")
Back to Journals » The Application of Clinical Genetics » Volume 12
TRIM5α 136Q, CCR5 Promoter 59029G And CCR264I Alleles Impact The Progression Of HIV In Children And Adolescents
Authors Dambaya B, Nkenfou CN, Mekue L, Této G, Ngoufack N, Ambada G, Flobert N, Colizzi V, Alexis N
Received 14 February 2019
Accepted for publication 27 May 2019
Published 7 November 2019 Volume 2019:12 Pages 203—211
DOI https://doi.org/10.2147/TACG.S205335
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Béatrice Dambaya,1,2 Céline Nguefeu Nkenfou,1,3 Linda Mekue,1,4 Georges Této,1 Nicole Ngoufack,1,2 Georgia Ambada,1,2 Njiokou Flobert,1 Vittorio Colizzi,5 Ndjolo Alexis1,6
1Chantal BIYA International Reference Centre for Research on HIV/AIDS Prevention and Management (CBIRC), Yaoundé, Cameroon; 2Department of Animal Biology, Faculty of Sciences, University of Yaounde I, Yaoundé, Cameroon; 3Department of Biological Sciences, Higher Teachers’ Training College, University of Yaounde I, Yaoundé, Cameroon; 4Department of Biochemistry, Faculty of Sciences, University of Dschang, Dschang, Cameroon; 5Department of Immunology, University of Rome Tor Vergata, Rome, Italy; 6Department of Ear, Nose and Throat, Faculty of Medicine and Biomedical Sciences, University of Yaounde I, Yaoundé, Cameroon
Correspondence: Céline Nguefeu Nkenfou
Higher Teacher Training College, University of Yaounde I, P.O. BOX 47, Yaounde, Cameroon
Tel +675573519
Email [email protected]
Background: Children show various degrees of vulnerability regarding HIV infection and disease progression. This disparity presents challenges for the follow-up of infected children. Here we investigated reasons behind this variability focusing on some host-related HIV genes.
Methods: We screened 570 Cameroonian children and adolescents, aged 1 to 19 years old. Among them, 137 were followed over 4 years, from 2010 to 2015. Upon signing a proxy consent, children and adolescents were classified according to their age, CD4 count, viral load and clinical symptoms as long-term non-progressors (LTNP), slow progressors (SP) and rapid progressors (RP). Their blood was collected every 6 months and used for biological and host genetic polymorphism analyses. Five genes were genotyped: Trim5α (R136Q), CCR5 promoter 59029G, CCR2-64I, SDF 3ʹA and CCR5-Δ32. Exposed non-infected (HEU) and unexposed HIV negative children (HNEU) were recruited as control groups.
Results: Among the 5 genes studied, the protective allele of Trim5α (R136Q) was present in all LTNP and in 72.34% and 2.56% of SP and RP, respectively (p<0.0001). The CCR5 promoter 59029G/G was also more present in LTNP and SP than in RP (p=0.02; p=0.04). The protective CCR2-64I homozygous genotype was almost absent in all groups, only the heterozygous genotype was present with a significant difference between RP vs SP (p=0.0001), and SP vs LTNP (p=0.0002). The CCR2-∆32 was completely absent either as homozygous or heterozygous genotype. It was a monomorphic allele. SDF 3ʹA was almost present as homozygous wild-type genotype in our study population and was associated neither to disease acquisition nor to disease progression.
Conclusion: Among the 5 genes described in the study, Trim 5α (R136Q), CCR5 promoter 59029G and CCR2V64I alleles were associated to the progression of HIV infection in children and adolescents.
Keywords: aids related genes, infected children, disease progression
Introduction
Globally, it is known that the pathogenesis of HIV-1 infection presents some variability in the clinical outcome of people exposed to and infected with HIV virus, due to multiple factors. One of these factors is the variability in the host genetic constitution. Disease progression follows the same trends in children as in adults and two extreme cases have been for longtime characterized. Those who succumb within 2 years after the infection are called the rapid progressors (RP), others who survive for several years after the infection without treatment are long-term non-progressors (LTNP); they show minimal or no progression of disease with relatively normal CD4 count and low viral loads for longer than 8 years.1 They are also called elite controllers. Beside them there is also a group of slow progressors, call chronic or normal progressors. Since some years now, a new group of those exposed and non-infected is growing all around the world and deserves attention. HIV disease pathogenicity varies according to viral biological properties, host immune and host genetic responses.1,2 Several immunoregulatory genes termed AIDS restriction genes (ARGs), impact the entry of the virus into the host cell.2 Mutations present in the receptors or their ligands have been associated with the HIV infection.3 In the context of HIV-1 infection, restriction factors may help host cells in controlling viral replication.4 The tripartite interaction motif 5α (Trim5α) has been identified as part of the intrinsic immunity that protects human and non-human primates against retroviral infection.5,6 In human, it has been observed that Trim 5 alpha escape variants develop late infection in a proportion of HIV-1 infected individuals.7 It is known that the presence of mutations in C-C chemokine receptors CCR2, CCR5, and CXCR4 ligands SDF1 (stromal cell-derived factor 1) are associated with protection against HIV-1 infection and restriction to AIDS progression.8 CCR2 is an important entry co-receptor for HIV-1 infecting CD4+ host cells.9 There are a number of controversies over SDF1 genotypes and its association to HIV-1 infection,10 with the SDF1-3ʹA polymorphism, consisting of a G to A mutation at position 801.11 Genetic polymorphism of CCR5 wild type and CCR5-Δ32 is one of the best documented studies showing how genetic polymorphism can regulate the prevalence of disease in a population.12 Thus, individuals in various populations harboring CCR2V64I, CCR5-Δ32 and CCR5 promoter mutations are less susceptible to HIV-1 infection and progress much slowly to AIDS.
Our study aimed at identifying the distribution of five commonly reported ARGs, Trim 5α, CCR264I, CCR5-Δ32, CCR5 promoter and SDF1 3ʹA mutations and correlate them with HIV/AIDS disease progression in a cohort of HIV-1 vertically infected children.
