Back to Journals » Journal of Hepatocellular Carcinoma » Volume 10
Tumor Microenvironment Composition and Related Therapy in Hepatocellular Carcinoma
Authors Li Z, Zhang Z, Fang L, Zhao J, Niu Z, Chen H, Cao G
Received 24 August 2023
Accepted for publication 10 November 2023
Published 21 November 2023 Volume 2023:10 Pages 2083—2099
DOI https://doi.org/10.2147/JHC.S436962
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 7
Editor who approved publication: Prof. Dr. Imam Waked
Zishuai Li,1– 3,* Zihan Zhang,4,* Letian Fang,1– 3,* Jiayi Zhao,1– 3,* Zheyun Niu,4 Hongsen Chen,1– 3 Guangwen Cao1– 3
1Key Laboratory of Biological Defense, Ministry of Education, Second Military Medical University, Shanghai, 200433, People’s Republic of China; 2Shanghai Key Laboratory of Medical Bioprotection, Second Military Medical University, Shanghai, 200433, People’s Republic of China; 3Department of Epidemiology, Second Military Medical University, Shanghai, 200433, People’s Republic of China; 4Department of Epidemiology, Tongji University School of Medicine Tongji University, Shanghai, 200120, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Guangwen Cao, Department of Epidemiology, Second Military Medical University, 800 Xiangyin Road, Shanghai, 200433, People’s Republic of China, Tel/Fax +86 21-8187-1060, Email [email protected]
Abstract: Globally, primary liver cancer is the third leading cause of cancer death, and hepatocellular carcinoma (HCC) accounts for 75%– 95%. The tumor microenvironment (TME), composed of the extracellular matrix, helper cells, immune cells, cytokines, chemokines, and growth factors, promotes the immune escape, invasion, and metastasis of HCC. Tumor metastasis and postoperative recurrence are the main threats to the long-term prognosis of HCC. TME-related therapies are increasingly recognized as effective treatments. Molecular-targeted therapy, immunotherapy, and their combined therapy are the main approaches. Immunotherapy, represented by immune checkpoint inhibitors (ICIs), and targeted therapy, highlighted by tyrosine kinase inhibitors (TKIs), have greatly improved the prognosis of HCC. This review focuses on the TME compositions and emerging therapeutic approaches to TME in HCC.
Keywords: hepatocellular carcinoma, tumor microenvironment, immunotherapy, targeted therapy
Graphical Abstract:

Introduction
Primary liver cancer (PLC) was the sixth most common cancer and the third leading cause of cancer death globally in 2020, with approximately 906,000 new cases and 830,000 deaths.1 In China, PLC is the second leading cause of cancer death, and hepatocellular carcinoma (HCC) accounted for 93.0% of PLC, intrahepatic cholangiocarcinoma (ICC) accounted for 4.3%, and combined hepatocellular-cholangiocarcinoma (CHC) accounted for 1.6%; 84.4% of HCC is seropositive for hepatitis B surface antigen.2 Unlike other malignant tumors, HCC is highly malignant and has a higher mortality rate in individuals aged 40–65 than in people older than 65 years in China.3 Therapeutic interventions, including surgical measures (either resection or liver transplantation) paired with non-surgical strategies like transcatheter arterial chemoembolization, radiofrequency ablation, chemotherapy, radiotherapy, biologic treatments, and traditional Chinese medicine, remain the cornerstone of HCC management. Unfortunately, most patients do not get survival benefits, and patients with survival benefits also have the problem of rapid recurrence and metastasis.4,5
The tumor microenvironment (TME) plays a crucial role in the development of HCC, including cell proliferation, migration, invasion, epithelial–mesenchymal transition (EMT), immune escape, neovascularization, and treatment resistance.6 Characteristics of the TME include hypoxia,6 abnormal vascular proliferation,7 acidification,8 inflammation,9 and immunosuppression.10 Hepatitis B and/or C virus infection, alcoholism, metabolic dysfunction-associated steatotic liver disease (MASLD), obesity, and metabolic syndrome are the major reasons for HCC. Chronic HBV infection activates and maintains chronic non-resolving inflammation. HBV cccDNA forms double-stranded DNA (dsDNA) in the cytoplasm or forms cyclic extrachromosomal DNA (ecDNA) with human genomic DNA fragments. In the process of inflammation-cancer transition, dsDNA promotes the activation of non-classical nuclear factor-kappa B (NF-κB) and produces cytokines such as transforming growth factor beta-1 (TGF-β1), plasminogen activator inhibitor-1 (PAI1), and helper T cell 2 (Th2), which recruit inhibitory immune cells such as myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), regulatory T cells (Tregs), and tumor-associated neutrophils (TANs). These inhibitory immune cells inhibit the antiviral and anti-tumor immune activities of cytotoxic CD8+ T cells (CTLs), natural killer cells (NK cells), and dendritic cells (DCs) by secreting interleukin-10 (IL-10), TGF-β1 and producing reactive oxygen species (ROS), forming an immunosuppressive TME that promotes the evolution and development of HCC.11 The complex TME provides more options for the treatment of HCC.
Targeted therapy and immunotherapy toward TME have provided a promising avenue for advanced or metastatic HCC. Targeted therapy mainly includes multi-target tyrosine kinase inhibitors (like Lenvatinib, Regorafenib, and Sorafenib), vascular endothelial growth factor receptor (VEGFR) antagonists (such as Apatinib and Axitinib), and VEGF/VEGFR monoclonal antibodies (such as Bevacizumab and Ramelimumab).12 Various immune checkpoint inhibitors have been developed, with anti-programmed cell death protein 1 (PD-1) and anti-programmed cell death ligand 1 (PD-L1) agents showing significant potential as adjuvant treatments for early-stage HCC, leading to notable extensions in survival.13,14 These treatments not only achieve more durable and robust efficacy across genders but also confer significant benefits in terms of quality of life for patients.15 The complex TME promotes the evolution and development of HCC and also provides potential molecular targets for targeted therapy and immunotherapy in HCC. In this review, we focus on the compositions of TME, the interaction between TME and HCC cells, and various therapeutic approaches to TME, with a view to providing a reference for efficient treatments of HCC.
The Composition of TME in HCC
As a dynamic system, TME is closely related to the occurrence, development, and metastasis of HCC. TME mainly comprises extracellular matrix (ECM), helper cells, immune cells, cytokines, chemokines, and growth factors.16 Helper cells in TME mainly include cancer-associated fibroblasts (CAFs), hepatic stellate cells (HSCs), and vascular endothelial cells. The immune cells are TAMs, TANs, Tregs, inhibitory B cells, MDSCs, DCs, CTLs, and NK cells. CTLs, NK cells, DCs, and helper T cell 1 (Th1) cytokines participate in anti-tumor as well as anti-viral immunity and inhibit tumor metastasis; TAN, Treg, helper T cell 17 (Th17), M2-TAMs, CAFs, and Th2 cytokines facilitate immune escape and HCC metastasis.6 Cancer cells and immune cells express co-inhibitory receptors, including cytotoxic T cell-associated antigen 4 (CTLA4), programmed death 1 (PD-1), T cell immunoglobulin domain and mucin domain-3 (TIM-3), and lymphocyte activation gene 3 (LAG3), which maintain the stability and immunosuppressive characteristic of TME in HCC.17
Pro-Tumor Cells in TME
TAMs
TAMs play a “double-edged sword” role in the occurrence and development of HCC. M1-TAMs can kill tumor cells, while M2-TAMs promote tumor development. The polarization of TAM is deeply associated with TME, and the polarization of macrophages to M1 and M2 is reversible and adjustable. With the progression of HCC, M1-TAMs gradually polarize to M2-TAMs, and the increase in the number of M2-TAMs also indicates a poor prognosis.18 M1-TAMs are stimulated by interferon gamma (IFN-γ), granulocyte-macrophage colony stimulating factor (GM-CSF), or lipopolysaccharide (LPS). Cytokines secreted by Th2 cells and tumor cells, including interleukin (IL)-4, IL-10, IL-13, colony stimulating factor 1 (CSF1), chemokine (C-C motif) ligand 2 (CCL2), chemokine (C-X-C motif) ligand 12 (CXCL12), and connective tissue growth factor (CTGF), promote the polarization of macrophages to M2.19 The upregulation of Wnt2b expression in macrophages promotes the polarization of TAMs from M1 to M2 by activating the Wnt2b/β-catenin/c-Myc signaling pathway.20
As shown in Figure 1, the tumor-promoting effects of M2-TAMs mainly include: secreting cytokines (such as IL-6, CXCL8, and IL-10), blocking the inducible nitric oxide synthase (iNOS) pathway, reducing the synthesis of nitric oxide (NO);21 inhibiting the activation of CTLs and NK cells and reducing their killing effects on HCC cells by secreting IL-10 and TGF-β;22,23 promoting HCC cells neovascularization by secreting vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), IL-17, matrix metallopeptidase 2 (MMP2), and MMP9; promoting HCC cells invasion and metastasis by secreting of MMPs, cathepsins, and serine proteases;24 triggering the recruitment of Tregs to exert immunosuppressive effects by secreting CCL17, CCL18, CCL20 and CCL22; promoting immune evasion by secreting prostaglandin E2, MMP7 and other mediators.25 Hypoxia-inducible factor-1α (HIF-1α) produced by HCC promotes the release of IL-1β from TAMs, and IL-1β induces epithelial–mesenchymal transition (EMT) in HCC.26 Chemokine (C-C motif) ligand 15 (CCL15) is significantly enriched in the core of cancerous areas, which promotes an immunosuppressive microenvironment by recruiting and polarizing M2-TAMs.27 Low expression of cluster of differentiation 86+ (CD86+) TAMs and high expression of CD206+ TAMs were significantly associated with an aggressive tumor phenotype, poor overall survival (OS), and a shorter time to recurrence.28 Carbonic anhydrase XII (CA12) mediates TAMs survival in the acidic TME and induces TAMs to secrete C-C motif chemokine ligand 8 (CCL8), which promotes EMT and metastasis in HCC.29
 |
Figure 1 The complex network of interactions between cellular components in TME. In TME, pro-tumor cells promote angiogenesis, ECM remodeling, and immune escape via secreting various cytokines and chemokines, such as VEGF, MMPs, CCL17, CXCL20, and so on. Meanwhile, pro-tumor cells also inhibit the function of anti-tumor cells through the release of several cytokines, such as IL-10, NO, TGF-β, ROS, and so on. The imbalance between pro-tumor strength and anti-tumor strength forms an immunosuppressive TME, which significantly promotes the malignant progression as well as the recurrence, metastasis, and drug resistance of HCC. Abbreviations: IL-4, interleukin-4; IL-10, interleukin-10; CSF-1, colony-stimulating factor 1; CXCL12, chemokine (C-X-C motif) ligand 12; CTGF, connective tissue growth factor; CCL2, chemokine (C-C motif) ligand 2; CXCL8, chemokine (C-X-C motif) ligand 8; IL-6, interleukin-6; TGF-β, transforming growth factor-β; VEGF, vascular endothelial growth factor; CCL17, chemokine (C-C motif) ligand 17; CCL20, chemokine (C-C motif) ligand 20; MMPs, matrix metallopeptidases; IFN-γ, interferon-γ; LPS, lipopolysaccharide; TNF-α, tumor necrosis factor-α; NO, nitric oxide; BMP2, bone morphogenetic protein 2; IDO1, indoleamine 2,3-dioxygenase 1; ROS, reactive oxygen species; TIM-3, T cell immunoglobulin domain and mucin domain-3; HSCs, hepatic stellate cells; MSCs, mesenchymal stromal cells; SDF-1, stromal cell-derived factor-1; VM, vasculogenic mimicry; HGF, hepatocyte growth factor; STAT3, signal transducer and activator of transcription 3; c-MET/FRA1/HEY1, cellular-mesenchymal epithelial transition factor/FOS-related antigen 1/hes related family bHLH transcription factor with YRPW motif 1. |
