Back to Journals » OncoTargets and Therapy » Volume 8
Triple-negative breast cancer: new perspectives for targeted therapies
Authors Tomao F ![]() , Papa A, Zaccarelli E, Rossi L, Caruso D, Minozzi M, Vici P
, Papa A, Zaccarelli E, Rossi L, Caruso D, Minozzi M, Vici P ![]() , Frati L, Tomao S, Ricci F
, Frati L, Tomao S, Ricci F
Received 14 May 2014
Accepted for publication 19 August 2014
Published 16 January 2015 Volume 2015:8 Pages 177—193
DOI https://doi.org/10.2147/OTT.S67673
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Dr Faris Farassati
Federica Tomao,1 Anselmo Papa,2 Eleonora Zaccarelli,2 Luigi Rossi,2 Davide Caruso,2 Marina Minozzi,2 Patrizia Vici,3 Luigi Frati,4 Silverio Tomao2
1Department of Gynecology and Obstetrics, “Sapienza” University of Rome, Policlinico “Umberto I”, Rome, 2Department of Medico-Surgical Sciences and Biotechnologies, “Sapienza” University of Rome, Oncology Unit, Istituto Chirurgico Ortopedico Traumatologico, Latina, 3Division of Medical Oncology B, Regina Elena National Cancer Institute, Rome, Italy; 4Department of Molecular Medicine, “Sapienza” University of Rome, Policlinico “Umberto I”, Rome, Italy
Abstract: Breast cancer is a heterogeneous disease, encompassing a large number of entities showing different morphological features and having clinical behaviors. It has became apparent that this diversity may be justified by distinct patterns of genetic, epigenetic, and transcriptomic aberrations. The identification of gene-expression microarray-based characteristics has led to the identification of at least five breast cancer subgroups: luminal A, luminal B, normal breast-like, human epidermal growth factor receptor 2, and basal-like. Triple-negative breast cancer is a complex disease diagnosed by immunohistochemistry, and it is characterized by malignant cells not expressing estrogen receptors or progesterone receptors at all, and human epidermal growth factor receptor 2. Along with this knowledge, recent data show that triple-negative breast cancer has specific molecular features that could be possible targets for new biological targeted drugs. The aim of this article is to explore the use of new drugs in this particular setting, which is still associated with poor prognosis and high risk of distant recurrence and death.
Keywords: basal-like breast cancer, estrogen–progesterone receptors, gene-expression microarray, human epidermal growth factor receptor 2, chemotherapy, target therapy
Introduction
In 2014, about 235,030 new cases of breast cancer (BC) are expected in the USA, with 40,430 cancer-related deaths.1 In 2009, approximately 170,000 BC cases were of the triple-negative (TN) phenotype,2 accounting for 10%–20% of invasive BC.1–3
Triple-negative breast cancer (TNBC) is defined by its lack of estrogen-receptor (ER) and progesterone-receptor (PR) expression, along with the absence of human epidermal growth factor receptor 2 (HER2) overexpression or gene amplification.3 Nowadays, TNBC is one of the most attractive areas of research in oncology. Reasons for this scientific interest are the lack of a recognized target for molecular-oriented therapy, which makes TNBC a new orphan disease. Furthermore, TNBC is a biologically aggressive neoplasia with a strong association with distant recurrence, visceral metastases, and death when compared with other BC types.4 Recurrence often occurs within 3 years of diagnosis, while 5-year mortality rates appear to be increased after initial diagnosis.4 Moreover, metastatic setting implies a much shorter median time from relapse to death.5 Particularly, the median survival of advanced TNBC is at best 12 months, much less than the median duration of survival observed in other advanced BC subtypes.5
Considering the gene-expression analysis, it appears very clear that TNBC is a heterogeneous BC form, which shows only partial overlapping features with so-called basal-like (BL) breast cancer (BLBC).6–8 In fact, not all TN tumors are identified as BL by gene expression, and not all BL tumors are TN; the discordance rate between the two definitions is 20%–30%.9 More recently, subtyping of three large clinical trials (GEICAM/9906, MA.12, and MA.5) using the PAM50 quantitative real-time reverse transcription-polymerase chain reaction (qRT-PCR)-based assay revealed that approximately 30% of tumors identified as TN by central pathology review do not fall into the BL subtype category.10 Therefore, significant biological heterogeneity exists within the group of patients diagnosed with TN disease, with the BL subtype being undoubtedly the most frequently observed (about 75%).11
Very recently, Lehmann et al12 analyzed gene-expression profiles from 21 BC datasets and confirmed the concept that a significant heterogeneity within TNBC and BC tumors occurs. According to this analysis, there are six different TNBC subtypes: two BL (BL1 and BL2), an immunomodulatory, a mesenchymal (M), a mesenchymal stem-like, and a luminal androgen receptor.12
In this regard, investigators identified BC cell lines representative of each of these molecular subtypes with different sensitivities to various targeted therapies currently under clinical investigation, providing an attractive platform for future drug development in TNBC.13
These fascinating results constitute the basis for future approaches in TNBC, using both cytotoxic drugs not classically used in BC (platinum salts) or alternative schedules (dose dense), and new drugs (poly-ADP-ribose-plymerase-1, agents targeting the epidermal growth factor receptor (EGFR), multi-tyrosine-kinase inhibitors, or antiangiogenic agents).
The aim of this paper is, therefore, to discuss targeted agents with a possible activity against TNBC.
Perspectives in treatment of TNBC: targeting the molecular feature
Hormonal therapy
Recent guidelines dictate that a hormonal receptorial threshold <1% should be used;14 about this, the St Gallen expert group recommends adjuvant endocrine therapy for any detectable hormone receptor.15
Recent findings show that other isoforms of ERs could be present that are not detected by the current techniques; in this way BCs expressing some form of ERs are labeled “hormone-receptor negative” BCs.
Three forms of ERs, ERα, ERβ, and G-protein-coupled receptors (GPCRs), are currently known; they are co-expressed in a large fraction of normal and BC tissues. Particularly the first two, the classical steroid receptors, are localized not only in the plasma membrane,16 but also in the cytosol and/or nucleus; traditionally, they exert their effects at the genomic level. In recent years, however, a large number of reports has described membrane-associated ERs, such as GPCRs.17,18 Recent studies showed that rapid effects are mediated by these novel transmembrane Estrogen receptors, also known as G- protein coupled receptor 30 (GPR 30).19–21 Anyhow, ERs play important roles in the initiation and progression of estrogen-related BC.22
ERα in several studies seems to be responsible for governing estrogen-dependent growth, response to endocrine therapy,23 and prognosis in ERα-positive BC; ERβ seems to be an antagonist against ERα effects.24–28 According to some data, it seems that ERβ functions are different when it is co-expressed with ERα than when it is expressed alone.29,30 In the same way, GPR30 plays a role in the regulation of cellular growth, proliferation, and apoptosis.31 Moreover, effects mediated by GPR30 are maintained when classic ERs are absent or blocked. In addition, GPR30 is involved in drug resistance, implying that GPR30 may be an important therapeutic target for estrogen-related tumors. Blocking both GPR30 and classic ERs may be a better strategy for the treatment of estrogen-related tumors. However, despite the indications that GPR30 may represent a novel estrogen-binding receptor, a description of its ligand-binding properties appears to be lacking in the scientific literature.32 Interestingly, ER antagonists/selective estrogen-receptor modulators, such as tamoxifen and ICI182,780, have also been shown to bind GPR30, which is consistent with previous functional studies showing that these same compounds are agonists for GPR30.32,33 Moreover, with the recent report of a highly selective, nonsteroidal GPR30 agonist, future studies of GPR30 function should be greatly facilitated by this novel reagent.34 However, some breast cells express only ERα, whereas other breast cells express only ERβ. There are also breast cells that express neither ERα nor ERβ.35
Anyhow, the ER status in BC patients has been traditionally defined by the presence or absence of ERα; in fact, the ER-negative status of TNBC patients is regularly negative for ERα but not necessarily negative for ERβ;36 indeed, several studies have reported expression of ERβ in a substantial fraction of ERα-negative and TNBC patients. Gruvberger-Saal et al examined ERα and ERβ expression in patients treated with Tamoxifen (TAM) for 2 years, observing that ERβ was significantly associated with increased distant disease-free survival (DFS); furthermore, ERβ was an independent marker within the ERα-negative tumors.37 Importantly, ERβ1 positivity was significantly associated with better survival in patients with ERα-/PR-negative or triple negative (TN)-tumors. On this basis, ERβ1 and ERβ variants may play some roles in estrogen signaling and the pathogenesis of TNBC; however, this hypothesis was not confirmed by clinical and histopathological parameters.38
Furthermore, approximately 75% of all BCs are ERα positive, which is a good predictor of treatment response.39–41 The ER-α gene is composed of six functional domains encoded by eight exons that commonly produce a 66.2 kDa protein (ER-α66).42 Furthermore, ER-α66 is the isoform detected in clinical practice. There are at least two different isoforms of ER-α66 described in human BC:43 ER-α46 and ER-α36.44,45 The importance of these isoforms lies in the possibility that some TNBCs are expressing some truncated ERa receptors that could be a target for anti-estrogen therapy. Particularly, ER-α36 is a new isoform of ERs without the transcriptional activation domains of the classical ER-α66.46 ER-α36 has been shown to transduce the membrane-initiated steroid signaling cascade, and acts as a dominant-negative effector of estrogen-dependent and -independent transactivation, mediated by ER-α66.47,48 ER-α66 expression can also be detected in the cytoplasm of BC cells after long-term treatment with tamoxifen, coinciding with resistance to the drug.48
Moreover, Shi et al indicated the importance of ER-α36 in the development of endocrine resistance in a subgroup of invasive BC that exhibits co-expression of ER-α66 and ER-α36. The functional importance of ER-α36 is related to its non-genomic ER activities; according to this hypothesis, activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) signaling pathway plays a major role. The MAPK/ERK signaling pathway is activated in response to both estrogens and antiestrogens, which might be of particular importance for ER-α66-negative BC, since this subgroup might still respond to antiestrogen-based therapy.45 Previous experiments demonstrated that antiestrogens induce a stronger and more prolonged activation of the MAPK/ERK signaling pathway than the estrogens.45
The androgen cluster is a particular kind of BC showing the androgen receptor (AR) in a very similar way to ER + PgR + BC.49 Until 1970, androgen therapy was one of the strategies used to manage BC. Moreover, the presence of the AR has been well known for a long time; even while the clinical use of this knowledge has been abandoned, experimental studies have continued, showing that the AR is the most commonly expressed nuclear hormone receptor in BC.50–52 Recent studies suggest that 10%–35% of all TNBC presents an AR-positive gene-expression profile; moreover, these TNBCs are frequently apocrine BC51, even if some other experiences show no significantly different histopathological features between AR-positive and AR-negative BC.53
Anyhow, AR-targeting has been introduced recently as a novel therapeutic option in TNBC,54 and a Phase II trial of nonsteroidal antiandrogen treatment is ongoing in women with advanced AR-positive, ER-negative, PR-negative BC. The preliminary results of this trial suggest attractive benefit.55 On this basis, other trials are investigating the use of androgen inhibition in TNBC.