Materials And Methods
Patients
From a total of 570 Cameroonian children and adolescents screened, 91 HIV positive participants aged from 1 to 15 years old, fulfilling inclusion criteria (perinatally infected), were enrolled and further analyzed. At the time of their enrollment, biological data, CD4+ Tcells count, viral load and clinical symptoms served as the set point for their classification as RP, SP and LTNP groups. Thirty-one HIV exposed uninfected (HEU) and 46 HIV non-exposed uninfected (HNEU) children were recruited as control groups.
After assuring anonymity, written informed consent from parents and guardians was obtained for biological and clinical testing as well as for genetic polymorphisms' analyses. The time of onset of HIV infection was considered as the date of their birth, and the length of infection was their age. For children older than 18 months, HIV status was tested by the detection of HIV-1 antibodies using Determine HIV 1/2 test (Alere, 357 Matsuhidai, Matsuda-shi, Chiba, 270–2214 Japan) and confirmed using the Genie III HIV-1/HIV-2 test (Biorad 3, Bd Raymond Poincaré, 92,430 Marnes La Coquette, France). For children less than 18 months, Dried Blood Spot (DBS) samples were tested for the presence of HIV proviral DNA using Roche Amplicor HIV DNA version 1.5. The medical records of each child were examined for any retrospective clinical signs or opportunistic infections such as skin rash, zona, oral candidosis, chronic diarrhea, heavy cough, bronchopneumonia and pulmonary tuberculosis. These criteria added to their age at enrollment, and their CD4+ T cell counts and viral load were used to classify each of them in a specific group, either as RP, SP or LTNP. The inclusion criteria for LTNP were defined as asymptomatic over 10 years after infection/diagnosis, plasma HIV RNA levels below 2000 copies/mL for viremic controllers (VC) without any antiretroviral therapy (ART).13 Slow progressors (SP) were defined as children who were ART naives or initiated ART within 10 years after infection/diagnosis, with known HIV-1 infection for more than 5 years, viral load above 2000 copies/mL. Rapid progressors (RP) were defined as children with CD4 cell count <350 cells/mm3, on ART or not, or who died within 2 years.
DNA Extraction And Polymerase Chain Reaction
The Buffy coat and DBS were used as a source of genomic DNA, that was extracted using QiaAmp DNA mini kit (Qiagen S.A. 3 Avenue du Canada, LP 809, 91,974 Courtaboeuf Cedex, France), according to the manufacturer’s instructions. DNA concentration was measured by a nanodrop spectrophotometer.
The CCR5-Δ32, CCR5 promoter, CCR2-64I, SDF1-3ʹ A and Trim 5α genetic variants in participants were determined by PCR followed by RFLP detection using the specific primers and restriction endonucleases as described previously.14–16 Nevertheless, this original protocol was optimized during our study.
The amplification of CCR5-∆32 was done as follows: 1 cycle for 30 s at 94°C, followed by 40 cycles of 30 s, 30 s, and 1 min at 94°c, 50°c, and 72°c, respectively, followed by a final extension of 10 mins at 72°c.
CCR5-promoter gene amplification was done as follows: 3 mins at 94°c, followed by 40 cycles of 30 s, 30 s, and 45 s at 94°c, 60°c, and 72°c, respectively, and a final extension of 10 mins at 72°C.
CCR2 gene was amplified in one cycle of 30 s at 94°C, followed by 40 cycles of 30 s, 30 s, and 30 s at 95°C, 63°C, and 72°C, respectively, and a final extension of 10 mins at 72°C.
To detect SDF-1 gene, the amplification started with a denaturation step of one cycle of 3 mins at 94°C, followed by 40 cycles of 30 s, 30 s, and 30 s at 94°C, 58°C, and 72°C, respectively, and a final extension of 10 mins at 72°C.
The amplification of Trim 5α gene fragment was done using the following conditions: one cycle of 3 mins at 94°C, followed by 40 cycles of 30 s, 30 s and 1 mins at 94°C, 50°C and 72°C, respectively, and a final extension of 10 mins at 72°C.
The amplified fragments were run in agarose gel with variable percentage depending on the fragment size, previously stained with ethidium bromide, and visualized under ultraviolet light.
The above-mentioned gene fragments size and their specific primers,16–18 are presented in Table 1.
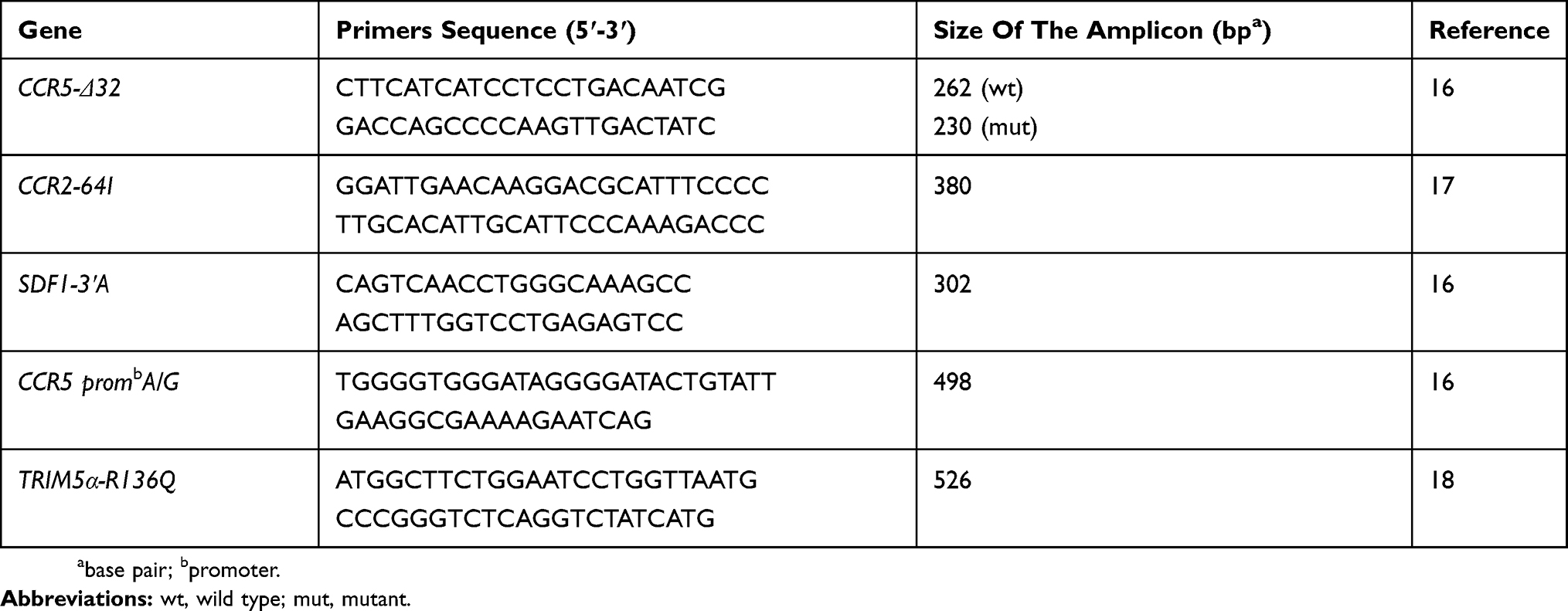 |
Table 1 Primers Used And Expected Fragments Size Of Studied Genes |
Genotypic Analyses
Genotyping was carried out with the use of restriction fragment length polymorphism (RFLP) method except for the CCR5-∆32 deletion based on the respective restriction enzyme sites in the 4 amplified (PCRs) products of CCR5 promoter, Trim 5α, CCR2 and SDF1 as described previously,16–18 (Table 2). The restriction enzymes used in this work were purchased from Thermo Fischer and used according to manufacturer instructions.
