Back to Journals » Neuropsychiatric Disease and Treatment » Volume 11
Triptolide attenuates cerebral ischemia and reperfusion injury in rats through the inhibition the nuclear factor kappa B signaling pathway
Authors Jin X, Ye F, Zhang J, Zhao Y, Zhou X, Wang Y
Received 2 February 2015
Accepted for publication 16 March 2015
Published 3 June 2015 Volume 2015:11 Pages 1395—1403
DOI https://doi.org/10.2147/NDT.S82052
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Wai Kwong Tang
Xiao-Qing Jin,1,2 Fei Ye,1 Jun-Jian Zhang,1 Yan Zhao,2 Xian-Long Zhou2
1Department of Neurology, 2Emergency Center, Zhongnan Hospital of Wuhan University, Wuhan, Hubei, People’s Republic of China
Abstract: Inflammation plays critical roles in the acute progression of the pathology of ischemic injury. Previous studies have shown that triptolide interferes with a number of pro-inflammatory mechanisms. In this study, we investigated whether triptolide has protective effects during acute cerebral ischemia/reperfusion (I/R) injury. Male Sprague Dawley rats received triptolide or vehicle at the onset of reperfusion following middle cerebral artery occlusion. Twenty-four hours after reperfusion, we evaluated neurological injuries, the expression of pro-inflammatory markers, and NF-κB activation. I/R rats treated with triptolide showed significantly better neurological deficit scores, decreased neural apoptosis, and reduced cerebral infarct volume and brain edema, and triptolide treatment suppressed the activation of NF-κB following I/R injury. Furthermore, the expression levels of pro-inflammatory cytokines at both the mRNA and protein levels were significantly decreased in rats receiving triptolide. These results indicate that the neuroprotective effects of triptolide during acute cerebral I/R injury are possibly related to the inhibition of both the NF-κB signaling pathway and inflammation.
Keywords: ischemic stroke, inflammation, rat model, NF-κB pathway
Introduction
Stroke is one of the most common causes of death and long-term disability in the adult population worldwide.1 Ischemic stroke, which is caused by a reduction in cerebral blood flow and can result in fatal brain damage, accounts for approximately 80% of all strokes.2 Thrombolysis and interventional recanalization are regularly applied approaches for the management of acute ischemic stroke.3,4 Although reperfusion leads to the temporary survival of cerebral cells in the ischemic region through the restoration of blood flow, these therapies have achieved limited clinical success because of their narrow therapeutic time window and the occurrence of reperfusion injury following recovery from ischemia.4 Ischemia reperfusion injury (IRI) is a complex disorder caused by oxidative damage, inflammation, and cerebral edema.5,6 In addition, both experimental and clinical evidence has indicated that inflammation is essential for the progression of cerebral IRI.7,8 Previous studies have suggested that treatment with anti-inflammatory agents could reduce tissue edema and improve outcome in stroke models.9
Triptolide, a bioactive ingredient extracted from Chinese medicinal plants, has been reported to exhibit anti-inflammatory activities in several disorders including arthritis,10 pulmonary hypertension,11 and traumatic brain injury.12 In vitro studies have also shown that triptolide is able to suppress the production of inflammatory cytokines in various cell lines.13,14 Those previous findings lend support to the possibility that triptolide might have anti-inflammatory and neuroprotective abilities when used to treat cerebral IRI. To date, no studies have investigated the possible protective effects of triptolide against cerebral IRI. In the present study, we used a rat middle cerebral artery occlusion (MCAO) model to investigate the protective effects of triptolide on cerebral IRI and to determine the possible mechanism for this effect.
Materials and methods
Animals and experimental protocol
This study was performed using specific pathogen-free (SPF) 7-week-old male Sprague-Dawley rats weighing 250–300 g. The animals were purchased from the Center for Animal Experiments, Zhongnan Hospital, Wuhan University (Wuhan, People’s Republic of China). This study was approved by the research committee of Wuhan University, and all animal experiments were conducted in accordance with the guidelines of the Wuhan University Animal Experimentation Committee. Animal experiments were conducted in accordance with the Guidelines for the Care and Use of Laboratory Animals of Wuhan University (Wuhan, Hubei, China). The Institutional Ethic Committee approved the animal study (20140210ZN11). A total of 100 rats were randomly divided into five groups (n=20 in each group): 1) sham group (SHAM); 2) ischemia and reperfusion group (IRI + Vehicle); and 3) three triptolide groups (IRI + TL, 3 subgroups, n=20 in each group). A single dose of triptolide (Figure 1A) (Sigma, Saint Louis, MO, USA, dissolved in pure dimethyl sulfoxide [DMSO]) was intraperitoneally (ip) injected at the onset of reperfusion in rats belonging to the three TL groups; a corresponding volume of vehicle (pure DMSO) was administered to the rats in the IRI group. The dosages used for triptolide administration (0.1 mg/kg, 0.5 mg/kg, and 5 mg/kg) were selected based upon our pilot study. At the conclusion of the reperfusion period, the neurological deficit scores were evaluated. The rats were then sacrificed for the collection of tissue samples.