TANs
TANs can be divided into subtype N1 (anti-tumor) and subtype N2 (pro-tumor).30 High levels of TANs infiltration and neutrophil/lymphocyte ratio (NLR) are correlated with the poor prognosis of HCC.31 LPS and IFN-γ promote N1 polarization, while TGF-β drives the acquisition of the N2 phenotype.32 N2-TANs predominantly produce chemokines such as CCL20 and CCL17, which recruit Tregs and TAMs to shape the immunosuppressive TME and promote HCC progression as well as sorafenib resistance.33 TANs secrete TNF-α and NO, kill CD8+ T cells, and inhibit their anti-tumor effect on HCC.31 TANs secrete high levels of bone morphogenetic protein 2 (BMP2) and TGF-β2 and induce the abnormal expression of microRNA −301b-3p in HCC cells, which promotes HCC cells to acquire “stem” characteristics. These cancer stem-like cells excessively produce chemokine (C-X-C motif) ligand 5 (CXCL5) and sustain elevated activity of the NF-κb signaling pathway. This, in turn, recruits even more TANs into the tumor, creating a vicious cycle that significantly exacerbates the malignancy of HCC (Figure 1).34
MDSCs
MDSCs represent a diverse group of immature myeloid cells known for their pronounced immunosuppressive capabilities.35 MDSCs mainly inhibit the antitumor function of CTLs and NK cells through arginase, iNOS, indoleamine 2,3-dioxygenase 1 (IDO1), reactive oxygen species (ROS), TGF-β, and IL-10.36 MDSCs can induce the differentiation and expansion of Tregs, deprive T cells of essential amino acids, promote oxidative stress, and promote the polarization of M2-TAMs and N2-TANs.37,38 MDSCs express galectin 9 and bind to TIM-3 on T cells to trigger T cell apoptosis.39 A specific subtype of MDSCs, characterized as CD11b+ CD33+ HLA-DR−, effectively hampers the proliferation of CD8+ T cells in HCC (Figure 1).40 Overexpression of ectonucleoside triphosphate diphosphohydrolase 2 (ENTPD2/CD39L1) mediated by HIF-1 converts extracellular ATP to 5′-AMP, which interdicts the differentiation of MDSCs.41 Upregulation of apolipoprotein B mRNA editing enzyme catalytic subunit 3B (APOBEC3B) in HCC inhibits global H3K27me3 abundance through interaction with polycomb repressor complex 2 (PRC2) and reduces H3K27me3 occupancy at the chemokine CCL2 promoter, thereby recruiting a large number of TAMs and MDSCs.42 G9a-mediated downregulation of solute carrier family 7 member 2 (SLC7A2) promotes CXCL1 secretion through the phosphatidylinositide 3-kinases (PI3K)/protein kinase B (PKB)/NF-κB pathway. Upregulation of SLC7A2 recruits MDSCs, which fosters a pro-tumor TME.43 Prior to the metastatic colonization of HCC cells into target tissues, cancer cells secrete several soluble factors, including CXCL17, G-CSF, osteopontin, CXCL12, TNF-α, TGF-β, and VEGF-A. These factors play key roles in the development of the immune system by influencing CD11b+/Gr-1+MDSC-mediated neo-angiogenesis, inducing inflammatory responses, remodeling the ECM, and recruiting immune cells, creating an immunosuppressed but nutrient-rich TME.44
CAFs
In HCC, CAFs mainly originate from HSCs and hematopoietic stem cells. Mesothelial cells, mesenchymal stromal cells (MSCs), and peritumoral fibroblasts can also be transformed into CAFs.45 In terms of their effects on cancer, CAFs can be broadly categorized into cancer-promoting CAFs (pCAFs) and cancer-restraining CAFs (rCAFs).46 The pCAFs enhance the “stemness” of CD24+ HCC by secreting hepatocyte growth factor (HGF) acting on the cellular-mesenchymal epithelial transition factor/FOS-related antigen 1/hes related family bHLH transcription factor with YRPW motif 1 (c-MET/FRA1/HEY1) signaling pathway47,48 or by secreting HGF and IL-6 acting on the phosphorylated signal transducer and activator of transcription 3 (STAT3)/Notch signaling pathway.49 HCC cells activate CAFs by secreting the exosome miR-1247-3p, which upregulates inflammatory factors such as IL-1, IL-6, and IL-8 and contributes to the resistance to sorafenib.50 Hypoxia also activates CAFs, induces angiogenesis, and promotes HCC cell proliferation by producing proangiogenic factors such as VEGF, MMP2, and MMP9.51 A peculiar phenomenon known as vasculogenic mimicry (VM) is observed when aggressive tumor cells mold themselves to establish vascular-like channels through ECM remodeling. This unique framework acts as an alternative blood supply route for malignant tumors. Freshly isolated CAFs from HCC specimens have shown the capability to maintain tumor blood supply. They achieve this by inducing VM formation in HCC cells, driven by the paracrine factors TGF-β and stromal cell-derived factor-1 (SDF-1).52,53 HCC cells and CAFs induce M2 polarization of TAMs. This process involves upregulating the mRNA expression of markers CD163 and CD206 in macrophages while concurrently diminishing IL-6 expression and secretion.54 CAFs promote the proliferation of Tregs by secreting TGF-β and CXCL13.55 CD36+ CAFs recruit CD33+ MDSCs by expressing macrophage migration inhibitory factor (MIF) via the lipid peroxidation/p38/CEBPs axis (Figure 1).56
Tregs
As a subset of CD4+ T cells, Treg is one of the pivotal factors in maintaining immune tolerance. Tumor-infiltrating Tregs promote tumor immune escape through both contact-dependent and contact-independent mechanisms. The proportion and absolute number of CD4+CD25+ T cells increased significantly in the vicinity of HCCs.57 Huh7 culture supernatant seems to promote the proliferation of CD4+CD25+ T cells.58 In the hypoxic TME, Tregs are significantly enriched and interact with type 2 conventional dendritic cells (cDC2), leading to the loss of antigen-presenting HLA-DR on cDC2.59 In naive T cells, CTLA-4 is localized in the intracellular space and expressed on the cell surface after receiving stimulation signals. Contrastingly, CTLA-4 is constitutively expressed in Tregs, which is associated with immunosuppressive functions.60 Long noncoding epidermal growth factor receptor (lnc-EGFR) specifically binds EGFR and promotes Tregs differentiation, which connects immunosuppressive states with HCC.61
Liver Cancer Stem Cells
Liver cancer stem cells (LCSCs), a class of cells capable of self-renewal and differentiation to form tumors, initiate HCC and are at the root of HCC recurrence.62 Intriguingly, the ECM modulates the expression of malignant markers in LCSCs, notably CD44 and CD133. ECM also plays a role in intercellular communication and acts as a catalyst for HCC.63 Multiple cytokines in TME (such as HIF-1α, BMP, OSM, MMP2, and MMP9) interact synergistically with ECM remodeling, EMT, and tumor neovascularization networks to activate Notch, TGF-β, STAT3, and Hedgehog signaling pathways, enhance LCSCs stemness and chemoresistance, and positively impact HCC progression.64–66 Endothelial cells secrete HIF-1α and VEGF, increase LOXL2 expression, induce EMT and VM formation, and synergize with ECM remodeling to promote LCSC maintenance and self-renewal.67,68 TAMs can activate the IL-6/STAT3 signaling pathway through the release of VEGF, MMP, the secretory proteins S100A9, TGF-β, and TNF-α, which, together with tumor neovascularization, alter LCSCs phenotype and function.69 TANs secrete BMP2 and TGFβ to induce LCSCs production, and LCSCs increase, in turn recruiting more TANs, creating positive feedback in HCC.70 Furthermore, CAFs indulge in ECM remodeling, primarily through collagen synthesis. They also release a cocktail of cytokines, including CCL2, CCL5, CXCL1, and IL-6. These factors activate Notch, TGF-β, Hedgehog, and STAT3 signaling pathways, significantly amplifying the stemness characteristics of LCSCs.71
Anti-Tumor Cells in TME
NK Cells
It is reported that 30–50% of the lymphocytes in the liver are NK cells, and the proportion of NK cells in the liver is five times higher than that in the spleen or peripheral blood.72 NK cells can directly release cytotoxic substances such as perforin and granzyme B or secrete cytokines such as TNF-α and IFN-λ to kill HCC cells. Another strategy employed by NK cells is antibody-dependent cell-mediated cytotoxicity (ADCC), which can also result in the destruction of HCC cells.73 However, HCC cells can circumvent the recognition and killing of NK cells in TME. Hypoxia-inducible gene 2 (HIG-2) promotes IL-10 release from HCC cells, activates the STAT3 signaling pathway of NKs, and inhibits the killing activity of NKs.74 HCC cells also evade the killing activity of NK cells by reducing the expression of tumor-associated antigens and increasing the expression of major histocompatibility complex class I (MHC-I)-related molecules, prompting the inactivation of NK cells.75
DCs
DCs can capture and phagocytose HCC cells and transfer them to lymph nodes while presenting tumor-associated antigens (TAAs) to T cells. T cells migrate and infiltrate into cancerous tissues, recognize T-cell receptors (TCRs) on HCC cells, and then bind to them to kill them.76 TNF-β and IL-12 induced by M1-TAM, IFN-γ produced by NKs, and IL-2 secreted by CTLs further activate T cells. Lysosome-associated membrane glycoprotein 3+ (LAMP3+) DCs are higher expressed in tumor tissues than paracancerous liver tissues with highly phagocytic activity. LAMP3+ DCs stimulate the immune response of CD8+T, CD4+ T cells, and NK cells in tumor-draining lymph nodes by receptor and ligand binding.77
Other Factors in TME
ECM
The ECM is a complex network comprising collagen, non-collagen fibers, elastin, proteoglycans, and aminoglycans. As HCC progresses, the ECM undergoes dynamic remodeling. While it serves as a physical shield, obstructing direct interaction between immune cells and tumor cells, the ECM also modulates the functions of immune cells.78 Research has identified six ECM-associated genes (namely SPP1, ADAMTS5, MMP1, BSG, LAMA2, and CDH1) that correlate with a dismal prognosis in HCC patients.79 ECM is gradually remodeled by cancer cells in TME, which facilitates tumor development and metastasis.80 MMPs are a class of zinc-dependent endoproteases that disrupt the ECM by destroying the structure of different proteins within it. Overexpression of different MMPs can lead to severe ECM catabolism as well as a significant elevation in EMT.80 CAFs have a central role in shaping the ECM. They deposit major ECM proteins like collagen, fibronectin, and laminin. Moreover, they release ECM-degrading enzymes, including MMPs, which not only bolster CAF migration but also expedite ECM degradation, setting the stage for tumor cell infiltration.81 HSCs are activated when the liver is damaged or subjected to extracellular stimuli. Activated HSCs participate in ECM remodeling by synthesizing ECM molecules and secreting MMPs.82
Growth Factors
HGF plays key roles in endothelial cell generation, tissue and organ regeneration, and cellular malignancy. Growth factor receptor-binding protein 2 (Grb-2), Grb2-associated binding protein 1 (Gab1), and EGFR promote HCC occurrence and progression by activating the C-stromal-epithelial transformation receptor (HGF/c-Met) axis.83 Insulin-like growth factor (IGF)/IGF-1 receptor (IGF-1R) signaling modulate stem cell differentiation and pluripotency during embryonic development. Dysregulated IGF/IGF1R signaling enhances tumor “stemness” and promotes drug resistance as well as tumor recurrence in hepatitis B virus (HBV)-related HCC (HBV-HCC).84 TGF-β maintains homeostasis in the normal liver, inhibits early-stage HCC, and promotes the progression of advanced HCC. TGF-β modulates the activity of various TME-associated cells (such as HSCs, CAFs, endothelial cells, and NK cells) and promotes the progression of HCC as well as the immune evasion of malignant cells.85 The functions and the targets of various growth factors are listed in Table 1.