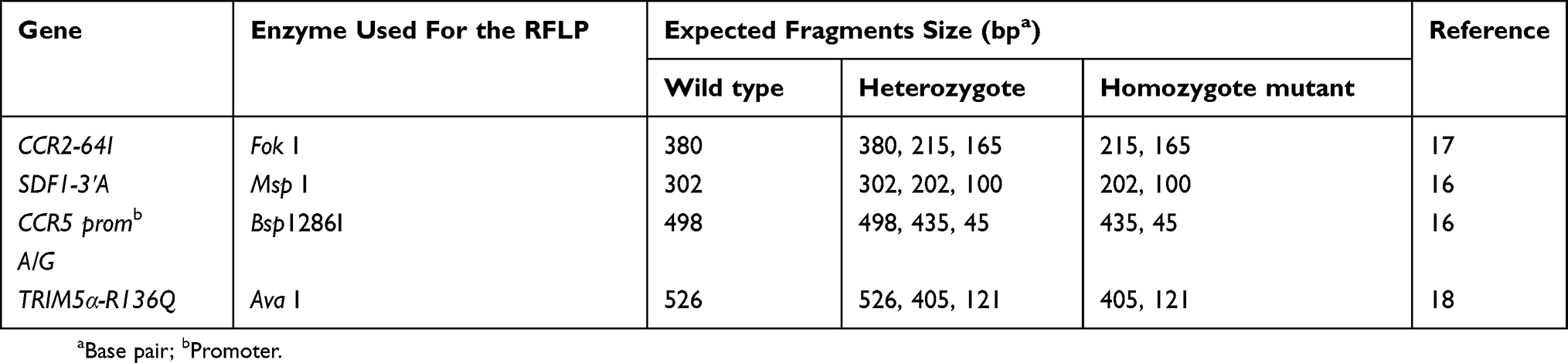 |
Table 2 Enzyme Used And Expected Fragments Size After Digestion |
Determination Of CD4 Counts
CD4+ T cells were quantified using a FACS Calibur flow cytometer [Becton Dickinson Immuno-cytometry System (BDIS), San Jose, CA, USA].
Determination Of HIV Viral Load
The HIV-1 viral load was determined from plasma by Abbott Real-Time HIV-1 assay (Abbott Molecular Diagnostics, Wiesbaden, Germany)19 with a detection limit of 40 copies/mL (1.6 log).
Statistical Analyses
The analyses were performed with the GraphPad Prism 6.0 software using nonparametric tests in all cases. Mann Whitney U-test was used to calculate p values and p <0.05 was considered statistically significant. When indicated, the data were reported as mean and confidence interval, or median and interquartile range (IQR). The allelic frequencies were calculated as (h + 2H)/2N, where H was the number of homozygous mutation genotypes, h was the number of heterozygous mutation genotypes and N was the total number of samples. The allele frequency was further analyzed by Hardy–Weinberg equilibrium (HWE). All alleles achieve HWE. The differences in the allele frequency of each genetic variant between and within the distinct group of HIV-1 seronegative and HIV-1 seropositive groups were determined by Chi-square or Fisher exact test when indicated.
Ethical Considerations
The National Ethics Committee reviewed the proposal for ethical consideration and approval was given under N°103/CNE/SE/2012. Proxy consent form was signed by parent before enrolment. As well, this study was conducted in accordance with the Declaration of Helsinki.
Results
During enrollment, 570 children were screened. Included in the study was a total of 168 children and adolescents classified as follows: 31 HEU, 46 HNEU, 39 RP, 47 SP and 5 LTNP.
These children and adolescents were followed up for 4 years. Among the 137 HIV perinatally acquired children, 40 (29.2%) were not yet on treatment, 97 (70.80%) were already taking drugs. After 4 years, 91 of these children were classified according to disease progression. Some, 46 (33.6%) HIV positive children were excluded as could not be classified in any of the groups. From the following, 5 LTNP were identified corresponding to 3.6% of the overall classified children and adolescents aged 11 to 15 years. The percentages of SP aged 7 to 15 years and RP aged 1 to 2 years, were, respectively, 28.46% and 34.30%. The Socio-demographic, immunologic and virologic characteristics of the study population are indicated in Table 3. Most of the children, 87.17% (35 out of 39), of the RP group were from the early infant diagnosis (EID) program, a PMTCT evaluation program of the Ministry of Public Health of Cameroon, and were for most of the time unfortunately already on stage C of CDC classification, characterized by severe clinical symptoms such as chronic diarrhea, heavy cough, oral candidiasis, skin rash, zona, bronchopneumonia, pulmonary tuberculosis. In the worst case, some were dead.