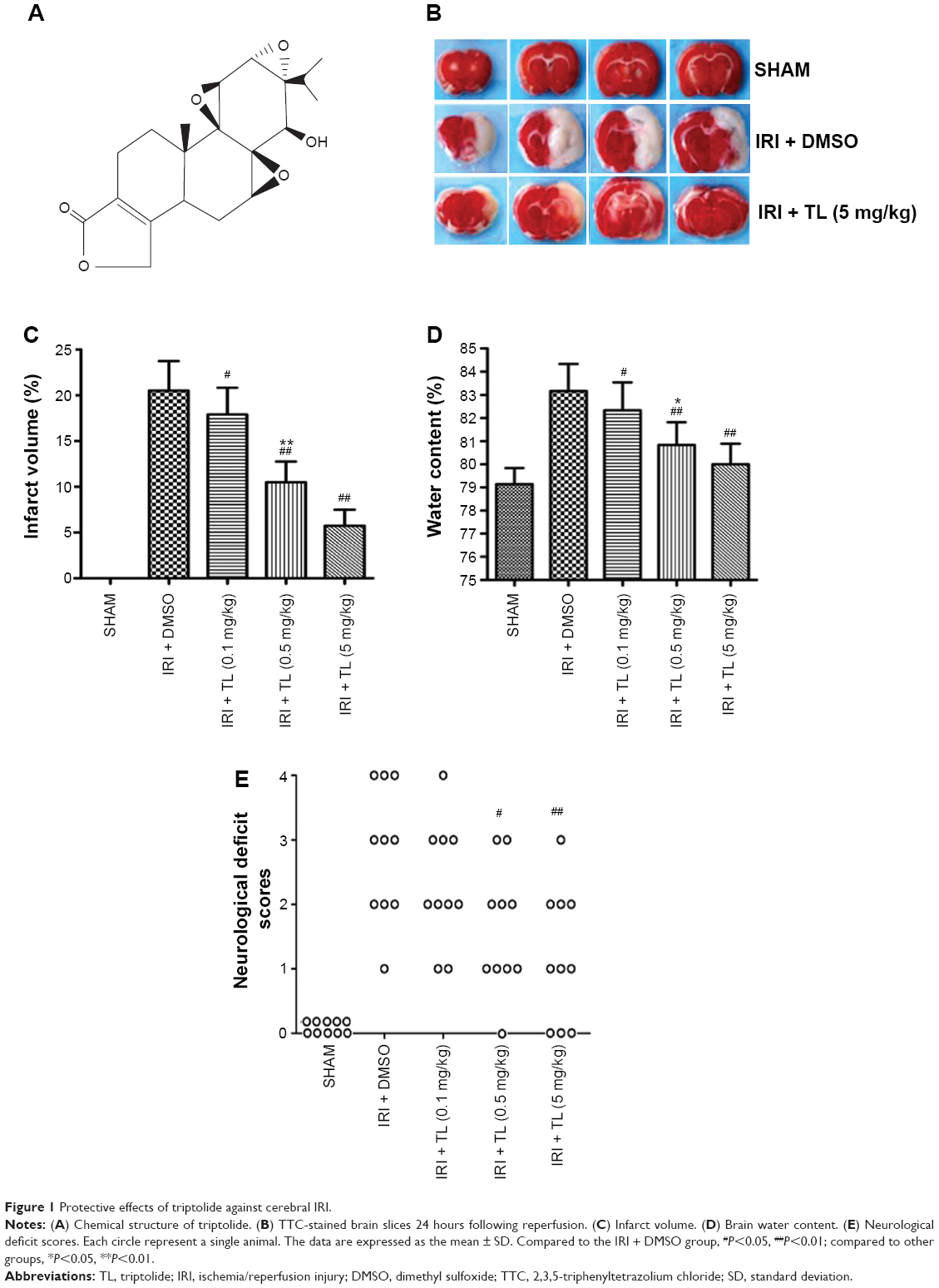 | Figure 1 Protective effects of triptolide against cerebral IRI. |
Animal model
The rat MCAO model has been established previously.15 Briefly, animals were anesthetized with an ip injection of pentobarbital sodium (50 mg/kg; Amresco, Cleveland, OH, USA) and were fixed in a supine position on a warming blanket to maintain their body temperature at 37°C–38°C. A 4/0 surgical nylon filament with a silicone-beaded tip was introduced into the right internal carotid artery (ICA) through the external carotid artery (ECA) to occlude the origin of the middle cerebral artery (MCA). After 2 hours of ischemia, the occlusion was released for a 24-hour reperfusion period. The arterial blood pressure (BP) and heart rate (HR) were monitored through the left femoral artery using a monitoring system (BL-420F; TaiMeng, Chengdu, People’s Republic of China). Arterial blood samples for blood gas analysis were collected through the left femoral artery using an i-STAT®1 analyzer (Abbott, Kyoto, Japan) prior to (baseline) and 15 minutes after the onset of MCAO (ischemia) and reperfusion (reperfusion).