Poly (adenosine diphosphate-ribose) polymerase inhibitors
Genomic integrity and cell survival are critically related to coordinated pathways of DNA repair.56 Poly (adenosine diphosphate-ribose) polymerase (PARP) enzymes – particularly the most abundant isoform, PARP1 – play a key role in these pathways by mediating the repair of single-strand DNA breaks via base-excision repair.57 Consequently, loss of PARP activity results in the accumulation of single-strand breaks, which are normally repaired by double-strand homologous recombination pathways that include the important tumor-suppressor proteins BRCA1 and BRCA2.58 It is well known that germ-line BRCA1 and BRCA2 mutations are associated with a high risk of oncogenesis for breast and ovarian cancers.59 TNBC shares clinical and pathological features with hereditary BRCA1-related BC; in sporadic TNBC, dysregulation of BRCA1 has been frequently observed together with other defects in homologous recombination pathways.60,61 A preclinical study showed that TNBC cells are more sensitive to PARP1 inhibitors when compared with non-TNBC cells, and that PARP inhibition acts synergically in association with gemcitabine and cisplatin in TN cells but not in luminal cancers.62 All this evidence provides a strong rationale for developing a new therapeutic approach to TNBC based on targeting the DNA-repair defects via PARP inhibition. Several PARP inhibitors, such as olaparib, veliparib, iniparib, niraparib, and rucaparib, are currently under early development in TNBC or BRCA-mutated BC.
Establishing which subtypes of BC are most indicated for PARP inhibitor treatment, and how to identify these, is a matter of strategic importance for further clinical development of this class of anticancer agents. Despite their extremely high therapeutic potential, more research on the biology of the numerous members of the PARP families and on their role in the molecular pathogenesis of BRCA1- and BRCA2-mutated is necessary before we can claim to understand the utility of the PARP inhibitors in clinical oncology.
Olaparib
Olaparib, an oral PARP inhibitor, demonstrated its antitumor activity in patients who were carriers of the BRCA1 or BRCA2 mutation in a Phase I trial.63 Moreover, olaparib has been investigated as a single agent in BRCA1- and BRCA2-mutated women with advanced BC in a Phase II trial.64 In this nonrandomized trial, patients were assigned to one of two sequential cohorts; in the first cohort oral olaparib at 400 mg twice daily was given continuously, while in the second one a lower dose of 100 mg twice daily was used. The primary endpoint was objective response rate (RR). All patients had received previous chemotherapy. The objective RR was confirmed in 11 of 26 patients in cohort 1 and six of 24 in cohort 2. The clinical benefit rate (CBR) was 52% for cohort 1 and 26% for cohort 2. Median progression-free survival (mPFS) was 5.7 months for cohort 1 and 3.8 months for cohort 2.64 Furthermore, olaparib toxicity was generally mild and manageable, with the most frequent adverse events (AEs) occurring in cohort 1 (fatigue, nausea, vomiting, and anemia). The results of this study provide an important proof-of-concept for PARP inhibition in BRCA-deficient BC; selecting patients with TNBC who are also BRCA deficient could isolate a patient population who may be more receptive to treatment with PARP inhibitors. It is interesting to observe that there is no reported activity for olaparib against non-BRCA-mutant or unreported BRCA status.65
In another Phase II, multicenter, open-label, non-randomized study, women with advanced high-grade serous and/or undifferentiated ovarian carcinoma or TNBC were enrolled and received olaparib 400 mg twice a day.66 Patients were stratified according to whether they had a BRCA1 or BRCA2 mutation or not. The primary endpoint was objective RR. In the TNBC cohort, no RECIST (Response Evaluation Criteria In Solid Tumors) validated responses were reported at the first stage, so this cohort was closed.
A Phase I study evaluated the safety, tolerability, and efficacy of olaparib in combination with paclitaxel in patients with metastatic TNBC (mTNBC).67 Patients who had received ≤1 prior cytotoxic regimens for mTNBC were treated continuously with olaparib plus weekly paclitaxel. Nineteen patients received treatment. Patients in cohort 2 received olaparib and paclitaxel at the same doses and schedule as patients in cohort 1. For patients in cohort 2, upon first occurrence of grade ≥2 neutropenia, the paclitaxel dose was omitted or delayed; olaparib dosing was continued. Objective RR in cohort 1 was 3/9 patients and in cohort 2 was 4/10 patients.
This agent is currently undergoing clinical evaluation in combination with paclitaxel in a Phase II single-arm trial68 and in combination with carboplatin and/or paclitaxel in a Phase I study69 – both trials are in patients with mTNBC. Olaparib is also undergoing investigation in combination with cisplatin in a Phase I single-arm trial in neoadjuvant approach for locally advanced TNBC.70
Veliparib
Veliparib is an oral PARP1 and PARP2 inhibitor. In a single-arm Phase II trial, the activity of veliparib and temozolomide (TMZ) was tested in 41 metastatic BC patients (15 TNBC patients).71 TMZ has minimal activity in BC, which is probably due to the methylated DNA-repair adducts by the base-excision repair pathway.71 The primary endpoint in the study was overall RR. Best responses for 24 patients evaluable at the time of abstract submission included one complete response (CR), two partial responses (PRs), seven stable diseases (SDs), and 14 progressive diseases (PDs); 17 patients were not yet evaluable for response. Therefore, veliparib and TMZ may be a promising new strategy in mTNBC.
In a subsequent expansion cohort, first-line therapy for metastatic BC and prior PARP inhibitor therapy were allowed, and eligible patients were required to have a known deleterious BRCA1 or BRCA2 mutation identified from prior clinical testing.72 All patients received veliparib and TMZ. Twenty eligible patients were enrolled. Best response for the 20 patients evaluable at the time of abstract submission included three PRs, six SDs, eleven PDs, and a clinical benefit rate (CBR) of 45%. Combined with the initial cohort of eight known carriers from the original 41 patients, the total RR was 25% (7/28) and CBR was 50%. The RR was 40% (6/15) in patients without prior platinum treatment, and 9% (1/11) in patients with prior platinum treatment. The mPFS for the 20 BRCA carriers was 85 days, and among patients with prior platinum treatment compared with no prior platinum, the mPFS was 70 and 179 days, respectively.
Iniparib
Iniparib was initially developed as a PARP inhibitor, but recent data suggest that it does not show characteristics typical of this class and its exact mechanism of action remains to be elucidated.73 The cellular effects of iniparib include the induction of γ-H2AX foci (a marker of DNA damage) and cell-cycle arrest in the G2/M phase in tumor cell lines.74 Iniparib also potentiates the cell-cycle effects of DNA damaging modalities in tumor cell lines, and antiproliferative activity has been demonstrated in a TNBC-related cell line.75
Results of a Phase II trial (Table 1) in patients with mTNBC showed that adding iniparib to gemcitabine and carboplatin significantly improved CBR and progression-free survival (PFS) (P=0.01) compared with chemotherapy alone.76 The median overall survival (mOS) was also significantly improved in the arm containing iniparib (P=0.01). There were essentially no differences in AE incidence between the two treatment groups, suggesting that iniparib was very well tolerated.
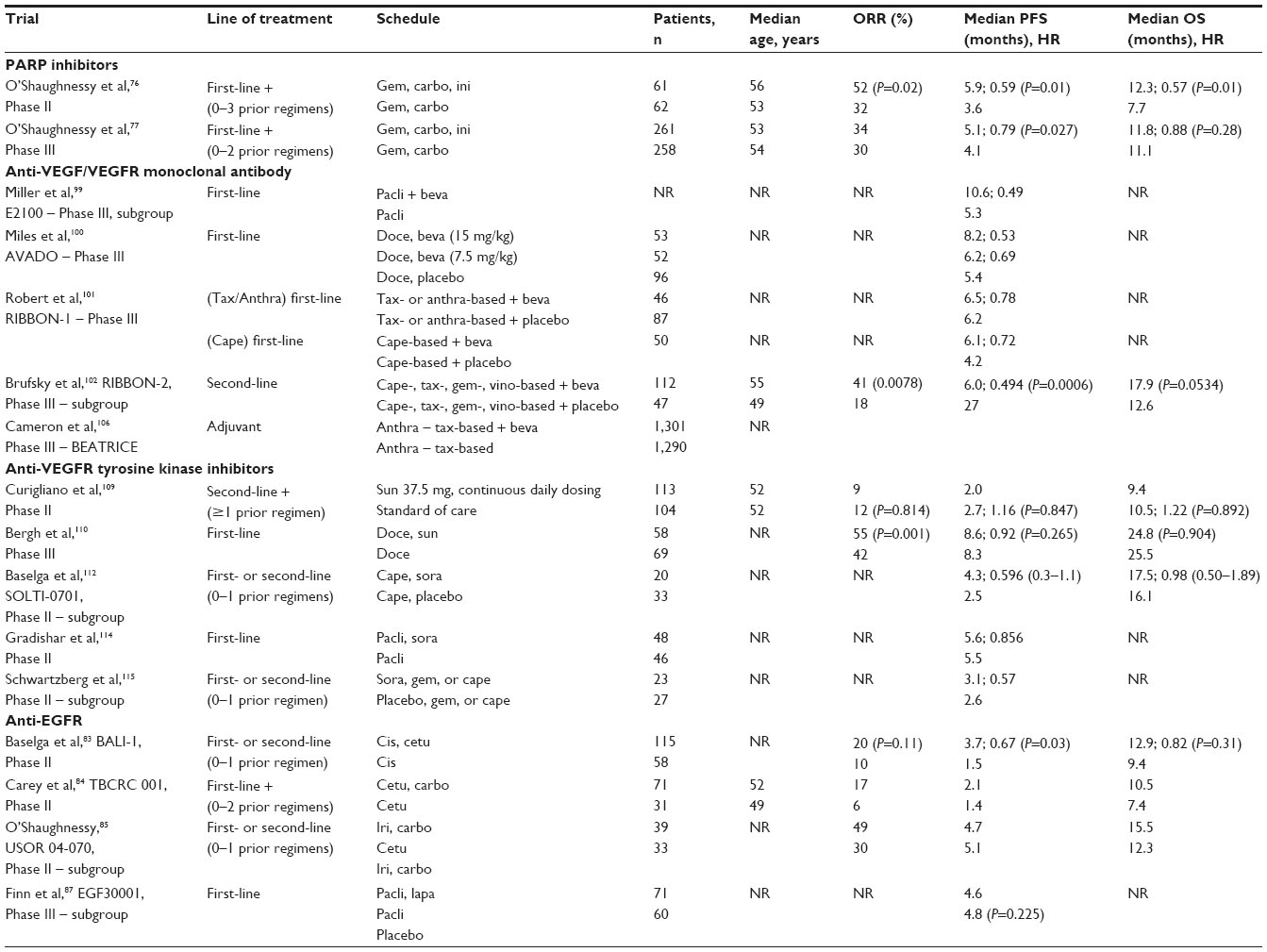 | Table 1 Trials about triple-negative breast cancer |
Results of a multicenter Phase III trial assessing the same iniparib combination in advanced TNBC failed to meet the primary study endpoints (Table 1).77
Ongoing clinical trials will further evaluate the benefits of weekly or biweekly iniparib in combination with gemcitabine and carboplatin in the advanced setting,78 iniparib and PF-01367338 in neoadjuvant and adjuvant settings, respectively, as well as other promising PARP inhibitors.79,80
Anti-EGFR
Different studies have indicated that EGFR is frequently overexpressed in TNBC and it is a negative prognostic factor,81,82 suggesting a potential role for EGFR-targeted therapies in this subset of patients. There are randomized data on EGFR inhibitors, including the monoclonal antibody cetuximab and the tyrosine-kinase inhibitors (TKIs) erlotinib and lapatinib in the management of TNBC.
