Back to Journals » Breast Cancer: Targets and Therapy » Volume 8
Triple-negative breast cancer: treatment challenges and solutions
Authors Collignon J, Lousberg L, Schroeder H, Jerusalem G
Received 10 January 2016
Accepted for publication 10 March 2016
Published 20 May 2016 Volume 2016:8 Pages 93—107
DOI https://doi.org/10.2147/BCTT.S69488
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Pranela Rameshwar
Joëlle Collignon,1 Laurence Lousberg,1 Hélène Schroeder,1 Guy Jerusalem,1,2
1Medical Oncology Department, CHU Sart Tilman Liege, Domaine Universitaire du Sart Tilman, Liege, Belgium; 2University of Liege, Liege, Belgium
Abstract: Triple-negative breast cancers (TNBCs) are defined by the absence of estrogen and progesterone receptors and the absence of HER2 overexpression. These cancers represent a heterogeneous breast cancer subtype with a poor prognosis. Few systemic treatment options exist besides the use of chemotherapy (CT). The heterogeneity of the disease has limited the successful development of targeted therapy in unselected patient populations. Currently, there are no approved targeted therapies for TNBC. However, intense research is ongoing to identify specific targets and develop additional and better systemic treatment options. Standard adjuvant and neoadjuvant regimens include anthracyclines, cyclophosphamide, and taxanes. Platinum-based CT has been proposed as another CT option of interest in TNBC. We review the role of this therapy in general, and particularly in patients carrying BRCA germ-line mutations. Available data concerning the role of platinum-based CT in TNBC were acquired primarily in the neoadjuvant setting. The routine use of platinum-based CT is not yet recommended by available guidelines. Many studies have reported the molecular characterization of TNBCs. Several actionable targets have been identified. Novel therapeutic strategies are currently being tested in clinical trials based on promising results observed in preclinical studies. These targets include androgen receptor, EGFR, PARP, FGFR, and the angiogenic pathway. We review the recent data on experimental drugs in this field. We also discuss the recent data concerning immunologic checkpoint inhibitors.
Keywords: triple-negative breast cancer, molecular subtype, platinum-based chemotherapy, targeted therapy, androgen receptor, BRCA1/2 mutation
A Letter to the Editor has been received and published for this article.
Introduction
In 2012, 1.7 million women worldwide were diagnosed with breast cancer (BC), and 521,900 women died from it.1 These statistics include all subtypes of BC, but it is well known that BC is not a homogeneous disease. Four major intrinsic subtypes have been identified by genomic studies: the luminal subtypes A and B, which express hormone receptor-related genes, basal-like (BL) BC, and HER2-positive BC.2,3
Triple-negative BC (TNBC) is a heterogeneous group characterized by the lack of expression of hormonal receptors and the absence of HER2 overexpression. The definition of negative estrogen receptor (ER) status by immunohistochemistry (IHC) is not concordant in the literature, with some definitions considering ER expression to be significant only if at least 10% of tumor cells express the receptors. However, the St Gallen guidelines,4 the American Society of Clinical Oncology,5 and the American College of Pathology5 have defined TNBC as BC with less than 1% of tumor cells expressing the ER and progesterone receptors via IHC.
TNBC represents approximately 15% of all BCs and is characterized by shorter overall survival and an early peak of distant recurrences at 3 years after diagnosis. The majority of deaths occur in the first 5 years following initial diagnosis. Late tumor recurrences are unusual with this BC subtype and recurrences generally are not observed after 8 years.6 TNBC has an aggressive clinical behavior, with a higher risk of both local and distant relapses that frequently present as visceral and/or brain metastases.7,8 TNBCs are frequently assimilated into the intrinsic subgroup of BCs that have been described in microarray-based expression profiling research as the BL molecular phenotype.2 However, not all TNBCs display a BL molecular phenotype on gene expression arrays. Indeed, 75% to 80% of TNBCs are actually BL cancers. Some markers that have been identified by IHC in tumor cells are also found in normal basal/myoepithelial cells of the breast, including high-molecular-weight basal cytokeratin 5/6 (CK5/6), CK14, B crystallin, CK17, epidermal growth factor receptor (EGFR) HER1, caveolin 1/2 (CAV1/2), vimentin, fascin, c-Kit, and P-cadherin. TNBC is less likely to express epithelial markers such as E-cadherin.9–11 Similarly, not all BL BCs are TNBC: up to 54% of cancers in the BL subgroup do not have a TN phenotype on IHC.11,12 Indeed, some BL cancers express ER or overexpress HER2. Currently, there is no international definition of TNBC/BL cancer, but the most appropriate approach to use in the absence of access to molecular profiling is a panel of four antibodies (ER, HER2, CK5/6, and EGFR HER1), which can best characterize BL tumors based on IHC criteria.9
In a large population-based study from the California Cancer Registry, TNBCs were significantly more frequent in women under the age of 40 years.13 TNBCs are also more frequent in women who are germ-line BRCA1/2 mutation carriers and in women of black race or Hispanic ethnicity.14 Up to 19.5% of patients suffering from TNBC present BRCA1/2 mutations.15,16 For this reason, it is recommended to test all women under the age of 60 years suffering from TNBC for BRCA1/2 mutations (National Comprehensive Cancer Network guidelines).17
Currently, only chemotherapy (CT) is routinely used as systemic treatment in patients with TNBC, although in some countries bevacizumab is still added to CT in advanced BC (ABC), even in the absence of any demonstrated overall survival benefit. However, intense research is ongoing to identify actionable targets. A large number of clinical trials are ongoing that aim to improve current treatment outcomes. Better knowledge of the biology of this BC subgroup will allow us to evaluate new specific treatment approaches dedicated to this hard-to-treat disease.
We first review the available data on molecular heterogeneity and BRCA1-associated TNBC/BL BC. Thereafter, we discuss the current treatment options and some promising new treatment approaches that include targeted treatments.
Understanding TNBC heterogeneity
Before molecular profiling confirmed the important heterogeneity in the biology of TNBC, clinical data had already indicated the existence of heterogeneous treatment responses and long-term outcomes. Some patients respond very well to neoadjuvant CT and present a pathologic complete response (pCR) at the time of surgery. Other patients present no response to neoadjuvant CT and suffer from early relapse after surgery.6,18,19 Unfortunately, predictive factors that allow the identification of patients who will present a pCR and those who will not benefit from CT at the time of diagnosis do not exist.