Measurement of cerebral infarct volume
At the end of the observation period, the rats were sacrificed, and their brains were collected. The assessment of infarct volume was carried out as previously described.16 In brief, brains were sectioned at 2 mm intervals and stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC, Amresco) for 15 minutes at 37°C. Images were digitalized, and the infarct areas were analyzed. The infarct volume is expressed as the percentage of the contralateral hemisphere.
Measurement of cerebral edema
Cerebral edema was evaluated by determining the water content of the brain 24 hours following reperfusion, as previously described17 with modifications (n=5 in each group). Briefly, the brains were immediately weighed to obtain the wet weight (WW) after removal and dried at 110°C overnight in an electric oven. The dried brains were weighed again to obtain the dry weight (DW). Brain water content was calculated as follows: water content (%) = [(WW − DW)/WW] ×100%.
TUNEL staining
Neuronal apoptosis was assessed by TUNEL staining using a commercial in situ cell death detection kit (Nanjing KeyGEN Biotech Co. Ltd, Nanjing, People’s Republic of China) according to the manufacturer's instructions. All sections were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen, Carlsbad, CA, USA). In each case, ten fields in the infarcted cortex were randomly selected for apoptotic cell counting in a blinded manner using an Olympus IX51 reflected light fluorescence microscope (Olympus, Japan), and the percentage of TUNEL positive cells (TUNEL/DAPI) was calculated.
Assessment of neurological deficit score
Neurological symptoms were assessed 24 hours after reperfusion using a neurological deficit score as previously described.18 The neurological deficit score ranges from 0 to 4 (0, forelimb flexion and body twisting when rats were suspended by the tail [no observable neurological deficit]; 1, rats failed to extend left forepaw; 2, rats circled to the left; 3, rats failed to move the left forepaw; and 4, rats could not walk).
Real-time PCR analysis
The expression of pro-inflammatory cytokines, including tumor necrosis factor alpha (TNF-α), interleukin beta (IL-β), and IL-6, in the peri-infarct cortical tissue was detected using real-time polymerase chain reaction (PCR). Tissue collection and preparation for the extraction of total RNA were carried out as previously described.19 Total RNA was extracted using TRI Reagent (Molecular Research Center, Inc., Cincinnati, OH, USA) and reverse-transcribed into cDNA according to the manufacturer’s instructions. The levels of TNF-α, IL-1β, and IL-6 mRNA expression were measured with SYBR green, and RT-PCR was performed using an ABI prism 7000 sequence detection system (ABI, Foster City, CA, USA). Expression values are shown as fold change relative to the control group. The following pairs of primers were used for PCR amplification: TNF-α primer (forward): ATCATCTTCTCAAAACTCGAGTGACAA; TNF-α primer (reverse): CTGCTCCTCTGCTTGGT; IL-1β primer (forward): GAAACAGCAATGGTCGGGAC; IL-1β primer (reverse): GAGACCTGACTTGGCAGAGG; IL-6 primer (forward): AACGAAAGTCAACTCCATCTG; IL-6 primer (reverse): GGTATCCTCTGTGAAGTCTCC.
