Back to Journals » OncoTargets and Therapy » Volume 7
Triple-negative breast cancer risk in women is defined by the defect of estrogen signaling: preventive and therapeutic implications
Authors Suba Z
Received 6 August 2013
Accepted for publication 8 November 2013
Published 23 January 2014 Volume 2014:7 Pages 147—164
DOI https://doi.org/10.2147/OTT.S52600
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Zsuzsanna Suba
National Institute of Oncology, Surgical and Molecular Tumor Pathology Centre, Budapest, Hungary
Abstract: Epidemiologic studies strongly support that triple-negative breast cancers (TNBCs) may be distinct entities as compared with estrogen receptor (ER)+ tumors, suggesting that the etiologic factors, clinical characteristics, and therapeutic possibilities may vary by molecular subtypes. Many investigations propose that reproductive factors and exogenous hormone use differently or even quite inversely affect the risk of TNBCs and ER+ cancers. Controversies concerning the exact role of even the same risk factor in TNBC development justify that the biological mechanisms behind the initiation of both TNBCs and non-TNBCs are completely obscure. To arrive at a comprehensive understanding of the etiology of different breast cancer subtypes, we should also reconsider our traditional concepts and beliefs regarding cancer risk factors. Malignancies are multicausal, but the disturbance of proper estrogen signaling seems to be a crucial risk factor for the development of mammary cancers. The grade of defect in metabolic and hormonal equilibrium is directly associated with TNBC risk for women during their whole life. Inverse impact of menopausal status or parity on the development of ER+ and ER- breast cancers may not be possible; these controversial results derive from the misinterpretation of percentage-based statistical evaluations. Exogenous or parity-associated excessive estrogen supply is suppressive against breast cancer, though the lower the ER expression of tumors, the weaker the anticancer capacity. In women, the most important preventive strategy against breast cancers – included TNBCs – is the strict control and maintenance of hormonal equilibrium from early adolescence through the whole lifetime, particularly during the periods of great hormonal changes.
Keywords: cancer prevention, infertility, insulin resistance, menopause, metabolic syndrome, type 2 diabetes
Introduction
Poorly differentiated triple-negative breast cancers (TNBCs) were first characterized in the literature in 2005 by the absence of steroid receptors for estrogen (ER) and progesterone (PR), as well as the lack of tyrosine kinase human epidermal growth factor receptor 2 (HER2).1 TNBC represents approximately 15%–20% of invasive breast cancers and has been strongly associated with younger age at diagnosis, family history of breast cancers, carrying type 1 mutation in breast cancer susceptibility gene (BRCA1), and African-American race of patients.2–6
Clinically, TNBCs exhibit fairly aggressive local growth and rapid progression and account for a high rate of early metastases, most commonly to visceral organs and the central nervous system.5,6 TNBCs are also characterized by diagnosis at a later stage and the poorest survival of patients as compared with cases of any other breast cancer types.2,7 Pathologic features are high grade, infiltrative spread, high rates of mitotic figures, and p53 mutations in addition to ER, PR, and HER2 negativity.8
Epidemiologic studies strongly support that TNBCs may be distinct entities as compared with steroid receptor positive tumors, suggesting that the etiologic factors, clinical features, and therapeutic possibilities of breast cancers may vary by molecular subtypes.9–14 Many literary data, however, refer to apparently common risk factors for TNBCs and non-TNBCs, such as metabolic syndrome (MS), type 2 diabetes, obesity, BRCA gene mutations, and the African-American race of women.6,15–18 Moreover, recent observations suggest that the stronger the risk factor for overall breast cancer, the higher the risk for development of TNBC type.6,19–22
By contrast, further investigations propose that reproductive factors and exogenous hormone use may differently or even quite inversely affect the risk of TNBCs and steroid receptor-positive cancers.11–13,23–25 Moreover, a puzzling question concerns the low overall breast cancer rates among young females, as opposed to the relatively high incidence rate, rapid progression, and poor outcome of TNBC at a young age.5,9,10 There are many controversies and inconsistencies concerning the exact role of even the same hormone-associated factor in TNBC development, justifying that the biologic mechanisms behind the initiation of both TNBCs and non-TNBCs are completely obscure.
The clinical and biologic significance of ER protein expression in breast cancers has been equivocally established,26 while the lack of PR protein has no additive prognostic value either in ER+ or ER− breast cancer cases.27 The role of HER2 protein expression in the progression of breast cancer seems to be paradigmatic. Retrospective clinical studies have suggested that patients with ER+ tumors are less likely to benefit from adjuvant tamoxifen therapy if their cancers are HER2+ than cases with HER2– tumors.28 Moreover, patients with ER+, HER2 overexpressing tumors exhibited higher rates of recurrence and mortality after tamoxifen therapy as compared with those who did not receive the agent.29
Conversely, lack of ERs is an equivocally crucial indicator of poor prognosis and fatal outcome in mammary cancer cases.7,10 These observations may raise the presumption that the available ERs in well-differentiated cancer cells supply the signaling pathways with tumor-killing capacity in the case of the sufficient exposure of their own specific ligands, estrogens. To arrive at a comprehensive understanding of the etiology of different breast cancer subtypes, we should also reconsider our traditional concepts and beliefs regarding cancer risk factors.
The purpose of the current study is to analyze in detail the provoking and defensive factors as players in mammary carcinogenesis based on the data of epidemiologic, clinical, experimental, biochemical, immunohistochemical, and genetic studies. Our aim is to clarify whether the different molecular subtypes of breast cancer also stand for etiological differences, or if there are quite common risk factors that differ only in intensity, exposure period, and coexistence while having a strengthening impact on each other.
Considering the puzzling processes behind TNBC development, the following questions are to be answered:
- What is the explanation for the conspicuous changes in TNBC incidence rate during the different periods of life in women?
- Is TNBC indeed a quite distinct entity, or is it a poorly differentiated variant of breast cancers induced by the same cancer risk factors?
- How can similar breast cancer risk factors modify or define the development of TNBCs and non-TNBCs?
- In what way can parity and exogenous hormone use affect breast cancer risk, and is there any possibility for their quite inverse impact on the development of TNBCs and non-TNBCs?
- What are the proposed new strategies for primary prevention and therapy of TNBC?
Difference in TNBC incidence rates between young and older women
Literary data support that young age is associated with an equivocally higher incidence rate of TNBCs as compared with older female cases.10,11 The defining age border for distinction of the two age groups of women is not identical. A population based study determines the age groups of breast cancer cases on the basis of the same principle, using mean age at menopause, or age around 50 years.2 Others define the critical threshold at a higher11 or lower age.7,10,12,30–32 In spite of the various age groupings, hormonally mediated risk factors are regarded as being essential in the apparently higher TNBC incidence rate (25%–49%) among young cases.
In a case control study, breast cancers in women less than 40 years of age exhibited a significantly higher rate of ER− tumors (33.8%) as compared with older cases (21.9%), suggesting a disproportional risk for poorly differentiated tumors in the young age group.7 Nevertheless, graphic representation of the raw numbers of age-dependent ER+ and ER− breast cancers clearly shows a close to twofold increase in the number of ER− tumors and an almost fourfold increase in the number of ER+ tumors with aging (Figure 1). Paradoxically, the percentage of ER− tumors exhibits a deceivingly inverse trend, decreasing in the older group of women, which is attributed to the excessive increase in ER+ cancers among these cases. Since all the statistical risk indices, such as odds ratios (ORs), hazard ratios (HRs), and incidence risk ratios are based on the percentage of TNBC cases, the majority of studies report on higher TNBC risk among young women, disregarding the low number of overall breast cancer cases.
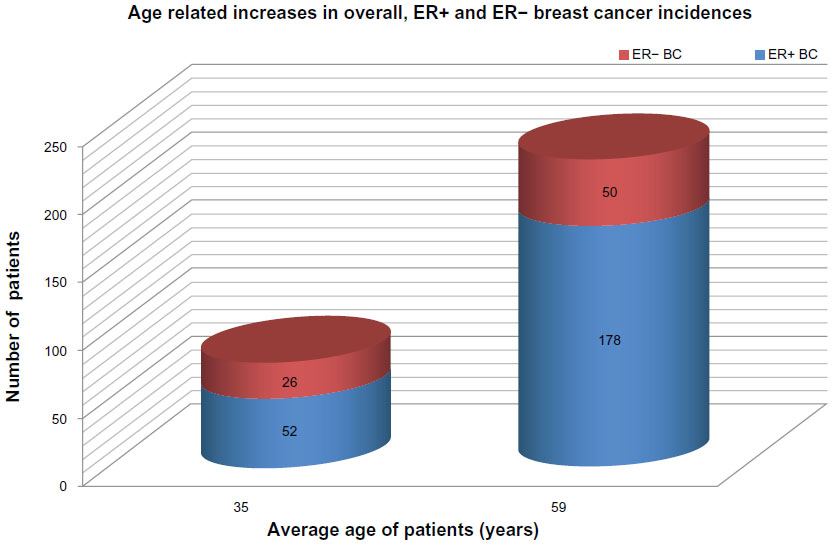 | Figure 1 Graphic representation of age-related increases in overall ER+ and ER− BC incidences. |
Considering all breast cancer cases, the incidence rate among young women is fairly low (23.9%), being much higher in older cases (76.1%).7 In young females, ER+ and ER− tumor incidences seem to be disproportionately suppressed by certain common factors as compared with older women. This suppression may be particularly strong against ER+ and weaker against ER− breast cancers. The relatively high percentage of surviving TNBCs among young cases may be attributed to the low incidence rate of more successfully eliminated ER+ cancers rather than to an excessive initiation of ER− tumors. The latter tumor types are submitted to only a moderate suppression.
What can the advantageous factor be in young women suppressing strongly the ER+ and moderately the ER− breast cancers? In premenopausal cases, healthy or slightly defective estrogen synthesis supplies the ligands for the available ERs of tumor cells. In the case of sufficient estrogen exposure, both the initiation and progression of ER+ breast cancers may be more effectively suppressed as compared with ER− tumors.33,34
Viscerally obese premenopausal women are hormonally challenged, and their increased breast cancer risk may be attributed to the decreased estrogen exposure.33 Among premenopausal obese women, hip circumference was found to be in direct association with moderately increased risk for ER+ tumor (HR =1.65) and highly increased risk for ER− breast cancer (HR =2.85).16 Stronger suppression of ER+ as compared with ER− breast cancers, even in young women with defective estrogen synthesis, may provide insight into the advances of estrogen signaling against more differentiated breast cancers. Nevertheless, estrogen also has alternative signaling pathways to overcome the ER− tumors (see Some aspects of the molecular mechanism of estrogen surveillance on cell proliferation), but the efficacy of such possibilities is weaker, reflected in the relatively high percentage of ER− tumor survival in young women.
The weakness of the anticancer capacity of estrogen level against ER− tumors may be justified by the fact that, of all the examined tumor markers, ER negativity was a crucial factor in defining biological aggressivity and fatal outcome in young breast cancer cases.7,10 Histochemical analysis of breast cancers with the same histologic grade exhibited lower expression of ERs and higher proliferative indices in premenopausal cases than in older women.10
Lack of intratumoral estrogen synthesis is associated with low differentiation of breast cancers, weak tumor regression, and poor prognosis.34 In young breast cancer cases (<40 years), locoregional control after breast-conserving surgery highlighted that the absence of CYP19-aromatase activity in removed tumor samples carried a highly significant risk for locoregional tumor recurrence.35
In conclusion, TNBC in young patients is not a distinct entity with mysteriously unique etiology. Low overall breast cancer incidence rates may be attributed to the healthy estrogen surveillance in the majority of young women. In young cases, tumor may develop as a consequence of usual, strong, or multiple risk factors associated with breast cancer. All cancer risk factors, such as positive family history, MS, type 2 diabetes, and BRCA gene mutation decrease the estrogen synthesis and/or damage the estrogen signaling in young cases; however, even the slightly defective estrogen surveillance may have stronger suppressive effect on ER+ breast cancers resulting in a disproportionately better survival of ER− tumors.