In a large randomized Phase II trial, 173 patients with mTNBC, who had received no more than one previous line of chemotherapy for metastatic disease, received cisplatin, with or without cetuximab (Table 1).83 The objective RR was 20% with cisplatin plus cetuximab and 10% with cisplatin alone (P=0.11). Cisplatin plus cetuximab resulted in a longer mPFS (P=0.032) and an approximately the same mOS (P=0.31) compared with cisplatin alone. Although the RR was doubled in the combination arm, it did not meet the prespecified primary endpoint of the study and was not found to be statistically significant (Table 1). Efforts are underway to identify those patients with mTNBC who benefit from cetuximab treatment, which may be correlated with lower expression of the α-crystallin B chain (encoded by the CRYAB gene), higher expression of phosphatase and tensin homologue (PTEN) and lack of KRAS expression in patients with BLBC.
In another randomized Phase II trial, 102 patients with mTNBC received cetuximab alone (arm 1) or cetuximab associated with carboplatin (arm 2).84 Two of 31 patients in arm 1 experienced PR and one had durable SD (CBR =10%). Of 71 patients treated in arm 2, one experienced CR and eleven experienced PR, for an overall RR of 17%. Ten had prolonged SD (CBR =31%). Patients treated with the combination, after progression while receiving single-agent cetuximab, had similar results (RR =16%; CBR =28%). Response was unrelated to previous lines of therapy. The mOS was 7.5 months for arm 1 and 10.4 months for arm 2.
The addition of cetuximab to irinotecan and carboplatin in first- and second-line metastatic BC patients in a Phase II trial resulted in improved RR among a subset of TNBC patients (objective RR 30% without cetuximab versus 49% with cetuximab).85 However, no improvements in either PFS or overall survival (OS) were apparent in the TNBC subgroup, and cetuximab combination resulted in a substantial increase in diarrhea compared with chemotherapy alone.
A randomized Phase II trial assessed the combination of erlotinib with carboplatin and docetaxel in the neoadjuvant treatment of TNBC patients and demonstrated promising activity with a pathological complete response (pCR) of 40%.86 In addition, patients were equally randomized between two erlotinib arms. Patients in arm 1 received erlotinib during all six cycles of chemotherapy and patients in arm 2 received erlotinib only during the last four cycles of chemotherapy, with only minimally increased toxicity. Five of 28 patients carried a BRCA mutation (two BRCA1 deleterious, two BRCA2 deleterious, one BRCA1 uncertain). The overall pCR rate was 39%. BRCA mutation status strongly correlated with pCR, with a rate of 100% in BRCA mutated patients, compared with 27% in those without BRCA mutation (P=0.006). Baseline EGFR and/or p53 expression of 10% was associated with a lower pCR rate in BRCA noncarriers (pCR: 12% vs 66%, P=0.025).
Retrospective data from two randomized Phase II trials demonstrated modest activity for gefitinib in combination with standard neoadjuvant chemotherapy, and a lack of activity for lapatinib in combination with paclitaxel in advanced TNBC patients.3,87
Ongoing prospective, randomized EGFR clinical trials, such as those investigating combinations of cetuximab with ixabepilone in both the early88 and advanced89 settings, will better define the role of these agents in TNBC.
Mammalian target of rapamycin inhibitors
Everolimus (RAD001) is an oral inhibitor of serine-threonine kinase mammalian target of rapamycin (mTOR), showing broad antitumor activities in preclinical models.90,91 An interesting study investigated the antitumor activity of everolimus in nine different TNBC cell lines.92 The study suggested that everolimus is a promising agent for the treatment of TNBCs, especially BLBC. Basal markers (EGFR and CK5/6) or cancer stem-cell markers (E-cadherin, snail, or twist) may be predictive markers of the response to everolimus in TNBCs.
Rapamycin potentiates the cytotoxicity of paclitaxel in cell lines with phosphoinositide 3-kinase (PI3K)/PTEN/Akt aberrations, suggesting that combination therapy may be effective in these tumors.93 In this light, a Phase II study investigated the role of everolimus added to standard neoadjuvant chemotherapy in patients with TNBC.94 Fifty patients were randomly assigned to 5-FU, epirubicin, and cyclophosphamide with docetaxel (FEC-T), or to FEC-T with everolimus 30 mg weekly. Twelve-week RR evaluated by ultrasounds were 29.6% vs 47.8%, respectively (P=0.075). There were no differences in RR at 24 weeks (P=0.27). The pCR rates after chemotherapy completion were 25.9% for FEC-T and 30.4% for FEC-T plus everolimus (P=0.76). In conclusion, adding everolimus to neoadjuvant paclitaxel showed a trend toward an improvement in the 12-week RR, but it did not improve the pCR rate in TNBC.
Another study evaluated the effects of co-inhibition of mTOR (using rapamycin) and EGFR (using lapatinib) in TNBC cell lines and nude mice models,95 showing that the combination therapy results in synergistic effects in TNBC models in vitro and suppresses TN tumor growth in vivo.
Anti-vascular endothelial growth factor/Vascular endothelial growth factor-receptor monoclonal antibodies
TNBC is a highly proliferative neoplasm that needs constant angiogenesis throughout all the phases of its development, invasion, and metastasis.96 Intra-tumoral expression of the vascular endothelial growth factor (VEGF) is significantly higher in TNBC than in non-TNBC, providing a biological rationale for targeting this pathway in the treatment of TNBC patients.97
The anti-VEGF monoclonal antibody bevacizumab (BV) has been shown to increase the RR and PFS of patients with metastatic BC when added to first-line chemotherapy in three randomized Phase III trials.99–101 A meta-analysis of TNBC subgroup data from these first-line Phase III trials revealed a 35% reduction in the risk of progression (hazard ratio [HR] =0.65) and a benefit in mPFS of 2.7 months (P<0.0001) when BV was added to chemotherapy regimens, compared with chemotherapy alone.98
An open-label, randomized, Phase III trial compared the efficacy and safety of paclitaxel with or without BV, as initial treatment for metastatic BC.99 Particularly, a retrospective subgroup analysis of the E2100 trial suggested that the addition of BV to paclitaxel reduced the risk of progression in first-line TNBC patients by 51% and doubled the mPFS.
The efficacy and safety of combining BV with docetaxel as first-line therapy for HER2-negative, locally recurrent, or metastatic BC was investigated in a three-arm, placebo-controlled, Phase III trial.100 In this study, patients were randomly assigned to docetaxel plus either placebo or BV 7.5 or 15 mg/kg every 3 weeks. In the TNBC subgroup, mPFS was 8.2 months in the docetaxel in combination with BV 15 mg/kg arm, and it was 6.2 and 5.4 months in the BV 7.5 mg/kg and placebo arms, respectively. The combination of BV with docetaxel had no major impact on the toxicity profile of docetaxel. Grade 3 to 4 AEs of special interest were more common in the BV arms than the placebo group, and most of the differences were attributable to hypertension, neutropenia, and febrile neutropenia. This randomized, double-blind study confirmed the clinical benefit of combining BV with a taxane, as previously reported in trial E2100. Taken together, these studies suggest that the combination of BV with taxane-based chemotherapy should be considered as an option for the first-line treatment of HER2-negative metastatic BC.
Moreover, Regimens in Bevacizumab for Breast Oncology (RIBBON)-1, a Phase III, randomized, and placebo-controlled trial, was designed to evaluate the efficacy and safety of BV in combination with chemotherapy regimens, including non-taxane, such as capecitabine, used as initial chemotherapy-based treatment of metastatic BC.101 In the capecitabine cohort, mPFS increased from 4.2 months in the placebo arm to 6.1 months in the BV arm. In the taxane/anthracyclines cohort, mPFS was 6.2 months in the placebo arm and 6.5 months in the BV arm. The results of the RIBBON-1 trial indicated no clear benefit for TNBC patients who added BV to chemotherapy.
Regarding the second line of treatment, the RIBBON-2 trial (Table 1) was designed to evaluate the efficacy and safety of combining BV with chemotherapeutic schedules commonly used for patients affected by metastatic BC who had received one previous cytotoxic regimen in the metastatic setting.102 In patients with TNBC, BV combined with chemotherapy resulted in a 3.3-month improvement in mPFS (P=0.0006), with a trend toward improved OS (P=0.0534).
BV has also been investigated in the neoadjuvant setting, and data from two large randomized clinical trials in patients with HER2-negative operable BC have been presented. The first, the GeparQuinto trial, evaluated the rate of pCR after neoadjuvant epirubicin, cyclophosphamide, and docetaxel chemotherapy with and without the addition of BV in patients with TNBC.103,104 The pCR rates were 27.9% without and 39.3% with BV (P=0.003).
In the second trial, the National Surgical Adjuvant Breast and Bowel Project (NSABP) B-40 study, patients were randomly assigned to receive neoadjuvant therapy consisting of docetaxel, docetaxel plus capecitabine, or docetaxel plus gemcitabine for four cycles, with all regimens followed by treatment with doxorubicin-cyclophosphamide for four cycles.104 Patients were also randomly assigned to receive or not to receive BV for the first six cycles of chemotherapy. The addition of BV significantly increased the rate of pCR; when this rate was examined according to hormone-receptor status, the effect of BV was more pronounced in the hormone-receptor-positive subset (15.1% without BV vs 23.2% with BV [P=0.007]), with a weaker effect in the hormone-receptor-negative subset (47.1% without BV vs 51.5% with BV [P=0.34]).
The differences in the results from the two trials could be attributed to the differences in the definition of HER2 negativity in the patients and to the different regimens that they received. Moreover, in the Phase 2 Cancer and Leukemia Group B (CALGB)/Alliance 40603 trial, a total of 454 women with operable stage II or III TNBC were randomly assigned to receive different schedules of treatment: standard neoadjuvant paclitaxel chemotherapy, including doxorubicin and cyclophosphamide, standard neoadjuvant therapy plus carboplatin, standard neoadjuvant therapy plus BV, or standard neoadjuvant therapy plus both carboplatin and BV.105 The pCR rate in breast tissue with or without neoadjuvant carboplatin was 60% versus 46% (odds ratio [OR] =1.76; P=0.0018); in the BV arm, pCR was 59% compared with 48% in the non-BV arm (OR =1.58; P=0.0089). When carboplatin and BV were used in combination against the chemotherapy backbone, the highest pCR rate was achieved, 67%, but the P-value for the carboplatin/BV interaction was 0.52, indicating the lack of a synergistic effect.