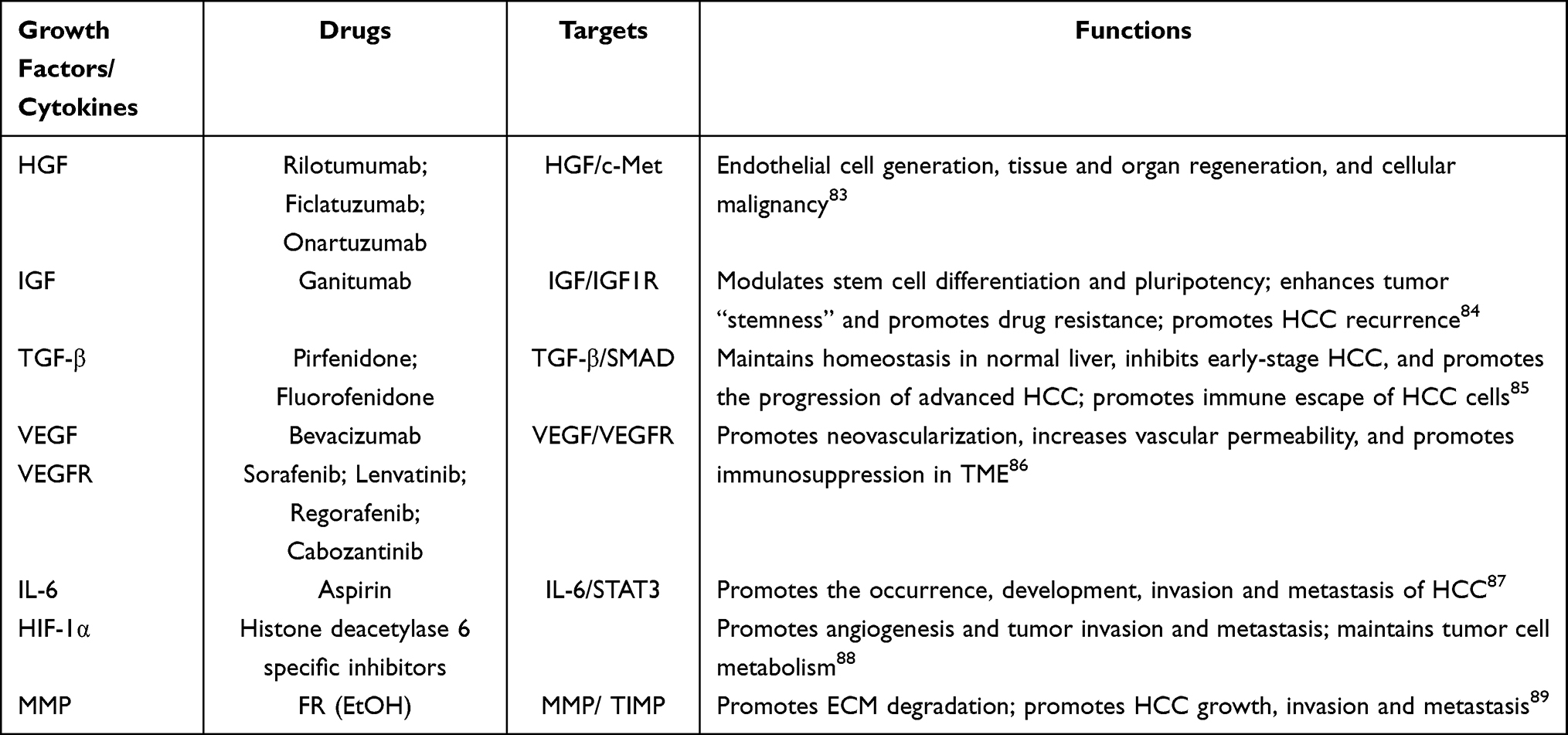 |
Table 1 The Functions and the Targets of Various Growth Factors in TME of HCC |
Additional Mediators in TME
Beyond the primary cellular constituents and factors of the TME, there exist several other potential mediators that significantly influence the environment. It was reported that upregulated expression of the 4-gene inflammatory signature (including lymphocyte-activating gene 3 (LAG-3), signal transducer and activator of transcription 1 (STAT1), CD8, and PD-L1) was associated with immunotherapy responses in HCC. However, only the percentage of LAG-3+ CD8+ cells was notably linked with the immune-checkpoint blockade (ICB) response. This indicates that the efficacy of ICB therapy in HCC can potentially be predicted by analyzing pre-treatment levels of LAG-3 and CD8 in the TME.90 Neuropilins (NRPs) are the receptors of multiple proteins involved in pivotal signaling pathways related to HCC progression. NRP1 and NRP2 were reported as potential therapeutic targets and biomarkers for HCC.91 Defined as the percentage of tumor cells within the TME, tumor purity was associated with the occurrence and development of HCC. Genes like AarF Domain Containing Kinase 3 (ADCK3), Hexokinase-3 (HK3), and palmitoyl-protein thioesterase 1 (PPT1) were associated with tumor purity in HCC. Elevated expression of ADCK3 and reduced expression of HK3 and PPT1 resulted in high levels of tumor purity and were related with a better prognosis.92 It was found that mucosal-associated invariant T (MAIT) cells secrete TNF to activate TNFR2 on regulatory T cells, which forms an immunosuppression TME in HCC.93 Furthermore, as a naturally occurring flavonoid compound, Oroxylin A has been demonstrated to hinder the progression of HCC by reshaping the immune landscape of the TME. Oroxylin A achieves this by inducing M1-like polarization of macrophages through the release of apoptosis-related extracellular vesicles. Additionally, it reduces the M2-like macrophage population while bolstering T cell infiltration within the TME.94
Molecular Bases of Immunotherapy and Targeted Therapy
As compared with adjacent tissue, HCC tissues express higher levels of immunosuppressive molecules such as PD-L1, CTLA4, LAG3, and TIM3.95 The expression of these molecules is negatively correlated with the infiltration of IFN-γ + T lymphocytes in TME. The blocking antibody of these inhibitory molecules can enhance the proliferation of CD4+ and CD8+ tumor-infiltrating T lymphocytes and the production of cytokines.96 In the microenvironment of chronic inflammation, the immune checkpoints CTLA4 and PD1 are upregulated. PD1 binds to PD-L1 to prevent TCR signaling, block T cell proliferation, and induce T cell exhaustion. Tregs constitutively express CTLA4 and block immune responses. CTLA4 binds CD80/CD86 and competes with CD28 to block T cell activation.97,98 Another molecule of significance is TIM3, which displays markedly increased expression in the peripheral blood monocytes and TAMs of individuals with HCC. The cytokine TGF-β plays a role in this elevation, as it encourages TIM3 expression and concurrently drives the alternative activation of macrophages.22
Over-expression of tumor-associated antigens (TAAs) and new tumor antigens formed in the process of somatic mutation in HCC cells stimulate specific T lymphocyte immune responses, which is the theoretical basis of tumor vaccine. TAAs mainly include tumor-overexpressed antigens (TERT, Wilms tumor antigen 1 (WT1)), carcinoembryonic antigens (AFP, glypican-3 (GPC3)), and cancer-testicular antigens (melanoma-associated antigens A (MAGE-A), synovial sarcoma X family member 2 (SSX-2), and New York esophageal squamous cell carcinoma 1 (NY-ESO-1)).99 Furthermore, there is an intriguing cell group referred to as cytokine-induced killer cells. These cells belong to the peripheral blood mononuclear cells and exhibit dual-surface markers: the T lymphocyte marker CD3 and the NK cell marker CD56. These cells, when subjected to activation via IFN, anti-CD3 antibody, and IL-2 in a controlled environment, gain a robust ability to target and kill a wide array of tumor cells. Impressively, they carry out this function with minimal side effects, making them an attractive option for potential therapeutic strategies.100
Targeting angiogenic factors and their related signal pathways has become a hot topic in the research on targeted therapy for HCC. Common HCC targets for targeted therapy include cellular-mesenchymal epithelial transition factor (c-Met), CD 24, CD 147, GPC3, fibroblast growth factor receptor (FGFR), VEGFR, EGFR, and mammalian target of rapamycin (mTOR).101 Signaling pathways involved in cell differentiation (Wnt), proliferation (EGF, IGF, HGF/C-MET, RAF/MEK/ERK), survival (Akt/m-TOR), and angiogenesis (VEGF, PDGF, FGF) contribute to HCC growth and metastasis and also provide potential molecular targets for targeted therapies in HCC.102
Immunotherapy and Targeted Therapy
Immunotherapy
Immunotherapy mainly includes immune checkpoint inhibitors (ICIs), tumor vaccines, and immune cell therapy. Nivolumab is the first ICI approved by the US Food and Drug Administration (FDA) for the treatment of HCC. An open-label, non-comparative, dose escalation and expansion trial (NCT01658878) was carried out to evaluate the efficacy of Nivolumab as second-line therapy for HCC, and it indicates that the objective response rate (ORR) was 20% and the disease control rate (DCR) was 64%.103 Recent trial (NCT02576509) indicates Nivolumab may be considered a therapeutic option for patients who are contraindicated with tyrosine kinase inhibitors (TKIs) and antiangiogenic agents.104 Tislelizumab is the first ICI for HCV-HCC, and its partial response (PR) rate was 17.6%, DCR was 76.4%, and progression-free survival (PFS) was 6.48 months (95% CI: 3.95–9.14).105 Camrelizumab is a PD-1 inhibitor made in China. It has been confirmed that Camrelizumab has a remarkable anti-tumor effect and tolerance to advanced solid tumors.106,107 In patients with previously sorafenib-treated advanced HCC, Avelumab demonstrated moderate efficacy and was well tolerated (NCT03389126).108 Tislelizumab also showed durable objective responses and acceptable tolerability in previously treated advanced HCC patients (NCT03419897).109 Studies on the application of other ICIs (such as Toripalimab, Sintilamab, Durvalumab, and Atezolizumab) in advanced HCC are under way (Table 2).