Hormonal factors affecting the changes in TNBC risk during the different life periods of women
Risk factors for breast cancer are usually evaluated when the tumors are clinically diagnosed. Nevertheless, cell kinetic studies of tumors demonstrated that the clinical appearance of solid breast cancers requires at least a period of 6–8 years from their initiation to the development of palpable size.36 Searching for the etiologic factors of breast cancer is difficult, since harmful effects at the estimated time of first mutation in the past might be more crucial than the momentary findings at the time of clinical diagnosis.
Great hormonal changes occurring during a woman’s life might strongly define the inclination to initiate any type of breast cancer, including TNBC (Table 1). The stronger the hormonal imbalance, characterized mainly by hyperinsulinism, hyperandrogenism, and low estrogen exposure, the higher the breast cancer risk, particularly for the poorly differentiated TNBC type.
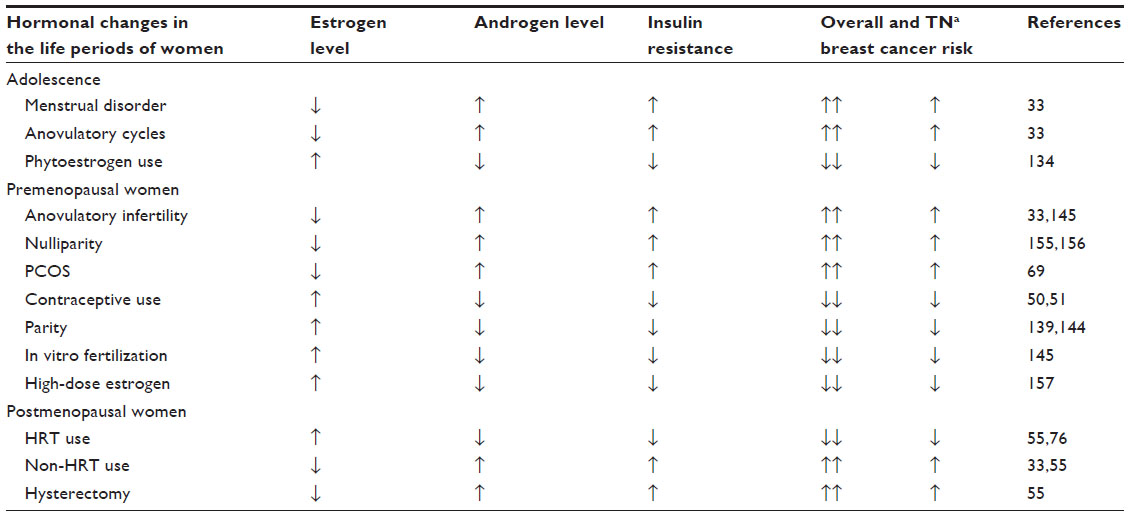 | Table 1 Lifelong changes in the levels of sex hormones and insulin resistance in women and their correlations with overall and TN breast cancer risk |
Three main phases seem to be particularly dangerous for breast cancer initiation during the life of women.33 Two of these are crucial periods inducing hormonal and metabolic storms, namely, adolescence (14–18 years) and the perimenopausal phase (45–55 years). Both periods present risks for overall breast cancer initiation if the biologic processes in the background become pathologic. The third, especially risky phase for breast cancer initiation is older age (over 60 years), when the hormonal and metabolic imbalance becomes stronger and the defense mechanisms against cancer initiation are debilitated.
The first challenges for the whole body of girls are pubertal changes, since the abrupt somatic and sexual development presents a real danger for the development of insulin resistance and the associated imbalance of male-to-female sexual hormone ratio.37,38 When a young girl inherits genetic or acquires somatic anomaly, such as glucose intolerance or obesity, overproduction of androgens will develop, at the expense of defective estrogen synthesis.33 This hormonal disturbance may induce insidiously symptom-free ovulatory failure or irregular anovulatory menstrual cycles.39 In severe cases, polycystic ovarian syndrome (PCOS) develops, which is the most prevalent hormonal alteration in young women.40
Adolescent hyperandrogenism is usually maintained until the early 30s and leads to definite or prolonged infertility, resulting in nulliparity or delayed childbirth.41,42 Low estrogen exposure and androgen excess at young age may provoke anovulatory infertility as well as initiation and even clinical appearance of breast, endometrial, and ovarian cancers in premenopause.33 Thus, reproductive failures and female cancer development have common sources: long-lasting hyperandrogenism, defective estrogen synthesis and the associated insulin resistance.
In healthy premenopausal women, breast cancer development is rare, as healthy ovulatory menstrual cycles are protective, and even a slightly or moderately defective estrogen synthesis may counteract cancer initiation.33 Increasing grades of insulin resistance associated with enhanced severity of sexual hormone imbalance, however, mean high risks for breast cancer development even in young cases and particularly for the initiation of poorly differentiated TNBC.
In cases of mild hyperinsulinemia and hyperandrogenism, preserved circulatory female sexual steroid levels and regular menstrual cycles may usually be enough to protect against breast tumor initiation. With aggravation of insulin resistance in young women, the associated moderate decrease in circulating estrogen level may cut off the ovulatory estrogen peak, resulting in anovulatory infertility. Even this slightly estrogen-deficient milieu may confer preferential risk for breast cancer as well as for endometrial and ovarian malignancies in anovulatory women. These female organs have the highest estrogen demand so as to preserve their structural and functional integrity.33,43
Among premenopausal cases, MS is associated with particular increase in the risk for ER−/PR− cancers and TNBCs6 parallel with decreasing estrogen exposure. In young premenopausal cases with type 2 diabetes, progressive insulin resistance and blocked aromatase activity inhibit the conversion of androgens to estrogen in both the ovaries and peripheral tissues.44 Low estrogen exposure is incapable of defending the cellular functions against strong insulin resistance and hyperandrogenism,33,45,46 resulting in early initiation and rapid clinical appearance of poorly differentiated, aggressive breast tumors, such as TNBCs.
Artificial cycles created by oral contraceptives (OCs) improves insulin resistance and provides substantial protection against endometrial and ovarian cancer in endangered anovulatory women.47,48 By contrast, the effect of OCs on breast cancer risk seems to be highly controversial. Increased risk was found to be confined to women who began pill use as teenagers or who had been pill users for a long time.49 Conversely, the relative risk was slightly decreased (0.9) for those who had previously used OCs50 and, in cases who had used OCs for at least 10 years, a markedly reduced risk for ER+ breast cancer was observed.51 Adequate selection of patients and controls having similar endogenous and familial risk factors should lead to the real evaluation of beneficial OC effect against breast cancer risk.33
Correlations between OC use and risk for different breast cancer subtypes are even much more controversial in young women. Although OCs may improve the hormonal equilibrium in both healthy and anovulatory women, there are fairly diverse hormonal statuses among OC nonuser control cases, which may affect the results concerning breast cancer risk. Considering the controversial findings, a stronger reduction of ER+ breast cancers51 and a weaker reduction, namely an increased percentage of ER− or triple-negative tumors,12,52 may be an actual result of long-lasting OC use.
The second risky period for breast cancer initiation may be the perimenopausal phase, when there is a slow or steep decline in ovarian female sexual steroid synthesis and the last menstrual cycle approaches. Breast cancer initiation is relatively frequent in hormonally challenged women between the ages of 45 and 55 years, and these tumors are predominantly hormone receptor-positive, which is attributed to the inequalities of the decreasing estrogen supply.43
In perimenopause, breast and other peripheral tissues may exhibit rapid compensatory hormone production for the completion of decreasing ovarian estrogen synthesis in an amount sufficient to kill or differentiate breast tumor cells initiated by chance. In this case, menopausal women are generally complaint-free and do not require medical help. In cases of defective hormonal adaptation to ovarian senescence the development of tissular estrogen synthesis in the breast may be late. This delay may result in tumor initiation, but later, the increasing extraovarian estrogen synthesis may promote killing or advantageous differentiation of early cancers.53 Women with transitorily insufficient estrogen levels frequently have strong menopausal complaints.
Postmenopausal hormone replacement therapy (HRT) is typically associated with highly differentiated, ER+ tumors, which are initiated in the late premenopausal or perimenopausal hormonal failure period, much earlier than the beginning of hormone treatment.54 Estrogen administration may help to differentiate the already existing subclinical tumors; however, the dose of hormone replacement is not always enough to kill them. The clinical diagnosis is always postmenopausal in case of tumors initiated in the perimenopausal phase due to the long latency period.
The breast cancer risk of women after hysterectomy may be high, attributed to the abrupt, shocking estrogen deprivation.43 A follow-up study on hysterectomized cases and controls justified the significantly lower risk of breast cancer in the estrogen-treated group of women.55
Breast cancers diagnosed in elderly women over 65 years are typically initiated in the postmenopausal period after 60 years of age. These patients are generally non HRT users and exhibit deepening estrogen deficiency and insulin resistance, even a high prevalence of type 2 diabetes and obesity. Both obesity and highly elevated fasting blood glucose level were found to be especially dangerous for mammary malignancies in elderly cases over 65 years of age.56,57 There is an increase in the ratio of TNBC among breast cancers in elderly cases, attributed to the low circulatory and tissular estrogen levels, hyperandrogenism, and associated insulin resistance. Since women from the elderly population are much fewer than middle-aged postmenopausal cases, pooled examinations of breast tumors diagnosed after menopause may give a blurred value with a predominance of hormone receptor-positive cases.
Taken together, the risk for breast cancer, and for TNBC in particular, is directly associated with the grade of defects in the metabolic and hormonal equilibrium during the whole life of women. Although breast cancer is multicausal, with diverse inherited and acquired etiologic factors, the lack of sufficient estrogen surveillance seems to be crucial risk for the development of mammary tumors, particularly for TNBCs in both young and older cases.
Common risk factors for ER+ and ER− breast cancers
The majority of breast cancer risk factors seem to be common for both ER+ and ER− tumors. Moreover, the stronger the risk factors, the higher the danger of TNBC development. The well-known risk factors of breast cancer, such as MS, type 2 diabetes, obesity, African-American race, and BRCA gene mutation all proved to be particularly strong for poorly differentiated, steroid receptor-negative tumors, such as TNBCs.6,18–22,58,59
MS and type 2 diabetes as risk factors for overall breast cancers, and TNBCs in particular
It is widely accepted that the higher the grade of insulin resistance with or without obesity in women, the higher the risk for the development of a more aggressive breast cancer.19,20,58 MS is a phase of insulin-resistant state characterized by a quartet of elevated fasting glucose, dyslipidemia, hypertension, and visceral obesity.60 Each of these symptoms alone is a risk factor for cancer; together, they mean a multiple risk.61,62
MS is associated with increased overall breast cancer risk, higher tumor aggressivity, and poorer prognosis.6,63,64 Positive correlations were found between MS and breast cancer incidence, due primarily to positive associations with serum levels of glucose and triglyceride, as well as diastolic blood pressure.65 Elevated fasting glucose level proved to be a significant risk for breast cancer in both pre- and postmenopausal women.66 Among breast cancer cases, 26% were considered obese, 16% hyperglycemic, 54% hypertensive, and 30% dyslipidemic.