In an adjuvant setting, the BEATRICE study, a randomized Phase III trial, assessed the addition of BV to chemotherapy for women with TNBC.106 Almost two-thirds of patients in both groups had node-negative disease. There was no difference between the chemotherapy-alone group and BV group in DFS (HR =0.87; 3-year invasive DFS =82.7% in the chemotherapy group versus 83.7% in the BV group; P=0.18). All secondary efficacy endpoints seemed to favor BV, but none was statistically significant.
While BV binds specifically to the ligand, vascular endothelial growth factor-A (VEGF-A), and all its isoforms, it has limited binding affinity for other vascular endothelial growth factor-receptor (VEGFR) ligands, VEGF-B, and VEGF-C. Newer antiangiogenic agents bind to VEGFR-2 and thus prevent all ligand binding to this target. Therefore, targeting VEGFR in this way could potentially lead to a more complete target inhibition and a more effective angiogenesis block. In this regard, the monoclonal antibody ramucirumab (IMC-1121B, ImClone) targeting VEGFR-2 is currently under investigation in combination with docetaxel in a Phase III clinical trial in patients with HER2-negative metastatic BC.107
Anti-VEGFR TKIs
Cell surface-receptor tyrosine kinases, including VEGFRs, are critical to angiogenesis, and the effects of two TKIs on endothelial cell proliferation have been evaluated in TNBC.
The anti-VEGFR TKIs sunitinib and sorafenib have shown some interesting degrees of activity in BC in clinical studies with substantial TNBC populations.
Sunitinib
Sunitinib was investigated in a preliminary Phase II trial as a single agent in patients with metastatic BC who had received prior anthracycline and taxane therapy. Neither steroid receptor nor HER2 status appeared to correlate with clinical response. Among the patients with TNBC, the RR was 15%, and 11% in the total population.108
Another randomized (Table 1), open-label Phase II trial was designed to test the hypothesis that the PFS of patients receiving sunitinib monotherapy (37.5 mg/day) on a continuous-daily-dosing schedule would be superior to that obtained with single-agent standard-of-care (SOC) chemotherapy in patients with previously treated TNBC. Almost all patients had received prior treatment containing an anthracycline and a taxane; approximately three-quarters of patients had received prior chemotherapy for advanced disease. Thirteen percent of patients received prior BV therapy in both arms. The mPFS was 2.0 months with sunitinib versus 2.7 months with SOC (P=0.888). Moreover, the mOS was not prolonged with sunitinib treatment compared with SOC (P=0.839).109
In a prospective and randomized Phase III study, patients with advanced BC were randomly assigned to open-label combination therapy (sunitinib and docetaxel) or monotherapy (docetaxel). There was no difference in mPFS (P=0.265) and mOS (P=0.904) between the two arms. The overall response rate (ORR) was significantly higher with the combination than with monotherapy (P=0.001). Duration of response was similar in both arms.110
Sorafenib
Sorafenib is a multi-kinase inhibitor with antiangiogenic/antiproliferative activity. Bianchi et al described the results of single-agent sorafenib in metastatic BC.111 Their study evaluated 52 patients demonstrating a 2% RR and a 13% SD at 6 months. The authors concluded that further investigation of single-agent sorafenib in this patient population is not recommended.
A randomized, double-blind, placebo-controlled Phase II trial assessed sorafenib with capecitabine for locally advanced or metastatic HER2-negative BC. Patients were randomly assigned to first- or second-line capecitabine with sorafenib or placebo.112 The results suggest a role for the combination of sorafenib and capecitabine in BC and supported a multinational, double-blind, randomized, placebo-controlled Phase III trial assessing the addition of sorafenib to first- or second-line capecitabine in advanced HER2-negative BC.113 In the NU07B1 trial, a multinational, double-blind, placebo-controlled Phase II screening trial, patients were randomized to paclitaxel in combination with placebo or sorafenib. There was no significant difference in OS between treatment arms (P=0.904), but the addition of sorafenib versus placebo to paclitaxel significantly increased median time to progression (8.1 vs 5.6 months, respectively; P=0.0343) and ORR (67% vs 54%, respectively; P=0.0468).114
Another double-blind, placebo-controlled Phase II study enrolled patients with locally advanced or metastatic HER2-negative BC and prior BV treatment. Patients were randomized to chemotherapy with sorafenib or placebo. The combination of sorafenib plus gemcitabine/capecitabine provided a clinically small but statistically significant improvement in PFS compared with placebo plus gemcitabine/capecitabine (HR =0.65; P=0.02). The mOS was 13.4 versus 11.4 months, respectively (HR =1.01; P=0.95).115
New small-molecule receptor TKIs such as apatinib116 and cediranib (in combination with olaparib)117 have reached Phase II clinical development in patients with TNBC.
Other target agents
7-hydroxystaurosporine
The majority of TNBCs carry mutations in TP53, a gene that encodes the tumor-suppressor protein p53 which is required for G1 checkpoint regulation in the presence of genotoxic stress.118,119
7-hydroxystaurosporine (UCN-01) was the first checkpoint kinase 1 inhibitor introduced in clinic, although it also inhibits several other serine-threonine protein kinases.120,121
In a Phase I study of UCN-01 and irinotecan in patients with refractory advanced solid tumor malignancies, two PR were observed in women with TNBC.122 Both tumors were deficient for p53. Therefore, it was hypothesized that UCN-01 and irinotecan would be an effective regimen in mTNBC. The goal of the Phase II trial was to further evaluate the association between irinotecan and UCN-1 in patients with mTNBC previously treated with anthracyclines and taxanes.123 A total of 25 patients were enrolled, and 22 were evaluated for response. No CRs were observed. One PR, lasting 24 weeks, occurred in a patient with liver and lung metastases. Tumor from this patient was found to be BL carrying a TP53 mutation. Two patients had prolonged SD that lasted for 37 and 28 weeks, respectively. The CBR and the OS were 12% and 11.3 months, respectively. Unfortunately, the study did not meet the predefined efficacy criteria for further evaluation. Among the 15 patients with sufficient tumor samples for TP53 mutation analysis, eight carried mutations in TP53. In this limited dataset, a significantly worse OS was observed in patients with mutations in TP53. In particular, the median OS was 5.5 months and 20.3 months in patients with and without TP53 mutation, respectively (P=0.004).
Bortezomib
Bortezomib is the first proteasome inhibitor to be approved for treatment for multiple myeloma and mantle-cell lymphoma.124,125 Particularly, bortezomib has been shown to block proteasome degradation of IκB, an inhibitor of nuclear factor-kappa B, and demonstrated remarkable antitumor activity against these hematological malignancies.126
Furthermore, preclinical studies have demonstrated an in vitro antitumor effect of bortezomib in BC models.127,128
Moreover, Tseng et al demonstrated that bortezomib induced significant apoptosis in TNBC cell lines but not in hormone-receptor-positive or HER2-overexpressing cells.129 This study reveals a novel mechanism by which bortezomib induces apoptosis in TNBC cells, that is CIP2A-dependent p-Akt downregulation. The data showed that 50/57 (87.7%) of tumor samples from TNBC patients presented variable CIP2A expression, and, as previously demonstrated, CIP2A expression has been shown to correlate with disease aggressiveness in BC.130
Dinaciclib
Dinaciclib (MK-7965, formerly SCH727965) is a novel, potent, small-molecule inhibitor of cyclin-dependent kinase (CDK) 1, CDK2, CDK5, and CDK9.
Previously, two Phase I dose-escalation trials of dinaciclib administered intravenously, weekly or every 3 weeks, were conducted in patients with advanced malignancies and showed some early evidence of clinical activity131,132
A Phase II trial was designed to assess the efficacy and safety of dinaciclib133 compared with standard doses of capecitabine in women with advanced BC. The median time to progression was 2.73 months with dinaciclib, compared with 4.17 months with capecitabine. The HR (dinaciclib/capecitabine) was 1.6. The estimated ORR following initial treatment with dinaciclib was 8% based on one confirmed PR among the 12 evaluable patients. Among the patients randomized to capecitabine, one patient achieved a PR for an estimated ORR of 7% among 14 evaluable patients. Although this trial did not select subpopulations of patients with BC on the basis of either histological or molecular markers, the confirmed and unconfirmed PRs achieved with dinaciclib occurred both in patients with ER-positive/ HER2-negative disease.
Panobinostat
Histone deacetylase inhibitors (HDACis) have emerged as a promising new class of multifunctional anticancer agents.134,135 HDACis have been linked to several downstream effects in tumor cell lines, such as cell-cycle arrest, induction of apoptosis, inhibition of angiogenesis, activation or inactivation of tumor-suppressor genes or oncogenes, and decreased invasion and metastases.134–136
Panobinostat (LBH589) is a potent pan-deacetylase inhibitor that can block multiple cancer-related pathways.137 HDACs can be subdivided into two groups: zinc-dependent (Class I, II, and IV) and zinc-independent (Class III).138 Panobinostat is a potent inhibitor with activity against Class I, II, and IV HDACi enzymes, suggesting true pan-HDAC activity.137
To date, panobinostat has demonstrated favorable clinical responses, with limited toxicity.139–141
In their study, Tate et al utilized four TNBC cell lines as models of TNBC growth and progression.142 In confirmation of other preclinical research,143–145 they found that panobinostat induced hyper-acetylation of histones H3 and H4, decreased proliferation and survival, and induced apoptosis and G2/M cell-cycle arrest.142 The panobinostat-induced effects on cell proliferation and survival appear to be TNBC cell specific as the ER-positive cell lines tested were unaffected at all doses tested; this result was contrary to previously published work, which reported that panobinostat significantly inhibited cell survival and induced cell death in ER-positive and ER-negative BC cell lines, though at a different time point.144,146
Dasatinib
Dasatinib, a potent oral inhibitor of the Src family of kinases, is approved for treating patients with Philadelphia chromosome-positive leukemias.147 Preclinical studies showed growth inhibition of BLBC cells by single-agent dasatinib more frequently than other phenotypes.147–149 Preclinical TNBC models also showed synergistic or additive dasatinib activity with chemotherapy,150 suggesting that dasatinib may provide clinical benefit in TNBC.
A prospective, open-label Phase II trial investigated the role of dasatinib in patients with locally advanced or mTNBC that had progressed after prior chemotherapy for advanced disease.151 Single-agent dasatinib showed limited efficacy in patients with TNBC. Overall, two of 43 patients (5%) achieved a RECIST PR, both occurring in the lymph nodes and lasting for 14 and 58 weeks, respectively. In addition, 11 of 43 patients (26%) experienced SD, including two lasting for 25 and 33 weeks. The mPFS was 8.3 weeks.