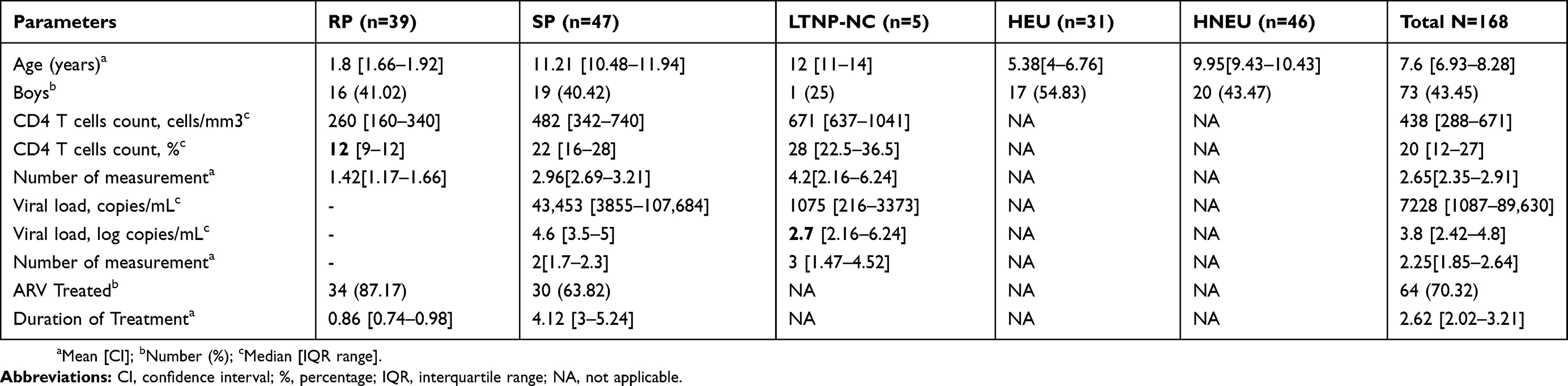 |
Table 3 Socio-Demographic, Immunologic And Virologic Characteristics Of The Study Population |
Genetic Variant Distribution Of The 5 Genes In The Study Population
In the overall population, the double mutation of Trim5 α was frequent (42.26%). The double mutation G/G of CCR5 promoter was less frequent at 20.83%, with 52.38% for heterozygous genotype. SDF3ʹA double mutation was absent, and the heterozygous genotype was present at 5.35%. Most of the participants (94.64%) were homozygous wild type for SDF 3ʹA. CCR5 delta 32 mutation was completely absent in the study population. These data are presented in Table 4.
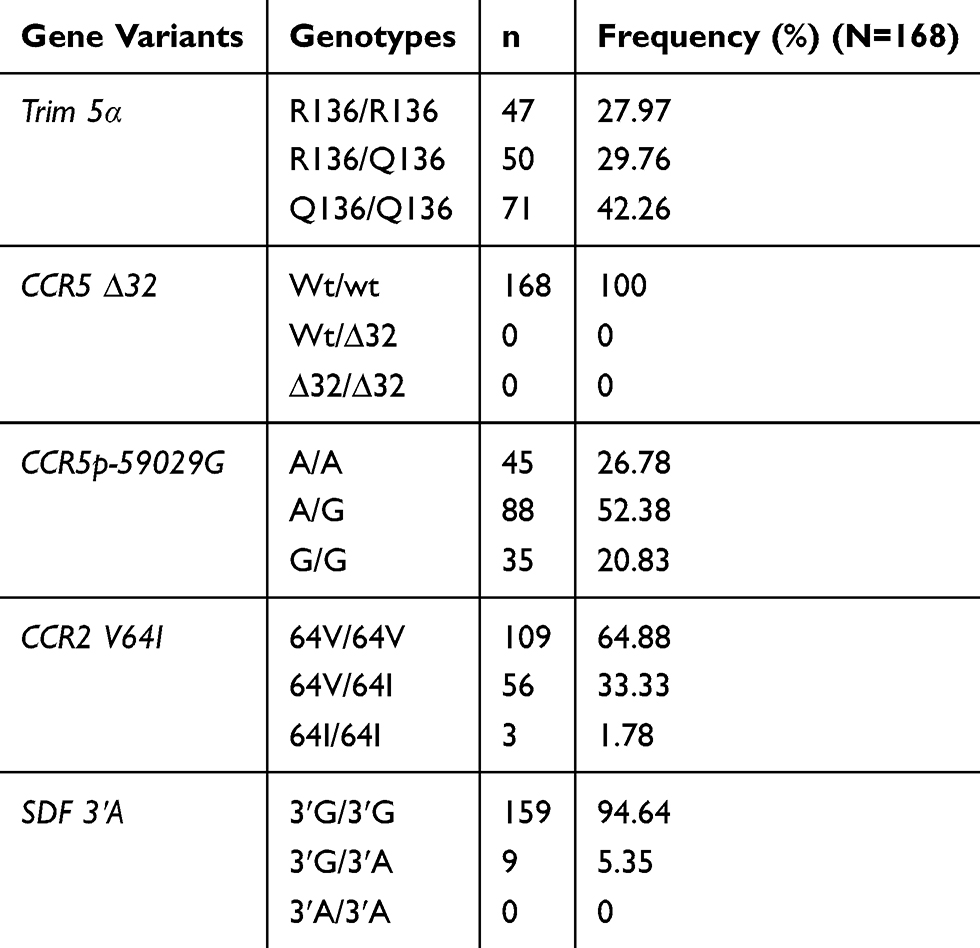 |
Table 4 Distribution Of TRIM 5α, CCR5∆32, CCR5 Promoter 59,029 A/G, CCR2-64I And SDF 3ʹA In The Study Population |
Distribution Of Genetic Variants In Infected Compared To Non-Infected Exposed Participants: Implication In Disease Acquisition
The distribution of various alleles and the frequencies of genotypes were compared between HIV infected participants and HIV exposed non-infected participants. A significant difference was observed between the two groups for Trim 5α R136Q, and CCR5 promoter 59,029 A/G. These alleles may be associated with HIV acquisition. CCR2V64I and SDF 3ʹA may not be associated with HIV infection. These data are summarized in Table 5.
