receiving crizotinib. Oncologist. 2020;25:702–711. doi:10.1634/theoncologist.2020-0088
51. Kohanbash G, Carrera DA, Shrivastav S, et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8+ T cell accumulation in gliomas. J Clin Invest. 2017;127(4):1425–1437. doi:10.1172/JCI90644
52. Peng W, Chen JQ, Liu C, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov. 2016;6(2):202–216. doi:10.1158/2159-8290.CD-15-0283
53. Zhao J, Chen AX, Gartrell RD, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med. 2019;25(3):462–469. doi:10.1038/s41591-019-0349-y
54. Tougeron D, Fauquembergue E, Rouquette A, et al. Tumor-infiltrating lymphocytes in colorectal cancers with microsatellite instability are correlated with the number and spectrum of frameshift mutations. Modern Pathology. 2009;22(9):1186–1195. doi:10.1038/modpathol.2009.80
55. Poon E, Mullins S, Watkins A, et al. The MEK inhibitor selumetinib complements CTLA-4 blockade by reprogramming the tumor immune microenvironment. J Immunother Cancer. 2017;5(1):63. doi:10.1186/s40425-017-0268-8
56. Kuske M, Westphal D, Wehner R, et al. Immunomodulatory effects of BRAF and MEK inhibitors: implications for Melanoma therapy. Pharmacol Res. 2018;136:151–159. doi:10.1016/j.phrs.2018.08.019
57. Brea EJ, Oh CY, Manchado E, et al. Kinase regulation of human MHC class I molecule expression on cancer cells. Cancer Immunol Res. 2016;4(11):936–947. doi:10.1158/2326-6066.CIR-16-0177
58. Jeong EH, Lee TG, Ko YJ, et al. Anti-tumor effect of CDK inhibitors on CDKN2A-defective squamous cell lung cancer cells. Cell Oncol. 2018;41(6):663–675. doi:10.1007/s13402-018-0404-6
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.