")
Back to Journals » Drug Design, Development and Therapy » Volume 16
Trimetazidine Reduces Cardiac Fibrosis in Rats by Inhibiting NOX2-Mediated Endothelial-to-Mesenchymal Transition
Authors Chen X, Xia X, Dong T, Lin Z, Du L, Zhou H
Received 1 February 2022
Accepted for publication 23 July 2022
Published 3 August 2022 Volume 2022:16 Pages 2517—2527
DOI https://doi.org/10.2147/DDDT.S360283
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Anastasios Lymperopoulos
Xingxing Chen,* Xue Xia,* Tiancheng Dong, Zhiwei Lin, Leilei Du, Hao Zhou
Department of Cardiology, the First Affiliated Hospital of Wenzhou Medical University, Wenzhou, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Hao Zhou, Department of Cardiology, the First Affiliated Hospital of Wenzhou Medical University, NanBai Xiang Avenue, Ouhai District, Wenzhou, 325000, People’s Republic of China, Tel +86 1 396 880 1939, Fax +86 577 555 79796, Email [email protected]
Purpose: Endothelial-to-mesenchymal transition (EndMT) is an important mechanism underlying cardiac fibrosis. The anti-ischemic drug trimetazidine (TMZ) is reportedly useful in ventricular remodeling and associated with NADPH oxidase (NOX) 2. This study aimed to investigate the possible effect of TMZ on cardiac fibrosis exerted via the inhibition of NOX2-mediated EndMT.
Methods: A cardiac fibrosis model was established in Sprague-Dawley rats through a subcutaneous injection of isoproterenol (ISO, 5 mg/kg/d). Echocardiographic parameters, myocardial fibrosis, NOX2 expression and EndMT were assessed. An in vitro model of EndMT was developed using human umbilical vein endothelial cells (HUVECs) via treatment with transforming growth factor-β (TGF-β) at 10 ng/mL for 24 h. HUVECs were administrated with TMZ or TMZ and lentivirus, the expression of EndMT and related proteins was observed by wound healing assay, immunoblotting, and immunofluorescence.
Results: Rats injected with ISO exhibited severe interstitial cardiac fibrosis and perivascular fibrosis, decreased left ventricular ejection fraction, and increased NOX activity. TMZ treatment mitigated cardiac fibrosis, ameliorated left ventricular dysfunction, and reduced NOX activity. In addition, TMZ effectively inhibited EndMT in ISO-treated rat hearts and TGF-β-treated HUVECs, as manifested by increased CD31 expression, decreased α-SMA expression, and suppressed cell migration. Compared with the control group, the expression of NOX2, nuclear factor-κB (NF-κB), and Snail was increased in vivo and in vitro but decreased with TMZ treatment. Furthermore, the overexpression of NOX2 by lentivirus abolished the protective effects of TMZ on TGF-β-induced EndMT.
Conclusion: TMZ may ameliorate EndMT and ISO-induced cardiac fibrosis through the NOX2/NF-κB/Snail pathway. The findings of the study may provide new insights into the potential role of TMZ in the pathophysiology of cardiac fibrosis.
Keywords: trimetazidine, endothelial-to-mesenchymal transition, cardiac fibrosis, NADPH oxidase 2
Introduction
Myocardial fibrosis, characterized as interstitial fibroblast proliferation and excessive collagen deposition, is the structural basis of myocardial stiffness and the key process of cardiac function transformation from the compensatory phase to heart failure.1 Thus, it is imperative to understand the mechanisms involved in inhibition of myocardial fibrosis.
Cardiac fibroblasts are generally the primary effector cells of fibrosis and have been reported to be partly derived from cardiac endothelial cells through the endothelial to mesenchymal transition (EndMT) process.2 EndMT is a process by which endothelial cells lose a portion of their cellular features and obtain the mesenchymal phenotype, including the loss of tight junctions and increased production of extracellular matrix. Recently, EndMT has been increasingly recognized as a vital process that contributes to various cardiovascular pathologies in adults, which include atherosclerosis, valvular heart disease, cardiac fibrosis, and myocardial infarction. EndMT reportedly promoted cardiac fibrosis, as evidenced by a lineage-tracking quantitative analysis that demonstrated that 27–35% of fibroblasts in cardiac fibrosis tissues are derived from endothelial cells.2 In the fibrotic hearts of patients with chronic kidney disease, nearly 20% of the fibroblasts were transformed via EndMT.3
Previous evidence suggests that the progression of cardiac remodeling and ventricular contractile dysfunction in patients with ischemic cardiomyopathy is a consequence of substrate metabolism disorder.4 Trimetazidine (TMZ) is an anti-anginal agent that reduces the oxidation rate of free fatty acids by inhibiting mitochondrial long-chain 3-ketoacyl CoA thiolase, which stimulates the heart to utilize glucose oxidation for energy supply and optimization of energy metabolism.5 Standard use of TMZ effectively relieves the clinical symptoms of patients with ischemic heart disease.6–8 TMZ has also been reported to improve cardiomyocytes’ tolerance to hypoxia without affecting the hemodynamics, left ventricular dimensions, and left ventricular ejection fraction (LVEF) in ischemic cardiomyopathy.9,10 Furthermore, TMZ exerts cardioprotection in non-ischemic etiologies such as idiopathic dilated cardiomyopathy and diabetic cardiomyopathy.11,12 Recent evidence shows that the early use of TMZ delays the incidence and development of cardiac fibrosis in rat models of diabetic cardiomyopathy,13 which suggests that the beneficial actions of TMZ on the heart may not only affect cardiomyocytes but also non-cardiomyocytes. Although some studies have evidence that TMZ inhibits cardiac fibrosis, its role as an anti-fibrotic through EndMT has not yet been elucidated.