Enzyme-linked immunosorbent assay
Cortical tissue was collected and homogenized 24 hours after reperfusion. The homogenate was centrifuged for 30 minutes at 10,000× g at 4°C. The protein concentration of the supernatant was detected with a BCA kit (Pierce, Rockford, IL, USA), and the levels of cytokines, including TNF-α, IL-1β, and IL-6, were determined using commercial Enzyme-linked immunosorbent assay kits (Abcam, Shanghai, People’s Republic of China).
Western blot analysis
Brain tissues were collected at the indicated time points for western blot assay. Cytosolic and nuclear extracts were prepared by the Meldrum method as previously described.20 Equal amounts of protein (50 μg) were resolved on 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels followed by electrophoresis and protein transfer to an Immobilon-P membrane (Millipore, Billerica, MA, USA). Membranes were incubated overnight with primary antibodies at 4°C, including anti-rat NF-κB p65 (#8242), IκB-α (#4814), phosphorylated IκB-α (#9246), Bcl-2 (#2870), and Bax (#2772) (Cell Signaling Technology, Beverly, MA, USA). After washing with TBS containing 0.05% Tween-20, the membranes were probed with horseradish peroxidase (HRP)-labeled secondary antibodies (Boster, Wuhan, People’s Republic of China), and the densities of the protein bands were analyzed using Quantity One software (Bio-Rad, Hercules, CA, USA). Protein levels were normalized to β-actin.
Statistical analysis
The data are expressed as the mean ± SD and were processed by the statistical analysis software SPSS version 18.0 (SPSS Inc., Chicago, IL, USA). The comparison of several means was performed using one-way and repeated-measure two-way analysis of variance followed by the Tukey–Kramer test to identify significant differences between groups. The Kruskal–Wallis test was used for the neurological deficit scores. A P-value <0.05 was considered significant.
Results
Physiological parameters during MCAO and reperfusion
Physiological parameters, including mean arterial pressure (MAP), HR, rectal temperature, arterial blood pH, PaO2 and PaCO2, during MCAO and reperfusion, are shown in Table 1. We observed no significant differences in MAP, HR, pH, PaO2, and PaCO2 at any time point before or during MCAO or during reperfusion.
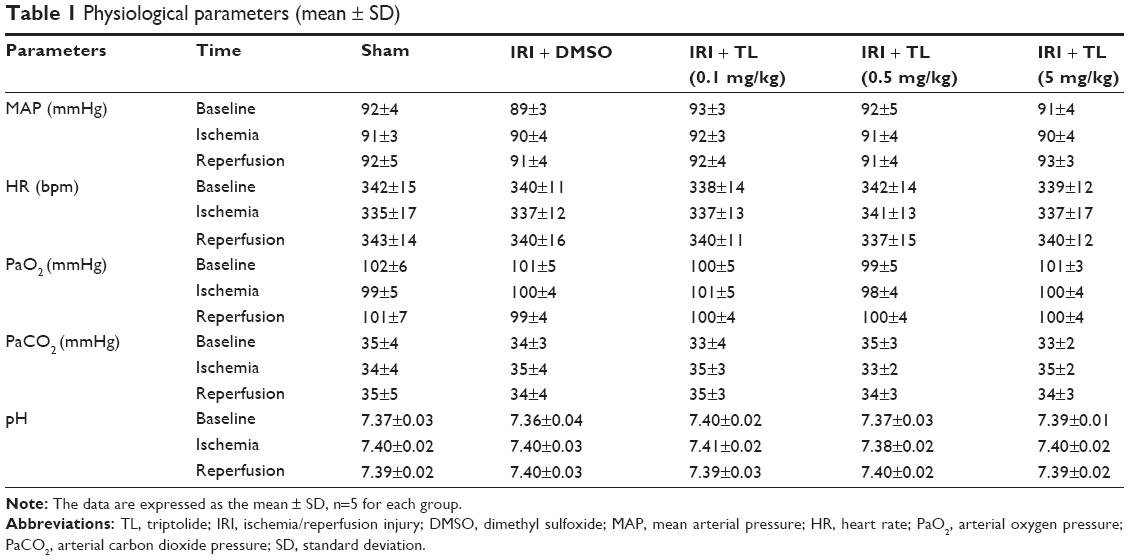 | Table 1 Physiological parameters (mean ± SD) |
Triptolide ameliorates MCAO-induced cerebral injury
Two hours of MCAO followed by a 24-hour reperfusion period induced an infarct volume of 21%±3% and a brain water content of 83%±2% in vehicle-treated (IRI + DMSO) rats. Treatment with triptolide significantly reduced the infarct volume in a dose-dependent manner (P<0.05) (Figure 1B and C). In addition, the brain water content was also reduced in rats treated with triptolide (P<0.05) (Figure 1D). At the conclusion of the observation period (24 hours following MCAO), rats in the IRI + DMSO group showed markedly greater neurological deficit scores (Figure 1E), while rats receiving triptolide (0.5 mg/kg and 5 mg/kg) showed a significant decrease in their neurological deficit (P<0.05).