The vast majority of TNBCs are high-grade invasive ductal carcinomas, but some rare cases are histologically different, such as adenoid cystic carcinoma, secretory carcinoma, medullary carcinoma, and metaplastic carcinoma. The prognosis depends on the TNBC pathological subtype.20–22
The Cancer Genome Atlas Research Network used six methods to analyze primary BCs: genomic DNA copy-number arrays, DNA methylation, exome sequencing, messenger-RNA arrays, microRNA sequencing, and reverse-phase protein arrays.23 Only in three genes did somatic mutations occur at a frequency higher than 10% across all BCs: TP53, PIK3CA, and GATA3. Specific mutations are more frequent in some BC subtypes. In TNBC/BL cancers, the most frequent findings were the loss of TP53, RB1, BRCA1, and PIK3CA.23 Known drivers, such as P53, PIK3CA, and PTEN, have the highest clonal frequencies, but at the time of diagnosis, some patients present low clonality, while others have a more extensive clonal evolution, illustrating further important heterogeneity in TNBC.24
Subtyping TNBC: clinical implications
More recently, gene expression profiling of 587 TNBCs identified six different subtypes: BL1 and BL2, an immunomodulatory (IM) subtype, a mesenchymal subtype, a mesenchymal stem like (MSL) subtype, and a luminal androgen receptor (LAR) subtype.25 The strengths of this study were to identify further the molecular drivers in corresponding cell-line models to provide preclinical platforms for the development of effective therapies (Table 1). For example, the authors showed that BL1 lines were the most sensitive to cisplatin and that the mesenchymal and MSL lines were most sensitive to the Abl/Src inhibitor dasatinib.20
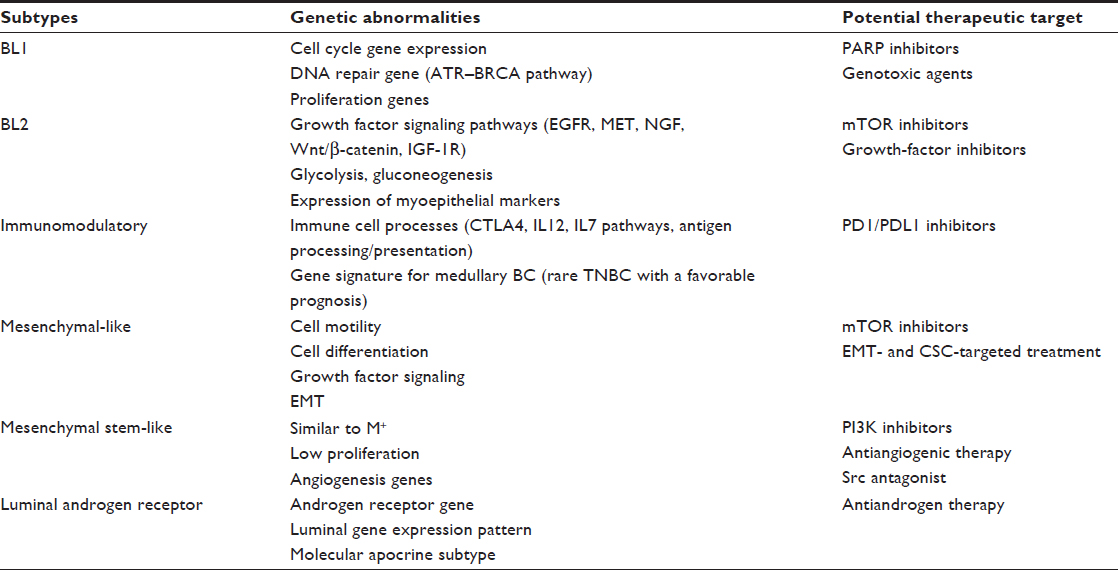 | Table 1 Genomic TNBC subtypes and potential therapeutic targets |
The same group used the intrinsic subtype tool to examine the composition of each TNBC subtype. The authors showed that all TNBC subtypes except MSL and LAR were composed primarily of the BL intrinsic subtype (BL1 [99%], BL2 [95%], IM [84%] and mesenchymal [97%]). The LAR subtype is classified as HER2 (74%) and luminal B (14%), and the MSL subtype includes BL (50%), normal-like (28%), and luminal B (14%).20
Other gene expression analyses have also defined a claudin-low tumor subtype.26 Molecular characterization showed that these tumors are enriched in epithelial-to-mesenchymal transition, immune system response, and stem-like features, but show low expression of luminal and proliferation-associated genes.27
Another group used genomic profiling of 198 TNBCs to identify four TNBC subtypes: LAR, mesenchymal, BL immunosuppressed, and BL immunoactivated.28 If compared with the results reported by Lehmann et al,25 LAR and mesenchymal tumors fall into the LAR and MSL subtypes.
The comprehensive analysis of TNBC can lead to the improved selection of study populations that have the highest probability of responding to specific treatments. For example, TNBC molecular subtypes may respond differently to CT. The clinical validity of the genomic classification was indeed confirmed by a retrospective analysis of the response to neoadjuvant CT. The overall pCR in this study was 28%. In the BL1 subtype, the pCR was the highest (51%),29 in comparison to 0 in the BL2 subtype and 10% in the LAR subtype, clearly showing the need to develop alternative treatments for some subgroups.
In the future, the evaluation of heterogeneity in TNBC and subtyping may lead to different therapeutic strategies. Potential targets and approaches include DNA damage and repair, immunomodulation, hormone receptor modulation, and signaling pathway inhibition.
Prospective trials will help us better understand the role of subtyping in the prediction of not only pCR but also long-term patient outcomes. New therapeutic strategies are needed for subgroups with the poorest therapeutic responses to standard CT.
DNA-damaging chemotherapy and DNA repair targets
BRCA1 mutation and “BRCAness”
Many external or internal agents, such as ultraviolet light, ionizing radiation, CT, and chemical substances or products of normal cellular metabolism, including oxidation and hydrolysis, affect double-stranded DNA. DNA repair mechanisms are important for maintaining the stability and integrity of the genome, and include nucleotide- and base-excision repair, homologous recombination, end joining, mismatch repair, and telomere metabolism.30,31 Inherited defects in one of these important genes can lead to cancer, as observed in the BRCA1/2 syndrome. DNA repair mechanisms are classified as the repair of single- or double-stranded damage. BRCA1 and BRCA2 are important proteins in the homologous recombination process when damage leads to breaks in both DNA strands. The proteins have also been implicated in other fundamental cellular processes, such as cell cycle control and transcription.32
BC in BRCA1 germ-line mutation carriers most often displays a TN phenotype, as indicated by IHC and genomic studies.33 Due to the similarity between sporadic TNBC and familial BRCA1 cancers, the concept of BRCAness has been developed.34–36 In sporadic cancers, BRCA1 is inactivated by an epigenetic mechanism: the aberrant methylation of cytosine residues in CpG dinucleotides.37 Aberrant methylation of the BRCA1 promoter is found in 11%–14% of sporadic BCs.35,38 In contrast, BRCA2 tumors lack a clear pathological phenotype.
Knowledge of DNA repair mechanism defects leads to some specific treatment approaches in TNBC. These tumors present potentially higher sensitivity to DNA-damaging agents, such as platinum salts. The concept of “synthetic lethality” is also tested in the clinic, with the development of drugs (poly ADP-ribose polymerase [PARP] inhibitors) that target single-stranded DNA repair when homologous recombination is defective in BRCA-mutant tumors or in BRCAness tumors.39 Several studies have attempted to find a biomarker of homologous recombination deficiency (HRD) with the aim of better predicting responders to PARP inhibitors and DNA-damaging chemotherapies.40–42
Platinum-based chemotherapy
Metastatic TNBC
The use of platinum compounds in metastatic BC was evaluated many years ago. Objective responses have been reported in ABC.43 Some retrospective analyses have also suggested the occurrence of increased survival with platinum-based CT in patients with advanced TNBC.44,45
A prospective Phase II study showed activity of platinum agents, including cisplatin (75 mg/m2/3 weeks) and carboplatin (area under the curve 6/3 weeks), in patients with metastatic TNBC, especially in patients with germ-line BRCA1/2 (gBRCA1/2) mutations.46 A total of 86 patients were enrolled. The overall response rate was 25.6%, but in patients with germ-line BRCA1/2 mutations, the response rate increased to 54.5%. Interestingly, using a measure of DNA repair function, those authors also identified patients without mutations who had the potential to benefit from platinum therapy. They used two HRD assays to characterize BRCA-like genomic instability: the HRD large-scale state transition assay and the HRD loss of heterozygosity assay. They observed that patients who presented higher values in these assays also responded better to platinum-based treatments, even in the absence of germ-line mutations.46
A prospective randomized trial comparing docetaxel with carboplatin in patients suffering from TNBC was presented at the 2014 San Antonio Breast Cancer Symposium (SABCS).47 Those authors observed similar results for unselected TNBC, but patients with BRCA1/2 mutations experienced a significantly higher response rate and improved progression-free survival (PFS) when receiving carboplatin in comparison to docetaxel.