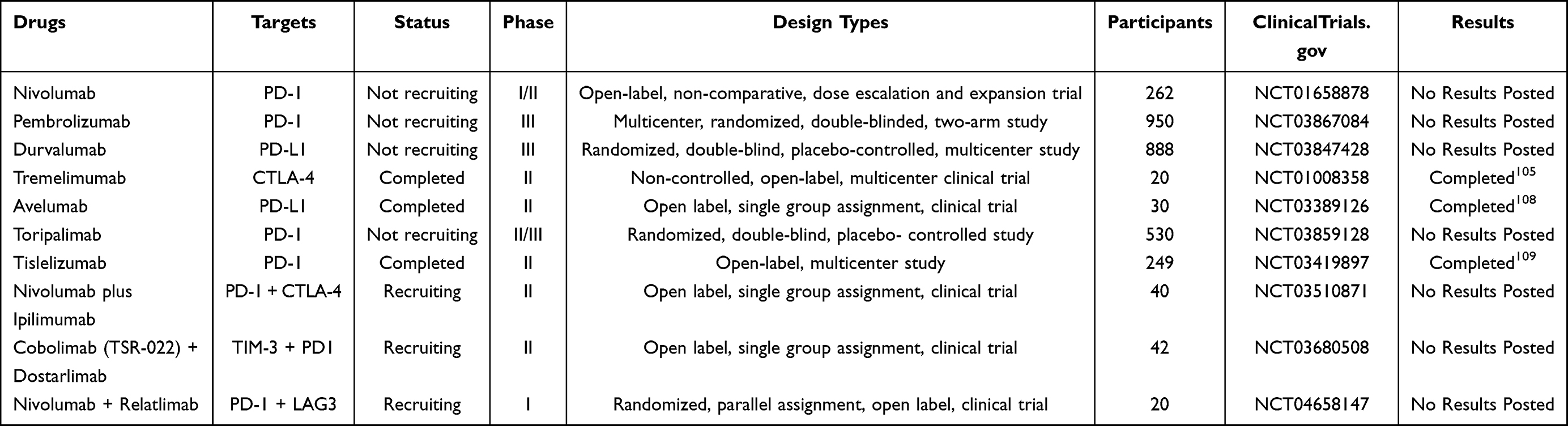 |
Table 2 Immune Checkpoint Inhibitors in HCC |
Tumor vaccine strategies encompass various approaches, including peptides, proteins, dendritic cells (DCs), and viral vector vaccines.110 A meta-analysis of 35 cohort studies indicates that the ORR of DCs vaccine (19%, 95% CI 11 to 29%) is significantly higher compared to the peptide vaccine (1%, 95% CI 0 to 5%).111 Additionally, HBV-related HCC may benefit more from tumor vaccines compared with hepatitis C virus-related HCC.111 The personalized neoantigen vaccines have gained approval as a safe and effective approach for preventing HCC recurrence.112 Immune cell therapy for HCC includes enhancing the activity of endogenous anti-HCC immune cells,113 inducing active anti-HCC immune cells in vitro,114 and genetically modifying HCC-specific immune cells (such as CAR-T cell therapy,115 and TCR-T cell therapy).116 The single-chain Fv recognizing TAAs and the immunoreceptor tyrosine-based activation motif are genetically reconstituted in vitro and then transfected into patient T cells, resulting in chimeric antigen receptor T (CAR-T) cells. In advanced HCCs expressing glypican-3 (GPC3), the intratumor injection of IL-7 and CCL19-secreting CAR-T cells led to complete tumor eradication after 30 days.117 The tumor-specific antigen receptor is introduced into T cells so that the patient’s T cells express the antigen-specific T cell receptor (TCR), that is, TCR-modified T cells (TCR-T). A phase I clinical trial using CAR-T cells targeting CD133 (CART-133) indicates that the survival time of CD133-positive and advanced metastatic HCC patients treated with CART-133 is significantly prolonged.118 A kind of HCV1406 TCR-T cell is constructed to treat HCV-HCC.119 CAR-T and TCR-T cells have great potential for the treatment of HCC, and related clinical trials are also under way (Table 3).
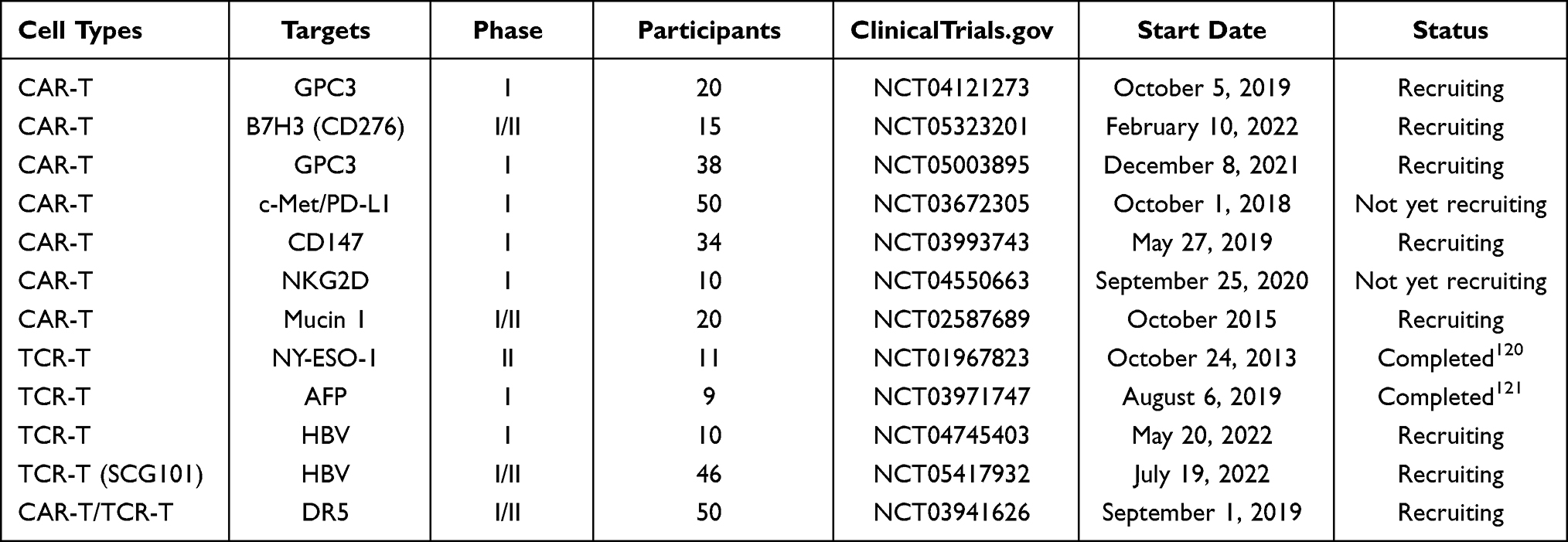 |
Table 3 CAR-T and TCR-T Cell Therapy |
Targeted Therapy
Molecular targeted drugs, such as sorafenib and Lenvatinib, have improved the survival of patients with advanced HCC who are ineligible for liver transplantation or resection.122,123 Sorafenib is an oral multi-target tyrosine kinase inhibitor (TKI) that inhibits tumor growth and angiogenesis, which extends overall survival (OS) by 2.8 months.124 A multicenter non-inferiority trial shows that the mOS of the Lenvatinib group and the Sorafenib group is 13.8 months and 12.3 months, respectively.125 Regorafenib is a second-line treatment for HCC with Sorafenib resistance, and it inhibits tumor angiogenesis, cell proliferation, and metastasis by altering TME and targeting multi-targets.126 Cabozantinib was approved by the FDA in January 2019 for the treatment of HCC. A double-blind clinical trial indicates that the mOS of the Cabozantinib group is 10.2 months and the median PFS is 5.2 months.127 Ramucirumab is an IgGl monoclonal antibody that binds to the extracellular region of vascular endothelial growth factor receptor 2 (VEGFR2), thus blocking the binding of VEGF to VEGFR and further preventing angiogenesis. The mOS of patients in the Ramucirumab group is 4.9 months longer than that in the placebo group, especially when AFP ≥ 400ng/mL, the mOS of patients in Ramucirumab group is prolonged by 8.6 months.128 Currently, Ramucirumab is recommended as a second-line treatment for advanced HCC patients with AFP levels ≥400ng/mL. Apatinib, another TKI, inhibits angiogenesis by targeting VEGFR2 and is recommended for second-line treatment in advanced HCC.129 Recently, small-molecule targeted drugs with more specific targets have been the focus of current clinical studies. MET-selective inhibitors (Tepotinib and Capmatinib) produce potent inhibition of advanced HCC with high MET while reducing off-target toxicity.130 The selective FGFR4 inhibitor Fisogatinib (also known as BLU-554)131 and the TGF-β receptor 1 inhibitor Galunisertib have also been found to be efficacious in advanced HCC.132 In addition, targeted combination therapies provide additional treatment options for patients with advanced HCC who cannot tolerate ICB therapy.
Antibody-Drug Conjugate (ADC) and Bispecific Antibody (BsAb) Therapy for HCC
ADCs contain monoclonal antibodies for targeted delivery and cytotoxic payloads for targeted destruction of malignant cells, allowing for selective delivery of cytotoxic drugs to tumor cells in the most appropriate manner.133 The transmembrane tight junction protein Claudin 6 (CLDN6) has been identified as a therapeutic target. An anti-CLDN6 monoclonal antibody conjugated with the cytotoxic agent (Mertansine) DM1 (CLDN6-DM1) has demonstrated potent antitumor effects, both as a standalone treatment and in combination with sorafenib.134 SHR-A1403 is a novel c-Met ADC consisting of an anti-c-Met monoclonal antibody conjugated with a novel cytotoxic microtubule inhibitor which showed significant anti-tumor activity in cancer cell lines, xenograft mouse models, and HCC PDX models.135 A humanized anti-c-Met antibody conjugated with oxaliplatin significantly improves cytotoxicity against c-Met-positive tumors.136
Bispecific antibodies (BsAb) combine the binding specificity of two different monoclonal antibodies, one activating receptors on killer effector cells and the other binding TAAs to initiate tumor cytotoxicity.137 In xenograft HCC models, GPC3/CD47 BsAb was superior to monotherapy or the combination of anti-CD47 and anti-GPC3 monoclonal antibodies.138 Tetravalent BsAb h8B-BsAb against GPC3 and CD3 antigens significantly induces tumor regression in HCC xenograft mouse models.139 In HBV-related HCC, anti-HBx/anti-CD3 BsAb was able to retarget effector cells in vitro and in vivo to lyse HBxAg-positive HCC cells.140
Immunotherapy Combined with Targeted Therapy for HCC
Anti-angiogenesis and ICIs are pivotal components of anti-HCC therapy. A large number of studies have confirmed that the combination of the two regimens is better than the single regimen, which can significantly increase the clinical benefits of HCC. Atezolizumab combined with Bevacizumab (“T+A” combination) is the first combination regimen that goes beyond the efficacy of sorafenib and has been approved by the FDA for first-line treatment of advanced HCC.141 Lenvatinib combined with pembrolizumab (“cola” combination) significantly prolongs the mOS of patients with unresectable HCC.142 In patients with resectable HCC, perioperative camrelizumab plus apatinib results in promising therapeutic efficacy.143 Table 4 lists several clinical trials related to combination therapy, which are expected to bring more treatment options to HCC.