In Ireland, MS was established in 39% of all newly diagnosed breast cancer cases and was associated with more aggressive tumor biology. Patients with advanced pathologic stage (II–IV) at diagnosis had MS in 51% of cases, whereas among early-stage cases, this rate was only 12%.64 A meta-analysis of 20 studies estimated a 20% increased risk of breast cancer for women with type 2 diabetes.67 A review of epidemiologic studies on the association between type 2 diabetes and breast cancer risk revealed moderate association, appearing to be more consistent among postmenopausal than premenopausal women.68
In young premenopausal women, a wide range of insulin-resistant states may occur, from mild hyperinsulinism to diabetes mellitus, which are associated with different stages of androgen excess as well as defective estrogen synthesis.33 PCOS in young cases is associated with anovulatory infertility and insulin resistance,40 and represents common risk for the cancers of highly hormone-dependent breast, endometrium, and ovary.69
In women with PCOS, OC administration improves anovulatory disorders, and has favorable impacts on carbohydrate and lipid metabolism as well.47 OC therapy creates a regular artificial cycle, ameliorates hirsutism and acne, and is protective against the development of endometrial carcinoma.48
In the mildly hyperandrogenic syndromes, only the ovulatory estrogen peak is missing, which results in occult anovulatory infertility and preferential cancer risk for the female organs with high estrogen demand.33,45,53 Nevertheless, the preserved, but slightly defective, estrogen level may be enough for the killing or differentiation of randomly initiated breast cancer cells. Accidental failures of estrogen defense in these cases yield biologically milder ER+ cancer development. Taken together, the lower incidence rates of breast cancers in young cases with low-grade insulin resistance may be attributed to the relatively preserved estrogen surveillance.33
In young women, another extremity of insulin resistance is advanced visceral obesity and/or type 2 diabetes conferring high breast cancer risk attributed to the concomitant hyperinsulinism, hyperandrogenism, defective aromatase activity, and failure of estrogen synthesis.33 Low estrogen supply cannot counteract the severe dysmetabolism and hyperandrogenism in premenopausal women, which explains the relatively increased incidence rate of poorly differentiated breast cancers, including TNBCs.
Postmenopausal aging in women seems to exhibit close correlation with an increased prevalence of MS.70 The risk of postmenopausal breast cancer was found to be significantly increased in cases of women with MS (OR =1.75), with the risk being much higher in those above the age of 70 years (OR =3.04).63 Breast cancer incidence in women diagnosed at or after the age of 65 years was strongly associated with highly elevated fasting blood glucose levels (≥7.0 mmol/L).57
MS and type 2 diabetes seem to be preferential risk factors for TNBC as compared with the ER+ breast cancer risk. In a sample of 176 individuals, 58% of TNBC patients exhibited the comorbid condition of MS as compared with 37% of the non-TNBC cases, using the MS criteria of the National Cholesterol Education Program.20
Insulin-resistant states, such as both MS and type-2 diabetes, are strong risk factors for breast cancer by several pathways, particularly for the poorly differentiated ER− tumors like TNBCs. Severe insulin resistance also disarms the cellular defense mechanisms by estrogen deprivation, and the defective estrogen surveillance allows easier escape for ER− tumors. The more advanced the insulin resistance, the higher the risk for TNBC development.
Obesity-mediated risk for overall breast cancer and TNBC
Both clinical and experimental evidence prove that obesity, particularly visceral fatty tissue deposition, leads to insulin resistance associated with diverse immunologic, metabolic, and hormonal alterations that mediate breast cancer risk.71,72
Distribution of fat deposition is thoroughly affected by male-to-female sexual steroid equilibrium.73 In young premenopausal obese women, the overall breast cancer risk is deceivingly moderate, as in the majority of these females, adipose tissue deposition is predominantly gluteofemoral, resulting in mild insulin resistance counteracted by the preserved hormonal cycle.33 By contrast, male-like central obesity and severe dysmetabolism in young obese women are associated with hyperandrogenism and a decrease in serum estradiol levels, particularly in the follicular phase of the cycle.74 Increased obesity-related and PCOS-associated breast cancer risk may be attributed particularly to this hormonal imbalance.33,45,75
In postmenopausal, estrogen-deficient obese women, the regional distribution of fat deposition near uniformly affects the visceral region in close correlation with severe dysmetabolism and high breast cancer risk.56 By contrast, HRT use in obese postmenopausal women equivocally reduces the incidence of breast cancer by means of the improvement of hormonal and metabolic balance.76,77
Body mass index (BMI) or body weight in kilograms reflect general adiposity and may not correctly refer to correlations among fat distribution, hormonal disorders, and overall breast cancer risk in young cases.78 In certain studies, BMI-defined obesity was erroneously reported as being inversely correlated with breast cancer risk in premenopausal women,56,79,80 which is indicative of the fact that general obesity is not always reliable in the estimation of tumor risk. By contrast, fat distribution measurements, such as hip (HC) and waist (WC) circumference and waist-to-hip ratio (WHR), give better information on abdominal fat accumulation and dysmetabolism-related cancer risk.81 Among obese young women, visceral obesity-related high WC and WHR values exhibited direct correlations with increased risk for premenopausal breast cancer.81–84
In young women with central obesity, the defects of estrogen synthesis and anovulatory infertility are strong risk factors for breast cancer; however, similar hormonal disorders are not rare, even among control cases with normal body weight.33 Strict selection of lean control women with healthy sex hormone equilibrium may equivocally justify the health advantage of normal body weight over obesity.
Slight or moderate insulin resistance in obese young women may be associated with still-sufficient estrogen signaling capable of killing the majority of developing ER+ tumors, but the more resistant, aggressive ER− cancers may survive, leading to a relative increase in their incidence rate. By contrast, strong insulin resistance in young obese women may counteract the partially preserved estrogen surveillance, which is advantageous for the increased prevalence of ER−/PR− tumors and TNBCs. In the meantime, the number of ER+ tumors may increase as well, attributed to the poor suppressive capacity of hormonal forces.
Considering these difficult correlations among obesity type, grade of insulin resistance, and level of estrogen surveillance with its different killing capacities in relation to ER+ and ER− cancers, one can understand the deceiving, apparently controversial clinical and epidemiologic findings.
BMI-defined general obesity is typically associated with increased TNBC risk, particularly in premenopausal women. In young cases, overweight and obesity seem to be in consistent direct correlation with the development of TNBC and other ER− types of breast tumors.9,15,17,84 Similarly, obesity and overweight are much more likely among young cases with TNBC as compared with cases with ER+/PR+ tumors.17,85
The impact of high BMI on ER+, as well as on non-TNBC tumors, is not uniform, depending on the hormonal status of obese women. In cases of a severe defect of hormonal surveillance, the ER+ cancer risk may show a somewhat milder increase as compared with ER− cancers.11,17 By contrast, when the estrogen defense is relatively preserved in obese young cases, ER+ tumor incidence rate may be largely suppressed.17,86,87
Each of the three circumference measurements for abdominal fat (WC, HC, and WHR) has been found to be statistically significantly associated with increased incidence of ER− breast cancer in premenopausal cases.22 HRs of ER− breast cancer for the highest versus lowest quintile of body fat distribution measures were 2.75 for WC, 2.40 for HC, and 1.95 for WHR. These correlations demonstrate that central obesity is strongly associated with increased risk for ER− breast cancers. In a further study, only HC was directly associated with increased breast cancer risk.16 After adjustment for BMI, both ER+/PR+ and ER−/PR− breast cancers showed directly but differently increased risk with central obesity (HR =1.65 and 2.65, respectively). These remarkable results show that, in premenopausal women, even visceral obesity-associated defective estrogen synthesis may be more suppressive for ER+/PR+ tumors than ER−/PR− ones.
In conclusion, obesity is a multifaceted disease associated with different grades of dangerous dysmetabolism and sexual hormone imbalance that promote breast cancer development. Obesity-associated defective estrogen surveillance allows easier escape for steroid receptor-negative tumors than for ER+ ones, resulting in conspicuous accumulation of ER− cancers and TNBCs in obese patients.
Role of light deficiency in disparities between African-American and white American women in TNBC incidence rate
Population-based data have demonstrated that African-American women develop breast cancer at an earlier age, which is diagnosed in a more advanced stage, and exhibits higher incidence rates for poorly differentiated ER− and TNBC types than white American women.9,59,88 TNBC incidence was found to be significantly higher in black women at all ages as compared with white women.89 Black race was strongly associated with ER−/PR− tumors as well, regardless of HER2 status.9 Tumor recurrence rate, metastatic spread, and mortality are all greater among black American women as compared with either Caucasian or Asian groups in North America.88,90–92
Epidemiologic results suggest that poor light exposure in northern regions is a marked cancer risk factor for inhabitants, and for women in particular.93 Among countries leading in the rank of female overall cancer morbidity in Europe, northern regions such as Denmark, Iceland, Norway, and Sweden are conspicuously highly represented.94 The risk of developing breast cancer is significantly high in Northern America and Europe, but fairly low in Asia and Africa. Incidence and mortality rates of breast cancer are five times higher in the United States than in Japan.95
Dark-skinned immigrants were found to have an excessive cancer risk as compared with the natives of northern adoptive countries, and the age at breast cancer diagnosis was found to be earlier.96,97 Excess cancer risk, rapid progression of poorly differentiated cancers, and worse prognosis in black-skinned American women may be conferred by poor natural light exposure and increased melatonin synthesis mediated by their pigmentation.93
Nowadays, melatonin is regarded as an anticancer agent, presumably being protective against hormone-dependent tumors due to its antiestrogenic impacts.98,99 Results from the Nurses’ Health Study cohort added substantial support for an inverse association between melatonin levels and breast cancer risk.100 Melatonin does indeed suppress the estrogen signaling pathways, as it interferes with the activation of nuclear ERs.101 Melatonin also inhibits the expression and activity of aromatase enzymes, which are responsible for estrogen synthesis and presumably, for the progression of ER+ breast cancers.102,103 Nevertheless, the well-documented antiestrogenic effects of melatonin administration do not justify its protective effect against cancers of the highly hormone-dependent female organs. By contrast, excessive melatonin exposure seems to be carcinogenic, as a result of the associated defective estrogen signaling, insulin resistance, hypothyroidism, and vitamin D deficiency.93,104
The increased prevalence of anovulatory disorders and early natural menopause before the age of 40 years in African-American women are manifest due to ovarian failure and are associated with a higher rate of all-cause and cancer-specific mortality.105 Both infertility and ovarian failure are high risk factors for breast cancer in young women, being congruent with the health disadvantages of African-American women.93
Melatonin excess in young, tumor-free African-American women is associated with obesity, hyperinsulinism, and high free testosterone level, resulting also in an increased breast cancer risk.106,107 African-American women with breast cancer exhibit MS, type 2 diabetes, and central obesity more frequently than white cases with similar tumors.108,109
A population-based study revealed wide-spread hypothyroidism among African-American women as compared with white women.110 As melatonin administration has been shown to suppress the thyroid function in animal experiments and clinical examinations,111,112 disproportional hypothyroidism in black-skinned American women may also be attributed to low light exposure. In a prospective study, hypothyroidism and low FT4 values exhibited a direct correlation with increased breast cancer risk.113
Correlations between the epidemiology of vitamin D deficiency, cancer incidence, and mortality were studied in the United States.114 The African-American population showed evidence of particularly widespread vitamin D deficiency. This observation suggests that adequate vitamin D replacement may be an important measure for reduction of race-related health disparities, including breast cancer incidence.115,116
In conclusion, the racial disadvantage of black-skinned American women in the incidence, progression, and outcome of TNBCs may largely be attributed to their defective estrogen signaling and further hormonal disturbances associated with insufficient light exposure.
