")
Back to Journals » OncoTargets and Therapy » Volume 11
TRIM32 overexpression improves chemoresistance through regulation of mitochondrial function in non-small-cell lung cancers
Authors Du Y, Zhang W, Du B, Zang S, Wang X, Mao X, Hu Z
Received 8 June 2018
Accepted for publication 21 August 2018
Published 5 November 2018 Volume 2018:11 Pages 7841—7852
DOI https://doi.org/10.2147/OTT.S176689
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Carlos E Vigil
Yaming Du,1,* Wei Zhang,1,* Binghui Du,1 Sheng Zang,1 Xinpeng Wang,1 Xin Mao,1 Zhansheng Hu2
1Department of Vascular Surgery, The First Affiliated Hospital of Jinzhou Medical University, Jinzhou, Liaoning, China; 2Department of Intensive Care Unit, The First Affiliated Hospital of Jinzhou Medical University, Jinzhou, Liaoning, China
*These authors contributed equally to this work
Background: TRIM32 is overexpressed in several human cancers. However, its expression pattern, biological characteristics and mechanisms in human non-small cell lung cancer (NSCLC) have not been reported.
Methods: We examined TRIM32 protein in 115 cases of NSCLC specimens. TRIM32 plasmid transfection and siRNA knockdown was carried out in NSCLC cell lines. AnnexinV/PI and JC-1 staining were performed to examine the change of apoptosis and mitochondrial membrane potential. Western blot was used to detect change of downstream proteins.
Results: We found that TRIM32 protein was upregulated in 69 cases and positively correlated with advanced TNM stage. TRIM32 overexpression also correlated with poor survival of NSCLC patients. Biological assays demonstrated that TRIM32 overexpression promoted while it depletion inhibited cell growth, colony formation and invasion. In addition, TRIM32 maintained NSCLC cell viability and reduced apoptosis when treated with cisplatin. JC-1 and CellRox staining demonstrated that TRIM32 could maintain mitochondrial membrane potential and reduce Reactive Oxygen Species (ROS) production after cisplatin treatment. Western blot analysis showed that TRIM32 overexpression downregulated caspase 3 cleavage and cytochrome c release. TRIM32 also positively regulated Bcl-2 protein expression and NF-κB signaling. Inhibition of NF-κB abolished the effects of TRIM32 on Bcl-2.
Conclusion: Taken together, our results indicated that TRIM32 is overexpressed in NSCLC and regulates cisplatin resistance, possibly through NF-κB and Bcl-2.
Keywords: NSCLC, TRIM32, NF-κB, apoptosis
Introduction
Lung cancer is one of the leading causes of all cancer-related deaths worldwide. Majority of the diagnosed lung cancer cases are non-small-cell lung cancers (NSCLCs).1 Despite many new clinical therapies aiming to improve patients’ survival, the prognosis of NSCLC remains poor. Growing evidence revealed that development of drug resistance plays a pivotal role in NSCLC progression. Therefore, identification of new biomarkers that can regulate chemoresistance is an important step toward the rational design of novel and effective NSCLC treatment.2
TRIM32, a tripartite motif (TRIM) family protein,3,4 was originally reported to regulate skeletal muscle stem cell differentiation and is necessary for normal adult muscle regeneration.5 TRIM32 was reported to be involved in carcinogenesis, with TRIM32 mRNA found to be highly expressed in skin cancer and head and neck cancer.6,7 TRIM32 is upregulated in hepatocellular carcinoma, gastric cancer, and breast cancer, where it serves as a prognostic predictor.8–10 Mechanistically, TRIM32 has E3 ubiquitinase activity and downregulates Abi2 to exert its oncogenic function.7 TRIM32 also negatively regulates tumor suppressor p53 to promote tumorigenesis.11 It was reported that TRIM32 promotes cell proliferation and invasion by activating beta-catenin signaling in gastric cancer.12 To date, the involvement of TRIM32 in NSCLC has not been reported. In the present study, we aimed to examine the clinical significance of TRIM32 in NSCLC tissues using immunohistochemistry. We further investigated the functions and molecular mechanisms of TRIM32 in NSCLC cell lines.
Materials and methods
Tissue samples
The present study was performed with the approval of the Ethics Committee of First Affiliated Hospital of Jinzhou Medical University. Participants provided written informed consent, and the study was performed according to the principles of the Declaration of Helsinki.
Immunohistochemistry
Immunostaining was performed using the Elivision kit from MaiXin (Fuzhou, China). After antigen retrieval in citrate buffer (pH 6.0) for 2 minutes in an autoclave, 0.3% H2O2 was used for 20 minutes. Sections were incubated with goat serum for 15 minutes. Then, the sections were incubated with TRIM32 antibody at 4°C overnight (1:200 dilution; Santa Cruz Biotechnology Inc., Dallas, TX, USA). Then, HRP conjugated polymers were applied to the sections for 30 minutes. 3,3′-Diaminobenzidine plus kit (MaiXin) was used to develop brown stain. Nuclear and cytoplasmic staining was considered as positive staining. For analysis of staining intensity, five views were examined per slide. The staining intensity was graded as 0 (none), 1 (weak), and 2 (moderate/strong). Percentage was graded as 1: 1%–25%, 2: 26%–50%, 3: 51%–75%, and 4: 76%–100%. Intensity and percentage scores were multiplied to give a final score of 0–8. TRIM32 was designated as low expression (score <4) or high expression (overexpression; score ≥4).
Cell culture and transfection
HBE, A549, H1299, H460, H358, H3255, H1975, and H2228 cell lines were obtained from American Type Culture Collection (Manassas, VA, USA). HBE was cultured using PRMI-1640 with 10% FBS and 5 ng/mL human recombinant EGF. A549 was cultured using F12K medium with 10% FBS. H1299, H460, H358, H3255, H1975, and H2228 cell lines were cultured in PRMI-1640 with 10% FBS under a temperature of 37°C. TRIM32 plasmid and its control empty vector (pCMV6-Entry) were obtained from Origene (Rockville, MD, USA). Transfection of plasmid was performed with Attractene transfection reagent (Qiagen, Hilden, Germany). Selection was performed using G418 for 2 weeks. shRNA sequences for TRIM32 and control shRNA were cloned into lentiviral vector pLKO. The shRNA sequence was ATAACTCCCTCAAGGTATATA. Lentiviruses were produced by co-transfection of 293T cells with packaging vectors pMD2.G and pspax2 using the calcium phosphate transfection method. After 48–72 hours of transfection, lentiviral supernatant was collected and concentrated to infect the cells. The infected cells were selected with 5 mg/mL puromycin for 2 weeks.