NADPH oxidase (NOX) enzymes play an important role in tissue remodeling as demonstrated by their involvement in epithelial to mesenchymal transition (EMT) and organ fibrosis.14–16 The primary function of NOX proteins, a family of heme-containing proteins, is to transport electrons from NADPH to oxygen, which results in the formation of reactive oxygen species (ROS).17 Previous studies have also elaborated on the role of NOX2 in the pathophysiology of allograft tubulointerstitial fibrosis mediated via EMT activation.18 TMZ prevents diabetes-induced left ventricular hypertrophy and fibrosis by inhibiting NOX2/TRPC3-induced oxidative stress.19 Thus, this study sought to investigate whether TMZ affects cardiac fibrosis through NOX2-mediated EndMT.
Materials and Methods
Animal Model
Male Sprague-Dawley rats (6-week-old) were obtained from the Shanghai Experimental Animal Center. The rats were randomly assigned to three groups (n = 10 per group): (1) control, (2) isoproterenol (ISO, 5 mg/kg/day), and (3) ISO + TMZ (15 mg/kg/day). The rats were subcutaneously injected with ISO (I6504, Sigma, St. Louis, MO, USA) for 7 days and then fed for another 14 days to establish experimental cardiac fibrosis. The control rats were similarly administered with an equal volume of saline. From day 1, daily gavage of TMZ (653322, Sigma, St. Louis, MO, USA) was performed for the ISO + TMZ group for 21 days, and the ISO group received the same volume of saline. On day 21, the rats were euthanized after subjecting them to echocardiography and the hearts were subsequently harvested. All experiments were approved by the Institutional Research Ethics Committee of Wenzhou Medical University and performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Echocardiography
The rats were anesthetized via the intraperitoneal injection of pentobarbital sodium (40 mg/kg). After the anterior chest depilation of the rats, the cardiac function was detected using a Sonos 5500 ultrasound machine (Philips, Bothell, WA, USA) with a 12 MHz linear array transducer. Two-dimensional, M-mode images were acquired in the parasternal long-axis view to measure the left ventricular end-diastolic diameter (LVEDD) and left ventricular end-systolic diameter. The results of LVEF and fractional shortening (FS) were obtained.
Histological Analysis
The heart tissues were fixed in 4% neutral formaldehyde and visualized via hematoxylin and eosin (HE) staining as well as Masson trichrome staining. Ventricle samples were embedded in paraffin, sectioned into 4-μm paraffin sections, and separately stained with HE and Masson trichrome (Solarbio Life Sciences, Beijing, China). The sectioned ventricle samples were examined via light microscopy. Collagen volume fraction (CVF) was calculated via quantitative morphometry with automated image analysis (Image-Pro Plus, Houston, TX, USA).
NOX Activity Assay
NOX activity was measured via lucigenin-enhanced chemiluminescence using a commercial kit (Nanjing JianCheng Bio, Jiangsu, China) according to the manufacturer’s instructions. Briefly, 0.1 g of tissue was accurately weighed, 1 mL of Reagent I and 10 µL of Reagent III were added, and the mixture was homogenized on ice. After centrifugation at 4°C for 5 min, the supernatant was transferred to another tube and centrifuged further at 4°C for 10 min. The obtained supernatant was demitochondrial cytoplasmic protein, and 200 µL of Reagent II and 2 µL of Reagent III were added and then sonicated to use the sample for the determination of NOX activity. The absorbance was detected at a wavelength of 600 nm using an automatic microplate reader (Multiskan MK3, Thermo scientific, USA).
Cell Culture and Lentivirus Transfection
Primary human umbilical vein endothelial cells (HUVECs) were purchased from the cell bank of the Chinese Academy of Science (Shanghai, China). The HUVECs were cultured in Dulbecco’s Modified Eagle Medium (Lonza, USA) with 10% fetal bovine serum (Gibco, NY, USA), 100 U/mL penicillin (Gibco, NY, USA), and 100 mg/mL streptomycin (Gibco, NY, USA). Transforming growth factor-β (TGF-β, 10 ng/mL; 100-21, PeproTech, New Jersey, USA) was used to induce EndMT. For the cells subjected to TMZ intervention, pretreatment was performed with TMZ for 1 h before TGF-β induction. The HUVECs were incubated at 37°C in a 5% CO2 incubator. Human recombinant lentivirus targeting NOX2 was purchased from Genechem Co., Ltd. (Shanghai, China). HUVECs were transfected with nontarget control lentivirus (lv-ctrl) or NOX2-targeted lentivirus (lv-NOX2) according to the manufacturer’s protocol. After 24-h transfection, the medium was changed, and the cells were preadministrated with TMZ (80 μM) for 1 h and then coincubated with TGF-β without medium change. In addition to the above, a new medium was replaced before each treatment.
Wound Healing Assay
The HUVECs were grown to confluence. A linear wound was made by scraping the cell monolayer with a p200 pipette tip, and the first image was photo-documented using the dish marker as the reference point. After scratching for 24 h, the cells were washed twice with phosphate buffer solution (PBS) to remove detached cells and debris. Then, the size of the injury was recorded at the indicated times. A minimum of three different wound field images were acquired and used for subsequent assessments.
Immunofluorescence Assay
Immunofluorescence staining was used to evaluate the EndMT of HUVECs. Briefly, ventricular myocardium sections or cell slides were prepared. Sections or slides were incubated with the primary antibodies against anti-CD31 (1:100; ab28364, Abcam) and anti-α-SMA (1:500; A5228, Sigma) at 4°C overnight. After washing thrice with PBS, the sections were incubated with secondary antibodies (1:500; Alexa Fluor 594 Goat Anti-Rabbit IgG H&L, 33112ES60; Alexa Fluor 594 Goat Anti-mouse IgG H&L, 33212ES60, YEASEN, Shanghai, China) for 1 h and then with DAPI for 5 min to stain the nuclei. Sections incubated with suitable isotype control primary antibodies and fluorescent-labeled secondary antibodies were used as negative controls.