Triptolide attenuates the production of pro-inflammatory cytokines
To determine the anti-inflammatory response following MCAO, we evaluated the expression of pro-inflammatory cytokines, including TNF-α, IL-1β, and IL-6, in ischemic brain tissue 24 hours following reperfusion. We found that the relative mRNA (Figure 2A) and protein (Figure 2B) expression of these cytokines were significantly elevated in vehicle-treated rats (P<0.05), while treatment with triptolide significantly attenuated the increase in TNF-α, IL-1β, and IL-6 mRNA and protein levels in a dose-dependent manner (P<0.05).
 | Figure 2 Effects of triptolide on (A) mRNA expression and (B) protein concentration of pro-inflammatory cytokines in ischemic brain tissue 24 hours following reperfusion. |
Triptolide prevents neuronal apoptosis following cerebral ischemia/reperfusion injury
DNA fragmentation following MCAO was determined by TUNEL staining (Figure 3A). TUNEL-positive cells were rarely detected in SHAM animals. Conversely, MCAO rats treated with vehicle showed a significant increase in the number of TUNEL-positive cells within the ischemic penumbra. Triptolide significantly reduced the percentage of apoptotic cells in a dose-dependent manner (P<0.05). We also evaluated the expression of the pro-apoptotic protein Bax (Figure 3B) and the anti-apoptotic protein Bcl-2 (Figure 3C) in ischemic brain tissue. MCAO-induced cerebral IRI led to a significant increase in Bax protein expression and a reduction in Bcl-2 expression when compared to sham-operated rats (P<0.05); however, triptolide reverted the expression of these two proteins to basal levels.
 | Figure 3 Anti-apoptotic effects of triptolide during MCAO-induced cerebral IRI. |
Triptolide inhibits the activation of NF-κB following MCAO-induced cerebral injury
The activation of NF-κB was evaluated in both the cytosol and nuclear fractions (Figure 4A). We found that the NF-κB p65 subunit was highly expressed in the cytosol but not in the nucleus of sham-operated rats. In MCAO rats, the ischemic tissue showed higher levels of NF-κB p65 in the nucleus compared to the cytosol, and this activation of NF-κB was inhibited by treatment with triptolide. In addition, we also determined the total protein levels of IκB-α and phosphorylation of IκB-α. Our results showed that, following cerebral IRI, the overall level of IκB-α protein was decreased along with an enhancement in IκB-α phosphorylation. Triptolide inhibited the I/R-induced phosphorylation and degradation of IκB-α in a dose-dependent manner (Figure 4B).
 | Figure 4 Effects of triptolide on NF-κB activation 24 hours after reperfusion. |
Discussion
The major finding of this study is that the administration of triptolide has protective effects against the cerebral IRI that is induced by MCAO. Treatment with triptolide improved neurological deficit scores, attenuated cerebral infarct volume, reduced brain edema, and decreased neuronal apoptosis and the production of pro-inflammatory cytokines including TNF-α, IL-1β, and IL-6. In addition, our results revealed that triptolide has marked inhibitory effects on the activation of NF-κB. These results suggest that the protective effects of triptolide against neuroinflammation are likely acting through the inhibition of the NF-κB signaling pathway.