Quantitative reverse transcriptase-PCR (qRT-PCR)
qRT-PCR was performed using SYBR Green master mix kit from Thermo Fisher Scientific (Waltham, MA, USA). PCR was performed using 7500 Real-Time PCR System (Thermo Fisher Scientific). GAPDH was used as the endogenous control. The fold change of target gene was calculated using the 2−ΔΔCt method.
Western blot
Proteins were separated by SDS-PAGE, transferred to polyvinylidene difluoride membrane (EMD Millipore, Billerica, MA, USA), and incubated overnight at 4°C with antibody against TRIM32 (1:700; Santa Cruz Biotechnology Inc.), p-IkB, IkB, p-AKT, p-ERK Bcl-2, cytochrome c, caspase 3, cleaved caspase 3 (1:1,000; Cell Signaling Technology, Danvers, MA, USA), and GAPDH (1:2,000; Santa Cruz Biotechnology Inc.). After incubation with HRP-linked anti-mouse/rabbit IgG (1:2,000; Santa Cruz Biotechnology Inc.) at 37°C for 2 hours, visualization was performed using enhanced chemiluminescence (Thermo Fisher Scientific) and DNR BioImaging Systems (DNR, Neve Yamin, Israel).
MTT assay and colony formation
For MTT assay, cells were plated in 96-well plates in a medium containing 10% FBS at ~5,000 cells per well. Then, 20 μL of 5 mg/mL MTT (thiazolyl blue) solution was added to each well and incubated for 4 hours at 37°C. The MTT formazan was dissolved in dimethyl sulfoxide. Then, the plate was measured at 490 nm using a plate reader.
For colony formation assay, cells were seeded in 6 cm culture plates and cultured in the incubator. After 2 weeks, the plates with cell colonies were stained using Giemsa. Colony number was counted.
Annexin V/propidium iodide analysis
Annexin V/propidium iodide staining kit from BD Biosciences was used to determine the rate of apoptosis according to the manufacturer’s protocol. Flow cytometry was performed using ACEA Flow Cytometer and NovoExpress software.
Matrigel invasion assay
Cell invasion assay was performed using a 24-well Transwell chamber coated with 20 μL Matrigel. Cell suspension with serum-free medium was transferred to the upper Matrigel chamber. Medium supplemented with 10% FBS was added to the lower chamber. After 18 hours, cells that invaded through the membrane were fixed and stained with hematoxylin.
JC-1 staining
The mitochondrial membrane potential (MMP) was detected by using JC-1 staining. Cells were harvested, washed with PBS, and incubated with 5 μM JC-1 (Cell Signaling Technology) for 30 minutes in the incubator. Then, the cells were washed and analyzed using ACEA flow cytometer, and data were analyzed using NovoExpress software (ACEA, San Diego, CA, USA).
Detection of ROS
ROS measurement was conducted using live cells with CellROX Deep Red reagent (Thermo Fisher Scientific) according to the manufacturer’s protocol. Cells were analyzed using ACEA flow cytometer (ACEA). Data were analyzed using NovoExpress software (ACEA).
Statistical analyses
SPSS version 17 for Windows was used for all statistical analyses. Chi-squared test was used to evaluate possible correlations between TRIM32 overexpression and clinicopathologic factors. The Kaplan–Meier method was used to estimate the probability of patient survival, and differences in the survival of subgroups of patients were compared by Mantel’s log-rank test. The Cox regression model was used for multivariate analysis. Student’s t-test was used to compare data between control and transfected cells. P<0.05 was considered to be statistically significant difference.