Western Blotting Analysis
The ventricular tissue was lysed in radioimmunoprecipitation assay buffer and then centrifuged at 4°C. The supernatant was collected, and protein concentrations were determined using the BCA protein assay (Solarbio Life Sciences, Beijing, China). Equal amounts of sample (80 μg) were separated using 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and then transferred to polyvinylidene fluoride membranes (pore size, 0.45 μm; Millipore, USA). Blots were blocked with 5% skimmed milk in Tris-buffered saline with 0.1% Tween 20 (TBST) and incubated with diluted primary antibodies overnight at 4°C, which was followed by incubation with horseradish peroxidase-conjugated secondary antibodies. Bands were obtained using a BioRad gel image analysis system (BioRad, Hercules, CA, USA) and analyzed using Image-Pro Plus (Media Cybernetics, Houston, TX, USA). The following antibodies were used: anti-CD31 (1:1000; ab28364, Abcam), anti-α-SMA (1:1000; ab5694, Abcam), anti-NOX2 (1:1000; ab80508, Abcam), Snail (1:1000; 3879, Cell Signaling Technology), and nuclear factor-κB (NF-κB) p65 (1:1000; 8242, Cell Signaling Technology).
Statistical Analysis
Data are presented as the mean ± standard deviation and were analyzed using the SPSS 20.0 software. Normality of the data was determined by the Kolmogorov–Smirnov and Shapiro–Wilk test in SPSS software. One-way analysis of variance was used to compare more than two groups. A p-value of <0.05 was considered statistically significant.
Results
TMZ Mitigated Cardiac Fibrosis and Improved Heart Function in ISO-Induced Rats
The study aimed to clarify the cardioprotective benefits of TMZ by improving cardiac fibrosis in rats with ISO-induced heart failure. The hearts of ISO-induced rats showed extensive collagen deposition; the extent of interstitial fibrosis was significantly higher in ISO-induced hearts than in control hearts (Figure 1A). ISO-induced hearts also exhibited a higher CVF than control hearts (Figure 1B), which indicates the successful establishment of a cardiac fibrosis model using ISO. Furthermore, ISO induction resulted in the deterioration of cardiac function, as demonstrated by increased LVEDD, decreased LVEF, and left ventricular FS (Figure 2A and B). TMZ markedly improved ISO-induced left ventricular fibrosis, LVEF, and left ventricular FS.
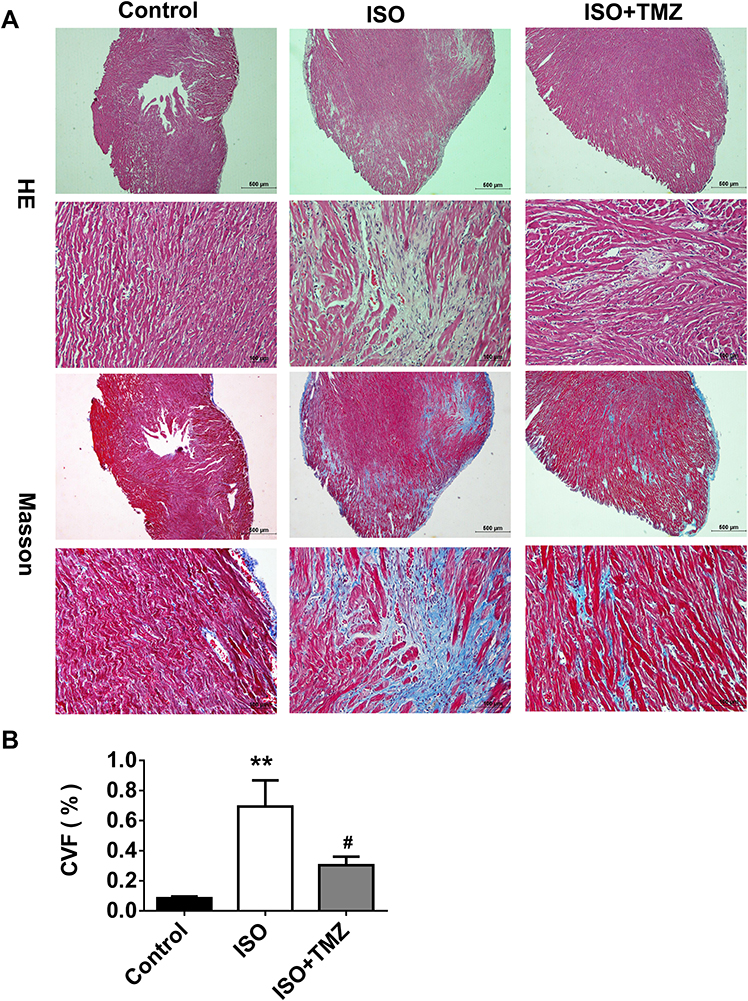 |
Figure 1 TMZ reduces ISO-induced cardiac fibrosis in rats. (A) HE staining and Masson staining of the rat cardiac tissues (n = 4 per group). (B) Analysis of collagen volume fraction (CVF) according to Masson staining results (n = 4 per group). Data are presented as mean ± standard deviation. **P < 0.01 vs Control; #P < 0.05 vs ISO. Abbreviation: ISO, isoproterenol. |
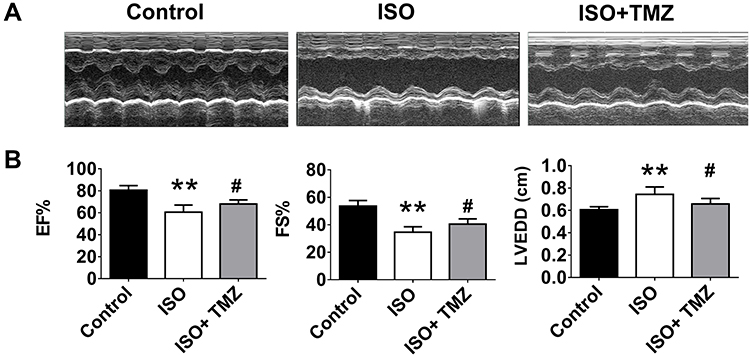 |
Figure 2 Echocardiographic assessment of cardiac function. (A) Representative M-mode echocardiographic photos. (B) Analysis of left ventricular function (EF, ejection fraction; FS, fractional shortening) and cardiac dimension (LVEDD, left ventricular end-diastolic internal diameter), n = 4 per group. Data are presented as mean ± standard deviation. **P < 0.01 vs Control; #P<0.05 vs ISO. Abbreviation: ISO, isoproterenol. |
TMZ Inhibited EndMT in ISO-Induced Hearts and TGF-β-Treated HUVECs
The results of the immunofluorescence and immunoblotting showed the occurrence of EndMT in the ISO group with the characteristic downregulation of endothelial-specific CD31 expression and upregulation of fibroblast-specific α-SMA expression (Figure 3A and B). Notably, TMZ administration reversed the changes caused by ISO induction.