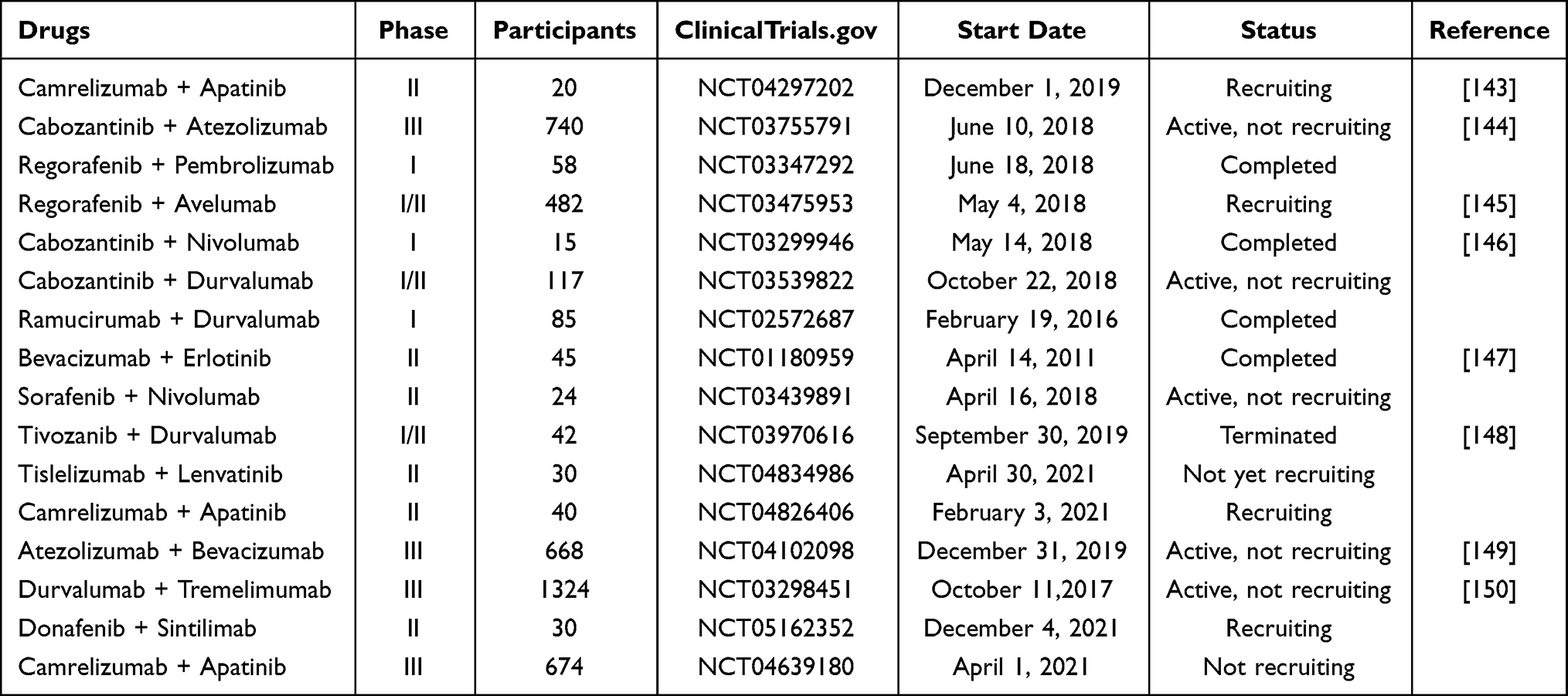 |
Table 4 Immunotherapy Combined with Targeted Therapy for HCC |
Future Directions
Cell resistance to targeted drugs is a major challenge for targeted therapy for HCC. Multi-target drug combinations can reduce the dose of a single drug while maintaining or enhancing antitumor activity and reducing the occurrence of adverse drug reactions and drug-resistant mutations.151 Furthermore, the identification of new therapeutic targets is of paramount importance, as there is currently a shortage of targeted drugs for dominant mutational drivers in HCC, such as TERT promoter mutations,152 CTNNB1 mutations,153 and TP53 mutations.154 The long-term effectiveness and durability of responses to immunotherapies remain areas of uncertainty. Identifying biomarkers and molecular signatures that reliably predict responses to specific immunotherapeutic agents is imperative for personalized treatment approaches. Tumor mutation load,155 circulating tumor cells,156 and circulating tumor DNA157 are both promising candidate biomarkers. In addition, the development of advanced immunomodulatory agents, such as CAR-T, TCR-T, therapeutic vaccines, and oncolytic viruses, holds promise for achieving deeper and more durable responses in a subset of HCC patients.
Conclusion
HCC is characterized by its aggressive and advanced-stage presentation upon diagnosis, posing significant challenges to curative interventions. TME is a complex immune system that plays an important role in promoting tumor cell proliferation, metastasis, and immune evasion. Notably, targeted therapy and immunotherapy following traditional chemotherapy, radiotherapy, and surgical treatment have achieved remarkable efficacy in advanced HCC by reprogramming the immunosuppressive TME. However, the application of these therapies is limited by the inter-individual and even intra-tumoral heterogeneity of HCC, as well as the absence of reliable biomarkers for predicting therapeutic responses, drug resistance, and immune tolerance. In the future, new therapeutic targets, reliable predictive biomarkers, and more rational and efficient combination therapy regimens will greatly improve the survival of HCC patients, especially those with advanced HCC. A better understanding of the interactions between TME and HCC cells is essential for developing novel effective therapeutic approaches for HCC.
Abbreviations
APOBEC3B, apolipoprotein B mRNA editing enzyme catalytic subunit 3B; ADC, Antibody-drug conjugate; BsAb, Bispecific antibody; CAFs, cancer-associated fibroblasts; CCL2, chemokine (C-C motif) ligand 2; CTL, cytotoxic CD8+T cells; CTLA4, cytotoxic T cell associated antigen 4; CXCL12, chemokine (C-X-C motif) ligand 12; CXCL5, chemokine (C-X-C motif) ligand 5; DCs, dendritic cells; ECM, extracellular matrix; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; HCV, hepatitis C virus; HSCs, hepatic stellate cells; ICIs, immune checkpoint inhibitors; IFN-γ, interferon gamma; IL-6, interleukin-6; LAG3, lymphocyte activation gene 3; LPS, lipopolysaccharide; MDSCs, myeloid-derived suppressor cells; MHR, major hydrophilic region; NAFLD, non-alcoholic fatty liver disease; NF κ B, nuclear factor κ B; NK cells, natural killer cells; PD-1, programmed death 1; PLC, primary liver cancer; TAMs, tumor associated macrophages; TANs, tumor associated neutrophils; Th1, helper T cell 1; TIM-3, T cell immunoglobulin domain and mucin domain-3; TKIs, tyrosine kinase inhibitors; TME, tumor microenvironment; Tregs, regulatory T cells; iNOS, inducible nitric oxide synthase; NO, nitric oxide; VEGF, vascular endothelial growth factor; PDGF, platelet-derived growth factor; MMP, matrix metallopeptidase; HIF-1 α, Hypoxia inducible factor 1 α; EMT, epithelial–mesenchymal transition; OS, overall survival; NLR, neutrophil/lymphocyte ratio; TGF-β, transforming growth factor-beta; BMP2, bone morphogenetic protein 2; IDO1, indoleamine 2,3-dioxygenase 1; ROS, reactive oxygen species; PRC2, poly-comb repressor complex 2; SLC7A2, solute carrier family 7 member 2; TAAs, tumor-associated antigens; GPC3, glypican-3; NY-ESO-1, New York esophageal squamous cell carcinoma 1; EGFR, epidermal growth factor receptor; mTOR, mammalian target of rapamycin; JAK/STAT, janus kinase/signal transducer and activator of transcription; RFA, radiofrequency ablation; ORR, objective response rate; DCR, disease control rate; PFS, progression-free survival; CAR-T, chimeric antigen receptor T; TCR-T, T cell receptor-modified T cells; dsDNA, double stranded DNA; ecDNA, extrachromosomal DNA.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the GWV-10.1-XK17/3-year public health program of Shanghai Municipal Health Commission (GC), the National Key Basic Research Program of China [grant number 2015CB554006 (GC)], and the National Natural Science Foundation of China [grant numbers 91529305 (GC), 81520108021 (GC), 81673250 (GC)].
Disclosure
The authors declare no conflicts of interest in this work.
References
1. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN ESTIMATES OF INCIDENCE AND MORTALITY WORLDWIDE FOR 36 CANCERS IN 185 COUntries. CA Cancer J Clin. 2021;71(3):209–249. doi:10.3322/caac.21660
2. Lin J, Zhang H, Yu H, et al. Epidemiological characteristics of primary liver cancer in Mainland China from 2003 to 2020: a Representative Multicenter Study. Front Oncol. 2022;12:906778. doi:10.3389/fonc.2022.906778
3. Jiang D, Zhang L, Liu W, et al. Trends in cancer mortality in China from 2004 to 2018: a nationwide longitudinal study. Cancer Commun. 2021;41(10):1024–1036. doi:10.1002/cac2.12195
4. Vogel A, Meyer T, Sapisochin G, Salem R, Saborowski A. Hepatocellular carcinoma. Lancet. 2022;400(10360):1345–1362. doi:10.1016/S0140-6736(22)01200-4
5. Rizzo A, Ricci AD, Brandi G. Systemic adjuvant treatment in hepatocellular carcinoma: tempted to do something rather than nothing. Future Oncol. 2020;16(32):2587–2589. doi:10.2217/fon-2020-0669
6. Lodetti Zangrandi G, Tirpanlar D, Pastore M, Soldani C, Lleo A, Raggi C. Tumor microenvironment highlighting tumor-associated macrophages and immune cells. Hepatoma Res. 2023;9:32. doi:10.20517/2394-5079.2023.32
7. Boedtkjer E, Pedersen SF. The acidic tumor microenvironment as a driver of cancer. Annu Rev Physiol. 2020;82(1):103–126. doi:10.1146/annurev-physiol-021119-034627
8. Yi M, Jiao D, Qin S, Chu Q, Wu K, Li A. Synergistic effect of immune checkpoint blockade and anti-angiogenesis in cancer treatment. Mol Cancer. 2019;18(1):60. doi:10.1186/s12943-019-0974-6