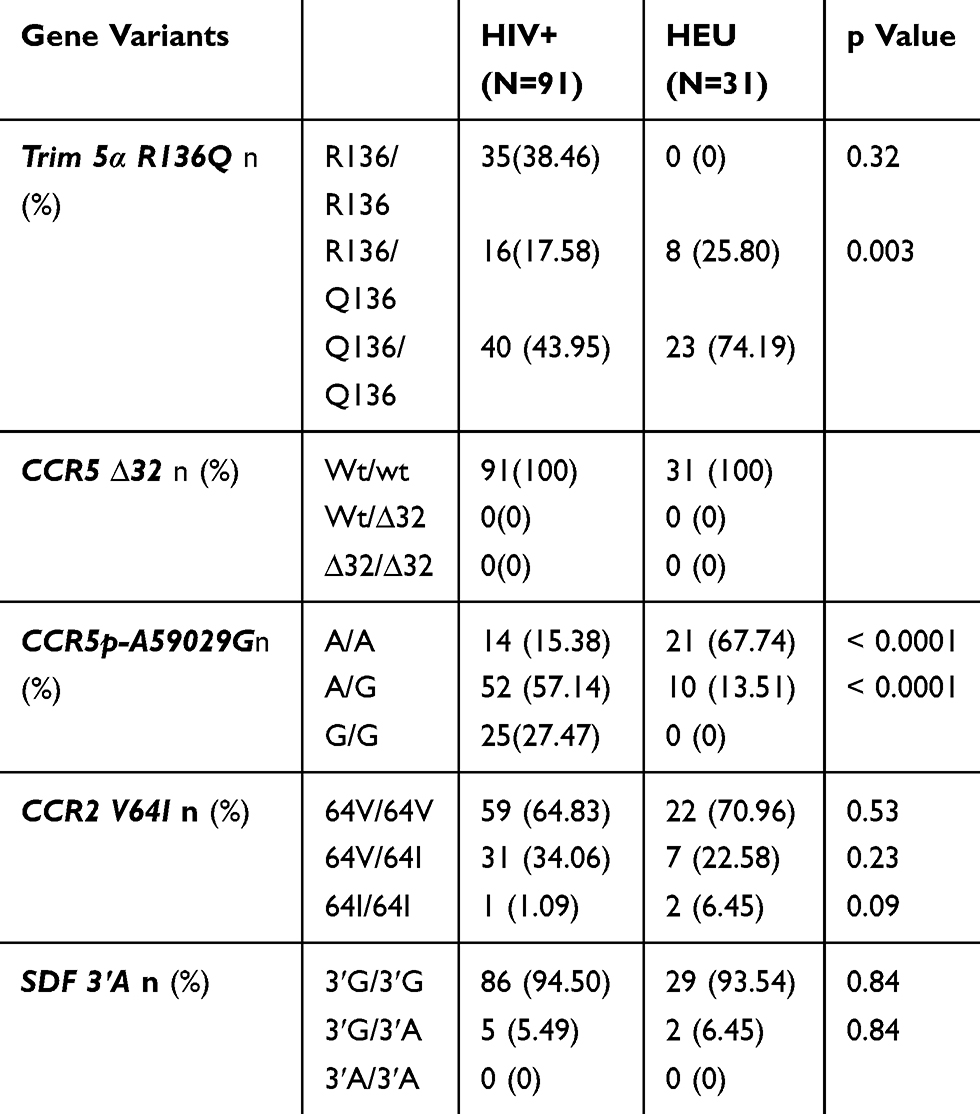 |
Table 5 Prevalence Of Genotype Frequencies In HIV-1 Infected And Exposed Uninfected Children And Adolescents |
Distribution Of Genetic Variants According To Disease Progression
Allele’s distribution in the HIV-1 infected group according to disease progression is presented in Table 6.
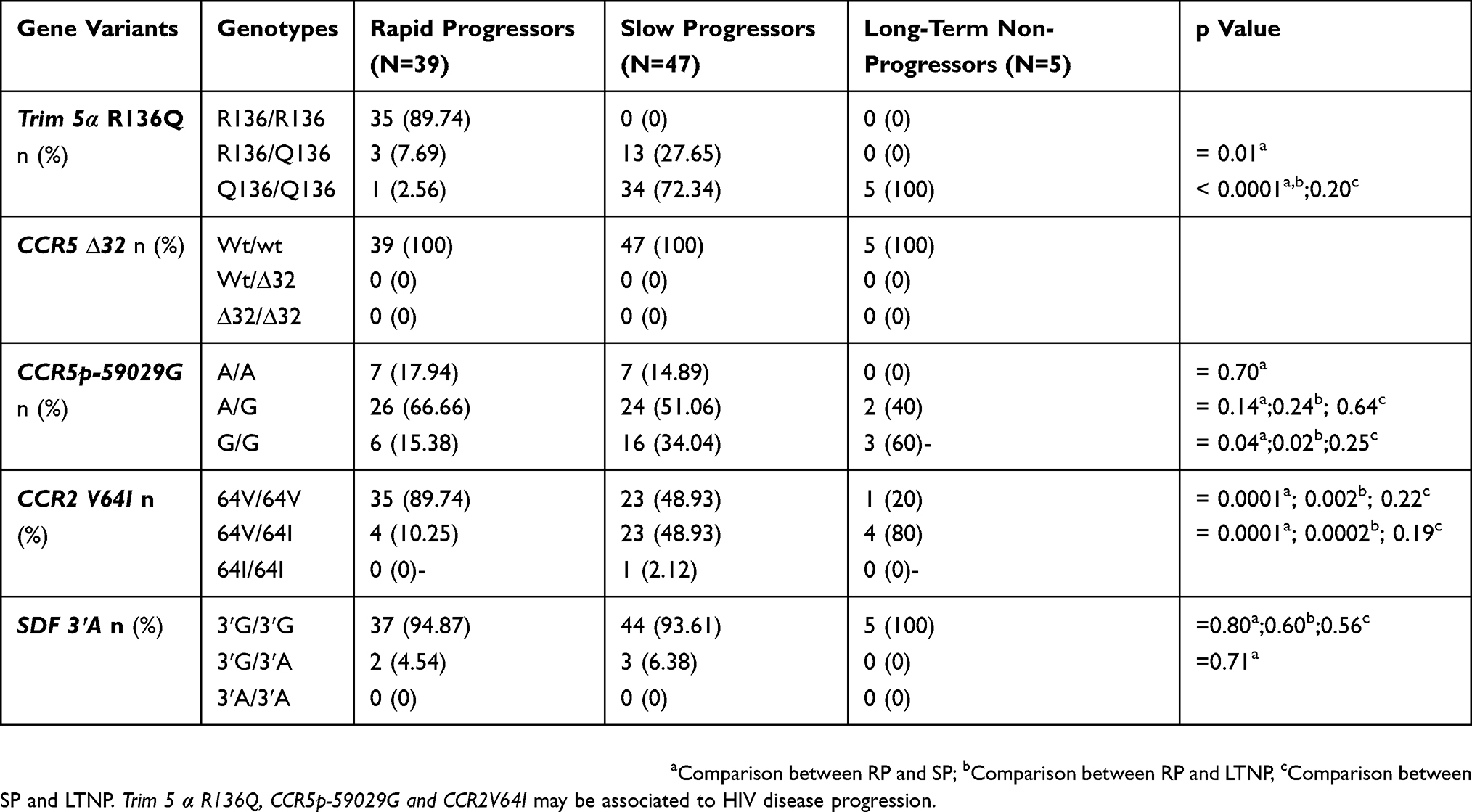 |
Table 6 Prevalence Of Genotype Frequencies In HIV-1 Infected Children And Adolescents According To Disease Progression |
For Trim 5α, the Q 136 polymorphism was observed with a minor allele frequency (MAF) of 0.06, 0.86 and 1 in RP, SP and LTNP groups, respectively. Among the 39 patients belonging to the RP group, 89.74% were homozygous wild type. No homozygous wild type was identified among SP and LTNP groups. The heterozygous (R136/Q136) genotype was observed in 7.69% of RP and 27.65% of SP with a significant difference (p=0.01) between the two groups. Notably, all LTNP were homozygous for the protective allele (Q136/Q136). The homozygous form of this protective allele was observed at 2.56% and 72.34%, respectively, in RP and SP. A significant difference (p<0.0001) was observed between RP and the two other groups (SP and LTNP).
The homozygous protective genotype (G/G) of CCR5 promoter 59029G was present in 15.38%, 34.04% and 60%, respectively, in RP, SP and LTNP groups. A significant difference was observed between RP and SP (p=0.04) and between RP and LTNP (p=0.02). The heterozygous genotype (A/G) was identified in 66.66%, 51.06% and 40% of RP, SP and LTNP groups, respectively.
The heterozygous genotype CCR2V64I was found in 10.25%, 48.93% and 80% in RP, SP and LTNP groups, respectively, with a significant difference between RP and SP (p=0.0001) and SP and LTNP (p=0.0002) groups. Overall, only one patient of the SP group was identified with the homozygous genotype of the protective allele (64I/64I).
The SDF3ʹA homozygous protective allele (3ʹA/3ʹA) was completely absent in our study group. The homozygous wild type form (3ʹG/3ʹG) was more represented in all the three groups at 94.87%, 93.61% and 100% of RP, SP and LTNP, respectively.
The overall analysis of genotype data did not show any deviation from the Hardy–Weinberg expected frequency, the χ2 tests showed that all genes' loci in HIV infected or uninfected are in equilibrium (P>0.05). The observed genotype frequencies had no significant difference from the frequencies expected in each group, indicating that the five alleles are effectively in genetic equilibrium as shown in Table 6.
Discussion
At the time of the study, 29.2% of the study population was not yet on treatment, which was in the era of selective treatment depending on the CD4+T lymphocytes count. Nevertheless, it has been shown that some children were able to remain asymptomatic for many years without taking ART.20–24 The percentage of 3.6% of LTNP obtained in our study confirmed the preliminary existing data of the presence of controllers in Cameroonian pediatric HIV infected population.21 This rate of LTNP