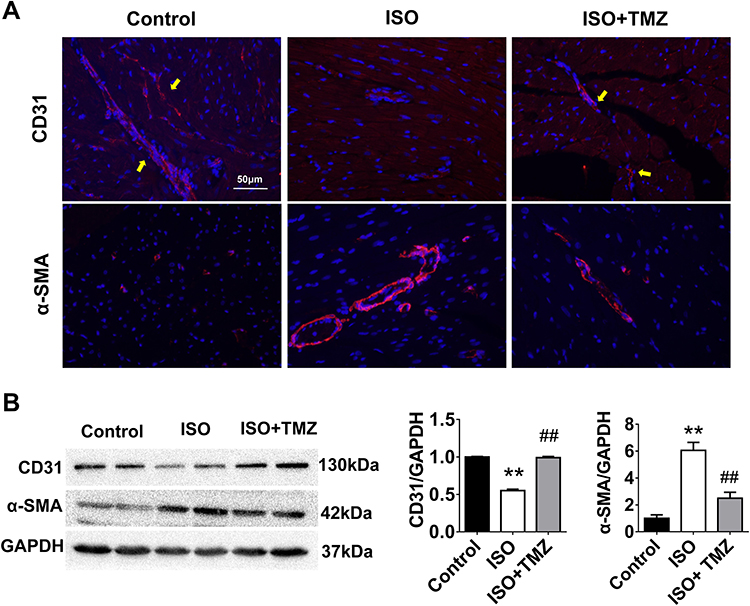 |
Figure 3 TMZ inhibits EndMT in the hearts of ISO-induced rats. (A) Immunofluorescence staining of EndMT markers (n = 4 per group). CD31 (endothelial marker, yellow arrow) and α-SMA (mesenchymal marker) are stained in red. Nuclei are stained with DAPI (blue). Scale bar, 50 μm. (B) Protein expression of CD31 and α-SMA measured by Western blotting analysis (n = 4 per group). Data are presented as mean ± standard deviation. **P < 0.01 vs Control; ##P < 0.01 vs ISO. Abbreviation: ISO, isoproterenol. |
To further investigate the effects of TMZ on EndMT, whether TMZ administration reduced TGF-β-induced EndMT in HUVECs was explored. HUVECs exposed to TGF-β exhibited elevated α-SMA levels and reduced CD31 levels on immunoblotting and immunofluorescent experiments compared with the control. TMZ treatment significantly downregulated α-SMA expression and upregulated CD31 expression (Figure 4A and B). The wound healing assay further showed that the percentage of wound healing distance between cell scratches in the TGF-β group was significantly higher than that in the control group, whereas the wound healing distance in the TGF-β + TMZ group was lower than that in the TGF-β group (Figure 5A and B).
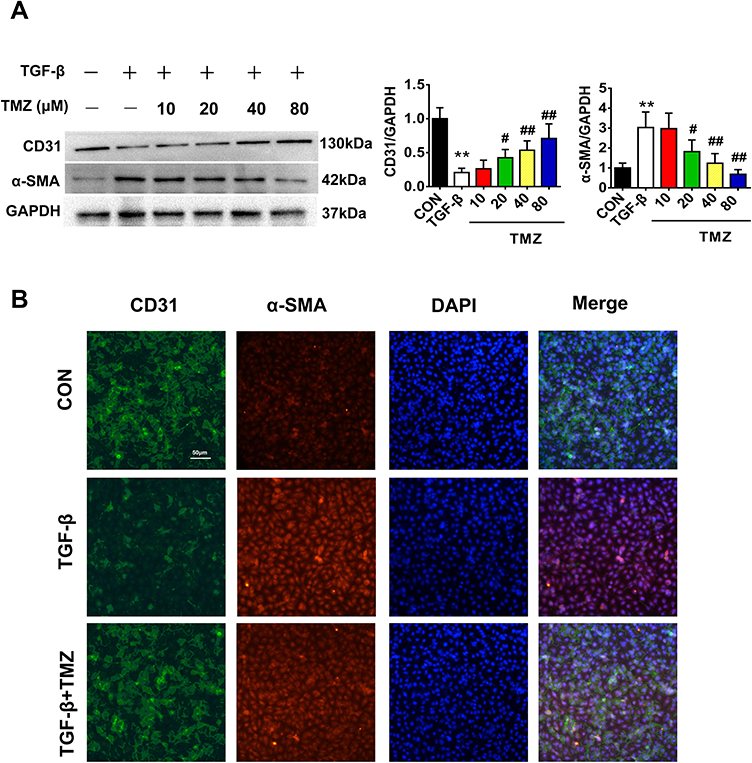 |
Figure 4 TMZ mitigates TGF-β-induced EndMT in HUVECs. (A) Western blotting analysis of the protein expression of CD31 and α-SMA (n = 4 per group). (B) Immunofluorescence staining of EndMT markers (n = 3 per group). Red, α-SMA; green, CD31; and nucleus, blue. Scale bar, 50 μm. Data are presented as mean ± standard deviation. **P < 0.01 vs CON; #P < 0.05 vs TGF-β; ##P < 0.01 vs TGF-β. Abbreviation: TGF-β, transforming growth factor-β. |
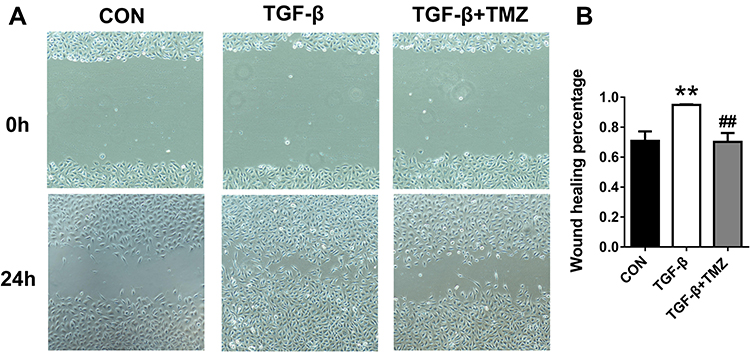 |
Figure 5 TMZ inhibits the migration of TGF-β-treated HUVECs. (A) Representative migration images of HUVECs. (B) Analysis of wound healing percentage was performed at 24 h. Data are presented as mean ± standard deviation. **P < 0.01 vs CON; ##P < 0.01 vs TGF-β. Abbreviation: TGF-β, transforming growth factor-β. |
NOX2 Participates in the Regulation of EndMT by TMZ
This study speculated that TMZ may regulate EndMT through the NOX2 pathway. Initially, ISO infusion for 7 days was noted to upregulate NOX activity in the left ventricular myocardium. NOX activity was significantly higher in ISO-induced hearts than in control hearts (ISO 113.85 ± 13.22 vs control 45.19 ± 8.11 U/mg prot; P < 0.01). NOX activity was significantly lower in the ISO + TMZ group than in the ISO group (77.40 ± 8.88 vs 113.85 ± 13.22 U/mg prot, respectively; P < 0.01). TMZ inhibited the increase of ISO-induced NOX activity. Moreover, the expression of NOX2 as well as NF-κB and Snail was also significantly increased in ISO-treated rats but decreased with TMZ treatment (Figure 6A).