9. Wang SZ, Lee SD, Sarkar D, et al. Immunological characterization of hepatocellular carcinoma. Hepatoma Res. 2021;7:6.
10. Sipos F, Műzes G. Cancer stem cell relationship with pro-tumoral inflammatory microenvironment. Biomedicines. 2023;11(1):189. doi:10.3390/biomedicines11010189
11. Liu W, Deng Y, Li Z, et al. Cancer evo-dev: a theory of inflammation-induced oncogenesis. Front Immunol. 2021;12:768098. doi:10.3389/fimmu.2021.768098
12. Huang A, Yang XR, Chung WY, Dennison AR, Zhou J. Targeted therapy for hepatocellular carcinoma. Signal Transduct Target Ther. 2020;5(1):146. doi:10.1038/s41392-020-00264-x
13. Rizzo A, Ricci AD. Challenges and future trends of hepatocellular carcinoma immunotherapy. Int J Mol Sci. 2022;23(19):11363. doi:10.3390/ijms231911363
14. Santoni M, Rizzo A, Kucharz J, et al. Complete remissions following immunotherapy or immuno-oncology combinations in cancer patients: the MOUSEION-03 meta-analysis. Cancer Immunol Immunother. 2023;72(6):1365–1379. doi:10.1007/s00262-022-03349-4
15. Santoni M, Rizzo A, Mollica V, et al. The impact of gender on the efficacy of immune checkpoint inhibitors in cancer patients: the MOUSEION-01 study. Crit Rev Oncol Hematol. 2022;170:103596. doi:10.1016/j.critrevonc.2022.103596
16. Hinshaw D, Shevde L. The tumor microenvironment innately modulates cancer progression. Cancer Res. 2019;79(18):4557–4566. doi:10.1158/0008-5472.CAN-18-3962
17. Jin H, Qin S, He J, et al. New insights into checkpoint inhibitor immunotherapy and its combined therapies in hepatocellular carcinoma: from mechanisms to clinical trials. Int J Biol Sci. 2022;18(7):2775–2794. doi:10.7150/ijbs.70691
18. Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20. doi:10.1016/j.immuni.2014.06.008
19. Zhou D, Luan J, Huang C, Li J. Tumor-associated macrophages in Hepatocellular carcinoma: friend or Foe? Gut Liver. 2021;15(4):500–516. doi:10.5009/gnl20223
20. Jiang Y, Han Q, Zhao H, Zhang J. Promotion of epithelial-mesenchymal transformation by hepatocellular carcinoma-educated macrophages through Wnt2b/β-catenin/c-Myc signaling and reprogramming glycolysis. J Exp Clin Cancer Res. 2021;40(1):13. doi:10.1186/s13046-020-01808-3
21. Wan S, Zhao E, Kryczek I, et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology. 2014;147(6):1393–1404. doi:10.1053/j.gastro.2014.08.039
22. Yan W, Liu X, Ma H, et al. Tim-3 fosters HCC development by enhancing TGF-β-mediated alternative activation of macrophages. Gut. 2015;64(10):1593–1604. doi:10.1136/gutjnl-2014-307671
23. Song S, Yuan P, Li P, Wu H, Lu J, Wei W. Dynamic analysis of tumor-associated immune cells in DEN-induced rat hepatocellular carcinoma. Int Immunopharmacol. 2014;22(2):392–399. doi:10.1016/j.intimp.2014.07.007
24. Liu G, Yin L, Ouyang X, Zeng K, Xiao Y, Li Y. M2 macrophages promote HCC cells invasion and migration via miR-149-5p/MMP9 signaling. J Cancer. 2020;11(5):1277–1287. doi:10.7150/jca.35444
25. Li C, Jiang P, Wei S, Xu X, Wang J. Regulatory T cells in tumor microenvironment: new mechanisms, potential therapeutic strategies and future prospects. Mol Cancer. 2020;19(1):116. doi:10.1186/s12943-020-01234-1
26. Zhang J, Zhang Q, Lou Y, et al. Hypoxia-inducible factor-1α/interleukin-1β signaling enhances hepatoma epithelial-mesenchymal transition through macrophages in a hypoxic-inflammatory microenvironment. Hepatology. 2018;67(5):1872–1889. doi:10.1002/hep.29681
27. Wang YF, Yuan SX, Jiang H, et al. Spatial maps of hepatocellular carcinoma transcriptomes reveal spatial expression patterns in tumor immune microenvironment. Theranostics. 2022;12(9):4163–4180. doi:10.7150/thno.71873
28. Dong P, Ma L, Liu L, et al. CD86⁺/CD206⁺, diametrically polarized tumor-associated macrophages, predict hepatocellular carcinoma patient prognosis. Int J Mol Sci. 2016;17(3):320. doi:10.3390/ijms17030320
29. Ning WR, Jiang D, Liu XC, et al. Carbonic anhydrase XII mediates the survival and prometastatic functions of macrophages in human hepatocellular carcinoma. J Clin Invest. 2022;132(7):e153110. doi:10.1172/JCI153110
30. Fridlender ZG, Sun J, Kim S, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2”. TAN Cancer Cell. 2009;16(3):183–194. doi:10.1016/j.ccr.2009.06.017
31. Arvanitakis K, Mitroulis I, Germanidis G. Tumor-associated neutrophils in Hepatocellular carcinoma pathogenesis, prognosis, and therapy. Cancers. 2021;13(12):2899. doi:10.3390/cancers13122899
32. Fridlender ZG, Albelda SM. Tumor-associated neutrophils: friend or foe? Carcinogenesis. 2012;33(5):949–955. doi:10.1093/carcin/bgs123
33. Zhou SL, Zhou ZJ, Hu ZQ, et al. Tumor-associated neutrophils recruit macrophages and T-regulatory cells to promote progression of Hepatocellular carcinoma and resistance to sorafenib. Gastroenterology. 2016;150(7):1646–58.e17. doi:10.1053/j.gastro.2016.02.040
34. Zhou SL, Yin D, Hu ZQ, et al. A positive feedback loop between cancer stem-like cells and tumor-associated neutrophils controls Hepatocellular carcinoma progression. Hepatology. 2019;70(4):1214–1230. doi:10.1002/hep.30630
35. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9(3):162–174. doi:10.1038/nri2506
36. Joshi S, Sharabi A. Targeting myeloid-derived suppressor cells to enhance natural killer cell-based immunotherapy. Pharmacol Ther. 2022;235:108114. doi:10.1016/j.pharmthera.2022.108114
37. Lu C, Rong D, Zhang B, et al. Current perspectives on the immunosuppressive tumor microenvironment in hepatocellular carcinoma: challenges and opportunities. Mol Cancer. 2019;18(1):130. doi:10.1186/s12943-019-1047-6
38. Salmaninejad A, Valilou SF, Soltani A, et al. Tumor-associated macrophages: role in cancer development and therapeutic implications. Cell Oncol. 2019;42(5):591–608. doi:10.1007/s13402-019-00453-z
39. Yang R, Sun L, Li CF, et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat Commun. 2021;12(1):832. doi:10.1038/s41467-021-21099-2
40. Zhou J, Liu M, Sun H, et al. Hepatoma-intrinsic CCRK inhibition diminishes myeloid-derived suppressor cell immunosuppression and enhances immune-checkpoint blockade efficacy. Gut. 2018;67(5):931–944. doi:10.1136/gutjnl-2017-314032
41. Chiu DK, Tse AP, Xu IM, et al. Hypoxia inducible factor HIF-1 promotes myeloid-derived suppressor cells accumulation through ENTPD2/CD39L1 in hepatocellular carcinoma. Nat Commun. 2017;8(1):517. doi:10.1038/s41467-017-00530-7
42. Wang D, Li X, Li J, et al. APOBEC3B interaction with PRC2 modulates microenvironment to promote HCC progression. Gut. 2019;68(10):1846–1857. doi:10.1136/gutjnl-2018-317601
43. Xia S, Wu J, Zhou W, et al. SLC7A2 deficiency promotes hepatocellular carcinoma progression by enhancing recruitment of myeloid-derived suppressors cells. Cell Death Dis. 2021;12(6):570. doi:10.1038/s41419-021-03853-y
44. Zhao Y, Wu T, Shao S, Shi B, Zhao Y. Phenotype, development, and biological function of myeloid-derived suppressor cells. Oncoimmunology. 2016;5(2):e1004983. doi:10.1080/2162402X.2015.1004983
45. Yin Z, Dong C, Jiang K, et al. Heterogeneity of cancer-associated fibroblasts and roles in the progression, prognosis, and therapy of hepatocellular carcinoma. J Hematol Oncol. 2019;12(1):101. doi:10.1186/s13045-019-0782-x
46. Mizutani Y, Kobayashi H, Iida T, et al. Meflin-positive cancer-associated fibroblasts inhibit pancreatic carcinogenesis. Cancer Res. 2019;79(20):5367–5381. doi:10.1158/0008-5472.CAN-19-0454
47. Lau EY, Lo J, Cheng BY, et al. Cancer-associated fibroblasts regulate tumor-initiating cell plasticity in Hepatocellular Carcinoma through c-Met/FRA1/HEY1 signaling. Cell Rep. 2016;15(6):1175–1189. doi:10.1016/j.celrep.2016.04.019
48. Li Y, Wang R, Xiong S, et al. Cancer-associated fibroblasts promote the stemness of CD24(+) liver cells via paracrine signaling. J Mol Med. 2019;97(2):243–255. doi:10.1007/s00109-018-1731-9
49. Xiong S, Wang R, Chen Q, et al. Cancer-associated fibroblasts promote stem cell-like properties of hepatocellular carcinoma cells through IL-6/STAT3/Notch signaling. Am J Cancer Res. 2018;8(2):302–316.
50. Fang T, Lv H, Lv G, et al. Tumor-derived exosomal miR-1247-3p induces cancer-associated fibroblast activation to foster lung metastasis of liver cancer. Nat Commun. 2018;9(1):191. doi:10.1038/s41467-017-02583-0
51. Zhou Y, Ren H, Dai B, et al. Hepatocellular carcinoma-derived exosomal miRNA-21 contributes to tumor progression by converting hepatocyte stellate cells to cancer-associated fibroblasts. J Exp Clin Cancer Res. 2018;37(1):324. doi:10.1186/s13046-018-0965-2
52. Caligiuri A, Parola M, Marra F, Cannito S, Gentilini A. Cholangiocarcinoma tumor microenvironment highlighting fibrosis and matrix components. Hepatoma Res. 2023;9:30.
53. She Q, Hu S, Pu X, Guo Q, Mou C, Yang C. The effect of hepatocellular carcinoma-associated fibroblasts on hepatoma vasculogenic mimicry. Am J Cancer Res. 2020;10(12):4198–4210.
54. Chen S, Morine Y, Tokuda K, et al. Cancer‑associated fibroblast‑induced M2‑polarized macrophages promote hepatocellular carcinoma progression via the plasminogen activator inhibitor‑1 pathway. Int J Oncol. 2021;59(2):59. doi:10.3892/ijo.2021.5239
55. Cheng JT, Deng YN, Yi HM, et al. Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis. 2016;5(2):e198. doi:10.1038/oncsis.2016.7
56. Zhu GQ, Tang Z, Huang R, et al. CD36(+) cancer-associated fibroblasts provide immunosuppressive microenvironment for hepatocellular carcinoma via secretion of macrophage migration inhibitory factor. Cell Discov. 2023;9(1):25. doi:10.1038/s41421-023-00529-z
57. Yang XH, Yamagiwa S, Ichida T, et al. Increase of CD4+ CD25+ regulatory T-cells in the liver of patients with hepatocellular carcinoma. J Hepatol. 2006;45(2):254–262. doi:10.1016/j.jhep.2006.01.036
58. Cao M, Cabrera R, Xu Y, et al. Hepatocellular carcinoma cell supernatants increase expansion and function of CD4(+)CD25(+) regulatory T cells. Lab Invest. 2007;87(6):582–590. doi:10.1038/labinvest.3700540
59. Suthen S, Lim CJ, Nguyen PHD, et al. Hypoxia-driven immunosuppression by Treg and type-2 conventional dendritic cells in HCC. Hepatology. 2022;76(5):1329–1344. doi:10.1002/hep.32419
60. Krummel MF, Allison JP. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J Exp Med. 1996;183(6):2533–2540. doi:10.1084/jem.183.6.2533
61. Jiang R, Tang J, Chen Y, et al. The long noncoding RNA lnc-EGFR stimulates T-regulatory cells differentiation thus promoting hepatocellular carcinoma immune evasion. Nat Commun. 2017;8:15129. doi:10.1038/ncomms15129