BRCA gene mutations induce particularly high TNBC risk by defective estrogen signaling
Inherited mutations in BRCA1 or BRCA2 genes predispose to breast, ovarian, and other cancers. Their ubiquitously expressed protein products are implicated in processes fundamental to all cells, including DNA repair and recombination, checkpoint control of cell cycle, and transcription.117 BRCA gene mutations lead to a defect of DNA double-strand break repair through homologous recombination. Disruption of BRCA proteins in mutation carriers can induce susceptibility to specific types of cancer.118
Incidence of hereditary breast and ovarian cancers reveals close correlation with BRCA mutations.119 BRCA1/BRCA2 mutations are responsible for 3%–8% of all breast cancer cases, but 30%–40% of familial cases. Ten percent of patients with ovarian cancer have a genetic predisposition. About 80% of families with a history of ovarian cancer have mutations in the BRCA1 and 15% in the BRCA2 genes. These correlations suggest strong parallelism between cancer initiations of the breast and ovaries, as these organs have similarly high estrogen demand and are fairly vulnerable in cases of defective estrogen signaling.46
Among women with germline BRCA1 mutation, almost 50% of breast cancers are triple negative, presenting with a high grade histologically.120,121 Among women with breast cancer, TNBC was established in 57.1% of BRCA1 mutation-positive and in 23.3% of BRCA2 mutation-positive cases, but in only 13.8% of BRCA– women.18
Strong correlation between BRCA mutations and high TNBC risk proposes certain mediators between germline mutations and the risk for poorly differentiated breast cancers. All justified risk factors for TNBC development seem to be in close correlation with estrogen loss or defective estrogen signaling, as well as further associated hormonal disorders. The question arises whether BRCA mutations may lead to breast cancer and particularly, TNBC development by the defect of estrogen signaling or by quite different pathways.
In BRCA1 gene-deficient human ovarian cancer cells, estradiol-mediated transcriptional activity of ERs has exhibited a relative decrease,122 while ERα showed an unexpected ligand-independent transcriptional activity that was not observed in BRCA1-proficient cells.123 Increased estrogen-independent and defective estrogen-dependent stimulations of ERs in BRCA1-deficient tumor cells suggest that high cancer risk may be attributed to the defect of ligand-activated ER signal. Ligand-independent activation of ERα in tumor cells seems to be a compensatory mechanism of omnipotent estrogen signaling in emergency situations. Excessive estrogen administration emerges as a breakthrough of the blockage of ligand-activated ER signaling in BRCA mutation carriers.
Correlations between BRCA1 mutation and low response to fertility treatments have been examined, as both germline mutations in BRCA genes and anovulatory infertility are associated with high susceptibility for breast and ovarian cancers.124 In BRCA1 mutation-positive women, the low response rate of ovaries to fertility treatment was significantly increased (33.3%) as compared with BRCA1 mutation-negative patients (3.3%). These results support the assumption that BRCA1 mutations are associated with defective estrogen signaling reflected by increased rate of ovulatory failure.
In women with BRCA1/2 mutation, earlier age at natural menopause (under the age of 40 years) was observed significantly more frequently than among unaffected cases (P<0.001).125,126 The high risk of premature ovarian failure among BRCA1/2 carriers reflects the disturbances in estrogen synthesis or ER signaling pathways. Disorders of estrogen signaling may at least partially confer the risk of tumors associated with BRCA1/2 mutation.33,46
Hyperestrogenism (71.7 pg/mL) was observed in BRCA2 mutation carrier patients as compared with the estrogen levels of women with BRCA1 mutations (45.5 pg/mL) and cases without BRCA mutation (38.5 pg/mL).127 Estrogen overproduction may be a contraregulatory effect against defective estrogen signaling in BRCA2 mutation carriers, mediating their markedly lower cancer risk compared with the high risk of BRCA1 mutation carriers. These observations justify the possibility of breast cancer prevention by high-dose estrogen administration in BRCA1/2 mutation cases.
Compensatory, excessive estrogen synthesis was also described in a single publication on a male patient presenting total lack of ERα function.128 Despite his elevated estrogen level, the patient exhibited glucose intolerance, hyperinsulinemia, obesity, and premature coronary artery disease attributed to the completely missing ERα signaling. By contrast, in BRCA gene mutation carriers, the elevated estrogen levels associated with high parity, the artificial hormonal cycle created by OCs, and estrogen administration may decrease the high cancer risk. Parity in BRCA1 mutation carriers has been shown to significantly reduce the risk for ovarian cancer;129 moreover, the risk was reduced with each additional full-term pregnancy in women with germline mutation.130 Furthermore, parity with highly elevated estrogen level also seemed to be protective against TNBC, similarly against the predominant ER+ tumors.23
Use of OCs was found to highly reduce the risk of ovarian cancers in women with both BRCA1 (OR =0.56) and BRCA2 mutations (OR =0.39).129 Ovarian cancer risk decreased with each year of long-term contraceptive use in women carrying BRCA1 or BRCA2 mutations.131 A protective effect of OC use was established as chemoprevention against ovarian cancers in young women with BRCA mutations, whereas the OC-associated risk of breast cancer in BRCA mutation carriers seemed to be heterogeneous, with inconsistent results.132 Nevertheless, the use of OCs for at least 12 months has been associated with strongly decreased breast cancer risk for BRCA1 mutation carriers (OR =0.22), but not for cases with BRCA2 mutation.133
Consumption of phytoestrogen-rich foods, such as soy, has been demonstrated as a preventive measure against breast cancer. Soy consumption may be beneficial in early life, before puberty or during adolescence.134 In animal experiments, prepubertal administration of 17β-estradiol reduced the later risk of breast cancer by inducing a persistent upregulation of the BRCA1 gene.135
Defective estrogen synthesis and/or disorders in ER signaling play significant roles in the development of diverse insulin-resistant states.46,136 In BRCA mutation carrier women, breast cancer development is frequently associated with high BMI and type 2 diabetes.137,138 These observations support the close correlation between defective estrogen signaling and insulin resistance in the development of breast cancer.45
In conclusion, BRCA1 and BRCA2 mutations seem to increase the breast cancer risk, particularly that of TNBC, by defective estrogen signaling. Upregulation of these genes by means of increased estradiol or phytoestrogen exposure may be a promising measure against mammary carcinogenesis.
Correlation between reproductive history and the risk of different breast cancer subtypes
Correlations between reproductive capacity and breast cancer risk have represented the greatest challenge for epidemiologists for a long time. Revolutionary molecular characterization of breast cancer subtypes has yielded further paradigms and contradictions.
The apparently controversial correlations between parity and the development of breast cancer subtypes suggest that hormonally mediated factors might be differently or quite inversely related to the development of ER+ and ER− breast cancers.13,24,25
It has been hypothesized that risk for ER+ breast cancer is positively associated with a woman’s lifetime exposure to endogenous ovarian hormones; thus, parity may strongly reduce the risk by decreasing the number of ovulatory cycles over a lifetime.139,140 By contrast, as TNBCs are ER−, risk factors operating through hormonal mechanisms are presumed to be less important in the etiology of these tumors as compared with ER+ cancers.13
Multiparity in women and risk for malignancies at several sites exhibit a strong inverse correlation.141–143 High parity shows a tumor protective effect, even against the cancers of highly hormone-dependent female organs, including overall breast cancer and endometrial and ovarian tumors.139,144 In anovulatory patients, a significantly decreased overall cancer risk was reported after ovulation induction and in vitro fertilization-assisted childbirth, mainly due to a lower-than-expected incidence of breast cancer.145
In experimental animals, pregnancy-equivalent high estrogen administration could consequently prevent the development of transplanted or chemically induced mammary tumors.146–149 In ovariectomized female mice, alcohol consumption and obesity enhanced the growth of experimental mammary tumors, while estrogen supplementation triggered the loss of body fat, improved insulin sensitivity, and suppressed tumor growth.149,150
Parity, and particularly multiparity are associated with a strongly decreased risk of the predominant ER+ breast cancer type.9,13,24,25,85 Among parous women, even the number of births was inversely associated with the risk of ER+ breast cancer (HR =0.88);151 among women who had had at least four pregnancies, a fairly decreased risk of ER+ breast cancer (OR =0.55) was observed.23 Conversely, TNBC incidence exhibited an apparently unchanged ratio in parous women,9,12,23 whereas in certain studies, an increased risk of TNBC was reported in multiparous cases.14,152,153 In a recent study, the number of births was found to be directly associated with the risk of TNBC.13
Nulliparity is generally in correlation with anovulatory disorders, thus these hormone-deficient cases may be regarded as opposite extremes as compared with multiparous women.33 Delayed first childbirth may also be associated with prolonged defective estrogen synthesis and ovulatory failures. Postpubertal sexual hormone imbalances are frequently associated with definite or prolonged fertility disorders, resulting in nulliparity and delayed first childbearing154 and inducing increased overall breast cancer risk among premenopausal women.155,156 High overall breast cancer risk in correlation with defective estrogen synthesis and anovulatory disorders demonstrates the role of physiologic estrogen level in preservation of mammary health.33 Administration of pregnancy-mimicking estrogen and progesterone doses to nulliparous women seems to be a useful strategy for protection against breast cancer.157 Nevertheless, women may remain nulliparous or may delay childbirth for social or professional reasons as well; there are, however, no reports on the alterations of breast cancer risk in these special groups.
It was suggested that nulliparity plays an inverse role in the risks of ER+ breast cancer and TNBC as compared with multiparity.24 Nulliparous status of women was associated with a 35% higher risk of ER+ breast cancer (HR =1.35), but with a 39% lower risk for TNBC (HR =0.61).13 Delayed first childbirth was also directly associated with risk for ER+ cancers, but showed no remarkable effect on the risk of TNBCs.15
Considering the apparently contradictory results, if women undertake more childbirths, they may be exposed to a stronger risk of developing TNBC; conversely, if they remain nulliparous, they are exposed to higher risk for ER+ cancers. So what should they do?
In multiparous women, good fertility-associated estrogen supply and excessive estrogen levels during pregnancies strongly and equivocally reduce the development of overall breast cancers and the predominant ER+ tumors in particular. A plausible explanation is that estrogen, being the specific ligand for ERs, may preferentially block the development and progression of ER+ cancers; however, its killing capacity against ER− cancers is slower and weaker, as the specific tumor receptors are missing. In estrogen-rich milieu, a fairly decreased number and percentage of ER+ tumors is associated with a moderately decreased number and an unchanged or deceivingly increased percentage of ER− breast cancers.
By contrast, in nulliparous, hormonally challenged women, the weakness of estrogen surveillance results in enhanced overall breast cancer risk. The insufficient estrogen supply has a defective killing capacity, even against the predominant, hormone-sensitive ER+ breast cancer cells, resulting in an increased number and percentage of surviving ER+ tumors. The survival possibility for hormonally weakly controlled ER− cancers, such as TNBCs, may disproportionately improve and their raw number may be somewhat increased, while the relative number (percentage) decreases.
For women who are afraid of developing breast cancer, it is plausible to choose parity either by natural means or by in vitro fertilization to prevent the development of both TNBC- and non-TNBC-type tumors.