Another study demonstrated a significant reduction in cell viability and induction of apoptosis in TNBC cell lines using the three-drug combination of dasatinib, cetuximab, and cisplatin compared with the dual combination of cetuximab and cisplatin.152
Wnt/Frizzled
Wnt signaling is a key oncogenic pathway in multiple cancers and regulates cell proliferation, migration, and differentiation.153,154 Wnt proteins, which are secreted glycoproteins, bind to the low-density lipoprotein receptor-related protein 5/6 (LRP5/6) and Frizzled (FZD), a seven-pass transmembrane receptor protein, to activate the Wnt/β-catenin signaling pathway.155 In the absence of Wnts, β-catenin is sequestered in a complex, which results in the ubiquitination and the subsequent degradation of β-catenin by the 26S proteasome. Conversely, when Wnt proteins are secreted from cells, they can form a ternary complex with the FZD and the LRP5/6 receptors, which results in the stabilization of cytosolic β-catenin. The β-catenin then translocates into the nucleus where it induces the expression of downstream target genes that regulate cell cycle, growth, and progression.156,157 Recently, two studies demonstrated that Wnt/β-catenin signaling activation is preferentially found in a subgroup of invasive TNBCs and is associated with a poor clinical outcome.158,159 This suggests that the Wnt/β-catenin signaling pathway plays an important role in TNBC development and progression.160
Multiple attempts to develop antibodies against key Wnt co-receptors, including LRP6 and FZD, have been reported,161–163 with varying degrees of success.
A O-acyltransferase Porcupine (PORCN) inhibitor, IWP2, has been reported to show good potency and specificity in inhibiting Wnt signaling in vitro.164,165 During the biosynthesis of Wnt ligands, Wnt undergoes posttranslational acylation (palmitoylation) that is mediated by PORCN, a membrane-bound oacyltransferase.166,167 PORCN is specific and dedicated to Wnt posttranslational acylation, which is required for subsequent Wnt secretion.168 Loss of PORCN leads to inhibition of Wnt ligand-driven signaling activities in knockout mouse models.168,169
A Phase I dose-escalation study of oral LGK974 is currently ongoing. The primary purpose of this study is to find the recommended dose of LGK974 that can be safely given to adult patients with malignancies (also patients with TNBC) dependent on Wnt ligands for whom no effective standard treatment is available.170
Discussion
TNBC is a heterogeneous disease at molecular, pathologic, and clinical levels. Stratification of TNBC into subclasses, using new markers, will identify new screening methods, prognostic factors, methodologies, and perhaps targets for personalized therapies. A lot of new targeted therapies are actually under study, but the efforts are not reaching the hoped results.
Anyhow, bevacizumab showed encouraging results in several trials, as suggested by the meta-analysis of O’Shaughnessy et al.98 Unfortunately, mPFS advantage was not confirmed also by an advantage in mOS; this may be due to the several currently available post-progression therapies. However, bevacizumab is a well-known drug with a manageable toxicity profile, so it could be useful to further evaluate its action in this difficult patient setting. Meanwhile other anti-VEGF therapies, like TKIs, which have good results in other diseases, have shown all their limits in TNBC; therefore, because TNBC is only a part of a larger BC population, results have been limited to subgroup analysis in the majority of clinical trials.
Regarding the anti-EGFR therapies, there was a strong rationale for their use in TNBC treatment, particularly because of the correlation between EGFR hyper-expression and negative prognosis in this disease. Furthermore, in some trials cetuximab showed a positive trend to an OS advantage in this setting, even if those trials enrolled only a small number of patients. From these results, it seems that the subpopulation of TNBC appears to be more sensitive to these therapies; moreover, many efforts are under way to identify TNBC patients who benefit from cetuximab treatment, which may be correlated with lower expression of alpha-crystallin B chain (encoded by the CRYAB gene), higher expression of PTEN homologue, and lack of KRAS expression in patients with BLBC.
In addition, erlotinib and gefitinib showed some actions in TNBC, but the experiences are too small to consider these results conclusive at all; furthermore, it seems that BRCA-mutated TNBC achieves a better response with erlotinib. The mTOR inhibitor everolimus also needs further evaluation to clarify its real action in this clinical setting.
On the basis of preclinical studies, there was a lot expected from PARP inhibitors. Until now, olaparib has not been studied in a Phase III trial, while iniparib activity, which in a Phase II study seemed very promising, was not confirmed in the Phase III study. However, the BRCA-mutated population seems to be the target of this drug family, so future clinical trials should be based on this class of TNBC.
Other target agents, even if based on fascinating theories, at the moment have not shown impressive achievement in the Phase I and Phase II trials in which they have been evaluated. Further investigations are needed, above all in translational studies involving the evaluation of predictive response biomarkers.
Anyhow, expression profiling and genomic studies are changing our view about the molecular biology of BC, which is currently considered as a group of distinct diseases from the molecular point of view. Indeed, although the important role of genomic studies – for example the assessment of p53 status, HER2 amplification, BRCA1 and 2 deletions, and epigenetic and chromosomal modifications – most of the studies regarding the molecular signature of BC are actually related to the expression profile of tumors (ie, the messenger RNA or protein level). The morphologic features of tumor cells have long been validated for the clinical classification of BC and are regularly used as a “gold standard” to ascertain prognostic outcome in patients. Identification of molecular markers, such as expression of the receptors for estrogen and progesterone and HER2, has played an important role in determining targets for the development of efficacious drugs for treatment, and has also offered additional predictive value for the therapeutic assessment of patients with BC. Currently, no routine specific diagnostic procedure exists for this subtype, and the patients’ management is similar to that of other subtypes regarding prevention, prognostic assessment, and treatment.
However, the detailed molecular characterization of TNBC is ongoing, both to better understand the different biology and clinical outcome, and to identify specific diagnostic, prognostic, and therapeutic targets. In particular, one important goal is, therefore, the identification of prognostic factors and markers to reliably select high- and low-risk subsets of patients with TN disease, in order to explore different biologically based treatments and to tailor the therapeutic approaches to the single patient.
The more we learn about TNBC, the more we can improve therapies; in recent years, medical research has been fully oriented toward the targeted therapies; we hope to reach also with TNBC what happened with HER2 in BC and with more uncommon tumors, like kidney cancer and gastrointestinal stromal tumor. To achieve the best for targeted therapy, it seems necessary to study in detail the molecular subtype present in TNBC in a such way as to avoid that a therapy against a particular molecular feature is studied in a population in which that trait is absent.
However, TNBC is clearly a complex disease; indeed the genetic heterogeneity is present not only in differently affected individuals, but also among tumors occurring at different sites within the same patient. As such, it is likely that its biology involves multiple redundancies and pathway cross-talk. If only one pathway is selectively inhibited, the efficacy of the therapeutic strategy would likely be undermined by activation of a compensatory pathway. Therefore, it is not surprising that, to date, not a single targeted therapy has been approved for the treatment of TNBC, for which cytotoxic chemotherapy remains the standard treatment. Combining two or more targeted agents may be required for a more rational and optimal approach to TNBC treatment. Alternatively, one technique that may show promise is gene therapy; in particular, gene transfer should correct abnormal genetic functions in cancer cells. Furthermore, these potential improvements are burdened with technical difficulties, such as insufficient infectivity or inadequately broad biodistribution of the transfer vector.
Conclusion
Important results have been achieved with antiangiogenic drugs, although much still needs to be improved. Perspectives on new therapeutic molecules are most interesting, in particular the PARP inhibitors. The overall impact of these agents on patient survival has not been as great as expected, probably because the molecular basis of this illness needs to be better understood so that treatment can be more appropriately tailored.
At the current time, we believe that the systematic evaluation of the predictive value of the genomic alterations is critically important for further progress in this field; for this reason, there is the need for tissue collection in the post-neoadjuvant and metastatic settings for a better understanding of the relevant pathways that are associated with TNBC pathogenesis and therapeutic resistance.
Disclosure
The authors declare no conflicts of interest in this work.
References
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. Cancer J Clin. 2014;64(1):9–29. | |
Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363(20):1938–1948. | |
Rakha EA, El-Sayed ME, Green AR, Lee AH, Robertson JF, Ellis IO. Prognostic markers in triple-negative breast cancer. Cancer. 2007;109(1):25–32. | |
Anders CK, Carey LA. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin Breast Cancer. 2009;9 Suppl 2:S73–S81. | |
Dent R, Trudeau M, Pritchard KI, et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res. 2007;13(15 Pt 1):4429–4434. | |
Dawood S, Broglio K, Esteva FJ, et al. Survival among women with triple receptor-negative breast cancer and brain metastases. Ann Oncol. 2009;20(4):621–627. | |
Ghasemi R, Grassadonia A, Tinari N, et al. Tumor-derived microvesicles: the metastasomes. Medical Hypotheses. 2013;80(1):75–82. | |
Weigelt B, Baehner FL, Reis-Filho JS. The contribution of gene expression profiling to breast cancer classification, prognostication and prediction: a retrospective of the last decade. J Pathol. 2010;220(2):263–280. | |