Results
TRIM32 is upregulated in NSCLC and correlated with poor prognosis
To explore the clinical significance of TRIM32 in human NSCLCs, we checked its protein expression in 115 cases of specimens using immunohistochemistry. Negative/weak staining was observed in normal bronchial epithelial tissues (Figure 1A). In lung cancer tissues, TRIM32 showed high cytoplasmic and nuclear expression in 69 out of 115 NSCLC specimens (Figure 1B–D). We analyzed the correlation of TRIM32 expression and clinical characteristics. Our results demonstrated that TRIM32 overexpression was significantly associated with advanced TNM stage (P=0.0193), as well as nodal metastasis (P=0.0212), as shown in Table 1.
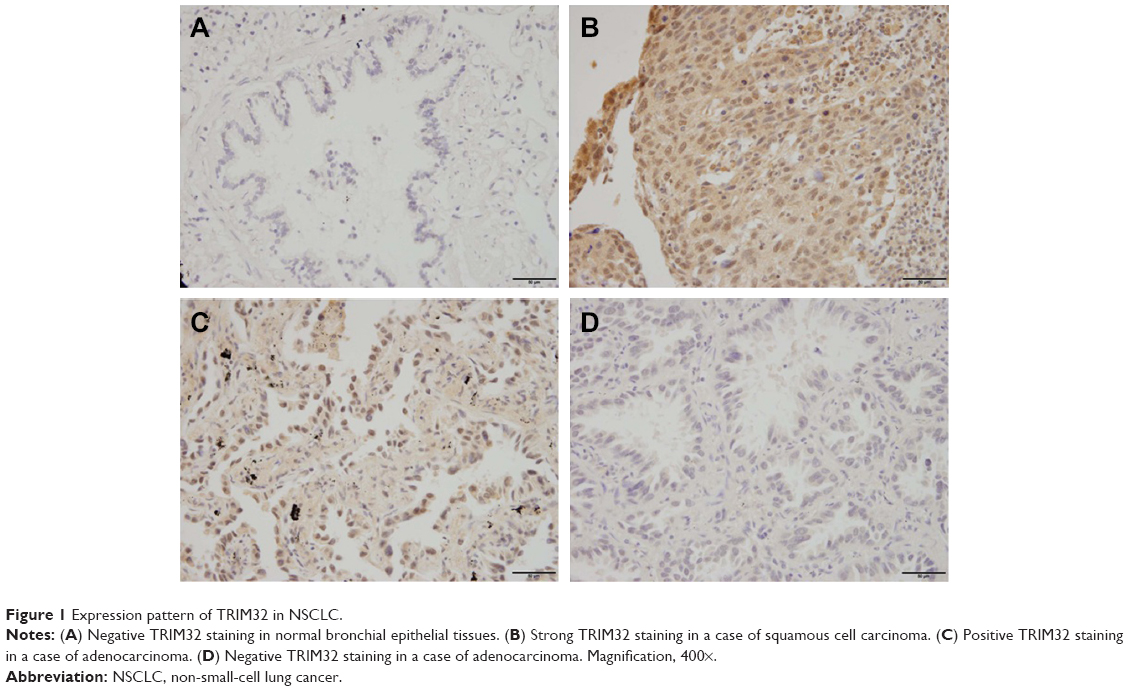 | Figure 1 Expression pattern of TRIM32 in NSCLC. |
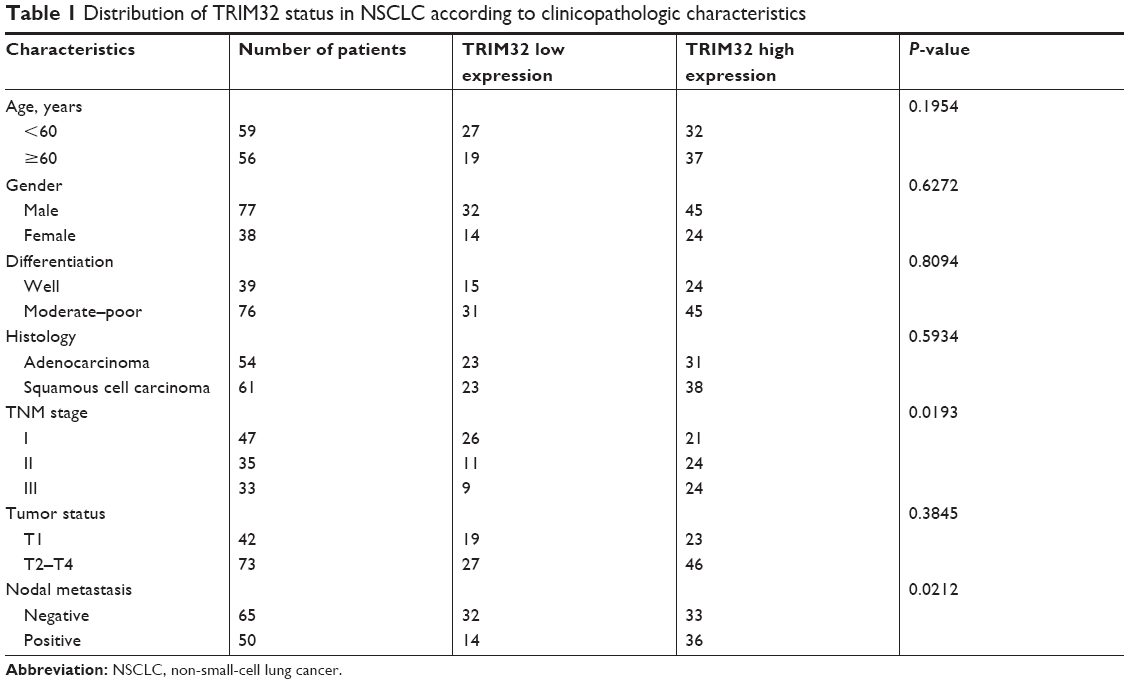 | Table 1 Distribution of TRIM32 status in NSCLC according to clinicopathologic characteristics |
The Kaplan–Meier analysis showed that patients with elevated TRIM32 expression had shorter overall survival than those with low TRIM32 expression (log-rank test, P=0.002; Figure 2A). Univariate analysis revealed that TNM stage and TRIM32 expression were statistically correlated with patients’ survival, which were further subjected to a multivariate Cox proportional hazards model. The results indicated that TNM stage and TRIM32 were independent and significant factors for prognosis (Table 2). The clinical relevance of TRIM32 was further validated using external datasets from The Cancer Genome Atlas. The 488 cases of NSCLC data were split into high-expression and low-expression groups (upper 65% was defined as high expression and lower 35% as low expression). We found a negative correlation between overall patient survival and TRIM32 expression using the Kaplan–Meier plot, and the log-rank test P-value was 0.032 (Figure 2B). Taken together, these data suggest that TRIM32 overexpression is an indicator of malignant phenotype and poor prognosis of NSCLC patients after curative resection.
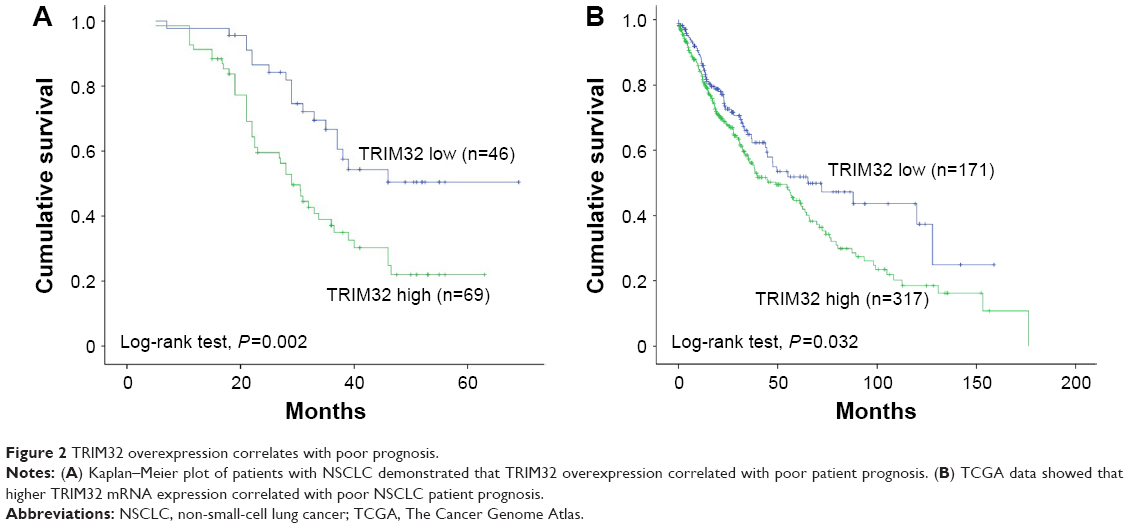 | Figure 2 TRIM32 overexpression correlates with poor prognosis. |
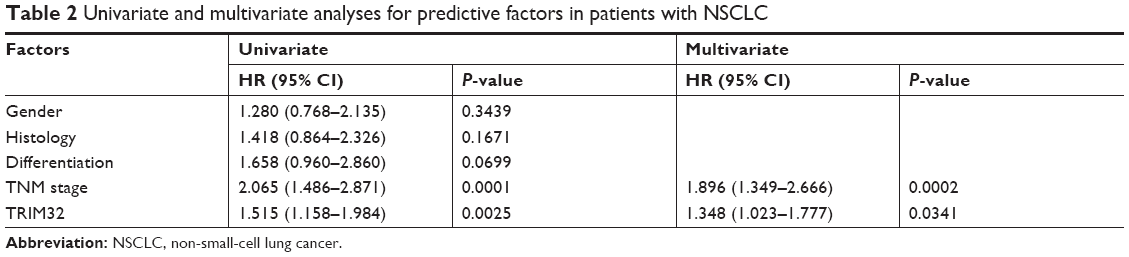 | Table 2 Univariate and multivariate analyses for predictive factors in patients with NSCLC |
TRIM32 is increased in NSCLC cell lines
Next we used Western blot and qRT-PCR to check TRIM32 protein and mRNA in normal bronchial epithelial HBE cell line and seven cancer cell lines including A549, H1299, H460, H358, H3255, H1975, and H2228 (Figure 3A). The results showed that TRIM32 expression was higher in lung cancer cell lines (especially H1299 and H2228) than in HBE. We chose A549 cells for TRIM32 plasmid transfection and H1299 cells for shRNA infection. Selection was performed by G418 (A549) and puromycin (H1299). Plasmid and shRNA efficiency was confirmed by PCR and Western blot (Figure 3B).