Some aspects of the molecular mechanism of estrogen surveillance on cell proliferation
The classic genomic mechanism of estrogen binding activates ERs in the nucleus, and they act as transcriptional modulators in the promoter region of target genes. ERs can also regulate gene expression without direct binding to DNA through interaction with transcription factor proteins in the nucleus.158 Estrogen action also has nongenomic signaling cascades through cell membrane-associated ERs.159 Finally, genomic and nongenomic pathways of ER signaling converge on the target genes.
Two receptor isoforms, ERα and ERβ, which belong to the steroid-thyroid hormone nuclear receptor supergene family, have been identified.160,161 By means of a thorough interplay, ERα and ERβ regulate the metabolic processes, growth, and proliferation of mammalian cells. They may oppose each other’s activities, eliciting sometimes opposite reactions in the presence of estradiol.162 Agonistic and antagonistic crosstalk of ERs and their thorough interaction with other hormonal and growth factor signals ensure that estrogen orchestrates the gene regulation of cell proliferation with high safety.158,159
Physiologic equilibrium of estradiol-induced mitotic activity and apoptotic cell death was observed in mouse mammary cell line, HC11, the cells of which expressed both ERα and ERβ, and showed no proliferative activity.163 Embryonic development demonstrates the importance of the omnipotent actions of estrogen. High estrogen level in the fetoplacental unit may ensure the safety of explosion-like cell proliferation, the silencing of mitotic activity, or even apoptotic cell death, if it is necessary.34
As estrogen and ER signals are essential in harmonizing the regulation of all cellular functions, sufficient hormone exposure and available intact receptors are indispensable for the health of mammalian.46 Separated activation or blocking of each ER isoform may produce thorough alterations and disturbances. It is quite impossible to interfere with or even improve this highly controlled safeguard of estrogen on cellular mechanisms.
It is a well-known fact that the higher the proliferative activity of a cell population, the stronger the danger of mutagenic failures and tumor initiation. Following conception, 17β-estradiol level increases exponentially from a 0.1 ng/mL level in the follicular phase of cycle up to a 30 ng/mL level at term, which means a 300-fold elevation.164 During pregnancy, an extreme increase in the estrogen supply of the fetoplacental unit serves as an exquisite safeguard for the abundant, explosion-like cell proliferation of embryonic structures. If estrogens would have even the slightest carcinogenic capacity, tumor birth would be a typical event instead of childbirth, due to the overwhelming estrogen supply in pregnancy.
In animal experiments, the administration of high doses of estradiol before or after carcinogen treatment was equally protective against mammary carcinogenesis.165 Different mechanisms have emerged as explanations for hormone-induced refractoriness to carcinogenesis. Target cells in the mammary gland may become non-susceptible to carcinogens by excessive hormone treatment through DNA protection,165 or chemically initiated tumor cells may undergo differentiation.166 As a further possibility, the apoptotic tumor cell-killing capacity of excessive estradiol administration may also reduce mammary cancer incidence.167
There are literary data on the supposed roles of membrane-associated ER signaling pathways in human cancer induction,168 particularly in breast cancer cases.169,170 Interactions of estrogen and growth factor receptors (GFRs) in various cancers have been regarded as a revelation of the cancer-provoking effect of estrogen. Nevertheless, the presumed synergistic carcinogenic capacity of ERs and GFRs would mean a constant danger for cancer initiation without contraregulatory impact. In the epithelial cells of the human breast, both growth inhibition and growth stimulation by estradiol were observed depending on the rate and activity of ERs and GFRs.171
In malignancies, a dynamic inverse relationship was revealed between the expressions of GFRs and membrane-associated ERs. Excessive epithelial growth factor administration on the human breast cancer cell line MCF-7 resulted in persistent decreases in ERα protein concentration in estradiol binding sites and in ERα gene transcription.172 Alternatively, high estrogen dose could inhibit lung carcinogenesis by reducing the level of insulin-like growth factor-I, which is a potent mitogenic agent for several malignant tumors, including lung and breast cancers.19,173
The recognition of counteractions between GFRs and ERs may explain how a successful ER blockage by tamoxifen treatment may induce overwhelming GFR predominance, resulting in fatal outcome of the disease. In patients with ER+/HER2+ tumors, higher recurrence and mortality rates were observed after tamoxifen therapy as compared with those who did not receive the agent.29
There are plausible possibilities for the anticancer effect of estrogen, even on ER− cancers. In apparently ER− breast tumor cells, inhibition of growth factor signaling yielded a potential restoration of ER expression.174 Abundant estradiol administration may also counteract the growth factor signaling, and the concomitant restoration of ER expression yields the possibility for the apoptotic killing of tumor cells. This may be one of the possible secrets of the antitumor capacity of pregnancy-equivalent estrogen levels, even in cases of ER− breast cancer cells.
In summary, pregnancy-associated high estrogen level is protective against the initiation and progression of all breast cancer subtypes; however, the intensity of this defense strongly depends on the molecular subtype of the tumors: the higher the ER expression of the cancer, the stronger the tumor-suppressive effect of estrogen.
Exogenous estrogen and antiestrogen treatment of breast cancer
High-dose estrogen was administered as endocrine therapy for postmenopausal women with breast cancer prior to the introduction of tamoxifen, the first antiestrogenic compound, in the 1970s.175 Clinical trials were conducted for the comparison of the results of synthetic estrogen, diethylstilbestrol, and tamoxifen treatment in women with advanced or recurrent breast cancer.176,177 It was established that tumor responses were similar, but the advantage of tamoxifen over synthetic estrogen was equivocal, because of the low incidence rate of toxic side effects. Following these observations, the use of estrogen therapy was almost completely abandoned.
Tamoxifen was the first compound designated as an antiestrogen, or, more exactly, a selective ER modulator that binds to ERs so as to inhibit their signaling pathways. It is widely used as adjuvant therapy for ER+ breast cancers; however, the benefits of antiestrogen are not equivocal in cancer therapy.178 Even among women with ER+ breast cancers, only 40%–50% of patients exhibit primarily successful tumor regression;179 later, a large proportion of earlier responsive breast cancers may acquire secondary resistance during tamoxifen therapy.180 Moreover, tamoxifen may elicit common side effects that are occasionally life threatening, such as stroke, pulmonary emboli, and malignancies at certain sites, attributed to the anomalous estrogen agonist activities of this compound.181
Another group of antiestrogens is known as aromatase inhibitors. These compounds prevent the activity of P450 aromatase, which converts steroid precursors and androgens to estrogen, causing estrogen deprivation in both healthy tissues and tumors.178 These also have severe side effects, however, related to estrogen depletion. Introduction of aromatase inhibitors successfully avoided the presumed estrogen agonist activities of selective ER modulators; however, toxicity aside, their anticancer effects were not reliable.178
Failures and controversies of antiestrogen therapy lead “back to the future”, and the old-fashioned high-dose estrogen treatment in patients with advanced breast cancer was demonstrated again to be effective.182 Moreover, breast cancer cases with acquired resistance to antiestrogen therapy were successfully treated, even by lower doses of estradiol.167,183,184 The anticancer mechanism of estrogen therapy remains unknown. A number of authors consider that estrogen may become an apoptotic trigger instead of a survival signal for tumor cells after many years of estrogen deprivation.184–186
Conclusion
The questions posed in this paper can be answered as follows:
- The grade of defect in metabolic and hormonal equilibrium is directly associated with TNBC risk for women during the different periods of their lives.
- TNBC is not a distinct entity with unique etiology, but rather its development is mediated by usual cancer risk factors.
- Common but strong and/or multiple breast cancer risk factors may provoke the development of poorly differentiated tumors, TNBCs in particular.
- Exogenous or parity-associated excessive estrogen supply is defensive against all breast cancer subtypes, but ER− tumors and TNBCs are much more resistant. Inverse impact of parity on ER+ and ER− breast cancers is not possible; erroneous results derive from the misinterpretation of statistical evaluations based exclusively on the percentage of cancer subtypes.
- The most important preventive strategy against breast cancers in women – including TNBCs – is the strict control of hormonal equilibrium from early adolescence throughout the whole life, particularly during the periods of great hormonal changes. Screening of symptom-free anovulatory disorders, cycle irregularities, and infertility, as well as estrogen substitution of these hormonal defects, may all be important methods in breast cancer prevention. In regard to inherited inclinations for breast cancer, exogenous hormone treatment and parity are indicated. In the peri- and postmenopausal periods, menopausal complaints and tumor risk factors such as hysterectomy, obesity, MS, and type 2 diabetes, are absolute indications for estrogen substitution therapy.
Effective breast cancer therapy requires a complete conversion, as worldwide, administration of antiestrogen compounds for breast cancer treatment has yielded thorough disappointment. Nevertheless, publications on successful, high-dose estradiol treatment of advanced breast cancer cases are increasing in number, with encouraging results.
Widespread estrogen use in the primary prevention and therapy of breast cancer may help to eradicate this dreadful, torturing disease.
Disclosure
The author reports no conflicts of interest in this work.