Early TNBC
The rate of pCR after anthracycline- and taxane-based neoadjuvant CT is higher in TNBC (±30%) than in luminal BC. In addition, patients presenting pCR after neoadjuvant CT generally have a better prognosis compared to patients who do not present pCR at the time of surgery.48,49 Unfortunately, patients suffering from TNBC who present residual disease after neoadjuvant CT have very poor outcomes. New therapies should be evaluated in patients who present residual disease after neoadjuvant CT. Retrospective research has suggested improved outcomes in terms of pCR when cisplatin is added to the neoadjuvant treatment. Byrski et al50 published a retrospective analysis of 6,903 patients, including 102 patients with the germ-line BRCA1 mutation. The highest pCR rate was reported in germ-line BRCA1-mutation carriers who received neoadjuvant cisplatin therapy: 24% of the BRCA1-mutation carriers had pCR, but in the subgroup that received cisplatin, a much higher rate of 83% pCR rate was observed.50
Five randomized studies evaluated the addition of carboplatin to standard neoadjuvant therapy51–55 (Table 2). Experimental arm 1 from the I-SPY 2 trial showed an increased rate of pCR in patients with TNBC if standard CT (paclitaxel/anthracycline–cyclophosphamide) was combined with carboplatin and the PARP inhibitor veliparib. A confirmatory Phase III trial is underway.55 The GeparSixto52 and CALGB 4060353 trials showed increased rates of pCR with carboplatin, but toxicities were also more frequent and more significant if carboplatin was added. More dose reductions and early study discontinuations occurred due to these toxicities. The incidence of grade 3–4 hematological toxicities almost doubled. Data on late toxicities are missing, because the median follow-up period was only 3 years. Recently, at the 2015 SABCS, improved overall survival for patients receiving carboplatin was reported in the GeparSixto trial.56 A subsequent Phase III trial evaluating two dose-dense regimens is recruiting participants (GeparOcto, NCT02125344).57 In contrast, in the CALGB 40603 trial, despite significantly higher pCR rates, neither carboplatin nor bevacizumab showed improved event-free or overall survival. In addition, not all Phase II trials evaluating the use of carboplatin have shown improvements in pCR.54
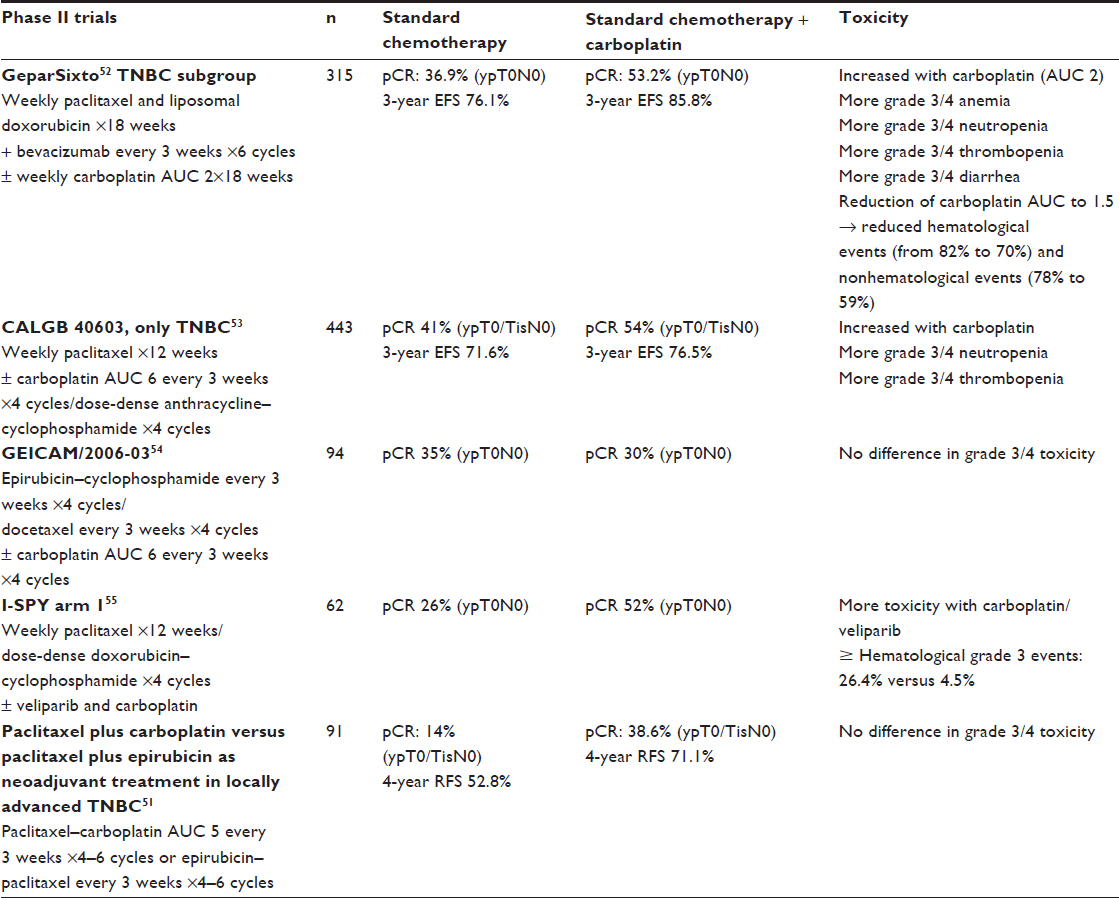 | Table 2 Carboplatin-based chemotherapy in neoadjuvant treatment: randomized Phase II results |
Currently, platinum compounds are not included in the guidelines for the treatment of early TNBC, but their role should be discussed in some specific cases, such as patients with a higher risk of relapse or requiring rapid disease control. Some recommend the use of carboplatin only for patients with known BRCA mutations,58 but the available data are conflicting, as illustrated by the GeparSixto trial, which showed better outcomes when using carboplatin in patients with TNBC, independently of germ-line BRCA status. Interestingly, the GeparQuinto trial showed a statistically higher pCR rate with a classical sequential anthracycline–taxane regimen and a trend for better disease-free survival (hazard ratio 0.64, P=0.06) in the subgroup of patients with BRCA mutations (82 of 471 patients).59 These results suggest higher chemosensitivity and better prognosis in patients with germ-line BRCA mutations, even without platinum compounds. Participation in clinical trials is recommended in the neoadjuvant and postneoadjuvant setting in the absence of pCR to better define the optimal standard systemic therapy for TNBC (Table 3).
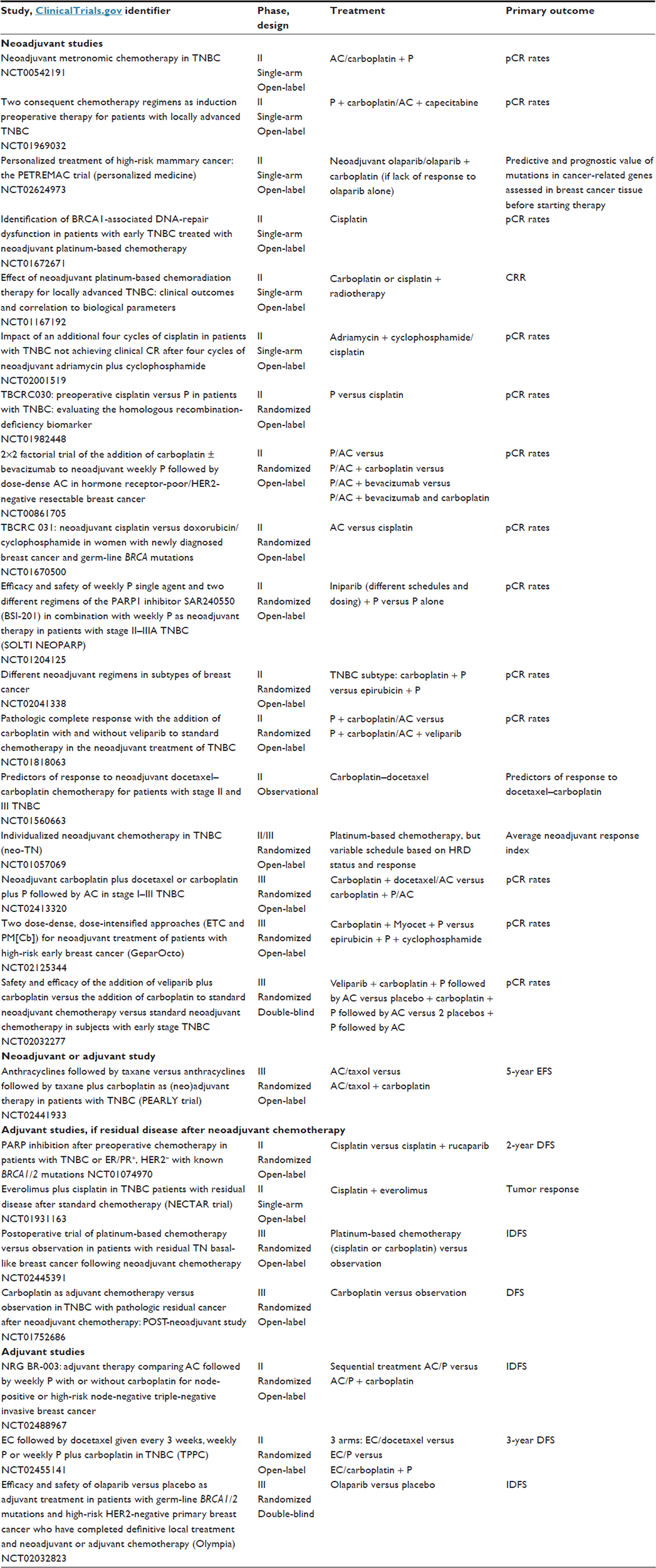 | Table 3 Trials in progress in the neoadjuvant and adjuvant settings for TNBC |
The potential role of PARP inhibitors in TNBC