NF-κB is a transcription factor that controls the expression of target genes involved in cell proliferation, apoptosis, and inflammation, and it is known to be highly activated in inflammatory disease states such as traumatic brain injury and cerebral ischemia.21,22 It has been shown previously that the activation of NF-κB following MCAO-induced cerebral injury was accompanied by the elevated expression of pro-inflammatory cytokines including TNF-α, IL-1β, and IL-6.23 These cytokines are produced by diverse cell types and serve as mediators of inflammation.24 In fact, these pro-inflammatory cytokines, including TNF-α, IL-1β, and IL-6, could also stimulate NF-κB activation in inflammatory diseases.25 In this study, we confirmed that both the mRNA and protein expression levels of TNF-α, IL-1β, and IL-6 were increased following cerebral IRI and that this increased production of pro-inflammatory cytokines was attenuated by the administration of triptolide. These findings are consistent with previous studies reporting the anti-inflammatory effects of triptolide.10–14
NF-κB was activated at the very early stage of MCAO-induced cerebral injury. In an animal MCAO model, the increased binding of NF-κB DNA was detected after 30 minutes of reperfusion.26 In addition, mice deficient in the p50 subunit of NF-κB showed a significant reduction in infarct volume in stroke models, suggesting that NF-κB plays a detrimental role in the response to cerebral ischemia.27 Moreover, the selective inhibition of NF-κB in neurons significantly reduced the infarct size and the number of TUNEL-positive cells following MCAO.28 However, the outcomes of direct NF-κB inhibition on focal cerebral ischemic injury are inconsistent, as Hill et al have shown that the inhibition of MCAO-induced NF-κB using diethyldithio carbamate in rats increased cell death.15 Previous studies have suggested that triptolide is absorbed very rapidly after oral administration in rats; the time to reach maximal plasma concentration ranges from 10.0 to 19.5 minutes with a half-life of 16.8–50.6 minutes.29,30 In this study, only a single dose of triptolide was injected ip at the onset of reperfusion. Thus, the short half-life of triptolide might reduce its bioactivity. Regardless, our results still show that the administration of triptolide at the onset of reperfusion significantly suppressed the translocation of NF-κB and the phosphorylation of IκB-α following MCAO. Time course studies have suggested that NF-κB is activated within 3 hours following MCAO-induced injury in the rat brain.23 It is possible that triptolide inhibits the early activation of NF-κB following cerebral ischemic injury. However, additional studies are needed to clarify the mechanism and to assess the best therapeutic window for triptolide following cerebral IRI. In addition, the administration of triptolide significantly reduced the percentage of TUNEL-positive cells following MCAO. Thus, the protective effects of triptolide against cerebral IRI are possibly related to its inhibitory effects on the activation of NF-κB.
Conclusion
Our results suggest that the administration of triptolide following cerebral ischemia reduces infarct volume and apoptosis and improves neurological function by inhibiting the activation of NF-κB and the release of pro-inflammatory cytokines in the brain. Triptolide might be a promising therapeutic agent for the prevention and/or treatment of cerebral IRI.
Acknowledgments
The authors would like to thank the Center for Animal Experiments, Wuhan University (Wuhan, Hubei, People’s Republic of China) for assistance. This study was financially supported by the Emergency Diagnostic and Therapeutic Center of Central China.
Disclosure
The authors declare no conflicts of interest in this work.
References
Donnan GA, Fisher M, Macleod M, Davis SM. Stroke. Lancet. 2008; 371:1612–1623. | ||
Durukan A, Tatlisumak T. Acute ischemic stroke: overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol Biochem Behav. 2007;87:179–197. | ||
Jauch EC, Saver JL, Adams HP Jr, et al. Guidelines for the early management of patients with acute ischemic stroke: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2013;44:870–947. | ||
Molina CA, Alvarez-Sabín J. Recanalization and reperfusion therapies for acute ischemic stroke. Cerebrovasc Dis. 2009;27:S162–S167. | ||
Huang J, Upadhyay UM, Tamargo RJ. Inflammation in stroke and focal cerebral ischemia. Surg Neurol. 2006;66:232–245. | ||