62. Lv D, Chen L, Du L, Zhou L, Tang H. Emerging regulatory mechanisms involved in liver cancer stem cell properties in Hepatocellular carcinoma. Front Cell Dev Biol. 2021;9:691410. doi:10.3389/fcell.2021.691410
63. Lam KH, Ma S. Noncellular components in the liver cancer stem cell niche: biology and potential clinical implications. Hepatology. 2023;78(3):991–1005. doi:10.1002/hep.32629
64. Guo Y, Xiao Z, Yang L, et al. Hypoxia‑inducible factors in hepatocellular carcinoma (Review). Oncol Rep. 2020;43(1):3–15. doi:10.3892/or.2019.7397
65. Jing L, Ruan Z, Sun H, et al. Epithelial-mesenchymal transition induced cancer-stem-cell-like characteristics in hepatocellular carcinoma. J Cell Physiol. 2019;234(10):18448–18458. doi:10.1002/jcp.28480
66. Chen H, Nio K, Yamashita T, et al. BMP9-ID1 signaling promotes EpCAM-positive cancer stem cell properties in hepatocellular carcinoma. Mol Oncol. 2021;15(8):2203–2218. doi:10.1002/1878-0261.12963
67. Zhao X, Sun B, Liu T, et al. Long noncoding RNA n339260 promotes vasculogenic mimicry and cancer stem cell development in hepatocellular carcinoma. Cancer Sci. 2018;109(10):3197–3208. doi:10.1111/cas.13740
68. Dongre A, Weinberg RA. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat Rev Mol Cell Biol. 2019;20(2):69–84. doi:10.1038/s41580-018-0080-4
69. Wei R, Zhu WW, Yu GY, et al. S100 calcium-binding protein A9 from tumor-associated macrophage enhances cancer stem cell-like properties of hepatocellular carcinoma. Int J Cancer. 2021;148(5):1233–1244. doi:10.1002/ijc.33371
70. Maniotis AJ, Folberg R, Hess A, et al. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol. 1999;155(3):739–752. doi:10.1016/S0002-9440(10)65173-5
71. Zhao Z, Bai S, Wang R, et al. Cancer-associated fibroblasts endow stem-like qualities to liver cancer cells by modulating autophagy. Cancer Manag Res. 2019;11:5737–5744. doi:10.2147/CMAR.S197634
72. Yoshida Y, Yoshio S, Yamazoe T, et al. Phenotypic characterization by single-cell mass cytometry of human intrahepatic and peripheral NK cells in patients with Hepatocellular carcinoma. Cells. 2021;10(6):1495. doi:10.3390/cells10061495
73. Mahgoub S, Abosalem H, Emara M, Kotb N, Maged A, Soror S. Restoring NK cells functionality via cytokine activation enhances cetuximab-mediated NK-cell ADCC: a promising therapeutic tool for HCC patients. Mol Immunol. 2021;137:221–227. doi:10.1016/j.molimm.2021.07.008
74. Cui C, Fu K, Yang L, et al. Hypoxia-inducible gene 2 promotes the immune escape of hepatocellular carcinoma from nature killer cells through the interleukin-10-STAT3 signaling pathway. J Exp Clin Cancer Res. 2019;38(1):229. doi:10.1186/s13046-019-1233-9
75. Huang CF, Wang SC, Chang WT, et al. Lower protein expression levels of MHC class I chain-related gene A in hepatocellular carcinoma are at high risk of recurrence after surgical resection. Sci Rep. 2018;8(1):15821. doi:10.1038/s41598-018-34155-7
76. Argenziano ME, Montori M, Scorzoni C, Benedetti A, Marzioni M, Maroni L. The role of tumor microenvironment in cholangiocarcinoma. Hepatoma Res. 2023;9:9. doi:10.20517/2394-5079.2022.98
77. Zhang Q, He Y, Luo N, et al. Landscape and dynamics of single immune cells in Hepatocellular Carcinoma. Cell. 2019;179(4):829–45.e20. doi:10.1016/j.cell.2019.10.003
78. Deligne C, Midwood KS. Macrophages and extracellular matrix in breast cancer: partners in crime or protective allies? Front Oncol. 2021;11:620773. doi:10.3389/fonc.2021.620773
79. Tang H, You T, Sun Z, Bai C, Wang Y. Extracellular matrix-based gene expression signature defines two prognostic subtypes of Hepatocellular Carcinoma with different immune microenvironment characteristics. Front Mol Biosci. 2022;9:839806. doi:10.3389/fmolb.2022.839806
80. Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, Werb Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat Commun. 2020;11(1):5120. doi:10.1038/s41467-020-18794-x
81. Cigliano A, Strain AJ, Cadamuro M. Signaling and molecular networks related to development and inflammation involved in CCA initiation and progression. Hepatoma Res. 2023;9(14):15. doi:10.20517/2394-5079.2023.09
82. Li Z, Wang F, Li Y, et al. Combined anti-hepatocellular carcinoma therapy inhibit drug-resistance and metastasis via targeting “substance P-hepatic stellate cells-hepatocellular carcinoma” axis. Biomaterials. 2021;276:121003. doi:10.1016/j.biomaterials.2021.121003
83. Wang H, Rao B, Lou J, et al. The function of the HGF/c-met axis in Hepatocellular carcinoma. Front Cell Dev Biol. 2020;8:55. doi:10.3389/fcell.2020.00055
84. Ngo MT, Jeng HY, Kuo YC, et al. The role of IGF/IGF-1R signaling in Hepatocellular carcinomas: stemness-related properties and drug resistance. Int J Mol Sci. 2021;22(4):1931. doi:10.3390/ijms22041931
85. Gonzalez-Sanchez E, Vaquero J, Férnandez-Barrena MG, et al. The TGF-β pathway: a pharmacological target in Hepatocellular carcinoma? Cancers. 2021;13(13):3248. doi:10.3390/cancers13133248
86. Han L, Lin X, Yan Q, et al. PBLD inhibits angiogenesis via impeding VEGF/VEGFR2-mediated microenvironmental cross-talk between HCC cells and endothelial cells. Oncogene. 2022;41(13):1851–1865. doi:10.1038/s41388-022-02197-x
87. Xu J, Lin H, Wu G, Zhu M, Li M. IL-6/STAT3 is a promising therapeutic target for Hepatocellular carcinoma. Front Oncol. 2021;11:760971. doi:10.3389/fonc.2021.760971
88. Salman S, Meyers DJ, Wicks EE, et al. HIF inhibitor 32-134D eradicates murine hepatocellular carcinoma in combination with anti-PD1 therapy. J Clin Invest. 2022;132(9):e156774. doi:10.1172/JCI156774
89. Cui Q, Wang X, Zhang Y, Shen Y, Qian Y. Macrophage-derived MMP-9 and MMP-2 are closely related to the rupture of the fibrous capsule of Hepatocellular carcinoma leading to tumor invasion. Biol Proced Online. 2023;25(1):8. doi:10.1186/s12575-023-00196-0
90. Cheung CCL, Seah YHJ, Fang J, et al. Immunohistochemical scoring of LAG-3 in conjunction with CD8 in the tumor microenvironment predicts response to immunotherapy in hepatocellular carcinoma. Front Immunol. 2023;14:1150985. doi:10.3389/fimmu.2023.1150985
91. Fernández-Palanca P, Payo-Serafín T, Méndez-Blanco C, et al. Neuropilins as potential biomarkers in hepatocellular carcinoma: a systematic review of basic and clinical implications. Clin Mol Hepatol. 2023;29(2):293–319. doi:10.3350/cmh.2022.0425
92. Zhao Y, Xu X, Wang Y, Wu LD, Luo RL, Xia RP. Tumor purity-associated genes influence hepatocellular carcinoma prognosis and tumor microenvironment. Front Oncol. 2023;13:1197898. doi:10.3389/fonc.2023.1197898
93. Zhou C, Sun BY, Zhou PY, et al. MAIT cells confer resistance to lenvatinib plus anti-PD1 antibodies in hepatocellular carcinoma through TNF-TNFRSF1B pathway [published online ahead of print, 2023 Sep 17]. Clin Immunol. 2023;256:109770. doi:10.1016/j.clim.2023.109770
94. Wang P, Cao J, Feng Z, et al. Oroxylin a promoted apoptotic extracellular vesicles transfer of glycolytic kinases to remodel immune microenvironment in hepatocellular carcinoma model. Eur J Pharmacol. 2023;957:176037. doi:10.1016/j.ejphar.2023.176037
95. Zhou G, Sprengers D, Boor PPC, et al. Antibodies against immune checkpoint molecules restore functions of tumor-infiltrating T cells in Hepatocellular Carcinomas. Gastroenterology. 2017;153(4):1107–19.e10. doi:10.1053/j.gastro.2017.06.017
96. Hilmi M, Neuzillet C, Calderaro J, Lafdil F, Pawlotsky JM, Rousseau B. Angiogenesis and immune checkpoint inhibitors as therapies for hepatocellular carcinoma: current knowledge and future research directions. J Immunother Cancer. 2019;7(1):333. doi:10.1186/s40425-019-0824-5
97. Llovet JM, Castet F, Heikenwalder M, et al. Immunotherapies for hepatocellular carcinoma. Nat Rev Clin Oncol. 2022;19(3):151–172. doi:10.1038/s41571-021-00573-2
98. Makarova-Rusher OV, Medina-Echeverz J, Duffy AG, Greten TF. The yin and yang of evasion and immune activation in HCC. J Hepatol. 2015;62(6):1420–1429. doi:10.1016/j.jhep.2015.02.038
99. Hu Z, Ott PA, Wu CJ. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat Rev Immunol. 2018;18(3):168–182. doi:10.1038/nri.2017.131
100. Chan JD, Lai J, Slaney CY, Kallies A, Beavis PA, Darcy PK. Cellular networks controlling T cell persistence in adoptive cell therapy. Nat Rev Immunol. 2021;21(12):769–784. doi:10.1038/s41577-021-00539-6
101. Siracusano G, Tagliamonte M, Buonaguro L, Lopalco L. Cell surface proteins in Hepatocellular carcinoma: from bench to bedside. Vaccines. 2020;8(1):41. doi:10.3390/vaccines8010041
102. Dimri M, Satyanarayana A. Molecular signaling pathways and therapeutic targets in Hepatocellular carcinoma. Cancers. 2020;12(2):491. doi:10.3390/cancers12020491
103. El-Khoueiry AB, Sangro B, Yau T, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, Phase 1/2 dose escalation and expansion trial. Lancet. 2017;389(10088):2492–2502. doi:10.1016/S0140-6736(17)31046-2
104. Yau T, Park JW, Finn RS, et al. Nivolumab versus sorafenib in advanced hepatocellular carcinoma (CheckMate 459): a randomised, multicentre, open-label, Phase 3 trial. Lancet Oncol. 2022;23(1):77–90. doi:10.1016/S1470-2045(21)00604-5
105. Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with Hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81–88. doi:10.1016/j.jhep.2013.02.022
106. Fang W, Yang Y, Ma Y, et al. Camrelizumab (SHR-1210) alone or in combination with gemcitabine plus cisplatin for nasopharyngeal carcinoma: results from two single-arm, phase 1 trials. Lancet Oncol. 2018;19(10):1338–1350. doi:10.1016/S1470-2045(18)30495-9
107. Mo H, Huang J, Xu J, et al. Safety, anti-tumour activity, and pharmacokinetics of fixed-dose SHR-1210, an anti-PD-1 antibody in advanced solid tumours: a dose-escalation, phase 1 study. Br J Cancer. 2018;119(5):538–545. doi:10.1038/s41416-018-0100-3
108. Lee DW, Cho EJ, Lee JH, et al. Phase II Study of avelumab in patients with advanced Hepatocellular carcinoma previously treated with sorafenib. Clin Cancer Res. 2021;27(3):713–718. doi:10.1158/1078-0432.CCR-20-3094
109. Ren Z, Ducreux M, Abou-Alfa GK, et al. Tislelizumab in patients with previously treated advanced Hepatocellular carcinoma (RATIONALE-208): a multicenter, non-randomized, open-label, phase 2 trial. Liver Cancer. 2022;12(1):72–84. doi:10.1159/000527175