Based on the synthetic lethality concept, PARP inhibitors were developed for the treatment of cancers with specific DNA-repair deficits, such as TNBC with BRCA1/2 mutations and BRCAness TNBC.35 The first drug of this class, olaparib, was approved by the US Food and Drug Administration in December 2014 as a single-agent treatment for patients with deleterious or suspected deleterious germ-line BRCA-mutated advanced ovarian cancer who were treated with three or more prior lines of CT. In BC, several Phase I and II trials have shown antitumor activity in BRCA-mutated patients (Table 4).60–63 In the proof-of-concept Phase II trial,61 Tutt et al61 reported on two cohorts of patients with BRCA1/2-mutated ABC. In the first cohort (27 patients), which was assigned 400 mg twice daily, the objective response rate was 41% compared to 22% in the cohort (27 patients) treated with a lower dose (100 mg twice daily). Toxicities were mild, and included nausea and fatigue. Clinical trials are ongoing in patients with high-risk BRCA-mutated primary BC in the neoadjuvant and adjuvant settings.64
Androgen receptor and TNBC
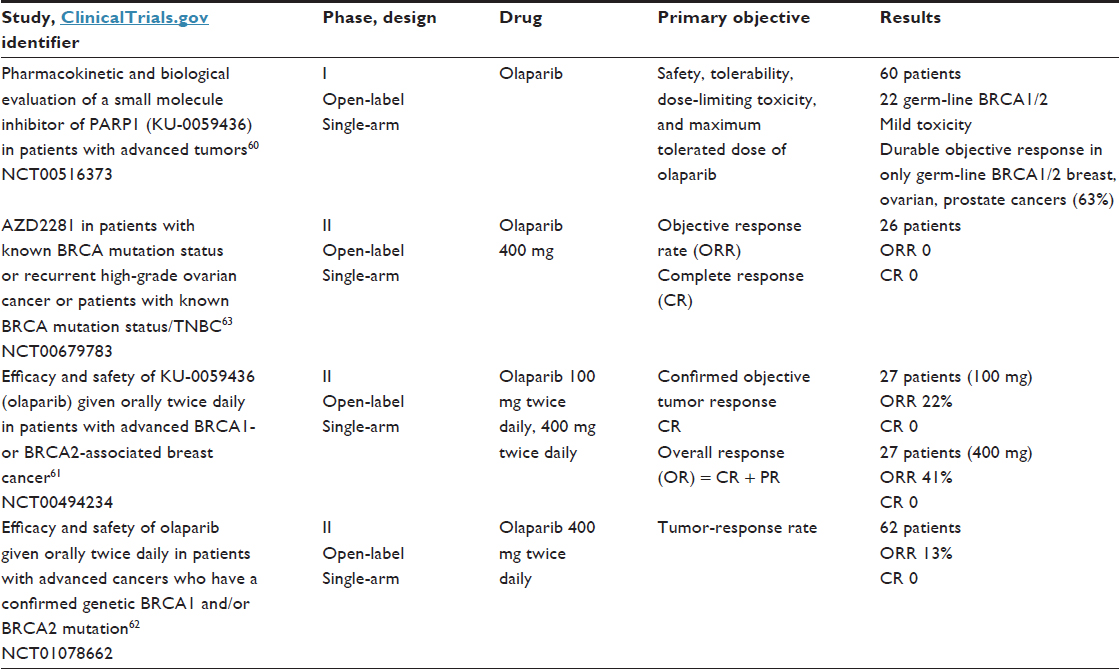 | Table 4 Results of trials with PARP inhibitors in triple-negative breast cancer |
The LAR subtype is characterized by luminal gene expression and is driven by the AR.25 The AR is expressed in normal and malignant breast tissue, and its prevalence is variable according to the subtype of BC. Approximately 10%–15% of TNBCs express the AR.65,66 The LAR subtype demonstrates some similarities with the apocrine subtype. Indeed, in this histologic subtype, the gene expression profile is highly correlated with the LAR subtype. These findings indicate that the LAR subtype includes BCs with apocrine histology.67,68 The function of the AR is less well understood in BC than in prostate cancer. In a paper by Doane et al,69 a cell line that recapitulated the molecular profile of the LAR subtype was identified. This cell line was used in preclinical models, and androgen-dependent growth was demonstrated in an estrogen-independent manner. This growth was inhibited by an AR antagonist (flutamide). This study was the first proof of concept for androgen blockade in the LAR subtype.67,69
At present, results from two Phase II studies have been presented and showed a benefit for androgen blockade in this LAR subtype. The first study was conducted by Gucalp et al.70 That was a multicenter Phase II study that investigated the use of bicalutamide at a dose of 150 mg daily in AR-positive, ER- and PgR-negative metastatic BC (26 patients). The majority of patients were HER2-negative. The AR was expressed in 12% of patients with ER/PgR-negative BC. This study showed a clinical benefit rate (CBR; = CR + partial response [PR] + stable disease >6 months) of 19% for bicalutamide. The median PFS was 12 weeks (comparable to single-agent or combination CT in TNBC). The treatment was well tolerated, with the most common side effects including fatigue, hot flashes, limb edema, and transaminase elevations.70
The second study evaluated the activity of the next-generation antiandrogen enzalutamide in advanced AR-positive TNBC. That study was a multicenter Phase II trial conducted in two stages. In stage 1, 26 patients were evaluated for the primary end point of the CBR at 16 weeks (CBR16; = CR + PR + stable disease at 16 weeks). These patients received enzalutamide at a dose of 160 mg orally daily. The stage 1 result was a CBR16 of 42% (95% confidence interval 24%–62%), including one CR and one PR.71 For the stage 2 study, 165 patients were screened, and 75 patients had AR IHC ≥10% and more than one postbaseline evaluation. Patients with TNBC had a median of one line of prior therapy. The data were presented at the American Society of Clinical Oncology’s 2015 meeting and showed a CBR16 of 35% and a median PFS of 14.7 weeks.72 Because of these results, interest in androgen blockade therapy in the LAR subtype is growing. A number of different trials are in the recruitment stage or are waiting for results (Table 5).