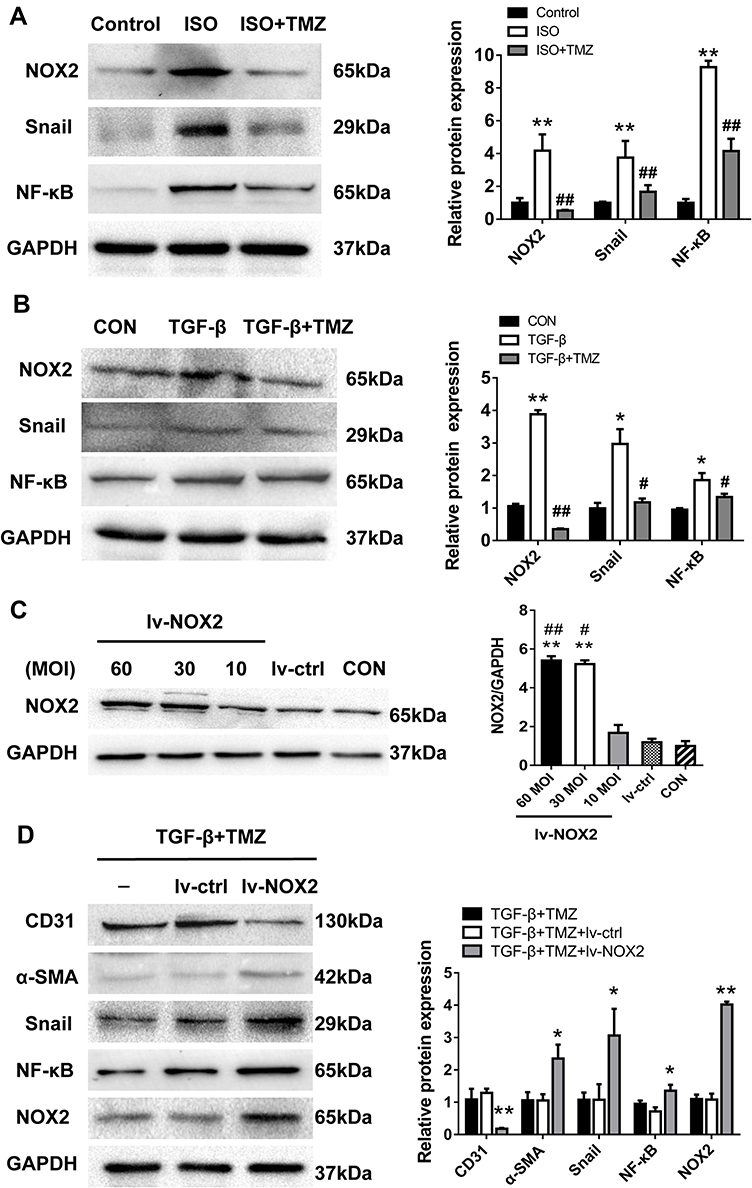 |
Figure 6 TMZ modulates EndMT via NOX2. (A) Western blotting analysis of the expression of NOX2, NF-κB, and Snail in ISO-treated rats (n = 3–4 per group). **P < 0.01 vs Control; ##P<0.01 vs ISO. ISO, isoproterenol. (B) Western blotting analysis of NOX2, NF-κB, and Snail expression in HUVECs (n = 4 per group). *P < 0.05 vs CON; **P < 0.01 vs CON; #P < 0.05 vs TGF-β; ##P < 0.01 vs TGF-β. (C) Western blotting analysis verified the efficiency of NOX2 overexpression (n = 3 per group). **P < 0.01 vs lv-ctrl; #P < 0.05 vs 10 MOI; ##P < 0.01 vs 10 MOI. (D) Western blotting analysis of the expression of NOX2, NF-κB, and Snail after NOX2 overexpression (n = 3–5 per group). Cells were transfected with nontarget (lv-ctrl) or NOX2-targeted lentivirus (lv-NOX2) for 24 h. Then, TMZ (80 μM) was added 1 h before the cells’ exposure to TGF-β. Data are presented as mean ± standard deviation. *P < 0.05 vs TGF-β + TMZ + lv-ctrl; **P < 0.01 vs TGF-β +TMZ + lv-ctrl. Abbreviation: TGF-β, transforming growth factor-β. |
In vitro, NOX2 expression was upregulated in TGF-β-treated HUVECs and downregulated with TMZ administration in the TGF-β group (Figure 6B). The HUVECs were transfected with lentivirus to overexpress NOX2 before TMZ administration. The results showed the upregulation of α-SMA expression and downregulation of CD31 expression in the TGF-β + TMZ + lv-NOX2 group compared with those in the TGF-β + TMZ + lv-ctrl group, which indicates that TMZ failed to reduce EndMT in NOX2-overexpressing HUVECs (Figure 6C and D). TMZ has previously been reported to inhibit pressure overload-induced cardiac fibrosis through NOX–ROS–connective tissue growth factor (CTGF) pathway.20 NOX2-dependent ROS generation may result in the activation of the mitogen-activated protein kinase (MAPK) and downstream transcription factor NF-κB. Compared with the control group, the expression of NF-κB and Snail was upregulated in TGF-β-treated HUVECs but was downregulated with TMZ administration (Figure 6B). The overexpression of NOX2 increased the protein levels of NF-κB and Snail (Figure 6D), as evidenced by the elevated expression of NF-κB and Snail in lv-NOX2-transfected HUVECs.