110. Butterfield LH. Cancer vaccines. BMJ. 2015;350:h988.
111. Han CL, Yan YC, Yan LJ, et al. Efficacy and security of tumor vaccines for hepatocellular carcinoma: a systemic review and meta-analysis of the last 2 decades. J Cancer Res Clin Oncol. 2023;149(4):1425–1441. doi:10.1007/s00432-022-04008-y
112. Cai Z, Su X, Qiu L, et al. Personalized neoantigen vaccine prevents postoperative recurrence in Hepatocellular carcinoma patients with vascular invasion. Mol Cancer. 2021;20(1):164. doi:10.1186/s12943-021-01467-8
113. Wang WC, Zhang ZQ, Li PP, et al. Anti-tumor activity and mechanism of oligoclonal hepatocellular carcinoma tumor-infiltrating lymphocytes in vivo and in vitro. Cancer Biol Ther. 2019;20(9):1187–1194. doi:10.1080/15384047.2019.1599663
114. Yu SJ, Ma C, Heinrich B, et al. Targeting the crosstalk between cytokine-induced killer cells and myeloid-derived suppressor cells in Hepatocellular carcinoma. J Hepatol. 2019;70(3):449–457. doi:10.1016/j.jhep.2018.10.040
115. Zhang Q, Zhang Z, Peng M, Fu S, Xue Z, Zhang R. CAR-T cell therapy in gastrointestinal tumors and hepatic carcinoma: from bench to bedside. Oncoimmunology. 2016;5(12):e1251539. doi:10.1080/2162402X.2016.1251539
116. Chen L, Qiao D, Wang J, Tian G, Wang M. Cancer immunotherapy with lymphocytes genetically engineered with T cell receptors for solid cancers. Immunol Lett. 2019;216:51–62. doi:10.1016/j.imlet.2019.10.002
117. Pang N, Shi J, Qin L, et al. IL-7 and CCL19-secreting CAR-T cell therapy for tumors with positive glypican-3 or mesothelin. J Hematol Oncol. 2021;14(1):118. doi:10.1186/s13045-021-01128-9
118. Wang Y, Chen M, Wu Z, et al. CD133-directed CAR T cells for advanced metastasis malignancies: a Phase I trial. Oncoimmunology. 2018;7(7):e1440169. doi:10.1080/2162402X.2018.1440169
119. Spear TT, Callender GG, Roszkowski JJ, et al. TCR gene-modified T cells can efficiently treat established hepatitis C-associated hepatocellular carcinoma tumors. Cancer Immunol Immunother. 2016;65(3):293–304. doi:10.1007/s00262-016-1800-2
120. Robbins PF, Morgan RA, Feldman SA, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29(7):917–924. doi:10.1200/JCO.2010.32.2537
121. Luo X, Cui H, Cai L, et al. Selection of a clinical lead TCR targeting alpha-fetoprotein-positive liver cancer based on a balance of risk and benefit. Front Immunol. 2020;11:623. doi:10.3389/fimmu.2020.00623
122. Ramadori G. C-kit expression in cancer cells or hematopoietic cells of the tumoral microenvironment: which is the basis for efficacy of TK inhibitors and immunotherapy in HCC? Hepatoma Res. 2021;7:69.
123. Feng ZY, Xu FG, Liu Y, et al. The immune microenvironment and progression of immunotherapy and combination therapeutic strategies for hepatocellular carcinoma. Hepatoma Res. 2021;7:3.
124. Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. doi:10.1056/NEJMoa0708857
125. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018;391(10126):1163–1173. doi:10.1016/S0140-6736(18)30207-1
126. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56–66. doi:10.1016/S0140-6736(16)32453-9
127. Abou-Alfa GK, Meyer T, Cheng AL, et al. Cabozantinib in patients with advanced and progressing Hepatocellular carcinoma. N Engl J Med. 2018;379(1):54–63. doi:10.1056/NEJMoa1717002
128. Kudo M, Hatano E, Ohkawa S, et al. Ramucirumab as second-line treatment in patients with advanced hepatocellular carcinoma: Japanese subgroup analysis of the REACH trial. J Gastroenterol. 2017;52(4):494–503. doi:10.1007/s00535-016-1247-4
129. Qin S, Li Q, Gu S, et al. Apatinib as second-line or later therapy in patients with advanced hepatocellular carcinoma (AHELP): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Gastroenterol Hepatol. 2021;6(7):559–568. doi:10.1016/S2468-1253(21)00109-6
130. Bouattour M, Raymond E, Qin S, et al. Recent developments of c-Met as a therapeutic target in hepatocellular carcinoma. Hepatology. 2018;67(3):1132–1149. doi:10.1002/hep.29496
131. Kim RD, Sarker D, Meyer T, et al. First-in-human Phase I study of fisogatinib (BLU-554) validates aberrant FGF19 signaling as a driver event in Hepatocellular carcinoma. Cancer Discov. 2019;9(12):1696–1707. doi:10.1158/2159-8290.CD-19-0555
132. Faivre S, Santoro A, Kelley RK, et al. Novel transforming growth factor beta receptor I kinase inhibitor galunisertib (LY2157299) in advanced hepatocellular carcinoma. Liver Int. 2019;39(8):1468–1477. doi:10.1111/liv.14113
133. Murali M, Kumar AR, Nair B, et al. Antibody-drug conjugate as targeted therapeutics against hepatocellular carcinoma: preclinical studies and clinical relevance. Clin Transl Oncol. 2022;24(3):407–431. doi:10.1007/s12094-021-02707-5
134. Kong FE, Li GM, Tang YQ, et al. Targeting tumor lineage plasticity in hepatocellular carcinoma using an anti-CLDN6 antibody-drug conjugate. Sci Transl Med. 2021;13(579):eabb6282. doi:10.1126/scitranslmed.abb6282
135. Yang CY, Wang L, Sun X, et al. SHR-A1403, a novel c-Met antibody-drug conjugate, exerts encouraging anti-tumor activity in c-Met-overexpressing models. Acta Pharmacol Sin. 2019;40(7):971–979. doi:10.1038/s41401-018-0198-0
136. Ma Y, Zhang M, Wang J, et al. High-affinity human anti-c-Met IgG conjugated to oxaliplatin as targeted chemotherapy for Hepatocellular carcinoma. Front Oncol. 2019;9:717. doi:10.3389/fonc.2019.00717
137. Hoseini SS, Cheung NV. Immunotherapy of hepatocellular carcinoma using chimeric antigen receptors and bispecific antibodies. Cancer Lett. 2017;399:44–52. doi:10.1016/j.canlet.2017.04.013
138. Du K, Li Y, Liu J, et al. A bispecific antibody targeting GPC3 and CD47 induced enhanced antitumor efficacy against dual antigen-expressing HCC. Mol Ther. 2021;29(4):1572–1584. doi:10.1016/j.ymthe.2021.01.006
139. Yu L, Huang N, Sun H, et al. Development of a tetravalent T-cell engaging bispecific antibody against glypican-3 for Hepatocellular carcinoma. J Immunother. 2021;44(3):106–113. doi:10.1097/CJI.0000000000000349
140. Liao Y, Tang Z, Liu K, et al. Preparation and application of anti-HBx/anti-CD3 bispecific monoclonal antibody (BsAb) retargeting effector cells for lysis of human hepatoma xenografts in nude mice. Oncol Rep. 1996;3(4):637–644.
141. Finn RS, Qin S, Ikeda M, et al. Atezolizumab plus bevacizumab in unresectable Hepatocellular Carcinoma. N Engl J Med. 2020;382(20):1894–1905. doi:10.1056/NEJMoa1915745
142. Finn RS, Ikeda M, Zhu AX, et al. Phase Ib study of lenvatinib plus pembrolizumab in patients with unresectable Hepatocellular Carcinoma. J Clin Oncol. 2020;38(26):2960–2970. doi:10.1200/JCO.20.00808
143. Xia Y, Tang W, Qian X, et al. Efficacy and safety of camrelizumab plus apatinib during the perioperative period in resectable hepatocellular carcinoma: a single-arm, open label, phase II clinical trial. J Immunother Cancer. 2022;10(4):e004656. doi:10.1136/jitc-2022-004656
144. Kelley RK, Rimassa L, Cheng AL, et al. Cabozantinib plus atezolizumab versus sorafenib for advanced hepatocellular carcinoma (COSMIC-312): a multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2022;23(8):995–1008. doi:10.1016/S1470-2045(22)00326-6
145. Cousin S, Cantarel C, Guegan JP, et al. Regorafenib-avelumab combination in patients with biliary tract cancer (REGOMUNE): a single-arm, open-label, phase II trial. Eur J Cancer. 2022;162:161–169. doi:10.1016/j.ejca.2021.11.012
146. Ho WJ, Zhu Q, Durham J, et al. Neoadjuvant cabozantinib and nivolumab converts locally advanced HCC into resectable disease with enhanced antitumor immunity. Nat Cancer. 2021;2(9):891–903. doi:10.1038/s43018-021-00234-4
147. Kaseb AO, Morris JS, Iwasaki M, et al. Phase II trial of bevacizumab and erlotinib as a second-line therapy for advanced hepatocellular carcinoma. Onco Targets Ther. 2016;9:773–780. doi:10.2147/OTT.S91977
148. Mahmood S, Li D, Lee A, et al. A multicenter, phase Ib/II, open-label study of tivozanib with durvalumab in advanced hepatocellular carcinoma (DEDUCTIVE). Future Oncol. 2022;18(40):4465–4471. doi:10.2217/fon-2022-0844
149. Qin S, Chen M, Cheng AL, et al. Atezolizumab plus bevacizumab versus active surveillance in patients with resected or ablated high-risk hepatocellular carcinoma (IMbrave050): a randomised, open-label, multicentre, phase 3 trial. Lancet. 2023;S0140-6736(23):01796–01798.
150. Song X, Kelley RK, Khan AA, et al. Exposure-response analyses of tremelimumab monotherapy or in combination with durvalumab in patients with unresectable Hepatocellular carcinoma. Clin Cancer Res. 2023;29(4):754–763. doi:10.1158/1078-0432.CCR-22-1983
151. Settleman J, Neto JMF, Bernards R. Thinking differently about cancer treatment regimens. Cancer Discov. 2021;11(5):1016–1023. doi:10.1158/2159-8290.CD-20-1187
152. Ningarhari M, Caruso S, Hirsch TZ, et al. Telomere length is key to hepatocellular carcinoma diversity and telomerase addiction is an actionable therapeutic target [published correction appears in J Hepatol. 2022 May;76(5):1242–1243]. J Hepatol. 2021;74(5):1155–1166. doi:10.1016/j.jhep.2020.11.052
153. Yu F, Yu C, Li F, et al. Wnt/β-catenin signaling in cancers and targeted therapies. Signal Transduct Target Ther. 2021;6(1):307. doi:10.1038/s41392-021-00701-5
154. Chen S, Wu JL, Liang Y, et al. Arsenic trioxide rescues structural p53 mutations through a cryptic allosteric site. Cancer Cell. 2021;39(2):225–239.e8. doi:10.1016/j.ccell.2020.11.013
155. McGrail DJ, Pilié PG, Rashid NU, et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann Oncol. 2021;32(5):661–672. doi:10.1016/j.annonc.2021.02.006
156. Winograd P, Hou S, Court CM, et al. Hepatocellular carcinoma-circulating tumor cells expressing PD-L1 are prognostic and potentially associated with response to checkpoint inhibitors. Hepatol Commun. 2020;4(10):1527–1540. doi:10.1002/hep4.1577
157. Kim AK, Hamilton JP, Lin SY, et al. Urine DNA biomarkers for hepatocellular carcinoma screening. Br J Cancer. 2022;126(10):1432–1438. doi:10.1038/s41416-022-01706-9
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Therapeutic Approaches to Penile Cancer: Standards of Care and Recent Developments
White J, Mason R, Lawen T, Spooner J, Faria KVM, Rahman F, Ramasamy R
Research and Reports in Urology 2023, 15:165-174
Published Date: 2 June 2023
Extensive-Stage Small-Cell Lung Cancer: Current Landscape and Future Prospects
Saida Y, Watanabe S, Kikuchi T
OncoTargets and Therapy 2023, 16:657-671
Published Date: 2 August 2023
Research Progress on Molecular Subtyping and Modern Treatment of Triple-Negative Breast Cancer
Tong L, Yu X, Wang S, Chen L, Wu Y
Breast Cancer: Targets and Therapy 2023, 15:647-658
Published Date: 24 August 2023
Breast Cancer: An Overview of Current Therapeutic Strategies, Challenge, and Perspectives
Wang J, Wu SG
Breast Cancer: Targets and Therapy 2023, 15:721-730
Published Date: 20 October 2023
Diagnosis, Prognosis, and Treatment of Triple-Negative Breast Cancer: A Review
Jie H, Ma W, Huang C
Breast Cancer: Targets and Therapy 2025, 17:265-274
Published Date: 17 March 2025