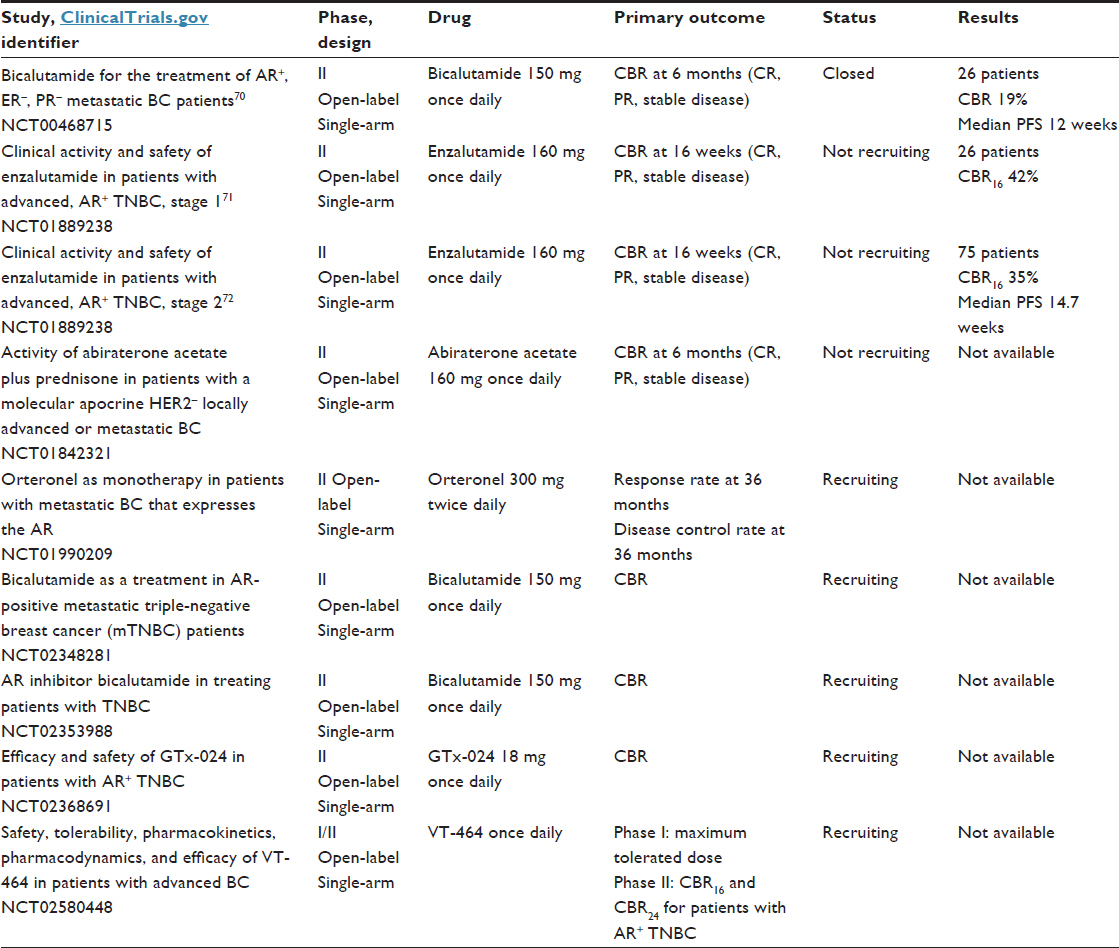 | Table 5 First results with antiandrogen therapy in TNBC and studies in progress |
Blocking the AR pathway is a promising approach in the treatment of metastatic patients with the LAR subtype of TNBC. As an example, we would like to mention an isolated clinical case in which a heavily pretreated woman with metastatic TNBC and AR expression achieved a complete clinical response after 4 months of treatment with the AR antagonist bicalutamide.73 Although this new targeted therapy is promising, more data are needed before it can be considered as a new validated treatment option. Future challenges related to the LAR subtype include understanding the role of the AR in tumorigenesis, understanding the escape mechanisms in AR-directed therapy, and discovering predictive biomarkers.74
Immune subtype and role of immunotherapy
BC was not previously considered to be an “immunogenic” malignancy. Nevertheless, accumulating evidence indicates the prognostic and predictive values of tumor-infiltrating lymphocytes (TILs) in BC.75,76 The degree of immune infiltration differs among BC subtypes. TIL levels are significantly higher in TNBC and HER2-positive BC.77–79 Hormone receptor-positive disease is the subtype associated with the least robust number of TILs. Recently, several studies confirmed the prognostic value of TILs in TNBC. The lymphocyte-predominant BC subtype, which contains high levels of TILs (>50%), is associated with improved disease-free and overall survival and pCR in the neoadjuvant setting.80–82
These findings suggest that immunomodulation could represent a new approach in the treatment of these aggressive BC subtypes.79 Our current understanding suggests that the immunogenic potential of TNBC is derived at least in part from its genetic instability and high mutation rate. Tumors from patients with TNBC are more likely than tumors from patients with other subtypes to exhibit chromosomal instability and potential mutations.83
TNBC is the subtype that is most frequently associated with TILs, but only a minority of TNBCs demonstrate a high number of TILs, suggesting that IM therapy could be necessary to promote immunorecognition and increase the adaptive immune infiltrate to levels adequate for a survival benefit in the majority of patients with this BC subtype. Patients with high levels of TILs at the time of diagnosis might benefit from the use of drugs that can enhance antitumoral immune responses.
Monoclonal antibodies have been developed to block specific immune-checkpoint proteins. Some of these antibodies have already been approved by the Food and Drug Administration for the treatment of metastatic melanoma. Three categories of these antibodies exist: antibodies that block CTLA4, PD1, or PDL1.
CTLA4 was the first immune checkpoint receptor to be targeted clinically. Two antibodies are known: ipilimumab and tremelimumab. Normally, after T-cell activation, CTLA4 is upregulated on the plasma membrane, where its function is to downregulate T-cell function through a variety of mechanisms, including preventing costimulation via CD28 and its ligand – B7. CTLA4 plays an essential role in maintaining normal immunologic homeostasis. Ipilimumab blocks CTLA4, and does not allow the T cell to interact with the receptor via CD28 on its cell surface.84
PD1 is also a negative regulator of T-cell activity that limits the activity of T cells at a variety of stages of the immune response when it interacts with its two ligands: PDL1 and PDL2. Unlike CTLA4, which is primarily believed to regulate immune responses early in T-cell activation, PD1 is primarily believed to inhibit effector T-cell activity in the effector phase within tissues and tumors.
Targeting PDL1 is a similarly promising approach to targeting PD1. However, targeting PDL1 may result in different biologic effects than targeting PD1. In addition to binding PD1, PDL1 is also believed to exert negative signals on T cells by interacting with B7. PDL1-blocking antibodies prevent this interaction, but PD1-blocking antibodies do not. Another slight difference is that PDL1 antibodies do not prevent PD1 from interacting with PDL2, although the effect of this interaction remains unknown. Nivolumab and pembrolizumab are antibodies that block PD1 on the surface of T cells and prevent those T cells from interacting with PDL1. Monoclonal antibodies that block PDL1 are being evaluated in clinical trials.84,85
Investigations evaluating the presence of PD1 on TILs and PDL1 on tumor cells in BC found that immune checkpoint proteins are upregulated in many BCs, particularly the TN subtype. The reported incidence of expression is highly variable. TILs expressing PD1 appear to be found more frequently in the TN subtype than in the other subtypes. In addition, the same pattern appears to occur for PDL1. These data support the study of immune checkpoint inhibitors in TNBC.86,87
Clinical trials of immunotherapy in TNBC