Discussion
Cardiac fibrosis is a common pathological manifestation and an important pathophysiological basis of many end-stage cardiovascular diseases.21 In this study, the effects of TMZ on ISO-induced cardiac fibrosis and its potential mechanisms were investigated. TMZ significantly reduced myocardial fibrosis and improved left ventricular dysfunction in ISO-induced rats. It also effectively inhibited EndMT in vivo and in vitro. NOX activity and NOX2 expression were upregulated in ISO-induced rats but downregulated in TMZ-treated rats. Moreover, the expression of NOX2 was upregulated in TGF-β-induced EndMT but was downregulated after TMZ treatment. The overexpression of NOX2 mitigated the protective effects of TMZ on EndMT. Furthermore, the expression of downstream NF-κB and Snail was downregulated wtih TMZ administration but upregulated with NOX2 overexpression. Together, these data suggest that TMZ protects against ISO-induced cardiac fibrosis by inhibiting EndMT and NOX2 activation.
TMZ is a clinically effective anti-ischemic drug that has been widely used in ischemic heart disease. Previous studies have reported that TMZ, at the cellular level, plays a direct protective role by altering the cellular aerobic metabolism, accelerating adenosine triphosphate production efficiency, and preserving mitochondrial structure and function. However, the cardioprotective benefit of TMZ is not limited to metabolism modulation; it also includes the improvement of myocardial apoptosis, fibrosis, autophagy, and inflammation. Previous clinical trials have demonstrated an increase in LVEF and a decrease in left ventricular volume in patients receiving TMZ for 6 months;8,22,23 moreover, the improvement in echocardiographic parameters suggests that TMZ helps ameliorate cardiac remodeling.
Previous evidence suggests that TMZ regulates local important factors (eg, Angiotensin II [Ang II] and TGF-β1) to inhibit myocardial fibrosis. Ang II can stimulate the secretion of various fibrogenic factors (eg, NOX2 and CTGF) in the myocardium, thus activating fibroblasts, increasing collagen synthesis, and accelerating myocardial fibrosis.24 Ang II also increases NOX2 release; hence, NOX2 is a crucial factor for Ang II–induced cardiac fibrosis. In a previous study, the induction of interstitial cardiac fibrosis by Ang II was completely inhibited in NOX2-knockout mice.25 TMZ prevented Ang II–induced myocardial fibrosis primarily through the NOX–ROS–CTGF signaling pathway and reduced ROS formation in cardiac fibroblast proliferation.20 In addition, the early administration of TMZ ameliorated myocardial fibrosis in diabetic cardiomyopathy by suppressing phosphorylation of extracellular signal-regulated kinase (ERK) and p38MAPK.13
The findings of our study also demonstrated that TMZ treatment ameliorated ISO-induced heart failure in rats by decreasing myocardial fibrosis, reducing collagen deposition, decreasing LVEDD, and improving left ventricular function. The results were consistent with those of the previous trials that showed improvement in left ventricular volume and contractility after TMZ administration. Furthermore, TMZ inhibited EndMT in ISO-induced hearts and TGF-β-treated HUVECs, which suggests that the anti-fibrosis effects of TMZ are associated with EndMT modulation. Similarly, a previous study showed that TMZ prevents renal dysfunction and tubulointerstitial fibrosis in diabetic rats by inhibiting EMT via the ROS and TGF-β/Smad pathways.26 To the best of our knowledge, this study is the first to report that TMZ improves cardiac fibrosis via EndMT regulation, which probes for further elucidation of the mechanisms underlying for the modulation of EndMT after TMZ administration.
Previous studies have suggested that endothelial NOX2 promotes interstitial cardiac fibrosis through EndMT and inflammation. Transgenic mice with endothelial-specific NOX2 overexpression develop severe cardiac fibrosis and enhanced EndMT compared with wild-type mice.27 A higher level of NOX2 expression was noted in ISO-induced hearts than in control hearts. The NOX activity was also found to be significantly increased in the ISO group. More definitive evidence showed that the expression of NOX2 was higher in TGF-β-induced EndMT than in control HUVECs. TMZ mitigated EndMT in cultured HUVECs and downregulated NOX2 expression. Furthermore, the overexpression of NOX2 weakened the inhibitory effects of TMZ on EndMT. These results strongly suggest that NOX2-mediated downregulation of EndMT is a vital mechanism in the amelioration of ISO-induced cardiac fibrosis with TMZ treatment.