Prat A, Perou CM. Deconstructing the molecular portraits of breast cancer. Mol Oncol. 2011;5(1):5–23. | |
Cheang MC, Martin M, Nielsen TO, et al. Quantitative hormone receptors, triple-negative breast cancer (TNBC), and molecular subtypes: a collaborative effort of the BIG-NCI NABCG. J Clin Oncol. 2012;30(Suppl):abstr 1008. | |
Prat A, Adamo B, Cheang MC, Anders CK, Carey LA, Perou CM. Molecular characterization of basal-like and non-basal-like triple-negative breast cancer. Oncologist. 2013;18(2):123–133. | |
Lehmann BD, Bauer JA, Chen X, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–2767. | |
Bertucci F, Finetti P, Cervera N, et al. How basal are triple-negative breast cancers? Int J Cancer. 2008;123(1):236–240. | |
Hammond ME, Hayes DF, Wolff AC, et al. American Society of Clinical Oncology/College Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol. 2010;28(16):2784–2795. | |
Goldhirsch A, Ingle JN, Gelber RD, Coates AS, Thürlimann B, Senn HJ; Panel members. Thresholds for therapies: highlights of the St Gallen International Expert Consensus on the primary therapy of early breast cancer 2009. Ann Oncol. 2009;20(8):1319–1329. | |
Levin ER. Plasma membrane estrogen receptors. Trends Endocrinol Metab. 2009;20():477–482. | |
Razandi M, Pedram A, Park ST, Levin ER. Proximal events in signaling by plasma membrane estrogen receptors. J Biol Chem. 2003;278(4):2701–2712. | |
Toran-Allerand CD, Guan X, MacLusky NJ, et al. ER-X: a novel, plasma membrane-associated, putative estrogen receptor that is regulated during development and after ischemic brain injury. J Neurosci. 2002;22(19):8391–8401. | |
Albanito L, Madeo A, Lappano R, et al. G protein-coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17beta-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer Res. 2007;67(4):1859–1866. | |
Prossnitz ER, Oprea TI, Sklar LA, Arterburn JB. The ins and outs of GPR30: a transmembrane estrogen receptor. J Steroid Biochem Mol Biol. 2008;109(3–5):350–353. | |
Maggiolini M, Vivacqua A, Fasanella G, et al. The G protein-coupled receptor GPR30 mediates c-fos up-regulation by 17beta-estradiol and phytoestrogens in breast cancer cells. J Biol Chem. 2004;279(26):27008–27016. | |
Govind AP, Thampan RV. Membrane associated estrogen receptors and related proteins: localization at the plasma membrane and the endoplasmic reticulum. Mol Cell Biochem. 2003;253(1–2):233–240. | |
Katzenellenbogen BS, Frasor J. Therapeutic targeting in the estrogen receptor hormonal pathway. Semin Oncol. 2004;31(1 Suppl 3):28–38. | |
Fox EM, Davis RJ, Shupnik MA. ERbeta in breast cancer – onlooker, passive player, or active protector? Steroids. 2008;73(11):1039–1051. | |
Lin CY, Ström A, Li Kong S, et al. Inhibitory effects of estrogen receptor beta on specific hormone-responsive gene expression and association with disease outcome in primary breast cancer. Breast Cancer Res. 2007;9(2):R25. | |
Chang EC, Frasor J, Komm B, Katzenellenbogen BS. Impact of estrogen receptor beta on gene networks regulated by estrogen receptor alpha in breast cancer cells. Endocrinology. 2006;147(10):4831–4842. | |
Paruthiyil S, Parmar H, Kerekatte V, Cunha GR, Firestone GL, Leitman DC. Estrogen receptor beta inhibits human breast cancer cell proliferation and tumor formation by causing a G2 cell cycle arrest. Cancer Res. 2004;64(1):423–428. | |
Williams C, Edvardsson K, Lewandowski SA, Ström A, Gustafsson JA. A genome-wide study of the repressive effects of estrogen receptor beta on estrogen receptor alpha signaling in breast cancer cells. Oncogene. 2008;27(7):1019–1032. | |
Murphy LC, Watson PH. Is oestrogen receptor-beta a predictor of endocrine therapy responsiveness in human breast cancer? Endocr Relat Cancer. 2006;13(2):327–334. | |
Skliris GP, Leygue E, Curtis-Snell L, Watson PH, Murphy LC. Expression of oestrogen receptor-beta in oestrogen receptor-alpha negative human breast tumours. Br J Cancer. 2006;95(5):616–626. | |
Filardo EJ, Quinn JA, Frackelton AR Jr, Bland KI. Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol Endocrinol. 2002;16(1):70–84. | |
Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology. 2005;146(2):624–632. | |
Filardo EJ, Quinn JA, Bland KI, Frackelton AR Jr. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. 2000;14(10):1649–1660. | |
Bologa CG, Revankar CM, Young SM, et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat Chem Biol. 2006;2(4):207–212. | |
Mann S, Laucirica R, Carlson N, et al. Estrogen receptor beta expression in invasive breast cancer. Hum Pathol. 2001;32(1):113–118. | |
Skliris GP, Leygue E, Watson PH, Murphy LC. Estrogen receptor alpha negative breast cancer patients: estrogen receptor beta as a therapeutic target. J Steroid Biochem Mol Biol. 2008;109(1–2):1–10. | |
Gruvberger-Saal SK, Bendahl PO, Saal LH, et al. Estrogen receptor beta expression is associated with tamoxifen response in ERalpha-negative breast carcinoma. Clin Cancer Res. 2007;13(7):1987–1994. | |
Treeck O, Lattrich C, Springwald A, Ortmann O. Estrogen receptor beta exerts growth-inhibitory effects on human mammary epithelial cells. Breast Cancer Res Treat. 2010;120(3):557–565. | |
Nadji M, Gomez-Fernandez C, Ganjei-Azar P, Morales AR. Immunohistochemistry of estrogen and progesterone receptors reconsidered: experience with 5,993 breast cancers. Am J Clin Pathol. 2005;123(1):21–27. | |
Levin ER, Pietras RJ. Estrogen receptors outside the nucleus in breast cancer. Breast Cancer Res Treat. 2008;108(3):351–361. | |
Allred DC. Issues and updates: evaluating estrogen receptor-alpha, progesterone receptor, and HER2 in breast cancer. Mod Pathol. 2010;23 Suppl 2:S52–S59. | |
Hirata S, Shoda T, Kato J, Hoshi K. Isoform/variant mRNAs for sex steroid hormone receptors in humans. Trends Endocrinol Metab. 2003;14(3):124–129. | |
Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF. A variant of estrogen receptor-{alpha}, hER-{alpha}36: transduction of estrogen- and antiestrogen-dependent membrane-initiated mitogenic signaling. Proc Natl Acad Sci U S A. 2006;103(24):9063–9068. | |
Shi L, Dong B, Li Z, et al. Expression of ER-α36, a Novel Variant of Estrogen Receptor α, and Resistance to Tamoxifen Treatment in Breast Cancer. JCO. 2009;27(21):3423–3429. | |
Pelekanou V, Notas G, Kampa M, et al. ERα36, a new variant of the ERα is expressed in triple negative breast carcinomas and has a specific transcriptomic signature in breast cancer cell lines. Steroids. 2012;77(10):928–934. | |
Wang Z, Zhang X, Shen P, Loggie BW, Chang Y, Deuel TF. Identification, cloning, and expression of human estrogen receptor-alpha36, a novel variant of human estrogen receptor-alpha66. Biochem Biophys Res Commun. 2005;336(4):1023–1027. | |
Weigelt B, Horlings HM, Kreike B, et al. Refinement of breast cancer classification by molecular characterization of histological special types. J Pathol. 2008;216(2):141–150. | |
Bratthauer GL, Lininger RA, Man YG, Tavassoli FA. Androgen and estrogen receptor mRNA status in apocrine carcinomas. Diagn Mol Pathol. 2002;11(2):113–118. | |
Park S, Koo J, Park HS, et al. Expression of androgen receptors in primary breast cancer. Ann Oncol. 2010;21(3):488–492. | |
Isola JJ. Immunohistochemical demonstration of androgen receptor in breast cancer and its relationship to other prognostic factors. J Pathol. 1993;170(1):31–35. | |
Niemeier LA, Dabbs DJ, Beriwal S, Striebel JM, Bhargava R. Androgen receptor in breast cancer: expression in estrogen receptor-positive tumors and in estrogen receptor-negative tumors with apocrine differentiation. Mod Pathol. 2010;23(2):205–212. | |
Nahleh Z. Androgen receptor as a target for the treatment of hormone receptor-negative breast cancer: an unchartered territory. Future Oncol. 2008;4(1):15–21. | |
Sutton LM, Cao D, Sarode V, et al. Decreased androgen receptor expression is associated with distant metastases in patients with androgen receptor-expressing triple-negative breast carcinoma. Am J Clin Pathol. 2012;138(4):511–516. | |
Gucalp A, Traina TA. Triple-negative breast cancer: role of the androgen receptor. Cancer J. 2010;16(1):62–65. | |
Memorial Sloan-Kettering Cancer Center. Bicalutamide in treating patients with metastatic breast cancer. In: ClinicalTrials.gov [website on the Internet]. Bethseda, MD: US National Library of Medicine; 2007 [updated October 31, 2013]. Available from: http://clinicaltrials.gov/show/NCT00468715. NLM identifier: NCT00468715. Accessed October 1, 2014. | |
Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411(6835):366–374. | |
Dantzer F, de La Rubia G, Ménissier-De Murcia J, Hostomsky Z, de Murcia G, Schreiber V. Base excision repair is impaired in mammalian cells lacking Poly(ADP-ribose) polymerase-1. Biochemistry. 2000;39(25):7559–7569. | |
McCabe N, Turner NC, Lord CJ, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006;66(16):8109–8115. | |
Levy-Lahad E, Friedman E. Cancer risks among BRCA1 and BRCA2 mutation carriers. Br J Cancer. 2007;96(1):11–15. | |
Turner NC, Reis-Filho JS, Russell AM, et al. BRCA1 dysfunction in sporadic basal-like breast cancer. Oncogene. 2007;26(14):2126–2132. | |
Tommiska J, Bartkova J, Heinonen M, et al. The DNA damage signalling kinase ATM is aberrantly reduced or lost in BRCA1/BRCA2-deficient and ER/PR/ERBB2-triple-negative breast cancer. Oncogene. 2008;27(17):2501–2516. | |
Hastak K, Alli E, Ford JM. Synergistic chemosensitivity of triple-negative breast cancer cell lines to poly(ADP-Ribose) polymerase inhibition, gemcitabine, and cisplatin. Cancer Res. 2010;70(20):7970–7980. | |
Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–134. | |
Tutt A, Robson M, Garber JE, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376(9737):235–244. | |
Khan OA, Gore M, Lorigan P, et al. A phase I study of the safety and tolerability of olaparib (AZD2281, KU0059436) and dacarbazine in patients with advanced solid tumours. Br J Cancer. 2011;104(5):750–755. | |
Gelmon KA, Tischkowitz M, Mackay H, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12(9):852–861. | |
Dent RA, Lindeman GJ, Clemons M, et al. Phase I trial of the oral PARP inhibitor olaparib in combination with paclitaxel for first- or second-line treatment of patients with metastatic triple-negative breast cancer. Breast Cancer Res. 2013;15(5):R88. | |
AstraZeneca. Phase I/II study of AZD2281 given in combination with paclitaxel in metastatic triple negative breast cancer. In: ClinicalTrials.gov [website on the Internet]. Bethseda, MD: US National Library of Medicine; 2008 [updated September 16, 2014]. Available from: http://clinicaltrials.gov/show/NCT00707707. NLM identifier: NCT00707707. Accessed October 1, 2014. | |
AstraZeneca. Study to assess the safety and tolerability of a parp inhibitor in combination with carboplatin and/or paclitaxel. In: ClinicalTrials.gov [website on the Internet]. Bethseda, MD: US National Library of Medicine; 2007 [updated July 23, 2014]. Available from: http://clinicaltrials.gov/show/NCT00516724. NLM identifier: NCT00516724. Accessed October 1, 2014. | |