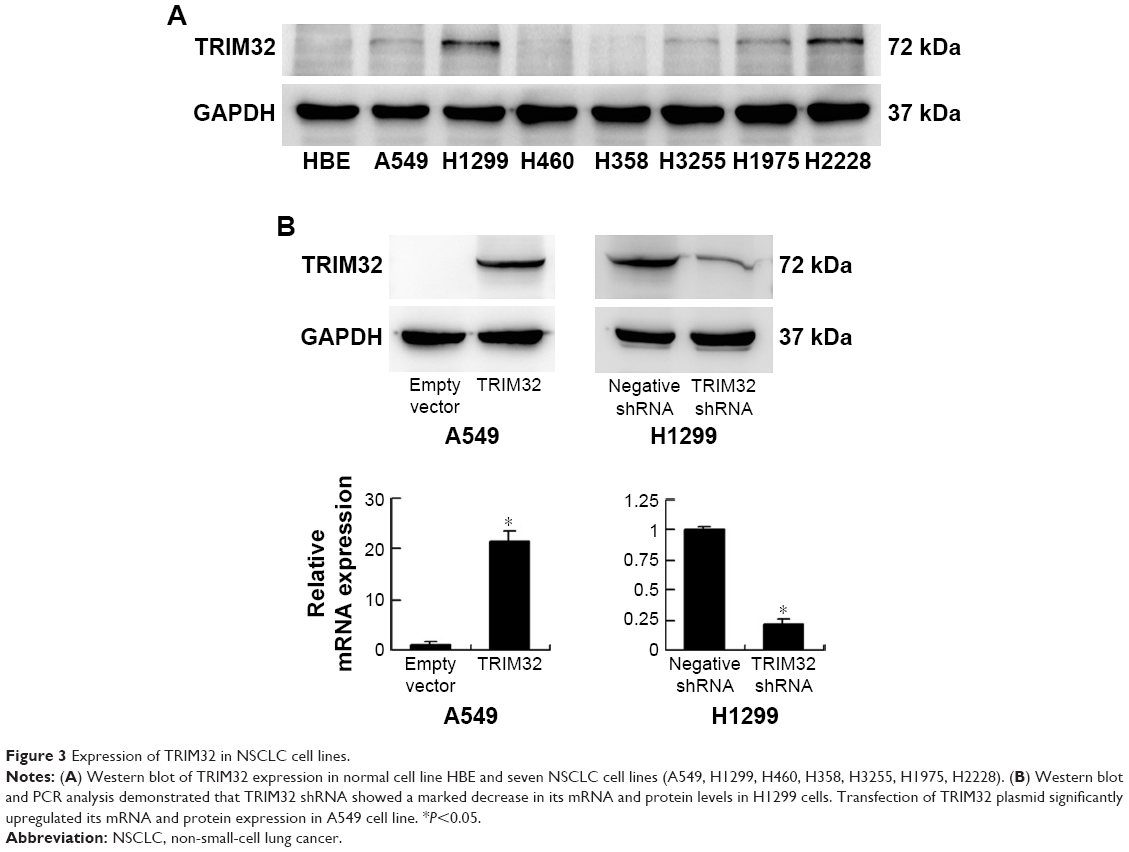 | Figure 3 Expression of TRIM32 in NSCLC cell lines. |
TRIM32 regulates cell proliferation, colony formation, and invasion
MTT and colony formation assay showed that TRIM32 overexpression increased A549 cell proliferation, while TRIM32 shRNA decreased H1299 cell proliferation (Figure 4A). As shown in Figure S1, TRIM32 also promoted H460 cell proliferation, while TRIM32 shRNA decreased H1975 cell proliferation. Colony formation ability demonstrated that TRIM32 upregulated the colony formation ability of A549 cells, while TRIM32 depletion downregulated the colony formation ability of H1299 cells (Figure 4B). Since TRIM32 overexpression correlated with positive nodal status, we evaluated if TRIM32 could influence cancer invasion. As shown in Figure 4C, TRIM32 overexpression significantly increased invading cell number, while its depletion downregulated H1299 cell invasion.