References
Brenton JD, Carey LA, Ahmed AA, Caldas C. Molecular classification and molecular forecasting of breast cancer: ready for clinical application? J Clin Oncol. 2005;23(29):7350–7360. | |
Bauer KR, Brown M, Cress RD, Parise CA, Caggiano V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: a population-based study from the California cancer Registry. Cancer. 2007;109(9):1721–1728. | |
Morris GJ, Naidu S, Topham AK, et al. Differences in breast carcinoma characteristics in newly diagnosed African-American and Caucasian patients: a single-institution compilation compared with the National Cancer Institute’s Surveillance, Epidemiology, and End Results database. Cancer. 2007;110(4):876–884. | |
Lund MJ, Trivers KF, Porter PL, et al. Race and triple negative threats to breast cancer survival: a population-based study in Atlanta, GA. Breast Cancer Res Treat. 2009;113(2):357–370. | |
Boyle P. Triple-negative breast cancer: epidemiological considerations and recommendations. Ann Oncol. 2012;23 Suppl 6:vi7–vi12. | |
Davis AA, Kaklamani VG. Metabolic syndrome and triple-negative breast cancer: a new paradigm. Int J Breast Cancer. 2012; 2012:809291. | |
Hartley MC, McKinley BP, Rogers EA, et al. Differential expression of prognostic factors and effect on survival in young (< or =40) breast cancer patients: a case-control study. Am Surg. 2006;72(12):1189–1194. | |
Carey L, Winer E, Viale G, Cameron D, Gianni L. Triple-negative breast cancer: disease entity or title of convenience? Nat Rev Clin Oncol. 2010;7(12):683–692. | |
Trivers KF, Lund MJ, Porter PL, et al. The epidemiology of triple-negative breast cancer, including race. Cancer Causes Control. 2009;20(7):1071–1082. | |
Talley LI, Grizzle WE, Waterbor JW, Brown D, Weiss H, Frost AR. Hormone receptors and proliferation in breast carcinomas of equivalent histologic grades in pre- and postmenopausal women. Int J Cancer. 2002;98(1):118–127. | |
Gaudet MM, Press MF, Haile RW, et al. Risk factors by molecular subtypes of breast cancer across a population-based study of women 56 years or younger. Breast Cancer Res Treat. 2011;130(2):587–597. | |
Dolle JM, Daling JR, White E, et al. Risk factors for triple-negative breast cancer in women under the age of 45 years. Cancer Epidemiol Biomarkers Prev. 2009;18(4):1157–1166. | |
Phipps AI, Chlebowski RT, Prentice R, et al. Reproductive history and oral contraceptive use in relation to risk of triple-negative breast cancer. J Natl Cancer Inst. 2011;103(6):470–477. | |
Yang XR, Sherman ME, Rimm DL, et al. Differences in risk factors for breast cancer molecular subtypes in a population-based study. Cancer Epidemiol Biomarkers Prev. 2007;16(3):439–443. | |
Phipps AI, Chlebowski RT, Prentice R, et al. Body size, physical activity, and risk of triple-negative and estrogen receptor-positive breast cancer. Cancer Epidemiol Biomarkers Prev. 2011;20(3):454–463. | |
Fagherazzi G, Chabbert-Buffet N, Fabre A, et al. Hip circumference is associated with the risk of premenopausal ER−/PR− breast cancer. Int J Obes (Lond). 2012;36(3):431–439. | |
Vona-Davis L, Rose DP, Hazard H, et al. Triple-negative breast cancer and obesity in a rural Appalachian population. Cancer Epidemiol Biomarkers Prev. 2008;17(12):3319–3324. | |
Atchley DP, Albarracin CT, Lopez A, et al. Clinical and pathologic characteristics of patients with BRCA-positive and BRCA-negative breast cancer. J Clin Oncol. 2008;26(26):4282–4288. | |
Davison Z, de Blacquière GE, Westley BR, May FE. Insulin-like growth factor-dependent proliferation and survival of triple-negative breast cancer cells: implications for therapy. Neoplasia. 2011;13(6):504–515. | |
Maiti B, Kundranda MN, Spiro TP, Daw HA. The association of metabolic syndrome with triple-negative breast cancer. Breast Cancer Res Treat. 2010;121(2):479–483. | |
Amadou A, Hainaut P, Romieu I. Role of obesity in the risk of breast cancer: lessons from anthropometry. J Oncol. 2013;2013:906495. | |
Harris HR, Willett WC, Terry KL, Michels KB. Body fat distribution and risk of premenopausal breast cancer in the Nurses’ Health Study II. J Natl Cancer Inst. 2011;103(3):273–278. | |
Ma H, Wang Y, Sullivan-Halley J, et al. Use of four biomarkers to evaluate the risk of breast cancer subtypes in the women’s contraceptive and reproductive experiences study. Cancer Res. 2010;70(2):575–587. | |
Yang XR, Chang-Claude J, Goode EL, et al. Associations of breast cancer risk factors with tumor subtypes: a pooled analysis from the Breast Cancer Association Consortium studies. J Natl Cancer Inst. 2011;103(3):250–263. | |
Palmer JR, Boggs DA, Wise LA, Ambrosone CB, Adams-Campbell LL, Rosenberg L. Parity and lactation in relation to estrogen receptor negative breast cancer in African American women. Cancer Epidemiol Biomarkers Prev. 2011;20(9):1883–1891. | |
Hammond ME, Hayes DF, Dowsett M, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol. 2010;28:2784–2795. | |
Hefti MM, Hu R, Knoblauch NW, et al. Estrogen receptor negative/progesterone receptor positive breast cancer is not a reproducible subtype. Breast Cancer Res. 2013;15:R68. | |
Yamauchi H, Stearns V, Hayes DF. When is a tumor marker ready for prime time? A case study of c-erbB-2 as a predictive factor in breast cancer. J Clin Oncol. 2001;19(8):2334–2356. | |
De Placido S, De Laurentiis M, Carlomagno C, et al. Twenty-year results of the Naples GUN randomized trial: predictive factors of adjuvant tamoxifen efficacy in early breast cancer. Clin Cancer Res. 2003;9(3):1039–1046. | |
Liedtke C, Hess KR, Karn T, et al. The prognostic impact of age in patients with triple-negative breast cancer. Breast Cancer Res Treat. 2013;138(2):591–599. | |
Zakhartseva LM, Gorovenko NG, Podolskaya SV, et al. Breast cancer immunohistochemical features in young women with BRCA 1/2 mutations. Exp Oncol. 2009;31(3):174–178. | |
Rudat V, El-Sweilmeen H, Fadel E, et al. Age of 40 years or younger is an independent risk factor for locoregional failure in early breast cancer: a single-institutional analysis in Saudi Arabia. J Oncol. 2012; 2012:370385. | |
Suba Z. Circulatory estrogen level protects against breast cancer in obese women. Recent Pat Anticancer Drug Discov. 2013;8(2):154–167. | |
Suba Z, editor. Failures and controversies of the antiestrogen treatment of breast cancer. Estrogen Prevention for Breast Cancer. Hauppauge, New York: Nova Science Publishers Inc; 2013:105–125. | |
Bollet MA, Savignoni A, De Koning L, et al. Tumor aromatase expression as a prognostic factor for local control in young breast cancer patients after breast-conserving treatment. Breast Cancer Res. 2009;11(4):R54. | |
Spratt JS, Meyer JS, Spratt JA. Rates of growth of human solid neoplasms: part I. J Surg Oncol. 1995;60:137–146. | |
Baer HJ, Colditz GA, Willett WC, Dorgan JF. Adiposity and sex hormones in girls. Cancer Epidemiol Biomarkers Prev. 2007;16(9):1880–1888. | |
Stoll BA. Teenage obesity in relation to breast cancer risk. Int J Obes Relat Metab Disord. 1998;22(11):1035–1040. | |
Polson DW, Wadsworth J, Adams J et al. Polycystic ovaries: a common finding in normal women. Lancet. 1988;1:870–872. | |
Bloomgarden ZT. Second World Congress on the Insulin Resistance Syndrome: mediators, pediatric insulin resistance, the polycystic ovary syndrome, and malignancy. Diabetes Care. 2005;28(8):1821–1830. | |
Apter D, Vihko R. Endocrine determinants of fertility: serum androgen concentrations during follow-up of adolescents into the third decade of life. J Clin Endocrinol Metab. 1990;71(4):970–974. | |
Sturgeon SR, Potischman N, Malone KE, et al. Serum levels of sex hormones and breast cancer risk in premenopausal women: a case-control study (USA). Cancer Causes Control. 2004;15(1):45–53. | |
Suba Z. Common soil of smoking-associated and hormone-related cancers: estrogen deficiency. Oncol Rev. 2010;4:73–87. | |
Stamataki KE, Spina J, Rangou DB, Chlouverakis CS, Piaditis GP. Ovarian function in women with non-insulin dependent diabetes mellitus. Clin Endocrinol (Oxf). 1996;45(5):615–621. | |
Suba Z. Interplay between insulin resistance and estrogen deficiency as co-activators in carcinogenesis. Pathol Oncol Res. 2012;18(2):123–133. | |
Suba Z. Low estrogen exposure and/or defective estrogen signaling induces disturbances in glucose uptake and energy expenditure. J Diabet Metab. 272, doi.4172/2155-6156.1000272. | |
ESHRE Capri Workshop Group. Ovarian and endometrial function during hormonal contraception. Hum Reprod. 2001;16(7):1527–1535. | |
Diamanti-Kandarakis E, Baillargeon J-P, Iuorno MJ, Jakubowicz DJ, Nestler JE. A modern medical quandary: polycystic ovary syndrome, insulin resistance, and oral contraceptive pills. J Clin Endocrinol Metab. 2003;88(5):1927–1932. | |
Deligeoroglou E, Michailidis E, Creatsas G. Oral contraceptives and reproductive system cancer. Ann N Y Acad Sci. 2003;997:199–208. | |
Marchbanks PA, McDonald JA, Wilson HG, et al. Oral contraceptives and the risk of breast cancer. New Engl J Med. 2002;346:2025–2032. | |
Bernstein L, Lacey JV Jr. Receptors, associations and risk factor differences by breast cancer subtypes: positive or negative? J Natl Cancer Inst. 2011;103(6):451–453. | |
Rosenberg L, Boggs DA, Wise LA, Adams-Campbell LL, Palmer JR. Oral contraceptive use and estrogen/progesterone receptor–negative breast cancer among African American women. Cancer Epidemiol Biomarkers Prev. 2010;19(8):2073–2079. | |
Suba Z, editor. Discovery of estrogen deficiency as common cancer risk factor for highly and moderately estrogen dependent organs. Estrogen Prevention for Breast Cancer. Hauppauge, New York: Nova Science Publishers Inc; 2013:1–22. | |
Dietel M, Lewis MA, Shapiro S. Hormone replacement therapy: pathobiological aspects of hormone-sensitive cancers in women relevant to epidemiological studies on HRT: a mini-review. Hum Reprod. 2005;20:2052–2060. | |
LaCroix AZ, Chlebowski RT, Manson JE, et al; WHI Investigators. Health outcomes after stopping conjugated equine estrogens among postmenopausal women with prior hysterectomy: a randomized controlled trial. JAMA. 2011;305:1305–1314. | |
La Vecchia C, Negri E, Franceschi S, et al. Body mass index and postmenopausal breast cancer: an age specific analysis. Br J Cancer. 1997;75(3):441–444. | |
Rapp K, Schroeder J, Klenk J, et al. Fasting blood glucose and cancer risk in a cohort of more than 140,000 adults in Austria. Diabetologia. 2006;49:945–952. | |
Colonna SV, Douglas Case L, Lawrence JA. A retrospective review of the metabolic syndrome in women diagnosed with breast cancer and correlation with estrogen receptor. Breast Cancer Res Treat. 2012;131(1):325–331. | |
Amirikia KC, Mills P, Bush J, Newman LA. Higher population-based incidence rates of triple-negative breast cancer among young African-American women: implications for breast cancer screening recommendations. Cancer. 2011;117(12):2747–2753. | |