NOX family members play important roles in ROS production in both phagocytic and non-phagocytic cells. ROS are generated by nonphagocytic NOXs that function as secondary messengers for various transcription factors, including NF-κB, p38MAPK, AP-1, and ERK1/2.17,28 Several inflammatory mediators such as proinflammatory cytokines, growth factors, oxidative stress, and toxins induce the conversion of endothelial cells into mesenchymal fibroblast-like cells that promote disease progression. NF-κB is a common player that drives EndMT.29 NF-κB expression was increased in TGF-β-treated HUVECS but decreased with TMZ treatment. The overexpression of NOX2 increased the expression of NF-κB. The expression trend of Snail was similar to that of NF-κB. TGF-β-induced EndMT is associated with strong upregulation of the expression of the transcriptional repressor Snail.30,31 Snail directly promotes the differentiation of endothelial cells into the mesenchymal phenotype; thus, enhanced Snail expression further validates the occurrence of EndMT. Previous studies have reported that the activation of NF-κB triggers Snail transcription and EMT induction by binding to its promoter.32,33 Therefore, the increased expression of NF-κB may activate downstream Snail expression and promote EndMT.
The role of EndMT in fibrosis has been confirmed by many studies, but there are also a few studies with different conclusions. Thomas Moore-Morris used new transgenic tracer mice to demonstrated that cardiac fibroblasts are rarely derived from EndMT during cardiac fibrosis induced by pressure overload.34 Another study found that epicardium-derived mesenchymal cells but not endocardial-derived mesenchymal cells were the major source of fibroblasts in tissues of endocardial fibroblastic hyperplasia.35 These results are inconsistent with previous studies published by Elisabeth M Zeisberg.2,36 Since there is no definitive standard for the diagnosis of EndMT, the disputes are mainly related to the use of different tracer animals, EndMT markers, time points and inducements.
This study has some limitations. In vivo experiments involving NOX2 overexpression or knockout were not performed in the present study. Some previous studies suggest that sex differences exist in ISO-induced cardiac fibrosis—male rats are more susceptible to cardiac fibrosis than female rats.37 In fact, sex-related differences are prevalent in cardiovascular disease risk, pathophysiology, disease manifestation, response to therapy, and prognosis.38 Thus, the effects of TMZ on cardiac fibrosis in female animals/women should be investigated further.
Conclusion
In summary, the study demonstrated that TMZ mitigated myocardial fibrosis and enhanced left ventricle contractility in ISO-induced heart failure in rats by inhibiting EndMT, perhaps partially through the NOX2/NF-κB/Snail pathway. The results provide experimental evidence to support the use of TMZ in the treatment of cardiac fibrosis; however, more in-depth studies are warranted in the future.
Acknowledgments
This research was supported by the Foundation for the Program of the Provincial Health Department of Zhejiang Province of China (2019RC051) and the Science and Technology Planning Project of Wenzhou Science & Technology Bureau of Zhejiang Province of China (Y20180144).
Disclosure
The authors confirm that there are no conflicts of interest in this work.
References
1. Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. 2014;71:549–1574. doi:10.1007/s00018-013-1349-6
2. Zeisberg EM, Tarnavski O, Zeisberg M, et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi:10.1038/nm1613
3. Charytan DM, Padera R, Helfand AM, et al. Increased concentration of circulating angiogenesis and nitric oxide inhibitors induces endothelial to mesenchymal transition and myocardial fibrosis in patients with chronic kidney disease. Int J Cardiol. 2014;176:99–109. doi:10.1016/j.ijcard.2014.06.062
4. Stanley WC, Recchi FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–1129. doi:10.1152/physrev.00006.2004
5. Kantor PF, Lucien A, Kozak R, et al. The anti-anginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long-chain 3-ketoacyl coenzyme A thiolase. Circ Res. 2000;86:580–588. doi:10.1161/01.RES.86.5.580
6. Li YY, Wang DW, Hu CX, et al. Efficacy and safety of adjunctive trimetazidine therapy for acute myocardial infarction: a systematic review and meta-analysis. Cardiology. 2016;135:188–195. doi:10.1159/000446640
7. Chazov EI, Lepakchin VK, Zharova EA, et al. Trimetazidine in Angina Combination Therapy-the TACT study: trimetazidine versus conventional treatment in patients with stable angina pectoris in a randomized, placebo-controlled, multicenter study. Am J Ther. 2005;12:35–42. doi:10.1097/00045391-200501000-00006
8. Napoli PD, Taccardi AA, Barsotti A. Long term cardioprotective action of trimetazidine and potential effect on the inflammatory process in patients with ischaemic dilated cardiomyopathy. Heart. 2005;91:161–165. doi:10.1136/hrt.2003.031310
9. Belardinelli R, Purcaro A. Effects of trimetazidine on the contractile response of chronically dysfunctional myocardium to low-dose dobutamine in ischaemic cardiomyopathy. Eur Heart J. 2001;22:2164–2170. doi:10.1053/euhj.2001.2653
10. Bertomeu-Gonzalez V, Bouzas-Mosquera A, Kaski JC. Role of trimetazidine in management of ischemic cardiomyopathy. Am J Cardiol. 2006;98:19–24. doi:10.1016/j.amjcard.2006.07.005
11. Wang WM, Tang QZ. Early administration of trimetazidine may prevent or ameliorate diabetic cardiomyopathy. Med Hypotheses. 2011;76:181–183. doi:10.1016/j.mehy.2010.09.012