is not far from the 3% observed from LTNP children of Spain. This was however less than the 9% attributed to an Italy pediatric12 and Ugandan25 study on LTNP, having in mind difficulty to have a consensus definition of LTNP all over the world.14,26–28 The classification of participants as SP, RP or LTNP has been published.21 In addition, among the LTNP adolescents, 4 were girls (80%) and 1 was a boy (20%). These results confirm the highest percentage of controllers among female infant as shown in the previous studies.20,29–36
Regarding host gene polymorphism, a significant difference was found in the distribution of 136Q allele of Trim 5α gene in the 3 groups of children ranging from RP, SP and LTNPs. This shift from arginine to glutamine at codon 136 of Trim 5 has also been observed in a study done on a different population in Kenya where this mutation conferred protection against HIV-1 in adult sex workers' cohort.37 In African-American individuals, the residue 136Q was also associated with protection against HIV-1 infection.38 This allele is involved in HIV infection as well as in HIV disease progression. It restricts non-infected cells from infection.
The protective allele of CCR5 promoter (as homozygous) distribution also varied from RP to SP and from RP to LTNP in our study. This allele may thus be associated with HIV-1 disease progression in Cameroonian children.
Looking at the distribution of CCR2 64I, a significant difference was noted in the overall population of HIV negative and positive children. According to disease progression, the same result was obtained in RP and SP groups and between RP and LTNP.
The frequency of the SDF1 variant (SDF1-3ʹA) has been shown to vary from 2% to 3% in our study population, no homozygous form of the protective allele was seen. There was no difference in the distribution of this allele neither in HIV infection nor in HIV disease progression. The percentages obtained in our study did not differ from that of the South Africans population (1% to 2%) but differ somehow from the 6% of African Americans,39,40 confirming the very low frequency of this protective allele in our study population.
As no case of protective CCR5-∆32 allele was observed in our study population, this strongly sustains earlier findings that the CCR5∆32 mutation is rare in Africans.41,42
Trim5α R136Q and CCR5p-59029G may be associated with HIV infection. Meanwhile, CCR2V64I and SDF 3ʹA may not be associated to HIV infection. Trim 5 α R136Q, CCR5p-59029G and CCR2V64I may be associated to HIV disease progression.
CCR5 is the main co-receptor for HIV transmission and thus play an important role in HIV acquisition and pathogenesis. Alleles, affecting the primary structure of CCR5 promoter may lead to nonfunctional receptors or otherwise influence AIDS progression. On the other hand, Trim5 is a host restriction factors belonging to the innate immune system that inhibits the replication of HIV-1 virus. Mutations occurring in this gene may affect HIV infectivity and thus HIV progression.