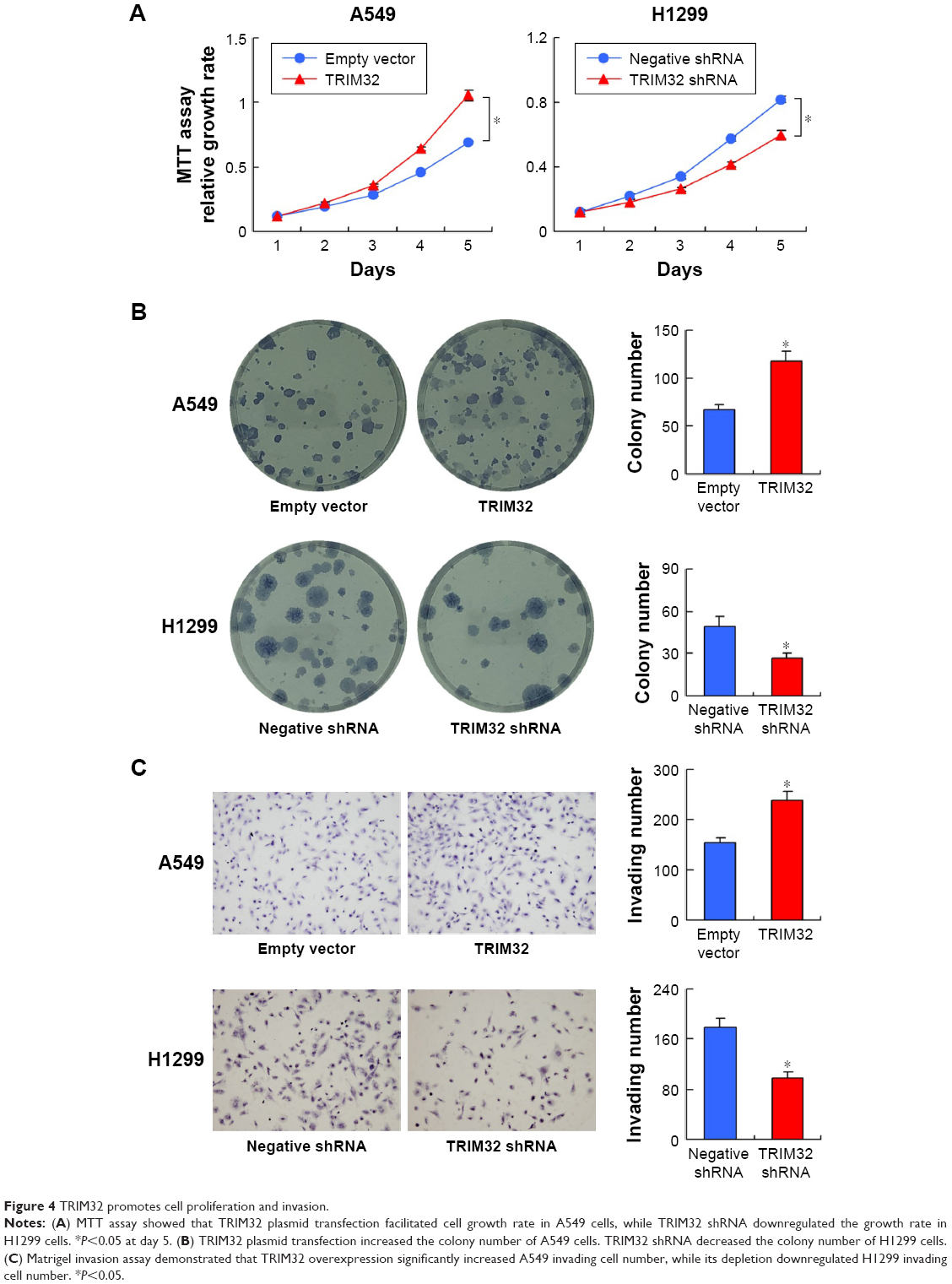 | Figure 4 TRIM32 promotes cell proliferation and invasion. |
TRIM32 mediates chemoresistance in NSCLC cells
To explore the role of TRIM32 in chemoresistance, we treated NSCLC cells with cisplatin and then performed MTT assay to examine cell viability. As shown in Figure 5A, TRIM32 overexpression upregulated A549 viability after treatment with different concentrations of cisplatin (0, 2.5, 5, 10 μg/mL). Cell viability was lower in H1299 cells infected with TRIM32 shRNA than in control cells after cisplatin treatment. As shown in Figure 5B, TRIM32 overexpression slightly downregulated the apoptosis rate in A549 cells without cisplatin treatment. The apoptosis rate was much lower in A549-TRIM32 cells than in control A549 cells after 24 hours of cisplatin (5 μg/mL) treatment. As for H1299 cells, the apoptosis percentage in H1299-shTRIM32 cells was significantly increased compared with the control cells after treatment with cisplatin. This was confirmed by the Western blot analysis (Figure 5C). These data indicate that TRIM32 mediated cisplatin resistance in NSCLC cells.
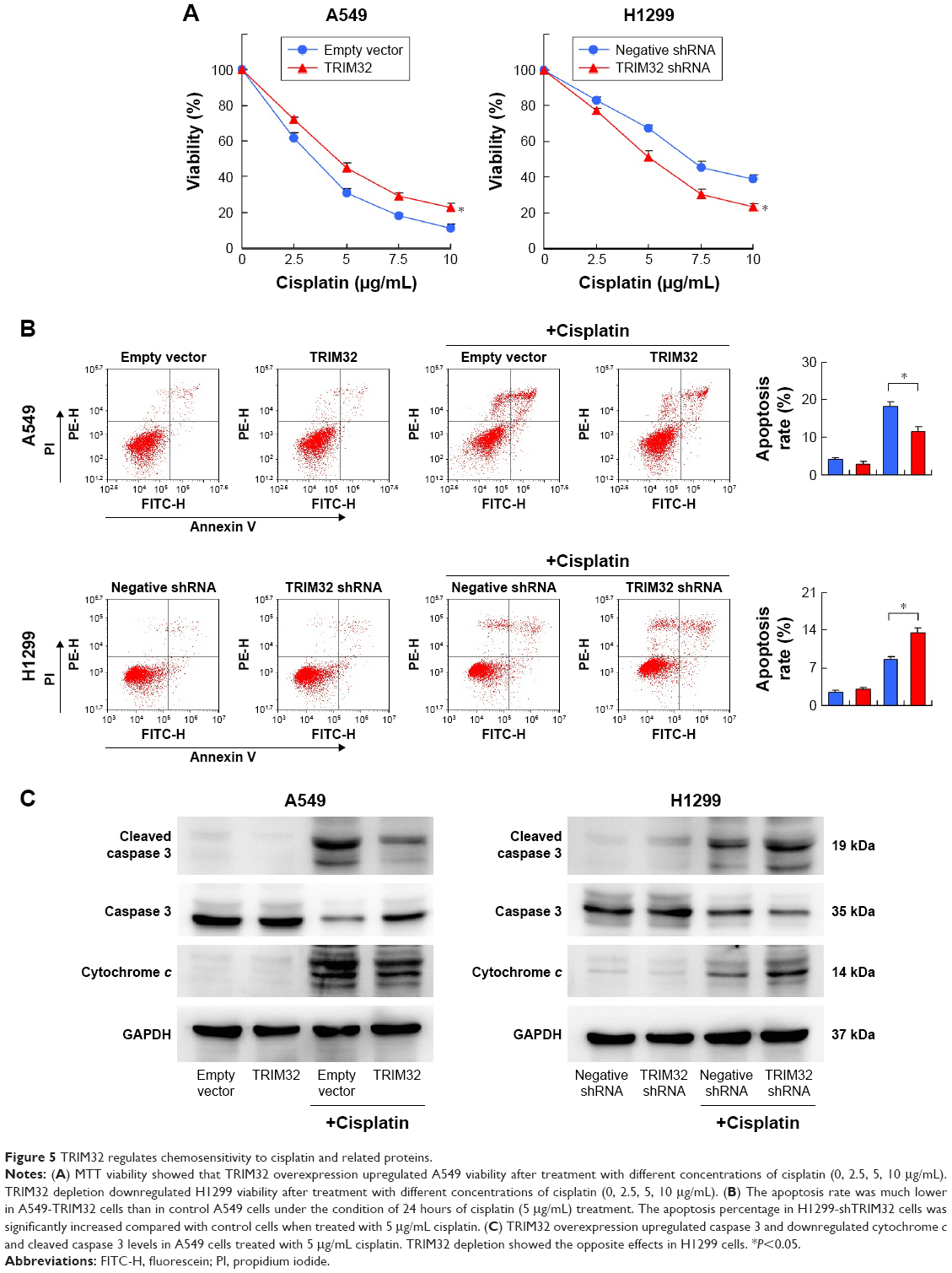 | Figure 5 TRIM32 regulates chemosensitivity to cisplatin and related proteins. |
TRIM32 regulates MMP and ROS production
Chemoresistance is closely related to mitochondrial function. Platinum-based chemotherapy has been shown to reduce the MMP in cancer cells, and cisplatin-resistant cell lines have elevated MMPs. Here, we determined if TRIM32 could regulate the MMP in NSCLC cells. JC-1 staining showed that TRIM32 overexpression downregulated the percentage of cells with green fluorescence, suggesting TRIM32 was able to maintain MMP in cisplatin-treated A549 cells. TRIM32 depletion showed the opposite effect in H1299-shTRIM32 cells (Figure 6A).