One of the first completed clinical trials of a PD1 monoclonal antibody (pembrolizumab) in TNBC was reported at the 2014 SABCS by Nanda et al.88 That was a Phase IB study that enrolled 32 patients with TNBC who had recurrent or metastatic disease (47% of whom had had more than three lines of previous CT). The participants were all PDL1-positive. Pembrolizumab was administered intravenously at a dose of 10 mg/kg every 2 weeks, and treatment could continue indefinitely as long as the patients were stable and their disease was not clearly progressing, as assessed by Response Evaluation Criteria in Solid Tumors version 1.1 every 8 weeks. Treatment with PD1 blockade was tolerable, with 56% of patients reporting an adverse event, but only 16% reporting grade 3–5 toxicity. Toxicity was essentially low-grade joint and muscle pain, fatigue, and nausea. One treatment-related death was caused by disseminated intravascular coagulation. Overall, 18.5% of 27 evaluable patients responded to pembrolizumab, with one (4%) CR, four (15%) PRs, and seven patients (26%) with stable disease. The median time to response was 18 weeks. The median PFS was just under 2 months. Three patients remained on pembrolizumab for at least 11 months.88
At the same meeting, initial data from a Phase I study of an anti-PDL1 monoclonal antibody (MPDL3280A, atezolizumab) in metastatic TNBC were also reported. Emens et al89 showed results from 12 patients with PDL1-positive disease. Grade 3–4 toxicities occurred in 8% of patients. Although immune-related adverse events have been reported with the use of immune checkpoint inhibitor agents, only one patient in this study demonstrated grade 2 pyrexia that was potentially attributable to immune activation. In general, immune-related adverse events occurred in a minority of patients. There were no toxicity-related deaths. Although over 90% of patients had been previously treated with more than two prior regimens and one-third of those enrolled had visceral metastases, the overall response rate was 33% in the nine patients who were evaluable for efficacy (one CR and two PRs). All responses were seen within the first 6 weeks of treatment.89
Recently, data presented at SABCS 2015 encouraged the evaluation of another anti-PDL1 agent (avelumab). The trial showed promising results in a subgroup of TNBC. For those patients with TNBC and PDL1 expression on immune cells, the clinical response with avelumab was 44.4% versus 2.6% in the absence of expression.90
All of these preliminary results are promising for the use of immuno-oncology agents in TNBC. The future challenge will be the identification of tumoral immune microenvironments that improve prognosis in BC. In this way, we hope to promote efficacious antitumoral immunity for all BCs. For example, in TNBC, controlling tumor growth with conventional chemotherapies in combination with immune checkpoint inhibitors could increase response rates.82,91 For patients with limited T-cell infiltration, vaccine priming before or concurrent with immune checkpoint inhibitors may also result in additional clinical benefits. Many studies of further immunotherapy in BC are ongoing or planned. We hope that the best is still to come with respect to this therapy in the field of TNBC (Table 6).92
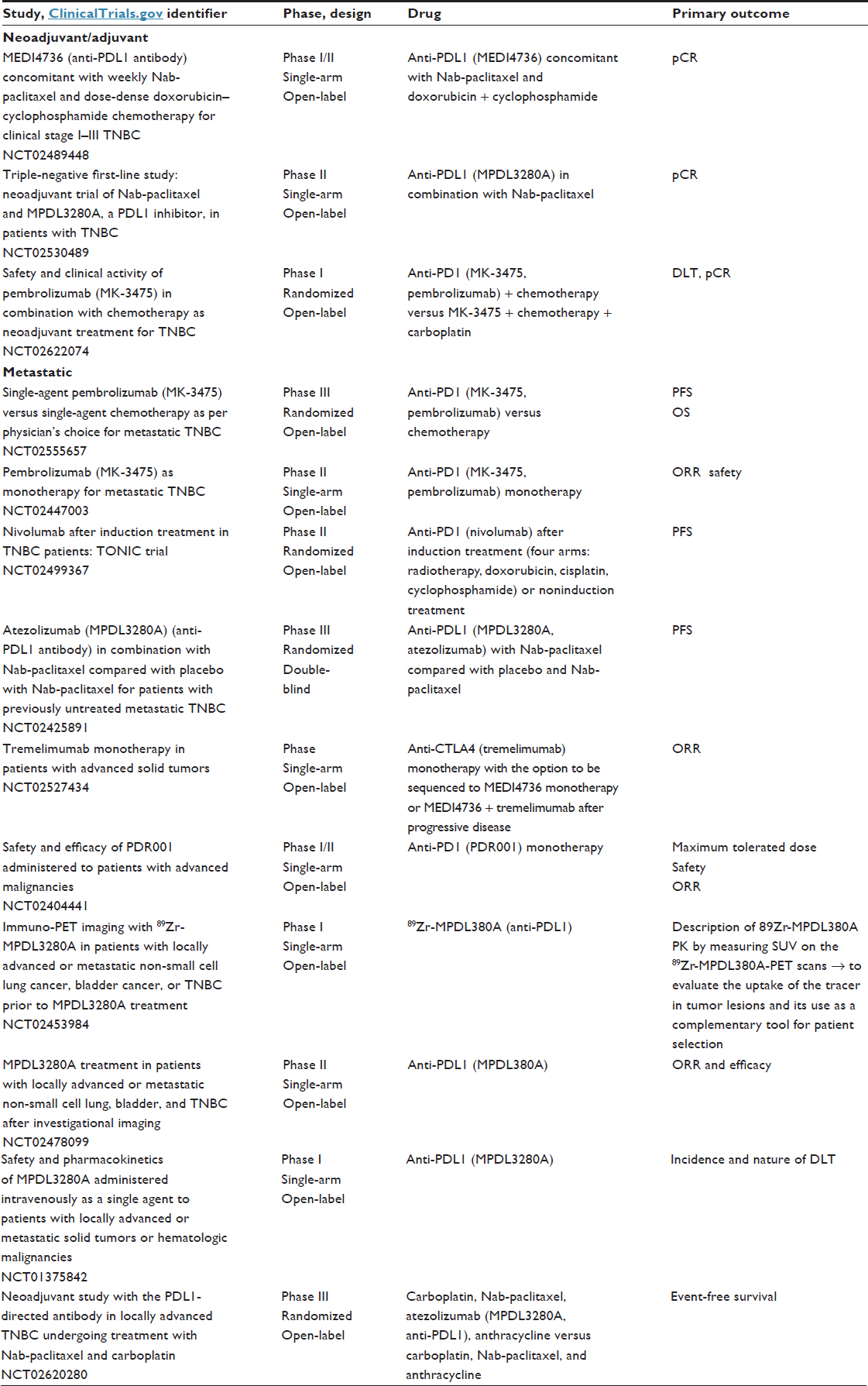 | Table 6 Immunotherapy trials in breast cancer |
Growth factor overexpression in TNBC
Different growth factors are overexpressed in TNBC, such as vascular endothelial growth factor (VEGF) and EGFR. Targeting these pathways showed only limited activity in unselected TNBC. For the VEGF pathway, different trials in patients with ABC and in the adjuvant setting did not show any benefit in overall survival, even in trials dedicated to TNBC, such as the BEATRICE trial.93 Similarly, for the EGFR pathway, the results have been disappointing. EGFR is overexpressed in more than 50% of TNBCs, but the rate of mutation is low (10%) and is found only in Asian populations.94 Trials with anti-EGFR therapies have suggested that EGFR overexpression is not correlated with the activity of anti-EGFR agents in TNBC.95,96 FGFRs may be a better target. FGFRs are expressed on many different cells, and regulate cell growth, survival, migration, and differentiation. In many cancer types, FGF signaling is implicated in oncogenic behavior. Targeting this pathway is a current area of drug development, not only primarily with tyrosine-kinase inhibitors but also with monoclonal antibodies that target FGFs/FGFRs and the FGF-ligand trap.97,98 In BC, only 9% of tumors have FGFR1 amplification and 4% have FGFR2 amplification.23,99 These tumors represent a very small population, but FGFRs could be an interesting target. Participation in clinical trials is crucial for improved evaluation of the potential of this new treatment strategy according to specific FGFR alterations.100
Conclusion and perspectives
Progress in the treatment of TNBC remains an important challenge. In clinical practice, we still use standard CT (anthracyclines and taxanes). Some data in favor of the use of platinum-based CT in TNBC are now available, particularly in BRCA-mutation carriers. Clinical research is focused on two main axes in the neoadjuvant setting: how to increase pCR and how to improve outcomes in patients with residual disease. New targeted treatments and immunotherapeutic drugs are under development. The challenge is to drive studies on more selected patient populations due to the importance of heterogeneity in TNBC. The most promising new approaches include immunotherapy with checkpoint inhibitors, PARP inhibitors, and AR inhibitors. Furthermore, active research to discover additional specific targets in TNBC is ongoing.101
Disclosure
GJ has received honoraria from Amgen, Pfizer, Novartis, Roche, and Celgene, and research grants from Novartis, Roche, and MSD. The other authors report no conflicts of interest in this work.