Reaven GM. Insulin resistance, cardiovascular disease, and the metabolic syndrome: How well do the emperor’s clothes fit? Diabetes Care. 2004;27(4):1011–1012. | |
Cowey S, Hardy RW. The metabolic syndrome: a high-risk state for cancer? Am J Pathol. 2006;169(5):1505–1522. | |
Doyle SL, Donohoe CL, Lysaght J, Reynolds JV. Visceral obesity, metabolic syndrome, insulin resistance and cancer. Proc Nutr Soc. 2012;71(1):181–189. | |
Rosato V, Bosetti C, Talamini R, Levi F, Negri E, La Vecchia C. [Metabolic syndrome and the risk of breast cancer]. Recenti Prog Med. 2011;102(12):476–478. Italian. | |
Healy LA, Ryan AM, Carroll P, et al. Metabolic syndrome, central obesity and insulin resistance are associated with adverse pathological features in postmenopausal breast cancer. Clin Oncol (R Coll Radiol). 2010;22(4):281–288. | |
Kabat GC, Kim M, Chlebowski RT, et al. A longitudinal study of the metabolic syndrome and risk of postmenopausal breast cancer. Cancer Epidemiol Biomarkers Prev. 2009;18(7):2046–2053. | |
Sieri S, Muti P, Claudia A, et al. Prospective study on the role of glucose metabolism in breast cancer occurrence. Int J Cancer. 2012;130(4):921–929. | |
Larsson SC, Mantzoros CS, Wolk A. Diabetes mellitus and risk of breast cancer: a meta-analysis. Int J Cancer. 2007;121(4):856–862. | |
Xue F, Michels KB. Diabetes, metabolic syndrome, and breast cancer: a review of the current evidence. Am J Clin Nutr. 2007;86(3):s823–s835. | |
Pierpoint T, McKeigue PM, Isaacs AJ, Wild SH, Jacobs HS. Mortality of women with polycystic ovary syndrome at long-term follow-up. J Clin Epidemiol. 1998;51(7):581–586. | |
Schneider JG, Tompkins C, Blumenthal RS, Mora S. The metabolic syndrome in women. Cardiol Rev. 2006;14(6):286–291. | |
Donohoe CL, Doyle SL, Reynolds JV. Visceral adiposity, insulin resistance and cancer risk. Diabetol Metab Syndr. 2011;3:12. | |
Rose DP, Vona-Davis L. Interaction between menopausal status and obesity in affecting breast cancer risk. Maturitas. 2010;66(1):33–38. | |
Spangenburg EE, Wohlers LM, Valencia AP. Metabolic dysfunction under reduced estrogen levels: looking to exercise for prevention. Exerc Sport Sci Rev. 2012;40(4):195–203. | |
Potischman N, Swanson CA, Siiteri P, Hoover RN. Reversal of relation between body mass and endogenous estrogen concentrations with menopausal status. J Natl Cancer Inst. 1996;88(11):756–758. | |
Suba Z, Kásler M. Interactions of insulin and estrogen in the regulation of cell proliferation and carcinogenesis. Orv Hetil. 2012;153(4):125–136. Hungarian. | |
Morimoto LM, White E, Chen Z, et al. Obesity, body size and risk of postmenopausal breast cancer: the Women’s Health Initiative (United States). Cancer Causes Control. 2002;13(8):41–51. | |
Ritte R, Lukanova A, Berrino F, et al. Adiposity, hormone replacement therapy use and breast cancer risk by age and hormone receptor status: a large prospective cohort study. Breast Cancer Res. 2012;14:R76. | |
Huang Z, Willett WC, Colditz GA, et al. Waist circumference, waist:hip ratio, and risk of breast cancer in the Nurses’ Health Study. Am J Epidemiol. 1999;150(12):1316–1324. | |
Franceschi S, Favero A, La Vecchia C, et al. Body size indices and breast cancer risk before and after menopause. Int J Cancer. 1996;67(2):181–186. | |
Kotlyarevska K, Wolfgram P, Lee JM. Is waist circumference a better predictor of insulin resistance than body mass index in US adolescents? J Adolesc Health. 2011;49(3):330–333. | |
Sonnenschein E, Toniolo P, Terry MB, et al. Body fat distribution and obesity in pre- and postmenopausal breast cancer. Int J Epidemiol. 1999;28(6):1026–1031. | |
Connolly BS, Barnett C, Vogt KN, Li T, Stone J, Boyd NF. A meta-analysis of published literature on waist-to-hip ratio and risk of breast cancer. Nutr Cancer. 2002;44(2):127–138. | |
Harvie M, Hooper L, Howell AH. Central obesity and breast cancer risk: a systematic review. Obes Rev. 2003;4(3):157–173. | |
Rose DP, Vona-Davis L. Influence of obesity on breast cancer receptor status and prognosis. Expert Rev Anticancer Ther. 2009;9(8):1091–1101. | |
Kwan ML, Kushi LH, Weltzien E, et al. Epidemiology of breast cancer subtypes in two prospective cohort studies of breast cancer survivors. Breast Cancer Res. 2009;11(3):R31. | |
Suzuki R, Orsini N, Saji S, Key TJ, Wolk A. Body weight and incidence of breast cancer defined by estrogen and progesterone receptor status – a meta-analysis. Int J Cancer. 2009;124(3):698–712. | |
Yang XR, Chang-Claude J, Goode EL, Couch FJ, Nevanlinna H, Milne RL. Associations of Breast Cancer Risk Factors with Tumor Subtypes: A Pooled Analysis from the Breast Cancer Association Consortium Studies. J Natl Cancer Inst. 2011;103(3):250–263. | |
Amend K, Hicks D, Ambrosone CB. Breast cancer in African-American women: differences in tumor biology from European-American women. Cancer Res. 2006;66:8327–8330. | |
Stead LA, Lash TL, Sobieraj JE, et al. Triple-negative breast cancers are increased in black women regardless of age or body mass index. Breast Cancer Res. 2009;11(2):R18. | |
Stark A, Stapp R, Raghunathan A, et al. Disease-free probability after the first primary ductal carcinoma in situ of the breast: a comparison between African-American and White-American women. Breast Cancer Res Treat. 2012;131:561–570. | |
Holmes L Jr, Opara F, Hossain J. A five-year breast cancer-specific survival disadvantage of African American women. Afr J Reprod Health. 2010;14:195–200. | |
DeSantis C, Naishadham D, Jemal A. Cancer statistics for African Americans, 2013. CA Cancer J Clin. 2013;63(3):151–166. | |
Suba Z. Light deficiency confers breast cancer risk by endocrine disorders. Recent Pat Anticancer Drug Discov. 2012;7(3):337–344. | |
Otto S. Cancer epidemiology in Hungary and the Béla Johan National Program for the decade of health. Pathol Oncol Res. 2003;9:126–130. | |
Montag A, Kumar V. The female genital system and breast. In: Kumar V, Abbas AK, Fausto N, Mitchell RN, editors. Robbins Basic Pathology. 8th ed. Philadelphia, PA: Saunders Elsevier; 2007:711–750. | |
Mousavi SM, Sundquist J, Hemminki K. Nasopharyngeal and hypopharyngeal carcinoma risk among immigrants in Sweden. Int J Cancer. 2010;127:2888–2892. | |
Hemminki K, Mousavi SM, Sundquist J, Brandt A. Does the breast cancer age at diagnosis differ by ethnicity? A study on immigrants to Sweden. Oncologist. 2011;16:146–154. | |
Viswanathan AN, Schernhammer ES. Circulating melatonin and the risk of breast and endometrial cancer in women. Cancer Lett. 2009;281:1–7. | |
Sánchez-Barceló EJ, Cos S, Mediavilla MD, Martínez-Campa C, González A, Alonso-González C. Melatonin-estrogen interactions in breast cancer. J Pineal Res. 2005;38:217–222. | |
Schernhammer ES, Hankinson SE. Urinary melatonin levels and postmenopausal breast cancer risk in the nurses’ health study cohort. Cancer Epidemiol Biomarkers Prev. 2009;18(1):74–79. | |
Cos S, González A, Martínez-Campa C, Mediavilla MD, Alonso-González C, Sánchez-Barceló EJ. Estrogen signaling pathway: a link between breast cancer and melatonin oncostatic actions. Cancer Detect Prev. 2006;30:118–128. | |
Gonzalez A, Cos S, Martinez-Campa C, et al. Selective estrogen enzyme modulator actions of melatonin in human breast cancer cells. J Pineal Res. 2008;45:86–92. | |
Knower KC, To SQ, Takagi K, et al. Melatonin suppresses aromatase expression and activity in breast cancer associated fibroblasts. Breast Cancer Res Treat. 2012;132(2):765–771. | |
Suba Z, editor. Insulin resistance, hyperinsulinemia and cancer risk. Estrogen Versus Cancer. Hauppauge, New York: Nova Science Publishers Inc.; 2009:19–40. | |
Li S, Rosenberg L, Wise LA, Boggs DA, Lavalley M, Palmer JR. Age at natural menopause in relation to all-cause and cause-specific mortality in a follow-up study of US black women. Maturitas. 2013;75(3):246–252. | |
Seo DC, Torabi MR. Racial/ethnic differences in body mass index, morbidity and attitudes toward obesity among US adults. J Natl Med Assoc. 2006;98(8):1300–1308. | |
Falkner B, Sherif K, Sumner A, Kushner H. Hyperinsulinism and sex hormones in young adult African Americans. Metabolism. 1999;48:107–112. | |
Rose DP, Haffner SM, Baillargeon J. Adiposity, the metabolic syndrome, and breast cancer in African-American and white American women. Endocr Rev. 2007;28:763–777. | |
Sarkissyan M, Wu Y, Vadgama JV. Obesity is associated with breast cancer in African-American women but not Hispanic women in South Los Angeles. Cancer. 2011;117:3814–3823. | |
Boucai L, Surks MI. Reference limits of serum TSH and free T4 are significantly influenced by race and age in an urban outpatient medical practice. Clin Endocrinol (Oxf). 2009;70:788–793. | |
Vriend J, Bertalanffy FD, Ralcewicz TA. The effects of melatonin and hypothyroidism on estradiol and gonadotropin levels in female Syrian hamsters. Biol Reprod. 1987;36:719–728. | |
Bellipanni G, Bianchi P, Pierpaoli W, Bulian D, Ilyia E. Effects of melatonin in perimenopausal women and menopausal women: a randomized and placebo controlled study. Exp Gerontol. 2001;36:297–310. | |
Kuijpens JL, Nyklíctek I, Louwman MW, Weetman TA, Pop VJ, Coebergh JW. Hypothyroidism might be related to breast cancer in post-menopausal women. Thyroid. 2005;15:1253–1259. | |
Giovannucci E. The epidemiology of vitamin D and cancer incidence and mortality: a review (United States). Cancer Causes Control. 2005;16:83–95. | |
Harris SS. Does vitamin D deficiency contribute to increased rates of cardiovascular disease and type 2 diabetes in African Americans? Am J Clin Nutr. 2011;93:1175S–1178S. | |
Grant WB, Peiris AN. Possible role of serum 25-hydroxyvitamin D in black-white health disparities in the United States. J Am Med Dir Assoc. 2010;11:617–628. | |
Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108:171–182. | |
Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. | |
Lux MP, Fasching PA, Beckmann MW. Hereditary breast and ovarian cancer: review and future perspectives. J Mol Med (Berl). 2006;84:16–28. | |
Lakhani SR, Van De Vijver MJ, Jacquemier J, et al. The pathology of familial breast cancer: predictive value of immunohistochemical markers estrogen receptor, progesterone receptor, HER-2, and p53 in patients with mutations in BRCA1 and BRCA2. J Clin Oncol. 2002;20(9):2310–2318. | |
Lee E, McKean-Cowdin R, Ma H, et al. Characteristics of triple-negative breast cancer in patients with a BRCA1 mutation: results from a population-based study of young women. J Clin Oncol. 2011;29(33):4373–4380. | |
Zheng L, Annab LA, Afshari CA, Lee WH, Boyer TG. BRCA1 mediates ligand-independent transcriptional repression of the estrogen receptor. Proc Natl Acad Sci U S A. 2001;98(17):9587–9592. | |
Fan S, Ma YX, Wang C, et al. Role of direct interaction in BRCA1 inhibition of estrogen receptor activity. Oncogene. 2001;20(1):77–87. | |