CCR2 is also a co-receptor of HIV virus. Reports on the role of CCR2 and HIV have been controversial. But in the present study, our data showed that CCR2 alleles are involved in disease progression, but not disease acquisition. In vitro CCR2 is rarely used as a co-receptor. This attest population-specific effects of chemokine receptor and ligand genes.43–45
Conclusion
Despite the limitation of our study, consisting in a small sample size of LTNP, we observed that the protective allele of Trim 5α was most frequent and more present in LTNP and SP and less in RP, followed by CCR5 promoter and CCR2. These 3 genes may be involved in controlling disease progression among Cameroonians. Trim5 α and CCR5 promoter may be involved in disease acquisition. Clinically, it may be interesting identifying children with susceptible genotypes in order to tailor their management. We prospect analyses with GWAS in order to identify other genes which can be associated to disease progression.
Acknowledgements
This study was approved and supported by funding from the International Society of Infectious Diseases (ISID) and CBIRC.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Dollfus C, Le Chenadec J, Faye A, et al. Long-term outcomes in adolescents perinatally infected with HIV-1 and followed up since birth in the French perinatal cohort (EPF/ANRS CO10). Clin Infect Dis. 2010;51:214–224. doi:10.1086/652976
2. Shah I, Nadiger M. Long term non progressors (LTNP) with vertically infected HIV children – a report from western India. Indian J Med Res. 2013;137:210–212.
3. O’Brien SJ, Nelson GW. Human genes that limit AIDS. Nat Genet. 2004;36565–36574.
4. Lederman MM, Penn-Nicholson A, Cho M. Mosier. Biology of CCR5 and its role in HIV infection and treatment. JAMA. 2006;296:815–826. doi:10.1001/jama.296.7.815
5. Ioannidis JP, Contopoulos-Ioannidis DG, Rosenberg PS, et al. Effects of CCR5-delta32 and CCR2-64I alleles on disease progression of perinatally HIV-1-infected children: an international meta-analysis. AIDS. 2003;17:1631–1638. doi:10.1097/00002030-200307250-00007
6. Wolf D, Goff SP. Host restriction factors blocking retroviral replication. Annu Rev Genet. 2008;42:143–163. doi:10.1146/annurev.genet.42.110807.091704
7. Stremlau M, Owens CM, Perron MJ, et al. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in old world monkeys. Nature. 2004;427:848–853. doi:10.1038/nature02343
8. Sayah DM, Sokolskaja E, Berthoux L, Luban J. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature. 2004;430:569–573. doi:10.1038/nature02777
9. Kootstra NA, Navis M, Beugeling C, van Dort KA, Schuitemaker H. The presence of the Trim5alpha escape mutation H87Q in the capsid of late stage HIV-1 variants is preceded by a prolonged asymptomatic infection phase. Aids. 2007;21:2015–2023. doi:10.1097/QAD.0b013e3282effa87
10. de Silva E, Stumpf MP. HIV and the CCR5-Delta32 resistance allele. FEMS Microbiol Lett. 2004;241:1–12. doi:10.1016/j.femsle.2004.09.040
11. Al-Abdulhadi SA, Al-Rabia MW. Linkage and haplotype analysis for chemokine receptors clustered on chromosome 3p21.3 and transmitted in family pedigrees with asthma and atopy. Ann Saudi Med. 2010;30:115–122. doi:10.4103/0256-4947.60516
12. Carrington M, Dean M, Martin MP, O’Brien SJ. Genetics of HIV-1 infection: chemokine receptor CCR5 polymorphism and its consequences. Hum Mol Genet. 1999;8:1939–1945. doi:10.1038/nm0798-786
13. Cherney RJ, Mo R, Meyer DT, et al. Discovery of disubstituted cyclohexanes as a new class of CC chemokine receptor 2 antagonists. J Med Chem. 2008;51:721–724. doi:10.1021/jm701488f
14. Mummidi S, Ahuja SS, Gonzalez E, et al. Genealogy of the CCR5 locus and chemokine system gene variants associated with altered rates of HIV-1 disease progression. Nat Med. 1998;4:786–793. doi:10.1038/nm0798-786
15. Casado C, Colombo S, Rauch A, et al. Host and viral genetic correlates of clinical definitions of HIV-1 disease progression. PLoS One. 2010;5:e11079. doi:10.1371/journal.pone.0011079
16. Kristiansen TB, Knudsen TB, Ohlendorff S, Eugen-Olsen J. A new multiplex PCR strategy for the simultaneous determination of four genetic polymorphisms affecting HIV-1 disease progression. J Immunol Methods. 2001;252:147–151. doi:10.1016/S0022-1759(01)00349-0
17. Magierowska M, Theodorou I, Debré P, et al. Combined genotypes of CCR5, CCR2, SDF1, and HLA genes can predict the long-term non progressor status in HIV-1-infected individuals. Blood. 1999;93:936–941.22. doi:10.1182/blood.V93.3.936
18. van Manen D, Rits M, Beugeling C, van Dort K, Schuitemaker H, Kootstra N. The effect of Trim5polymorphisms on the clinical course of HIV-1 infection. PLoSPathog. 2008;4(2):e18. doi:10.1371/journal.ppat.0040018
19. Winkler C, Modi W, Smith MW, et al. Genetic restriction of AIDS pathogenesis by an SDF-1 chemokine gene variant. ALIVE Study, Hemophilia Growth and Development Study (HGDS), Multicenter AIDS Cohort Study (MACS), Multicenter Hemophilia Cohort Study (MHCS), San Francisco City Cohort (SFCC). Science. 1998;279:389–393. doi:10.1126/science.279.5349.389
20. Mbida AD, Sosso S, Flori P, et al. Measure of viral load by using the abbott real-time HIV-1 assay on dried blood and plasma spot specimens collected in 2 rural dispensaries in Cameroon. J Acquir Immune Defic Syndr. 2000;52:9–16. doi:10.1097/QAI.0b013e3181aeccbc
21. Nkenfou CN, Temgoua ES, Dambaya B, et al. Characterization of asymptomatic children infected with the human immunodeficiency virus at birth. J AIDS Clin Res. 2014;6:405.