AstraZeneca. Phase I AZD2281/cisplatin in advanced solid tumour patients. In: ClinicalTrials.gov [website on the Internet]. Bethseda, MD: US National Library of Medicine; 2008 [updated August 4, 2014]. Available from: http://clinicaltrials.gov/show/NCT00782574. NLM identifier: NCT00782574. Accessed October 1, 2014. | |
Isakoff SJ, Overmoyer B, Tung NM, et al. A phase II trial of the PARP inhibitor veliparib (ABT888) and temozolomide for metastatic breast cancer. J Clin Oncol. 2010;28:(Abstr 1019). | |
Isakoff SJ, Overmoyer B, Tung NM, et al. A phase II trial expansion cohort of the PARP inhibitor veliparib (ABT888) and temozolomide in BRCA1/2 associated metastatic breast cancer. Cancer Res. 2011;71(Suppl 24):P3-16-05. | |
Mateo J, Ong M, Tan DS, Gonzalez MA, de Bono JS. Appraising iniparib, the PARP inhibitor that never was – what must we learn? Nat Rev Clin Oncol. 2013;10(12):688–696. | |
Ossovskaya V, Lim C, Schools G, Kalurupalle S, Roninson IB, Broude E. Cell cycle effects of iniparib, a PARP Inhibitor, in combination with gemcitabine and carboplatin in the MDA-MB-468(-) triple-negative breast cancer (TNBC) cell line. Cancer Res. 2010;70(Suppl 24):P5-06-09. | |
Ossovskaya V, Lim CU, Schools G, Kalurupalle S, Roninson IB, Broude E. The chemosensitizing properties of iniparib in combination with DNA-damaging agents in the MDA-MB-468(-) triple-negative breast cancer (TNBC) cell line. Cancer Res. 2011;71(Suppl 8):LB-401. | |
O’Shaughnessy J, Osborne C, Pippen JE, et al. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N Engl J Med. 2011;364(3):205–214. | |
O’Shaughnessy J, Schwartzberg LS, Danso MA, et al. A randomized phase III study of iniparib (BSI-201) in combination with gemcitabine/carboplatin (G/C) in metastatic triple-negative breast cancer (TNBC). J Clin Oncol. 2011;29(Suppl):abstr 1007. | |
Sanofi. Study of SAR240550 (BSI-201) in combination with gemcitabine/carboplatin, in patients with metastatic triple negative breast cancer. In: ClinicalTrials.gov [website on the Internet]. Bethseda, MD: US National Library of Medicine; 2010 [updated January 13, 2014]. Available from: http://clinicaltrials.gov/show/NCT01045304. NLM identifier: NCT01045304. Accessed October 1, 2014. | |
Sanofi. Two regimens of SAR240550/weekly paclitaxel and paclitaxel alone as neoadjuvant therapy in triple negative breast cancer patients (SOLTI NEOPARP). In: ClinicalTrials.gov [website on the Internet]. Bethseda, MD: US National Library of Medicine; 2010 [updated September 14, 2013]. Available from: http://clinicaltrials.gov/show/NCT01204125. NLM identifier: NCT01204125. Accessed October 1, 2014. | |
Hoosier Cancer Research Network. PARP inhibition for triple negative breast cancer (ER-/PR-/HER2-) with BRCA1/2 mutations. In: ClinicalTrials.gov [website on the Internet]. Bethseda, MD: US National Library of Medicine; 2010 [updated August 20, 2013]. Available from: http://clinicaltrials.gov/show/NCT01074970. NLM identifier: NCT01074970. Accessed October 1, 2014. | |
Corkery B, Crown J, Clynes M, O’Donovan N. Epidermal growth factor receptor as a potential therapeutic target in triple-negative breast cancer. Ann Oncol. 2009;20(5):862–867. | |
Sutton LM, Han JS, Molberg KH, et al. Intratumoral expression level of epidermal growth factor receptor and cytokeratin 5/6 is significantly associated with nodal and distant metastases in patients with basal-like triple-negative breast carcinoma. Am J Clin Pathol. 2010;134(5):782–787. | |
Baselga J, Gómez P, Greil R, et al. Randomized phase II study of the anti-epidermal growth factor receptor monoclonal antibody cetuximab with cisplatin versus cisplatin alone in patients with metastatic triple-negative breast cancer. J Clin Oncol. 2013;31(20):2586–2592. | |
Carey LA, Rugo HS, Marcom PK, et al. TBCRC 001: randomized phase II study of cetuximab in combination with carboplatin in stage IV triple-negative breast cancer. J Clin Oncol. 2012;30(21):2615–2623. | |
O’Shaughnessy J, Weckstein D, Vukelja S, et al. Preliminary results of a randomized phase II study of weekly irinotecan/carboplatin with or without cetuximab in patients with metastatic breast cancer. Breast Cancer Res Treat. 2007;106:S32. (Abstr 308, presented data – SABCS 2007). | |
Sharma P, Khan QJ, Kimler BF, et al. Results of a phase II study of neoadjuvant platinum/taxane based chemotherapy and erlotinib for triple negative breast cancer. Cancer Res. 2010;70(Suppl 24):P1-11-07. | |
Finn RS, Press MF, Dering J, et al. Estrogen receptor, progesterone receptor, human epidermal growth factor receptor 2 (HER2), and epidermal growth factor receptor expression and benefit from lapatinib in a randomized trial of paclitaxel with lapatinib or placebo as first-line treatment in HER2-negative or unknown metastatic breast cancer. J Clin Oncol. 2009;27(24):3908–3915. | |
The Methodist Hospital System. Neo-adjuvant study in triple negative breast cancer patients (ICE). In: ClinicalTrials.gov [website on the Internet]. Bethseda, MD: US National Library of Medicine; 2010 [updated February 18, 2014]. Available from: http://clinicaltrials.gov/show/NCT01097642. NLM identifier: NCT01097642. Accessed October 1, 2014. | |
Bristol-Myers Squibb. Randomized Phase II study of ixabepilone alone and ixabepilone plus cetuximab as first-line treatment for female subjects with triple negative locally advanced non-resectable and/or metastatic breast cancer. In: ClinicalTrials.gov [website on the Internet]. Bethseda, MD: US National Library of Medicine; 2008 [updated May 9, 2012]. Available from: http://clinicaltrials.gov/show/NCT00633464. NLM identifier: NCT00633464. Accessed October 1, 2014. | |
Lu CH, Wyszomierski SL, Tseng LM, et al. Preclinical testing of clinically applicable strategies for overcoming trastuzumab resistance caused by PTEN deficiency. Clin Cancer Res. 2007;13(19):5883–5888. | |
Marinov M, Ziogas A, Pardo OE, et al. AKT/mTOR pathway activation and BCL-2 family proteins modulate the sensitivity of human small cell lung cancer cells to RAD001. Clin Cancer Res. 2009;15(4):1277–1287. | |
Yunokawa M, Koizumi F, Kitamura Y, et al. Efficacy of everolimus, a novel mTOR inhibitor, against basal-like triple-negative breast cancer cells. Cancer Sci. 2012;103(9):1665–1671. | |
Meric-Bernstam F, Gonzalez-Angulo AM. Targeting the mTOR signaling network for cancer therapy. J Clin Oncol. 2009;27(13):2278–2287. | |
Gonzalez-Angulo AM, Akcakanat A, Liu S, et al. Open-label randomized clinical trial of standard neoadjuvant chemotherapy with paclitaxel followed by FEC versus the combination of paclitaxel and everolimus followed by FEC in women with triple receptor-negative breast cancer. Ann Oncol. 2014;25(6):1122–1127. | |
Liu T, Yacoub R, Taliaferro-Smith LD, et al. Combinatorial effects of lapatinib and rapamycin in triple-negative breast cancer cells. Mol Cancer Ther. 2011;10(8):1460–1469. | |
Schneider BP, Miller KD. Angiogenesis of breast cancer. J Clin Oncol. 2005;23(8):1782–1790. | |
Linderholm BK, Hellborg H, Johansson U, et al. Significantly higher levels of vascular endothelial growth factor (VEGF) and shorter survival times for patients with primary operable triple-negative breast cancer. Ann Oncol. 2009;20(10):1639–1646. | |
O’Shaughnessy J, Romieu G, Diéras V, Byrtek M, Duenne AA, Miles D. Meta-analysis of patients with triple-negative breast cancer (TNBC) from three randomized trials of first-line bevacizumab (BV) and chemotherapy treatment for metastatic breast cancer (MBC). Cancer Res. 2010;70(Suppl 24):P6-12-03. | |
Miller K, Wang M, Gralow J, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357(26):2666–2676. | |
Miles DW, Chan A, Dirix LY, et al. Phase III study of bevacizumab plus docetaxel compared with placebo plus docetaxel for the first-line treatment of human epidermal growth factor receptor 2-negative metastatic breast cancer. J Clin Oncol. 2010;28(20):3239–3247. | |
Robert NJ, Diéras V, Glaspy J, et al. RIBBON-1: randomized, double-blind, placebo-controlled, phase III trial of chemotherapy with or without bevacizumab for first-line treatment of human epidermal growth factor receptor 2-negative, locally recurrent or metastatic breast cancer. J Clin Oncol. 2011;29(10):1252–1260. | |
Brufsky A, Valero V, Tiangco B, et al. Impact of bevacizumab (BEV) on efficacy of second-line chemotherapy (CT) for triple-negative breast cancer (TNBC):analysis of RIBBON-2. J Clin Oncol. 2010;29(Suppl):abstr 1010. | |
Gerber B, Loibl S, Eidtmann H, et al; German Breast Group Investigators. Neoadjuvant bevacizumab and anthracycline-taxane-based chemotherapy in 678 triple-negative primary breast cancers; results from the geparquinto study (GBG 44). Ann Oncol. 2013;24(12):2978–2984. | |
Bear HD, Tang G, Rastogi P, et al. Bevacizumab added to neoadjuvant chemotherapy for breast cancer. N Engl J Med. 2012;366(4):310–320. | |
Sikov WM, Berry DA, Perou CM, et al. Impact of the addition of carboplatin (Cb) and/or bevacizumab (B) to neoadjuvant weekly paclitaxel (P) followed by dose-dense AC on pathologic complete response (pCR) rates in triple-negative breast cancer (TNBC):CALGB 40603 (Alliance). Cancer Res. 2013;73:S5-01. | |
Cameron D, Brown J, Dent R, et al. Adjuvant bevacizumab-containing therapy in triple-negative breast cancer (BEATRICE):primary results of a randomised, phase 3 trial. Lancet Oncol. 2013;14(10):933–942. | |
Eli Lilly and Company. Phase III study of docetaxel + ramucirumab or placebo in breast cancer. In: ClinicalTrials.gov [website on the Internet]. Bethseda, MD: US National Library of Medicine; 2008 [updated August 29, 2014]. Available from: http://clinicaltrials.gov/show/NCT00703326. NLM identifier: NCT00703326. Accessed October 1, 2014. | |
Burstein HJ, Elias AD, Rugo HS, et al. Phase II study of sunitinib malate, an oral multitargeted tyrosine kinase inhibitor, in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J Clin Oncol. 2008;26(11):1810–1816. | |
Curigliano G, Pivot X, Cortés J, et al. Randomized phase II study of sunitinib versus standard of care for patients with previously treated advanced triple-negative breast cancer. Breast. 2013;22(5):650–656. | |
Bergh J, Bondarenko IM, Lichinitser MR, et al. First-line treatment of advanced breast cancer with sunitinib in combination with docetaxel versus docetaxel alone: results of a prospective, randomized phase III study. J Clin Oncol. 2012;30(9):921–929. | |
Bianchi G, Loibl S, Zamagni C, et al. Phase II multicenter, uncontrolled trial of sorafenib in patients with metastatic breast cancer. Anticancer Drugs. 2009;20(7):616–624. | |
Baselga J, Segalla JG, Roché H, et al. Sorafenib in combination with capecitabine: an oral regimen for patients with HER2-negative locally advanced or metastatic breast cancer. J Clin Oncol. 2012;30(13):1484–1491. | |