Oktay K, Kim JY, Barad D, Babayev SN. Association of BRCA1 mutations with occult primary ovarian insufficiency: a possible explanation for the link between infertility and breast/ovarian cancer risks. J Clin Oncol. 2010;28(2):240–244. | |
Lin WT, Beattie M, Chen LM, et al. Comparison of age at natural menopause in BRCA1/2 mutation carriers with a non-clinic-based sample of women in northern California. Cancer. 2013;119(9):1652–1659. | |
Finch A, Valentini A, Greenblatt E, et al; Hereditary Breast Cancer Study Group. Frequency of premature menopause in women who carry a BRCA1 or BRCA2 mutation. Fertil Steril. 2013;99(6):1724–1728. | |
Kim J, Oktay K. Baseline E(2) levels are higher in BRCA2 mutation carriers: a potential target for prevention? Cancer Causes Control. 2013;24(3):421–426. | |
Smith EP, Boyd J, Frank GR, et al. Estrogen resistance caused by a mutation in the estrogen-receptor gene in a man. N Engl J Med. 1994;331:1056–1061. | |
McLaughlin JR, Risch HA, Lubinski J, et al; Hereditary Ovarian Cancer Clinical Study Group. Reproductive risk factors for ovarian cancer in carriers of BRCA1 or BRCA2 mutations: a case-control study. Lancet Oncol. 2007;8(1):26–34. | |
Antoniou AC, Rookus M, Andrieu N, et al. Reproductive and hormonal factors, and ovarian cancer risk for BRCA1 and BRCA2 mutation carriers: results from the International BRCA1/2 Carrier Cohort Study. Cancer Epidemiol Biomarkers Prev. 2009;18(2):601–610. | |
Whittemore AS, Balise RR, Pharoah PD, et al. Oral contraceptive use and ovarian cancer risk among carriers of BRCA1 or BRCA2 mutations. Br J Cancer. 2004;91(11):1911–1915. | |
Cibula D, Zikan M, Dusek L, Majek O. Oral contraceptives and risk of ovarian and breast cancers in BRCA mutation carriers: a meta-analysis. Expert Rev Anticancer Ther. 2011;11(8):1197–1207. | |
Milne RL, Knight JA, John EM, et al. Oral contraceptive use and risk of early-onset breast cancer in carriers and noncarriers of BRCA1 and BRCA2 mutations. Cancer Epidemiol Biomarkers Prev. 2005;14(2):350–356. | |
Adlercreutz HJ. Phytoestrogens and breast cancer. J Steroid Biochem Mol Biol. 2002;83(1–5):113–118. | |
Cabanes A, Wang M, Olivo S, et al. Prepubertal estradiol and genistein exposures up-regulate BRCA1 mRNA and reduce mammary tumorigenesis. Carcinogenesis. 2004;25(5):741–748. | |
Suba Z. Beneficial role of estrogen signaling in glucose homeostasis and energy expenditure. In: Johnson CC, Williams DB, editors. Glucose Uptake: Regulation, Signaling Pathways and Health Implications. Hauppauge, New York: Nova Science Publishers Inc; 2013. | |
Bordeleau L, Lipscombe L, Lubinski J, et al; Hereditary Breast Cancer Clinical Study Group. Diabetes and breast cancer among women with BRCA1 and BRCA2 mutations. Cancer. 2011;117(9):1812–1818. | |
Kotsopoulos J, Olopado OI, Ghadirian P, et al. Changes in body weight and the risk of breast cancer in BRCA1 and BRCA2 mutation carriers. Breast Cancer Res. 2005;7(5):R833–R843. | |
Key TJ. Hormones and cancer in humans. Mutat Res. 1995;333(1–2):59–67. | |
den Tonkelaar I, de Waard F. Regularity and length of menstrual cycles in women aged 41–46 in relation to breast cancer risk: results from the DOM-project. Breast Cancer Res Treat. 1996;38(3):253–258. | |
Wolpert BJ, Amr S, Ezzat S, et al. Estrogen exposure and bladder cancer risk in Egyptian women. Maturitas. 2010;67(4):353–357. | |
Teras LR, Patel AV, Rodriguez C, Thun MJ, Calle EE. Parity, other reproductive factors, and risk of pancreatic cancer mortality in a large cohort of US women (United States). Cancer Causes Control. 2005;16(9):1035–1040. | |
Bosetti C, Negri E, Franceschi S, Conti E, Levi F, Tomei F. Risk factors for oral and pharyngeal cancer in women: a study from Italy and Switzerland. Br J Cancer. 2000;82(1):204–207. | |
Britt K, Ashworth A, Smalley M. Pregnancy and the risk of breast cancer. Endocr Relat Cancer. 2007;14(4):907–933. | |
Källén B, Finnström O, Lindam A, Nilsson E, Nygren KG, Olausson PO. Malignancies among women who gave birth after in vitro fertilization. Hum Reprod. 2011;26(1):253–258. | |
Lai YC, Hamazaki K, Yoshizawa K, et al. Short-term pregnancy hormone treatment of N-methyl-N-nitrosourea-induced mammary carcinogenesis in relation to fatty acid composition of serum phospholipids in female Lewis rats. In Vivo. 2010;24(4):553–560. | |
Holcomb VB, Hong J, Núñez NP. Exogenous estrogen protects mice from the consequences of obesity and alcohol. Menopause. 2012;19(6):680–690. | |
Rajkumar L, Kittrell FS, Guzman RC, Brown PH, Nandi S, Medina D. Hormone-induced protection of mammary tumorigenesis in genetically engineered mouse models. Breast Cancer Res. 2007;9(1):R12. | |
Hong J, Holcomb VB, Kushiro K, Núñez NP. Estrogen inhibits the effects of obesity and alcohol on mammary tumors and fatty liver. Int J Oncol. 2011;39(6):1443–1453. | |
Nkhata KJ, Ray A, Dogan S, Grande JP, Cleary MP. Mammary tumor development from T47-D human breast cancer cells in obese ovariectomized mice with and without estradiol supplements. Breast Cancer Res Treat. 2009;114(1):71–83. | |
Phipps AI, Malone KE, Porter PL, Daling JR, Li CI. Reproductive and hormonal risk factors for postmenopausal luminal, HER-2-overexpressing, and triple-negative breast cancer. Cancer. 2008;113(7):1521–1526. | |
Millikan RC, Newman B, Tse CK, et al. Epidemiology of basal-like breast cancer. Breast Cancer Res Treat. 2008;109(1):123–139. | |
Xing P, Li J, Jin F. A case-control study of reproductive factors associated with subtypes of breast cancer in Northeast China. Med Oncol. 2010;27(3):926–931. | |
Velentgas P, Daling JR. Risk factors for breast cancer in younger women. J Natl Cancer Inst Monogr. 1994;(16):15–24. | |
Opdahl S, Alsaker MD, Janszky I, Romundstad PR, Vatten LJ. Joint effect of nulliparity and other breast cancer risk factors. Br J Cancer. 2011;105(5):731–736. | |
Newcomb PA, Trentham-Dietz A, Hampton JM, et al. Late age at full term birth is strongly associated with lobular breast cancer. Cancer. 2011;117(9):1946–1956. | |
Tsubura A, Uehara N, Matsuoka Y, Yoshizawa K, Yuri T. Estrogen and progesterone treatment mimicking pregnancy for protection from breast cancer. In Vivo. 2008;22(2):191–201. | |
Björnström L, Sjöberg M. Mechanisms of estrogen receptor signaling: convergence of genomic and nongenomic actions on target genes. Mol Endocrinol. 2005;19:833–842. | |
Segars JH, Driggers PH. Estrogen action and cytoplasmic signaling cascades. Part I: membrane-associated signaling complexes. Trends Endocrinol Metab. 2002;13:349–354. | |
Enmark E, Pelto-Huikko M, Grandien K, et al. Human estrogen receptor beta-gene structure, chromosomal localization, and expression pattern. J Clin Endocrinol Metab. 1997;82:4258–4265. | |
Nilsson S, Mäkelä S, Treuter E, et al. Mechanisms of estrogen action. Physiol Rev. 2001;81:1535–1565. | |
Lindberg MK, Movérare S, Skrtic S, et al. Estrogen receptor (ER)-beta reduces ERalpha-regulated gene transcription, supporting a “ying yang” relationship between ERalpha and ERbeta in mice. Mol Endocrinol. 2003;17:203–208. | |
Shyamala G, Ferenczy A. The nonresponsiveness of lactating mammary gland to estradiol. Endocrinol. 1982;110:1249–1256. | |
Lindberg BS, Johansson ED, Nilsson BA. Plasma levels of nonconjugated oestrone, oestradiol-17beta and oestriol during uncomplicated pregnancy. Acta Obstet Gynecol Scand Suppl. 1974;32:21–36. | |
Sivaraman L, Stephens LC, Markaverich BM, et al. Hormone-induced refractoriness to mammary carcinogenesis in Wistar-Furth rats. Carcinogenesis. 1998;19(9):1573–1581. | |
Grubbs CJ, Juliana MM, Whitaker LM. Short-term hormone treatment as a chemopreventive method against mammary cancer initiation in rats. Anticancer Res. 1988;8(1):113–117. | |
Leick MB, Jordan VC. Evolution of estrogen action in breast cancer: from culprit to killer. In: Suba Z, editor. Estrogen Prevention for Breast Cancer. - Hauppauge, New York: Nova Science Publishers Inc; 2012:127–149. | |
Henderson BE, Ross R, Bernstein L. Estrogens as a cause of human cancer. Cancer Res. 1988;48:246–253. | |
Marquez DC, Pietras RJ. Membrane-associated binding sites for estrogen contribute to growth regulation in human breast cancer cells. Oncogene. 2001;20:5420–5430. | |
Pietras RJ, Arboleda J, Wongvipat N, et al. HER-2 tyrosine kinase pathway targets estrogen receptor and promotes hormone independent growths in human breast cancer cells. Oncogene. 1995;10:2435–2446. | |
Lundholt BK, Briand P, Lykkesfeldt AE. Growth inhibition and growth stimulation by estradiol of estrogen receptor transfected human breast epithelial cell lines involve different pathways. Breast Cancer Res Treat. 2001;67:199–214. | |
Stoica A, Saceda M, Doraiswamy VL, Coleman C, Martin MB. Regulation of estrogen receptor-alpha gene expression by epidermal growth factor. J Endocrinol. 2000;165(2):371–378. | |
Marquez-Garban DC, Chen HW, Fishbein MC, Goodglick L, Pietras RJ. Estrogen receptor signaling pathways in human non-small cell lung cancer. Steroids. 2007;72:135–143. | |
Massarweh S, Schiff R. Resistance to endocrine therapy in breast cancer: exploiting estrogen receptor/growth factor signaling crosstalk. Endocr Relat Cancer. 2006;13 Suppl 1:S15–S24. | |
Ingle JN. Estrogen as therapy for breast cancer. Breast Cancer Res. 2002;4:133–136. | |
Cole MP, Jones CT, Todd ID. A new anti-oestrogenic agent in late breast cancer. An early clinical appraisal of ICI46474. Br J Cancer. 1971;25(2):270–275. | |
Ingle JN, Ahmann DL, Green SJ, et al. Randomized clinical trial of diethylstilbestrol versus tamoxifen in postmenopausal women with advanced breast cancer. N Engl J Med. 1981;304(1):16–21. | |
Lin NU, Winer EP. Advances in adjuvant endocrine therapy for postmenopausal women. J Clin Oncol. 2008;26:798–805. | |
Hayes DF. Tamoxifen: Dr Jekyll and Mr Hyde? J Natl Cancer Inst. 2004;96:895–897. | |
Osborne CK. Tamoxifen in the treatment of breast cancer. N Engl J Med. 1998;339:1609–1618. | |
Braithwaite RS, Chlebowski RT, Lau J, George S, Hess R, Col FN. Meta-analysis of vascular and neoplastic events associated with tamoxifen. J Gen Intern Med. 2003;18:937–947. | |
Lønning PE. Additive endocrine therapy for advanced breast cancer back to the future. Acta Oncol. 2009;48:1092–1101. | |
Ellis MJ, Gao F, Dehdashti F, et al. Lower-dose vs high-dose oral estradiol therapy of hormone receptor-positive, aromatase inhibitor-resistant advanced breast cancer: a phase 2 randomized study. JAMA. 2009;302:774–780. | |
Jordan VC, Ford LG. Paradoxical clinical effect of estrogen on breast cancer risk: a “new” biology of estrogen-induced apoptosis. Cancer Prev Res (Phila). 2011;4(5):633–637. | |
Maximov PY, Lewis-Wambi JS, Jordan VC. The paradox of oestradiol-induced breast cancer cell growth and apoptosis. Curr Signal Transduct Ther. 2009;4(2):88–102. | |
Lewis-Wambi JS, Jordan VC. Estrogen regulation of apoptosis: how can one hormone stimulate and inhibit? Breast Cancer Res. 2009;11(3):206. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.