Eltzschig HK, Eckle T. Ischemia and reperfusion-from mechanism to translation. Nat Med. 2011;17:1391–1401. | ||
Lakhan SE, Kirchgessner A, Hofer M. Inflammatory mechanisms in ischemic stroke: therapeutic approaches. J Transl Med. 2009;7:97. | ||
Shah IM, Macrae IM, Di Napoli M. Neuroinflammation and neuroprotective strategies in acute ischaemic stroke-from bench to bedside. Curr Mol Med. 2009;9:336–354. | ||
Kwon SW, Sohn EJ, Kim DW, et al. Anti-inflammatory effect of transduced PEP-1-heme oxygenase-1 in Raw 264.7 cells and a mouse edema model. Biochem Biophys Res Commun. 2011;411:354–359. | ||
Lin N, Liu C, Xiao C, et al. Triptolide, a diterpenoid triepoxide, suppresses inflammation and cartilage destruction in collagen-induced arthritis mice. Biochem Pharmacol. 2007;73:136–146. | ||
Faul JL, Nishimura T, Berry GJ, Benson GV, Pearl RG, Kao PN. Triptolide attenuates pulmonary arterial hypertension and neointimal formation in rats. Am J Respir Crit Care Med. 2000;162:2252–2258. | ||
Lee HF, Lee TS, Kou YR. Anti-inflammatory and neuroprotective effects of triptolide on traumatic brain injury in rats. Respir Physiol Neurobiol. 2012;182:1–8. | ||
Matta R, Wang X, Ge H, Ray W, Nelin LD, Liu Y. Triptolide induces anti-inflammatory cellular responses. Am J Transl Res. 2009;1:267–282. | ||
Lu Y, Liu Y, Fukuda K, Nakamura Y, Kumagai N, Nishida T. Inhibition by triptolide of chemokine, proinflammatory cytokine, and adhesion molecule expression induced by lipopolysaccharide in corneal fibroblasts. Invest Ophthalmol Vis Sci. 2006;47:3796–3800. | ||
Hill WD, Hess DC, Carroll JE, et al. The NF-kB inhibitor diethyldithiocarbamate (DDTC) increases brain cell death in a transient middle cerebral artery occlusion model of ischemia. Brain Res Bull. 2001;55:375–386. | ||
Arrick DM, Sun H, Mayhan WG. Influence of exercise training on ischemic brain injury in type 1 diabetic rats. J Appl Physiol. 1985;2012(113): 1121–1127. | ||
Chao X, Zhou J, Chen T, et al. Neuroprotective effect of osthole against acute ischemic stroke on middle cerebral ischemia occlusion in rats. Brain Res. 2010;1363:206–211. | ||
Murakami K, Kondo T, Kawase M, et al. Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J Neurosci. 1998;18:205–213. | ||
Patzer A, Zhao Y, Stöck I, Gohlke P, Herdegen T, Culman J. Peroxisome proliferator-activated receptorsgamma (PPARgamma) differently modulate the interleukin-6 expression in the peri-infarct cortical tissue in the acute and delayed phases of cerebral ischaemia. Eur J Neurosci. 2008;28:1786–1794. | ||
Meldrum DR, Shenkar R, Sheridan BC, Cain BS, Abraham E, Harken AH. Hemorrhage activates myocardial NFkB and increases tumor necrosis factor in the heart. J Mol Cell Cardiol. 1997;29:2849–2855. | ||
Nonaka M, Chen XH, Pierce JE, et al. Prolonged activation of NF-kappaB following traumatic brain injury in rats. J Neurotrauma. 1999;16:1023–1034. | ||
Stephenson D, Yin T, Smalstig EB, et al. Transcription factor nuclear factor-kappa B is activated in neurons after focal ischemia. J Cereb Blood Flow Metab. 2000;20:592–603. | ||
Berti R, Williams AJ, Moffett JR, et al. Real time PCR mRNA analysis of the inflammatory cascade associated with ischemia reperfusion brain injury. J Cereb Blood Flow Metab. 2002;22:1068–1079. | ||
Tuttolomondo A, Di Sciacca R, Di Raimondo D, Renda C, Pinto A, Licata G. Inflammation as a therapeutic target in acute ischemic stroke treatment. Curr Top Med Chem. 2009;9:1240–1260. | ||
Bonizzi G, Karin M. The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. | ||
Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M. NF-κB is activated and promotes cell death in focal cerebral ischemia. Nat Med. 1999;5:554–559. | ||
Nurmi A, Lindsberg PJ, Koistinaho M, et al. Nuclear factor-kappaB contributes to infarction after permanent focal ischemia. Stroke. 2004;35:987–991. | ||
Zhang W, Potrovita I, Tarabin V, et al. Neuronal activation of NF-κB contributes to cell death in cerebral ischemia. J Cereb Blood Flow Metab. 2005;25:30–40. | ||
Mao Y, Tao X, Lipsky PE. Pharmacokinetics of triptolide. Development and application of a high performance liquid chromatographic method for quantitation of triptolide in plasma. J Liq Chromatogr Relat Technol. 1999;22:1355–1366. | ||
Shao F, Wang G, Xie H, Zhu X, Sun J, A J. Pharmacokinetic study of triptolide, a constituent of immunosuppressive chinese herb medicine, in rats. Biol Pharm Bull. 2007;30:702–707. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.