Bayer. Phase III trial comparing capecitabine in combination with sorafenib or placebo in the treatment of locally advanced or metastatic HER2-negative breast cancer. In: ClinicalTrials.gov [website on the Internet]. Bethseda, MD: US National Library of Medicine; 2010 [updated September 25, 2014]. Available from: http://clinicaltrials.gov/show/NCT01234337. NLM identifier: NCT01234337. Accessed October 1, 2014. | |
Gradishar WJ, Kaklamani V, Sahoo TP, et al. A double-blind, randomised, placebo-controlled, phase 2b study evaluating sorafenib in combination with paclitaxel as a first-line therapy in patients with HER2-negative advanced breast cancer. Eur J Cancer. 2013;49(2):312–322. | |
Schwartzberg LS, Tauer KW, Hermann RC, et al. Sorafenib or placebo with either gemcitabine or capecitabine in patients with HER-2-negative advanced breast cancer that progressed during or after bevacizumab. Clin Cancer Res. 2013;19(10):2745–2754. | |
Fudan University. Study of apatinib in metastatic triple-negative breast cancer patients. In: ClinicalTrials.gov [website on the Internet]. Bethseda, MD: US National Library of Medicine; 2010 [updated December 1, 2014]. Available from: http://clinicaltrials.gov/show/NCT01176669. NLM identifier: NCT01176669. Accessed October 1, 2014. | |
National Cancer Institute. Cediranib maleate and olaparib in treating patients with recurrent ovarian, fallopian tube, or peritoneal cancer or recurrent triple-negative breast cancer. In: ClinicalTrials.gov [website on the Internet]. Bethseda, MD: US National Library of Medicine; 2010 [updated September 19, 2014]. Available from: http://clinicaltrials.gov/show/NCT01116648. NLM identifier: NCT01116648. Accessed October 1, 2014. | |
Shah SP, Roth A, Goya R, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012; 486(7403):395–399. | |
Kastan MB, Onyekwere O, Sidransky D, Vogelstein B, Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991;51(23 Pt 1):6304–6311. | |
Ma CX, Janetka JW, Piwnica-Worms H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol Med. 2011;17(2):88–96. | |
Busby EC, Leistritz DF, Abraham RT, Karnitz LM, Sarkaria JN. The radiosensitizing agent 7-hydroxystaurosporine (UCN-01) inhibits the DNA damage checkpoint kinase hChk1. Cancer Res. 2000;60(8):2108–2112. | |
Fracasso PM, Williams KJ, Chen RC, et al. A Phase 1 study of UCN-01 in combination with irinotecan in patients with resistant solid tumor malignancies. Cancer Chemother Pharmacol. 2011;67(6):1225–1237. | |
Ma CX, Ellis MJ, Petroni GR, et al. A phase II study of UCN-01 in combination with irinotecan in patients with metastatic triple negative breast cancer. Breast Cancer Res Treat. 2013;137(2):483–492. | |
Orlowski RZ, Kuhn DJ. Proteasome inhibitors in cancer therapy: lessons from the first decade. Clin Cancer Res. 2008;14(6):1649–1657. | |
Voorhees PM, Orlowski RZ. The proteasome and proteasome inhibitors in cancer therapy. Annu Rev Pharmacol Toxicol. 2006;46:189–213. | |
Orlowski RZ, Baldwin AS Jr. NF-kappaB as a therapeutic target in cancer. Trends Mol Med. 2002;8(8):385–389. | |
Cardoso F, Ross JS, Picart MJ, Sotiriou C, Durbecq V. Targeting the ubiquitin-proteasome pathway in breast cancer. Clin Breast Cancer. 2004;5(2):148–157. | |
Codony-Servat J, Tapia MA, Bosch M, et al. Differential cellular and molecular effects of bortezomib, a proteasome inhibitor, in human breast cancer cells. Mol Cancer Ther. 2006;5(3):665–675. | |
Tseng LM, Liu CY, Chang KC, Chu PY, Shiau CW, Chen KF. CIP2A is a target of bortezomib in human triple negative breast cancer cells. Breast Cancer Res. 2012;26;14(2):R68. | |
Côme C, Laine A, Chanrion M, et al. CIP2A is associated with human breast cancer aggressivity. Clin Cancer Res. 2009;15(16):5092–5100. | |
Mita MM, Mita AC, Moseley J, et al. A phase I study of the CDK inhibitor dinaciclib (SCH 727965) administered every 3 weeks in patients (pts) with advanced malignancies: final results. J Clin Oncol. 2011;29(Suppl):abstr 3080. | |
Nemunaitis J, Saltzman M, Rosenberg MA, et al. A phase I dose-escalation study of the safety, pharmacokinetics (PK), and pharmacodynamics (PD) of SCH 727965, a novel cyclin-dependent kinase inhibitor, administered weekly in subjects with advanced malignancies. J Clin Oncol. 2009;27:abstr 3535. | |
Mita M, Joy AA, Mita A, et al. Randomized phase II trial of the cyclin-dependent kinase inhibitor dinaciclib (MK-7965) versus capecitabine in patients with advanced breast cancer. Clin Breast Cancer. 2013;14(3):169–176. | |
Drummond DC, Noble CO, Kirpotin DB, Guo Z, Scott GK, Benz CC. Clinical development of histone deacetylase inhibitors as anticancer agents. Annu Rev Pharmacol Toxicol. 2005;45:495–428. | |
Liu T, Kuljaca S, Tee A, Marshall GM. Histone deacetylase inhibitors: multifunctional anticancer agents. Cancer Treat Rev. 2006;32(3):157–165. | |
Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26(37):5541–5552. | |
Atadja P. Development of the pan-DAC inhibitor panobinostat (LBH589):successes and challenges. Cancer Lett. 2009;280(2):233–241. | |
Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res. 2007;5(10):981–989. | |
Fukutomi A, Hatake K, Matsui K, et al. A phase I study of oral panobinostat (LBH589) in Japanese patients with advanced solid tumors. Invest New Drugs. 2012;30(3):1096–1106. | |
Drappatz J, Lee EQ, Hammond S, et al. Phase I study of panobinostat in combination with bevacizumab for recurrent high-grade glioma. J Neurooncol. 2012;107(1):133–138. | |
Lemoine M, Younes A. Histone deacetylase inhibitors in the treatment of lymphoma. Discov Med. 2010;10(54):462–470. | |
Tate CR, Rhodes LV, Segar HC, et al. Targeting triple-negative breast cancer cells with the histone deacetylase inhibitor panobinostat. Breast Cancer Res. 2012;14(3):R79. | |
Catalano MG, Pugliese M, Gargantini E, et al. Cytotoxic activity of the histone deacetylase inhibitor panobinostat (LBH589) in anaplastic thyroid cancer in vitro and in vivo. Int J Cancer. 2012;130(3):694–604. | |
Fortunati N, Catalano MG, Marano F, et al. The pan-DAC inhibitor LBH589 is a multi-functional agent in breast cancer cells: cytotoxic drug and inducer of sodium-iodide symporter (NIS). Breast Cancer Res Treat. 2010;124(3):667–675. | |
Palmieri D, Lockman PR, Thomas FC, et al. Vorinostat inhibits brain metastatic colonization in a model of triple-negative breast cancer and induces DNA double-strand breaks. Clin Cancer Res. 2009;15(19):6148–6157. | |
Rao R, Balusu R, Fiskus W, et al. Combination of pan-histone deacetylase inhibitor and autophagy inhibitor exerts superior efficacy against triple-negative human breast cancer cells. Mol Cancer Ther. 2012;11(4):973–983. | |
Brave M, Goodman V, Kaminskas E, et al. Sprycel for chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia resistant to or intolerant of imatinib mesylate. Clin Cancer Res. 2008;14(2):352–359. | |
Finn RS, Dering J, Ginther C, et al. Dasatinib, an orally active small molecule inhibitor of both the src and abl kinases, selectively inhibits growth of basal-type/“riple-negative” breast cancer cell lines growing in vitro. Breast Cancer Res Treat. 2007;105(3):319–326. | |
Huang F, Reeves K, Han X, et al. Identification of candidate molecular markers predicting sensitivity in solid tumors to dasatinib: rationale for patient selection. Cancer Res. 2007;67(5):2226–2238. | |
Pichot CS, Hartig SM, Xia L, et al. Dasatinib synergizes with doxorubicin to block growth, migration, and invasion of breast cancer cells. Br J Cancer. 2009;101():38–47. | |
Finn RS, Bengala C, Ibrahim N, et al. Dasatinib as a single agent in triple-negative breast cancer: results of an open-label phase 2 study. Clin Cancer Res. 2011;17(21):6905–6913. | |
Kim EM, Mueller K, Gartner E, Boerner J. Dasatinib is synergistic with cetuximab and cisplatin in triple-negative breast cancer cells. J Surg Res. 2013;185(1):231–239. | |
Polakis P. Wnt signaling in cancer. Cold Spring Harb Perspect Biol. 2012;4(5):pii:a008052. | |
Nusse R, Varmus H. Three decades of Wnts: a personal perspective on how a scientific field developed. EMBO J. 2012;31(12):2670–2684. | |
Liu J, Pan S, Hsieh MH, et al. Targeting Wnt-driven cancer through the inhibition of Porcupine by LGK974. Proc Natl Acad Sci U S A. 2013;110(50):20224–20229. | |
Polakis P. The many ways of Wnt in cancer. Curr Opin Genet Dev. 2007;17(1):45–51. | |
MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17(1):9–26. | |
Khramtsov AI, Khramtsova GF, Tretiakova M, Huo D, Olopade OI, Goss KH. Wnt/b-catenin pathway activation is enriched in basal-like breast cancers and predicts poor outcome. Am J Pathol. 2010;176(6):2911–2920. | |
Geyer FC, Lacroix-Triki M, Savage K, et al. β-Catenin pathway activation in breast cancer is associated with triple-negative phenotype but not with CTNNB1 mutation. Mod Pathol. 2011;24():209–231. | |
Howe LR, Brown AM. Wnt signaling and breast cancer. Cancer Biol Ther. 2004;3(1):36–41. | |
Gurney A, Axelrod F, Bond CJ, et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc Natl Acad Sci U S A. 2012;109(29):11717–11722. | |
Gong Y, Bourhis E, Chiu C, et al. Wnt isoform-specific interactions with coreceptor specify inhibition or potentiation of signaling by LRP6 antibodies. PLoS One. 2010;5:e12682. | |
Ettenberg SA, Charlat O, Daley MP, et al. Inhibition of tumorigenesis driven by different Wnt proteins requires blockade of distinct ligand-binding regions by LRP6 antibodies. Proc Natl Acad Sci U S A. 2010;107(35):15473–15478. | |
Chen B, Dodge ME, Tang W, et al. Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat Chem Biol. 2009;5(2):100–107. | |
Dodge ME, Moon J, Tuladhar R, et al. Diverse chemical scaffolds support direct inhibition of the membrane-bound O-acyltransferase porcupine. J Biol Chem. 2012;287(27):23246–23254. | |
Clevers H, Nusse R. Wnt/β-catenin signaling and disease. Cell. 2012;149(6):1192–1205. | |
Herr P, Hausmann G, Basler K. WNT secretion and signalling in human disease. Trends Mol Med. 2012;18(8):483–493. | |
Biechele S, Cox BJ, Rossant J. Porcupine homolog is required for canonical Wnt signaling and gastrulation in mouse embryos. Dev Biol. 2011;355(2):275–285. | |
Barrott JJ, Cash GM, Smith AP, Barrow JR, Murtaugh LC. Deletion of mouse Porcn blocks Wnt ligand secretion and reveals an ectodermal etiology of human focal dermal hypoplasia/Goltz syndrome. Proc Natl Acad Sci U S A. 2011;108(31):12752–12757. | |
Novartis Pharmaceuticals. A study of oral LGK974 in patients with malignancies dependent on wnt ligands. In: ClinicalTrials.gov [website on the Internet]. Bethseda, MD: US National Library of Medicine; 2011 [updated March 31, 2014]. Available from: http://clinicaltrials.gov/show/NCT01351103. NLM identifier: NCT01351103. Accessed October 1, 2014. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.