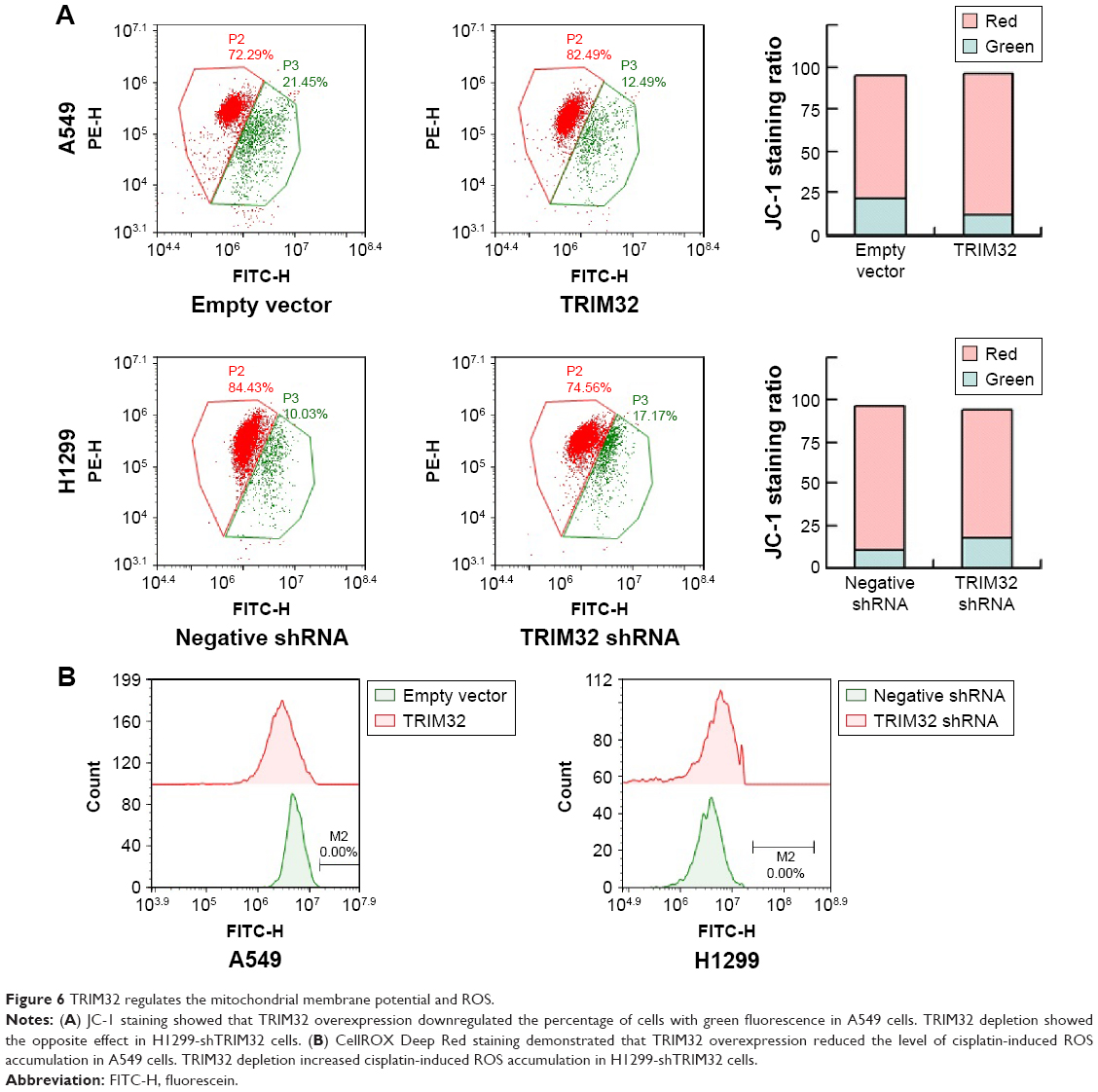 | Figure 6 TRIM32 regulates the mitochondrial membrane potential and ROS. |
ROS induced by chemotherapeutic agents is closely associated with mitochondrial function, cytochrome c release, and apoptosis. Using CellROX Deep Red staining, we were able to demonstrate that TRIM32 overexpression reduced the level of cisplatin-induced ROS accumulation in A549 cells. Accordingly, TRIM32 depletion increased cisplatin-induced ROS accumulation in H1299-shTRIM32 cells (Figure 6B).
TRIM32 regulates NF-κB and Bcl-2 in NSCLC cells
To elucidate the mechanism of TRIM32 on chemoresistance, we screened a panel of apoptosis-related proteins. As shown in Figure 5C, TRIM32 overexpression downregulated cytochrome c and cleaved caspase 3 levels in A549 cells. TRIM32 also upregulated Bcl-2 expression, which was reported to suppress mitochondrial apoptosis pathway (Figure 7A). On the other hand, H1299-shTRIM32 showed increased level of caspase 3 cleavage and cytochrome c release, with downregulation of Bcl-2 expression (Figures 5C and 7A). Bcl-2 has been reported as a downstream target of many signaling pathways. Additional screening revealed that TRIM32 could upregulate IkB phosphorylation status (Figure 7A). NF-κB inhibition with QNZ (300 nM) blocked IkB phosphorylation and attenuated TRIM32-induced Bcl-2 upregulation (Figure 7B).