22. Buchbinder SP, Katz MH, Hessol NA, O’Malley PM, Holmberg SD. Long-term HIV-1 infection without immunologic progression. AIDS. 1994;8:1123–1128. doi:10.1097/00002030-199408000-00014
23. Cao Y, Qin L, Zhang L, Safrit J, Ho DD. Virologic and immunologic characterization of long-term survivors of human immunodeficiency virus type 1 infection. N Engl J Med. 1995;332:201–208. 14. doi:10.1056/NEJM199501263320401
24. Easterbrook PJ. Non-progression in HIV infection. AIDS. 1994;8:1179–1182. doi:10.1097/00002030-199408000-00023
25. Lévy JA. HIV pathogenesis and long-term survival. AIDS. 1993;7:1401–1410. doi:10.1097/00002030-199311000-00001
26. Laeyendecker O, Redd AD, Lutalo T, et al. Frequency of long-term nonprogressors in HIV-1 seroconverters from Rakai, Uganda. J Acquir Immune Defic Syndr. 2009;52:316–319. doi:10.1097/QAI.0b013e3181bc08f5
27. Grabar S, Selinger-Leneman H, Abgrall S, et al. Prevalence and comparative characteristics of long-term nonprogressors and HIV controller patients in the French hospital data base on HIV. AIDS. 2009;23:1163–1169. doi:10.1097/QAD.0b013e32832b44c8
28. Mandalia S, Westrop SJ, Beck EJ, Nelson M, Gazzard BG, Imami N. Are long-term nonprogressors very slow progressors? Insights from the Chelsea and Westminster HIV cohort. PLoS One. 2012;7:e29844. doi:10.1371/journal.pone.0029844
29. Fellay J, Shianna K, Ge D, et al. A whole-genome association study of major determinants for host control of HIV-1. Science. 2007;317:944–947. doi:10.1126/science.1143767
30. Madec Y, Boufassa F, Porter K, Meyer L. Spontaneous control of viral load and CD4 cell count progression among HIV-1 seroconverters. AIDS. 2005;19:2001–2007. doi:10.1097/01.aids.0000194134.28135.cd
31. Dean M, Carrington M, Winkler C, et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia growth and development study, multicenter AIDS cohort study, multicenter hemophilia cohort study, san francisco city cohort, ALIVE study. Science. 1996;273:1856–1862. doi:10.1126/science.273.5283.1856
32. Eugen-Olsen J, Iversen AKN, Garred P, et al. Heterozygosity for a deletion in the CKR-5 gene leads to prolonged AIDS-free survival and slower CD4 T-cell decline in a cohort of HIV-seropositive individuals. AIDS. 1997;11:305–310. doi:10.1097/00002030-199703110-00007
33. Klein MR, Van Der Burg SH, Hovenkamp E, et al. Characterization of HLA-B57-restricted human immunodeficiency virus type 1 Gag- and RT-specific cytotoxic T lymphocyte responses. J Gen Virol. 1998;79:2191–2201. doi:10.1099/0022-1317-79-9-2191
34. Piacentini L, Biasin M, Fenizia C, Clerici M. Genetic correlates of protection against HIV infection: the ally within. J Intern Med. 2009;265:110–124. doi:10.1111/jim.2008.265.issue-1
35. Ruiz-Mateos E, Ferrando-Martinez S, Machmach K, et al. High levels of CD57+CD28−T-cells, low T-cell proliferation and preferential expansion of terminally differentiated CD4+ T-cells in HIV-elite controllers. Curr HIV Res. 2010;8:471–481. doi:10.2174/157016210793499268
36. Pereyra F, Jia X, McLaren PJ, et al. The major genetic determinants of HIV-1 control affect HLA class I peptide presentation. Science. 2010;330:1551–1557.
37. Miura T, Brumme CJ, Brockman MA, et al. HLA-associated viral mutations are common in human immunodeficiency virus type 1 elite controllers. J Virol. 2009;83:3407–3412. doi:10.1128/JVI.02459-08
38. Price H, Lacap P, Tuff J, et al. A TRIM5 alpha exon 2 polymorphism is associated with protection from HIV-1 infection in the Pumwani sex worker cohort. AIDS. 2010;24:1813–1821. doi:10.1097/QAD.0b013e32833b5256
39. Anzala AO, Ball TB, Rostron T, et al. CCR2-64I allele and genotype association with delayed AIDS progression in African women. University of Nairobi Collaboration for HIV Research. Lancet. 1998;351:1632–1633. doi:10.1016/S0140-6736(05)77688-1
40. Williamson C, Loubser SA, Brice B, et al. Allelic frequencies of host genetic variants influencing susceptibility to HIV-1 infection and disease in South African populations. AIDS. 2000;14:449–451. doi:10.1097/00002030-200003100-00020
41. Martinson JJ, Chapman NH, Rees DC, Liu YT, Clegg JB. Global distribution of the CCR5 gene 32-basepair deletion. Nat Genet. 1997;16:100–103. doi:10.1038/ng0597-100
42. Ramaley PA, French N, Kaleebu P, Gilks C, Whitworth J, Hill AV. HIV in Africa (communication arising): chemokine-receptor genes and AIDS risk. Nature. 2002;417:140. doi:10.1038/417140a
43. Hendel H, Henon N, Lebuanec H, et al. Distinctive effects of CCR5, CCR2, and SDF1 genetic polymorphisms in AIDS progression. J Acquir Immune Defic Syndr Hum Retrovirol. 1998;19:381–386. doi:10.1097/00042560-199812010-00009
44. Easterbrook PJ, Rostron T, Ives N, et al. Chemokine receptor polymorphisms and human immunodeficiency virus disease progression. J Infect Dis. 1999;180:1096–1105. doi:10.1086/jid.1999.180.issue-4
45. Ioannidis JP, Rosenberg PS, Goedert JJ, et al. Effects of CCR5-Δ32, CCR2–64I and SDF-1 3′A polymorphisms on HIV disease progression: an international meta-analysis of individual patient data. Ann Intern Med. 2001;135:782–795. doi:10.7326/0003-4819-135-9-200111060-00008
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.