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. | |
Perou CM, Sørlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–752. | |
Sørlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98(19):10869–10874. | |
Goldhirsch A, Ingle JN, Gelber RD, Coates AS, Thürlimann B, Senn HJ. Thresholds for therapies: highlights of the St Gallen International Expert Consensus on the primary therapy of early breast cancer 2009. Ann Oncol. 2009;20(8):1319–1329. | |
Wolff AC, Hammond ME, Schwartz JN, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. Arch Pathol Lab Med. 2007;131(1):18–43. | |
Dent R, Trudeau M, Pritchard KI, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13(15 Pt 1):4429–4434. | |
Irvin WJ Jr, Carey LA. What is triple-negative breast cancer? Eur J Cancer. 2008;44(18):2799–2805. | |
Lin NU, Claus E, Sohl J, Razzak AR, Arnaout A, Winer EP. Sites of distant recurrence and clinical outcomes in patients with metastatic triple-negative breast cancer: high incidence of central nervous system metastases. Cancer. 2008;113(10):2638–2645. | |
Nielsen TO, Hsu FD, Jensen K, et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res. 2004;10(16):5367–5374. | |
Rakha EA, Reis-Filho JS, Ellis IO. Basal-like breast cancer: a critical review. J Clin Oncol. 2008;26(15):2568–2581. | |
Reis-Filho JS, Tutt AN. Triple negative tumours: a critical review. Histopathology. 2008;52(1):108–118. | |
Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363(20):1938–1948. | |
Bauer KR, Brown M, Cress RD, Parise CA, Caggiano V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: a population-based study from the California cancer Registry. Cancer. 2007;109(9):1721–1728. | |
Carey LA, Perou CM, Livasy CA, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA. 2006;295(21):2492–2502. | |
Eiermann W, Bergh J, Cardoso F, et al. Triple negative breast cancer: proposals for a pragmatic definition and implications for patient management and trial design. Breast. 2012;21(1):20–26. | |
Gonzalez-Angulo AM, Timms KM, Liu S, et al. Incidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancer. Clin Cancer Res. 2011;17(5):1082–1089. | |
Daly MB, Pilarski R, Axilbund JE, et al. Genetic/familial high-risk assessment: breast and ovarian, Version 2.2015. J Natl Compr Canc Netw. 2016;14(2):153–162. | |
Liedtke C, Mazouni C, Hess KR, et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol. 2008;26(8):1275–1281. | |
Carey LA, Dees EC, Sawyer L, et al. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res. 2007;13(8):2329–2334. | |
Lehmann BD, Pietenpol JA. Identification and use of biomarkers in treatment strategies for triple-negative breast cancer subtypes. J Pathol. 2014;232(2):142–150. | |
Montagna E, Maisonneuve P, Rotmensz N, et al. Heterogeneity of triple-negative breast cancer: histologic subtyping to inform the outcome. Clin Breast Cancer. 2013;13(1):31–39. | |
Kandil D, Khan A. Triple negative breast carcinoma: the good, the bad and the ugly. Diagn Histopathol (Oxf). 2012;18(5):210–216. | |
Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. | |
Shah SP, Roth A, Goya R, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486(7403):395–399. | |
Lehmann BD, Bauer JA, Chen X, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–2767. | |
Herschkowitz JI, Simin K, Weigman VJ, et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007; 8(5):R76. | |
Prat A, Parker JS, Karginova O, et al. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12(5):R68. | |
Burstein MD, Tsimelzon A, Poage GM, et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin Cancer Res. 2015;21(7):1688–1698. | |
Masuda H, Baggerly KA, Wang Y, et al. Differential response to neoadjuvant chemotherapy among 7 triple-negative breast cancer molecular subtypes. Clin Cancer Res. 2013;19(19):5533–5540. | |
Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411(6835):366–374. | |
Hoeijmakers JH, Bootsma D. DNA repair: incisions for excision. Nature. 1994;371(6499):654–655. | |
Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108(2):171–182. | |
Metzger-Filho O, Tutt A, de Azambuja E, et al. Dissecting the heterogeneity of triple-negative breast cancer. J Clin Oncol. 2012;30(15):1879–1887. | |
Turner NC, Reis-Filho JS. Basal-like breast cancer and the BRCA1 phenotype. Oncogene. 2006;25(43):5846–5853. | |
Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4(10):814–819. | |
Lips EH, Mulder L, Oonk A, et al. Triple-negative breast cancer: BRCAness and concordance of clinical features with BRCA1-mutation carriers. Br J Cancer. 2013;108(10):2172–2177. | |
Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358(11):1148–1159. | |
Esteller M, Silva JM, Dominguez G, et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst. 2000;92(7):564–569. | |
McLornan DP, List A, Mufti GJ. Applying synthetic lethality for the selective targeting of cancer. N Engl J Med. 2014;371(18):1725–1735. | |
Schouten PC, Linn SC. Challenges in the use of DNA repair deficiency as a biomarker in breast cancer. J Clin Oncol. 2015;33(17):1867–1869. | |
Watkins JA, Irshad S, Grigoriadis A, Tutt AN. Genomic scars as biomarkers of homologous recombination deficiency and drug response in breast and ovarian cancers. Breast Cancer Res. 2014;16(3):211. | |
Timms KM, Abkevich V, Hughes E, et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014;16(6):475. | |
Sledge GW Jr, Roth BJ. Cisplatin in the management of breast cancer. Semin Oncol. 1989;16(4 Suppl 6):110–115. | |
Sirohi B, Arnedos M, Popat S, et al. Platinum-based chemotherapy in triple-negative breast cancer. Ann Oncol. 2008;19(11):1847–1852. | |
Staudacher L, Cottu PH, Diéras V, et al. Platinum-based chemotherapy in metastatic triple-negative breast cancer: the Institut Curie experience. Ann Oncol. 2011;22(4):848–856. | |
Isakoff SJ, Mayer EL, He L, et al. TBCRC009: a multicenter phase II clinical trial of platinum monotherapy with biomarker assessment in metastatic triple-negative breast cancer. J Clin Oncol. 2015;33(17):1902–1909. | |
Tutt A, Ellis P, Kilburn L, et al. The TNT trial: a randomized phase III trial of carboplatin (C) compared with docetaxel (D) for patients with metastatic or recurrent locally advanced triple negative or BRCA1/2 breast cancer [abstract]. 2014. Available from: http://www.abstracts2view.com/sabcs14/view.php?nu=SABCS13L_789. Accessed January 1, 2016. | |
Carey LA, Dees EC, Sawyer L, et al. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res. 2007;13(8):2329–2334. | |
Cortazar P, Zhang L, Untch M, et al. Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet. 2014;384(9938):164–172. | |
Byrski T, Gronwald J, Huzarski T, et al. Pathologic complete response rates in young women with BRCA1-positive breast cancers after neoadjuvant chemotherapy. J Clin Oncol. 2010;28(3):375–379. | |
Zhang P, Yin Y, Xu B, et al. Carboplatin plus paclitaxel compared with epirubicin plus paclitaxel as neoadjuvant chemotherapy for triple-negative breast cancer – a phase II clinical trial. Cancer Res. 2013;73:P3-14-07. | |
von Minckwitz G, Schneeweiss A, Loibl S, et al. Neoadjuvant carboplatin in patients with triple-negative and HER2-positive early breast cancer (GeparSixto; GBG 66): a randomised phase 2 trial. Lancet. 2014;15(7):747–756. | |
Sikov WM, Berry DA, Perou CM, et al. Impact of the addition of carboplatin and-or bevacizumab to neoadjuvant once-per-week paclitaxel followed by dose-dense doxorubicin an cyclophosphamide on pathologic complete response rates in stages II to III triple negative breast cancer: CALGB 40603 (Alliance). J Clin Oncol. 2015;33(1):13–21. | |
Alba E, Chacon JI, Lluch A, et al. A randomized phase II trial of platinum salts in basal-like breast cancer patients in the neoadjuvant setting: results from the GEICAM/2006-03, multicenter study. Breast Cancer Res Treat. 2012;136(2):487–493. | |
[No authors listed]. Positive results for drug combo in I-SPY 2 trial. Cancer Discov. 2014;4(2):OF2. | |
von Minckwitz G, Loibl S, Schneeweiss A, et al. Early survival analysis of the randomized phase II trial investigating the addition of carboplatin to neoadjuvant therapy for triple-negative and HER2-positive early breast cancer (GeparSixto). Cancer Res. 2016;76:S2–S4. | |