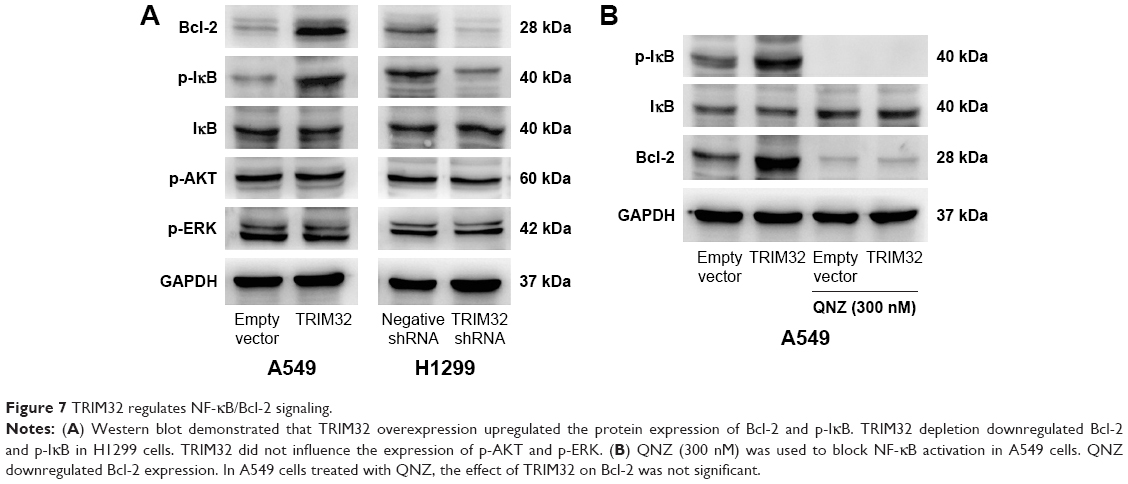 | Figure 7 TRIM32 regulates NF-κB/Bcl-2 signaling. |
Discussion
TRIM32 overexpression has been implicated in various cancers. However, its expression pattern in NSCLC remains unexplored. Our study demonstrated that TRIM32 was upregulated in NSCLC tissues compared with normal bronchial epithelium. Clinically, TRIM32 overexpression correlated with advanced TNM stage and nodal metastasis. Importantly, TRIM32 overexpression correlated with poor prognosis and serves as an independent predicting factor. To our knowledge, this is the first study showing the clinical significance of TRIM32 in NSCLC. Our result was also supported by The Cancer Genome Atlas data, analysis of which showed that high TRIM32 levels correlated with poor prognosis in 488 cases of NSCLC patients. The clinical significance of TRIM32 has also been implicated in other cancer types. TRIM32 upregulation has been originally indicated in skin carcinogenesis.6 TRIM32 overexpression correlated with poor prognosis of hepatocellular carcinoma, gastric cancer, and breast cancer.8–10 These reports were in accordance with our results, supporting TRIM32 as an oncoprotein and a predictor of malignant cancer progression.
Next we validated its biological roles in NSCLC cell lines. MTT and colony formation assays showed that TRIM32 promoted cell growth rate and colony formation ability. Matrigel invasion assay demonstrated that TRIM32 facilitated cell invasion. Accordingly, several reports showed that TRIM32 functioned as an oncoprotein by promoting cell proliferation and invasion in hepatocellular carcinoma, gastric and breast cancer cell lines.8–10 Furthermore, our data first demonstrated that TRIM32 could reduce cisplatin sensitivity and maintain MMP in NSCLC cells. NSCLC often exhibits resistance to platinum-based drugs, limiting their efficacy.13 Our results indicated that TRIM32 might be a promising target to circumvent resistance to platinum-based chemotherapy in NSCLC.
Mitochondria play a pivotal role during the process of apoptosis.14 Chemotherapeutic drugs, such as cisplatin, induce apoptosis partly through mitochondrial pathway.15–17 Downregulation of MMP could trigger apoptosis through mitochondria-dependent pathway, which releases cytochrome c with increased membrane permeability.18 Our data demonstrated that TRIM32 upregulated MMP compared with control. Platinum-based drugs could induce the formation of ROS.19 Normally ROS are a by-product of cell metabolism. However, excessive ROS could trigger apoptosis by altering the MMP and damaging the respiratory chain.20,21 Our data demonstrated that TRIM32 had a protective role in cisplatin-induced ROS formation. To our knowledge, this is the first study to show the protective roles of TRIM32 on MMP and ROS in cisplatin-treated NSCLC cells.
Mechanistically, TRIM32 was reported to mediate the ubiquitination and degradation of Abl-interactor 2, a tumor suppressor and a cell migration inhibitor.7 TRIM32 also interacts with p53 and promotes p53 degradation through ubiquitination.11 Our results indicated that TRIM32 upregulated Bcl-2, an important antiapoptosis protein, blocking mitochondrial apoptosis pathway, reducing ROS and maintaining MMP.22,23 Further analysis demonstrated that TRIM32 also activated NF-κB signaling pathway. Bcl-2 was reported as a downstream target of NF-κB. Their relationship has been reported in various cancer cells including NSCLC cells.24,25 Our data confirmed the link between NF-κB/Bcl-2 and TRIM32 using NF-κB inhibitor QNZ, suggesting the involvement of NF-κB/Bcl-2/mitochondiral function in TRIM32-induced chemoresistance.
In conclusion, the present study demonstrates that TRIM32 is overexpressed in human NSCLC and serves as a predictor for poor prognosis. TRIM32 mediates chemoresistance through regulation of mitochondrial function and NF-κB/Bcl-2 signaling pathway. Blockage of TRIM32 expression and function may potentiate cisplatin responsiveness in NSCLC.
Acknowledgments
This study was funded by Liaoning province reform of emphasis clinical diagnosis and treatment ability of youth project plan descriptions of the construction project (No LNCCC-D29-2015), Science and Technology Department of capability development plan to guide project in Liaoning province (No 20172255044), and the Natural Science Fund Guidance Project Plan Project in Liaoning Province (No 20170540336).