12. Jatain S, Kapoor A, Sinha A, et al. Metabolic manipulation in dilated cardiomyopathy: assessing the role of trimetazidine. Indian Heart J. 2016;68:803–808. doi:10.1016/j.ihj.2016.04.023
13. Zhang L, Ding WY, Wang ZH, et al. Early administration of trimetazidine attenuates diabetic cardiomyopathy in rats by alleviating fibrosis, reducing apoptosis and enhancing autophagy. J Transl Med. 2016;14:109. doi:10.1186/s12967-016-0849-1
14. Rhyu DY, Yang YQ, Ha H, et al. Role of reactive oxygen species in TGF-beta1-induced mitogen activated protein kinase activation and epithelial-mesenchymal transition in renal tubular epithelial cells. J Am Soc Nephrol. 2005;16:667–675. doi:10.1681/ASN.2004050425
15. Nabeebaccus A, Zhang M, Shah AM. NADPH oxidases and cardiac remodelling. Heart Fail Rev. 2011;16:5–12. doi:10.1007/s10741-010-9186-2
16. Kato K, Hecker L. NADPH oxidases: pathophysiology and therapeutic potential in age-associated pulmonary fibrosis. Redox Biol. 2020;33:101541. doi:10.1016/j.redox.2020.101541
17. Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi:10.1152/physrev.00044.2005
18. Djamali A, Vidyasagar A, Adulla M, Hullett D, Reese S. Nox-2 is a modulator of fibrogenesis in kidney allografts. Am J Transplant. 2009;9:74–82. doi:10.1111/j.1600-6143.2008.02463.x
19. Tang SG, Liu XY, Wang SP, Wang HH, Jovanović A, Tan W. Trimetazidine prevents diabetic cardiomyopathy by inhibiting Nox2/TRPC3-induced oxidative stress. J Pharmacol Sci. 2019;139:311–318. doi:10.1016/j.jphs.2019.01.016
20. Liu XH, Gai YL, Liu F, et al. Trimetazidine inhibits pressure overload-induced cardiac fibrosis through NADPH oxidase-ROS-CTGF pathway. Cardiovasc Res. 2010;88:150–158. doi:10.1093/cvr/cvq181
21. Czubryt MP, Hale TM. Cardiac fibrosis: pathobiology and therapeutic targets. Cell Signal. 2021;85:110066. doi:10.1016/j.cellsig.2021.110066
22. Fragasso G, Piatti PM, Monti L, et al. Short- and long-term beneficial effects of trimetazidine in patients with diabetes and ischemic cardiomyopathy. Am Heart J. 2003;146:E18–E25. doi:10.1016/S0002-8703(03)00415-0
23. Rosano GM, Vitale C, Sposato B, Mercuro G, Fini M. Trimetazidine improves left ventricular function in diabetic patients with coronary artery disease: a double-blind placebo-controlled study. Cardiovasc Diabetol. 2003;2:16. doi:10.1186/1475-2840-2-16
24. Lijnen P, Petrov V. Induction of cardiac fibrosis by aldosterone. J Mol Cell Cardiol. 2000;32:865–879. doi:10.1006/jmcc.2000.1129
25. Johar S, Cave AC, Narayanapanicker A, Grieve DJ, Shah AM. Aldosterone mediates angiotensin II-induced interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase. FASEB J. 2006;20:1546–1548. doi:10.1096/fj.05-4642fje
26. Yang Y, Wang Y, He ZW, et al. Trimetazidine inhibits renal tubular epithelial cells to mesenchymal transition in diabetic rats via upregulation of Sirt1. Front Pharmacol. 2020;11:1136. doi:10.3389/fphar.2020.01136
27. Murdoch CE, Chaubey S, Zeng L, et al. Endothelial NADPH oxidase-2 promotes interstitial cardiac fibrosis and diastolic dysfunction through proinflammatory effects and endothelial-mesenchymal transition. J Am Coll Cardiol. 2014;63:2734–2741. doi:10.1016/j.jacc.2014.02.572
28. Gill PS, Wilcox CS. NADPH oxidases in the kidney. Antioxid Redox Signal. 2006;8:1597–1607. doi:10.1089/ars.2006.8.1597
29. Maleszewska M, Moonen JR, Huijkman N, van de Sluis B, Krenning G, Harmsen MC. IL-1β and TGFβ2 synergistically induce endothelial to mesenchymal transition in an NFκB-dependent manner. Immunobiology. 2013;218:443–454. doi:10.1016/j.imbio.2012.05.026
30. Kokudo T, Suzuki Y, Yoshimatsu Y, Yamazaki T, Watabe T, Miyazono K. Snail is required for TGFbeta induced endothelial-mesenchymal transition of embryonic stem cell-derived endothelial cells. J Cell Sci. 2008;121:3317–3324. doi:10.1242/jcs.028282
31. Xu X, Tan X, Tampe B, Sanchez E, Zeisberg M, Zeisberg EM. Snail is a direct target of Hypoxia-inducible Factor 1α (HIF1α) in hypoxia-induced endothelial to mesenchymal transition of human coronary endothelial cells. J Biol Chem. 2015;290:16653–16664. doi:10.1074/jbc.M115.636944
32. Wu YD, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP. Stabilization of snail by NF-kappaB is required for inflammation-induced cell migration and invasion. Cancer Cell. 2009;15:416–428. doi:10.1016/j.ccr.2009.03.016
33. Tsubaki M, Komai M, Fujimoto S, et al. Activation of NF-κB by the RANKL/RANK system up-regulates snail and twist expressions and induces epithelial-to-mesenchymal transition in mammary tumor cell lines. J Exp Clin Cancer Res. 2013;32:62. doi:10.1186/1756-9966-32-62
34. Moore-Morris T, Guimarães-Camboa N, Banerjee I, et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J Clin Invest. 2014;24:2921–2934. doi:10.1172/JCI74783
35. Zhang H, Huang X, Liu K, et al. Fibroblasts in an endocardial fibroelastosis disease model mainly originate from mesenchymal derivatives of Epicardium. Cell Res. 2017;27:1157–1177. doi:10.1038/cr.2017.103
36. Xu X, Friehs I, Zhong Hu T, et al. Endocardial fibroelastosis is caused by aberrant endothelial to mesenchymal transition. Circ Res. 2015;116:857–866. doi:10.1161/CIRCRESAHA.116.305629
37. Peter AK, Walker CJ, Ceccato T, et al. Cardiac fibroblasts mediate a sexually dimorphic fibrotic response to β-adrenergic stimulation. J Am Heart Assoc. 2021;10:e018876. doi:10.1161/JAHA.120.018876
38. Bairey Merz CN, Shaw LJ, Reis SE, et al. Insights from the NHLBI-Sponsored Women’s Ischemia Syndrome Evaluation (WISE) Study: part II: gender differences in presentation, diagnosis, and outcome with regard to gender-based pathophysiology of atherosclerosis and macrovascular and microvascular coronary disease. J Am Coll Cardiol. 2006;47:S21–29. doi:10.1016/j.jacc.2004.12.084
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.