German Breast Group. A phase III trial comparing two dose-dense, dose-intensified approaches (ETC and PM[Cb]) for neoadjuvant treatment of patients with high-risk early breast cancer (GeparOcto). 2016. Available from: https://clinicaltrials.gov/ct2/show/NCT02125344. Accessed December 24, 2015. | |
Coates AS, Winer EP, Goldhirsch A, et al. Tailoring therapies – improving the management of early breast cancer: St Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2015. Ann Oncol. 2015;26(8):1533–1546. | |
Fasching PA, Loibl S, Eidtmann H, et al. BRCA mutations, therapy response and prognosis in the neoadjuvant GeparQuinto study. 2015. Available from: http://www.abstracts2view.com/sabcs15/view.php?nu=SABCS15L_822&terms=. Accessed January 1, 2016. | |
Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–134. | |
Tutt A, Robson M, Garber JE, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376(9737):235–244. | |
Kaufman B, Shapira-Frommer R, Schmutzler RK, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33(3):244–250. | |
Gelmon KA, Tischkowitz M, Mackay H, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12(9):852–861. | |
Sonnenblick A, de Azambuja E, Azim HA Jr, Piccart M. An update on PARP inhibitors – moving to the adjuvant setting. Nat Rev Clin Oncol. 2015;12(1):27–41. | |
Niemeier LA, Dabbs DJ, Beriwal S, Striebel JM, Bhargava R. Androgen receptor in breast cancer: expression in estrogen receptor-positive tumors and in estrogen receptor-negative tumors with apocrine differentiation. Mod Pathol. 2010;23(2):205–212. | |
Barton VN, D’Amato NC, Gordon MA, Christenson JL, Elias A, Richer JK. Androgen receptor biology in triple negative breast cancer: a case for classification as AR+ or quadruple negative disease. Horm Cancer. 2015;6(5–6):206–213. | |
Proverbs-Singh T, Feldman JL, Morris MJ, Autio KA, Traina TA. Targeting the androgen receptor in prostate and breast cancer: several new agents in development. Endocr Relat Cancer. 2015;22(3):R87–R106. | |
Farmer P, Bonnefoi H, Becette V, et al. Identification of molecular apocrine breast tumours by microarray analysis. Oncogene. 2005;24(29):4660–4671. | |
Doane AS, Danso M, Lal P, et al. An estrogen receptor-negative breast cancer subset characterized by a hormonally regulated transcriptional program and response to androgen. Oncogene. 2006;25(28):3994–4008. | |
Gucalp A, Tolaney S, Isakoff SJ, et al. Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic breast cancer. Clin Cancer Res. 2013;19(19):5505–5512. | |
Traina TA, O’Shaughnessy J, Nanda R, et al. Abstract P5-19-09: preliminary results from a phase 2 single-arm study of enzalutamide, an androgen receptor (AR) inhibitor, in advanced AR+ triple-negative breast cancer (TNBC). Cancer Res. 2015;75:P5-19-09. | |
Traina TA, Miller K, Yardley DA, et al. Results from a phase 2 study of enzalutamide (ENZA), an androgen receptor (AR) inhibitor, in advanced AR+ triple-negative breast cancer (TNBC). J Clin Oncol., 2015;(33 Suppl):1003. | |
Arce-Salinas C, Riesco-Martinez MC, Hanna W, Bedard PH, Warner E. Complete response of metastatic androgen receptor-positive breast cancer to bicalutamide: case report and review of the literature. J Clin Oncol. 2016;34(4):e21–e24. | |
Anestis A, Karamouzis MV, Dalagiorgou G, Papavassiliou AG. Is androgen receptor targeting an emerging treatment strategy for triple negative breast cancer? Cancer Treat Rev. 2015;41(6):547–553. | |
Loi S. Tumor-infiltrating lymphocytes, breast cancer subtypes and therapeutic efficacy. Oncoimmunology. 2013;2(7):e24720. | |
Loi S, Michiels S, Salgado R, et al. Tumor infiltrating lymphocytes are prognostic in triple negative breast cancer and predictive for trastuzumab benefit in early breast cancer: results from the FinHER trial. Ann Oncol. 2014;25(8):1544–1550. | |
Ibrahim EM, Al-Foheidi ME, Al-Mansour MM, Kazkaz GA. The prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancer: a meta-analysis. Breast Cancer Res Treat. 2014;148(3):467–476. | |
Adams S, Gray RJ, Demaria S, et al. Prognostic value of tumor-infiltrating lymphocytes in triple-negative breast cancers from two phase III randomized adjuvant breast cancer trials: ECOG 2197 and ECOG 1199. J Clin Oncol. 2014;32(27):2959–2966. | |
Loi S, Sirtaine N, Piette F, et al. Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02-98. J Clin Oncol. 2013;31(7):860–867. | |
Ono M, Tsuda H, Shimizu C, et al. Tumor-infiltrating lymphocytes are correlated with response to neoadjuvant chemotherapy in triple-negative breast cancer. Breast Cancer Res Treat. 2012;132(3):793–805. | |
Denkert C, Loibl S, Noske A, et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J Clin Oncol. 2010;28(1):105–113. | |
Denkert C, von MG, Brase JC, et al. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. J Clin Oncol. 2015;33(9):983–991. | |
Smid M, Hoes M, Sieuwerts AM, et al. Patterns and incidence of chromosomal instability and their prognostic relevance in breast cancer subtypes. Breast Cancer Res Treat. 2011;128(1):23–30. | |
Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol. 2015;33(17):1974–1982. | |
Homet MB, Ribas A. Anti-programmed cell death protein-1/ligand-1 therapy in different cancers. Br J Cancer. 2015;112(9):1421–1427. | |
Gatalica Z, Snyder C, Maney T, et al. Programmed cell death 1 (PD-1) and its ligand (PD-L1) in common cancers and their correlation with molecular cancer type. Cancer Epidemiol Biomarkers Prev. 2014;23(12):2965–2970. | |
Mittendorf EA, Philips AV, Meric-Bernstam F, et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol Res. 2014;2(4):361–370. | |
Nanda R, Chow LQ, Dees EC, et al. Abstract S1-09: a phase IB study of pembrolizumab (MK-3475) in patients with advanced triple-negative breast cancer. Cancer Res. 2015;75:S1–S9. | |
Emens LA, Braiteh FS, Cassier P, et al. Inhibition of PD-L1 by MPDL3280A leads to clinical activity in patients with metastatic triple-negative breast cancer. Cancer Res. 2015;75:PD1–PD6. | |
Dirix LY, Takacs I, Nikolinakos P, et al. Avelumab (MSB0010718C), an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: a phase IB JAVELIN solid tumor trial. Cancer Res. 2016;76:S1–S4. | |
Tung NM, Winer EP. Tumor-infiltrating lymphocytes and response to platinum in triple-negative breast cancer. J Clin Oncol. 2015;33(9):969–971. | |
Cimino-Mathews A, Foote JB, Emens LA. Immune targeting in breast cancer. Oncology (Williston Park). 2015;29(5):375–385. | |
Cameron D, Brown J, Dent R, et al. Adjuvant bevacizumab-containing therapy in triple-negative breast cancer (BEATRICE): primary results of a randomised, phase 3 trial. Lancet Oncol. 2013;14(10):933–942. | |
Teng YH, Tan WJ, Thike AA, et al. Mutations in the epidermal growth factor receptor (EGFR) gene in triple negative breast cancer: possible implications for targeted therapy. Breast Cancer Res. 2011;13(2):R35. | |
Masuda H, Zhang D, Bartholomeusz C, Doihara H, Hortobagyi GN, Ueno NT. Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res Treat. 2012;136(2):331–345. | |
Carey LA, Rugo HS, Marcom PK, et al. TBCRC 001: randomized phase II study of cetuximab in combination with carboplatin in stage IV triple-negative breast cancer. J Clin Oncol. 2012;30(21):2615–2623. | |
Turner N, Grose R. Fibroblast growth factor signalling: from development to cancer. Nat Rev Cancer. 2010;10(2):116–129. | |
Wesche J, Haglund K, Haugsten EM. Fibroblast growth factors and their receptors in cancer. Biochem J. 2011;437(2):199–213. | |
Turner N, Lambros MB, Horlings HM, et al. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene. 2010;29(14):2013–2023. | |
Abu-Khalaf MM, Mayer IA, Litten JB, et al. A phase 2, randomized, open-label, multicenter, safety and efficacy study of oral lucitanib in patients with metastatic breast cancer with alterations in the FGF pathway. Cancer Res. 2015;75:OT1-1-13. | |
Shu SK, Lin CY, He HS, et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016;529(7586):413–417. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.