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. | ||
Dong QZ, Wang Y, Tang ZP, et al. Derlin-1 is overexpressed in non-small cell lung cancer and promotes cancer cell invasion via EGFR-ERK-mediated up-regulation of MMP-2 and MMP-9. Am J Pathol. 2013;182(3):954–964. | ||
Malfavon-Borja R, Sawyer SL, Wu LI, Emerman M, Malik HS. An evolutionary screen highlights canonical and noncanonical candidate antiviral genes within the primate TRIM gene family. Genome Biol Evol. 2013;5(11):2141–2154. | ||
Meroni G. Genomics and evolution of the TRIM gene family. Adv Exp Med Biol. 2012;770:1–9. | ||
Nicklas S, Otto A, Wu X, et al. TRIM32 regulates skeletal muscle stem cell differentiation and is necessary for normal adult muscle regeneration. PLoS One. 2012;7(1):e30445. | ||
Horn EJ, Albor A, Liu Y, et al. RING protein Trim32 associated with skin carcinogenesis has anti-apoptotic and E3-ubiquitin ligase properties. Carcinogenesis. 2004;25(2):157–167. | ||
Kano S, Miyajima N, Fukuda S, Hatakeyama S. Tripartite motif protein 32 facilitates cell growth and migration via degradation of Abl-interactor 2. Cancer Res. 2008;68(14):5572–5580. | ||
Cui X, Lin Z, Chen Y, et al. Upregulated TRIM32 correlates with enhanced cell proliferation and poor prognosis in hepatocellular carcinoma. Mol Cell Biochem. 2016;421(1–2):127–137. | ||
Ito M, Migita K, Matsumoto S, et al. Overexpression of E3 ubiquitin ligase tripartite motif 32 correlates with a poor prognosis in patients with gastric cancer. Oncol Lett. 2017;13(5):3131–3138. | ||
Zhao TT, Jin F, Li JG, et al. TRIM32 promotes proliferation and confers chemoresistance to breast cancer cells through activation of the NF-κB pathway. J Cancer. 2018;9(8):1349–1356. | ||
Liu J, Zhang C, Wang XL, et al. E3 ubiquitin ligase TRIM32 negatively regulates tumor suppressor p53 to promote tumorigenesis. Cell Death Differ. 2014;21(11):1792–1804. | ||
Wang C, Xu J, Fu H, et al. TRIM32 promotes cell proliferation and invasion by activating β-catenin signalling in gastric cancer. J Cell Mol Med. 2018;22(10):5020–5028. | ||
Twentyman PR, Wright KA, Mistry P, Kelland LR, Murrer BA. Sensitivity to novel platinum compounds of panels of human lung cancer cell lines with acquired and inherent resistance to cisplatin. Cancer Res. 1992;52(20):5674–5680. | ||
Decaudin D, Marzo I, Brenner C, Kroemer G. Mitochondria in chemotherapy-induced apoptosis: a prospective novel target of cancer therapy (review). Int J Oncol. 1998;12(1):141–152. | ||
Guerra F, Arbini AA, Moro L. Mitochondria and cancer chemoresistance. Biochim Biophys Acta. 2017;1858(8):686–699. | ||
Kim JS, Lee JM, Chwae YJ, et al. Cisplatin-induced apoptosis in Hep3B cells: mitochondria-dependent and -independent pathways. Biochem Pharmacol. 2004;67(8):1459–1468. | ||
Zhao W, You CC, Zhuang JP, et al. Viability inhibition effect of gambogic acid combined with cisplatin on osteosarcoma cells via mitochondria-independent apoptotic pathway. Mol Cell Biochem. 2013;382(1–2):243–252. | ||
Chen X, Wong JY, Wong P, Radany EH. Low-dose valproic acid enhances radiosensitivity of prostate cancer through acetylated p53-dependent modulation of mitochondrial membrane potential and apoptosis. Mol Cancer Res. 2011;9(4):448–461. | ||
Zhang JG, Lindup WE. Role of mitochondria in cisplatin-induced oxidative damage exhibited by rat renal cortical slices. Biochem Pharmacol. 1993;45(11):2215–2222. | ||
Jing XB, Cai XB, Hu H, Chen SZ, Chen BM, Cai JY. Reactive oxygen species and mitochondrial membrane potential are modulated during CDDP-induced apoptosis in EC-109 cells. Biochem Cell Biol. 2007;85(2):265–271. | ||
Brozovic A, Ambriović-Ristov A, Osmak M. The relationship between cisplatin-induced reactive oxygen species, glutathione, and BCL-2 and resistance to cisplatin. Crit Rev Toxicol. 2010;40(4):347–359. | ||
Akl H, Vervloessem T, Kiviluoto S, et al. A dual role for the anti-apoptotic Bcl-2 protein in cancer: mitochondria versus endoplasmic reticulum. Biochim Biophys Acta. 2014;1843(10):2240–2252. | ||
Chong SJ, Low IC, Pervaiz S. Mitochondrial ROS and involvement of Bcl-2 as a mitochondrial ROS regulator. Mitochondrion. 2014;19(Pt A):39–48. | ||
Seo M, Nam HJ, Kim SY, Juhnn YS. Inhibitory heterotrimeric GTP-binding proteins inhibit hydrogen peroxide-induced apoptosis by up-regulation of Bcl-2 via NF-kappaB in H1299 human lung cancer cells. Biochem Biophys Res Commun. 2009;381(2):153–158. | ||
Chen PM, Cheng YW, Wu TC, Chen CY, Lee H. MnSOD overexpression confers cisplatin resistance in lung adenocarcinoma via the NF-κB/Snail/Bcl-2 pathway. Free Radic Biol Med. 2015;79:127–137. |
Supplementary material
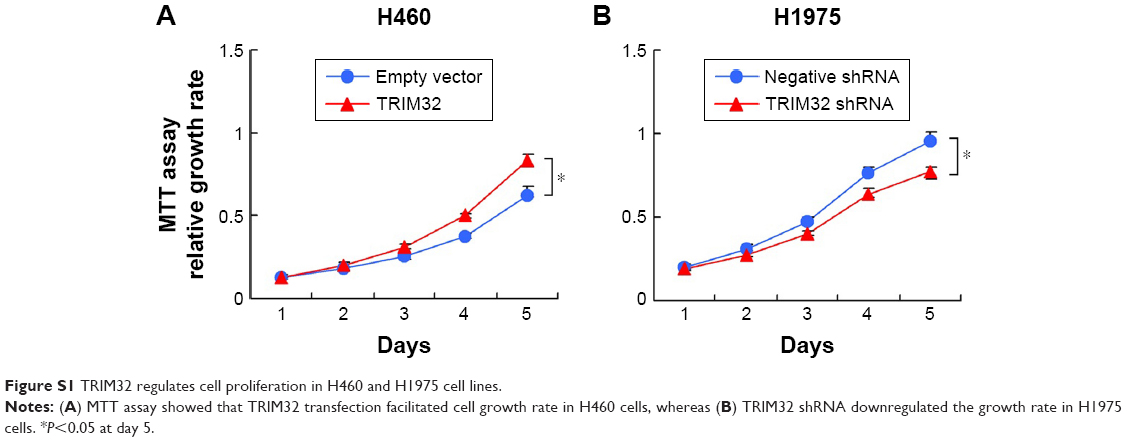 | Figure S1 TRIM32 regulates cell proliferation in H460 and H1975 cell lines. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.