Back to Journals » Journal of Pain Research » Volume 12
Trigeminal Autonomic Cephalalgias Manifested As The Only Initial Symptom Of Ehlers–Danlos Syndrome Type IV
Authors Chen MJ, Li HF ![]() , Mao S
, Mao S
Received 6 June 2019
Accepted for publication 6 November 2019
Published 27 November 2019 Volume 2019:12 Pages 3215—3220
DOI https://doi.org/10.2147/JPR.S218580
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Michael Überall
Mei-Jiao Chen, Hong-Fu Li, Shanying Mao
Department of Neurology, Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
Correspondence: Shanying Mao; Hong-Fu Li
Department of Neurology, Second Affiliated Hospital, Zhejiang University School of Medicine, 88 Jiefang Road, Hangzhou 310009, China
Email [email protected]; [email protected]
Abstract: Ehlers–Danlos syndrome (EDS) type IV is characterized by thin skin with visible veins, easy bruising, characteristic facial features, arterial and digestive complications, as well as rupture of the gravid uterus. It has never been previously reported that trigeminal autonomic cephalalgias (TACs) could manifest as the only initial symptom of EDS type IV. Here, we report a case of a 27-year-old man who presented atypical headache like TACs stimulated by right internal carotid artery dissection. About one month after his discharge, he suffered dissection of the right renal artery and splenic artery, in addition to partial infarction of the right kidney and spleen. Genetic testing revealed a novel splicing variant c.799-1G>A within COL3A1. He was ultimately diagnosed with Ehlers–Danlos syndrome type IV. This case expanded the genetic spectrum and clinical manifestation of EDS type IV and provided a significant implication for the diagnosis of EDS type IV when the initial symptom manifested as TACs, not the typical presentation of EDS type IV.
Keywords: trigeminal autonomic cephalalgias, internal carotid artery dissection, Ehlers–Danlos syndrome, whole exome sequencing, Sanger sequencing
Introduction
Ehlers–Danlos syndrome (EDS) type IV, the vascular type, is an autosomal dominant disorder caused by mutations in COL3A1.1–3 It is a rare connective tissue disease and the prevalence for all EDS is estimated at 1 in 10,000 to one in 25,000. EDS type IV accounts for 5–10% of EDS.1 Patients with vascular type are characterized by thin skin with visible veins, easy bruising, characteristic facial features, arterial and digestive complications, as well as rupture of the gravid uterus.1 Significant vascular complications or rupture of hollow organs usually lead to premature death in the third to fourth decade.2 Thus, early definite diagnosis of EDS type IV may help to lessen the harm of medical and surgical complications. However, when the initial symptoms do not present as typical forms, early definite diagnosis may face challenges. It is recognized that internal carotid artery dissection (ICAD) can be caused by EDS type IV, and symptomatic cluster headache (CH) attacks simulated by ICAD have been reported in many cases. However, it has never been reported that trigeminal autonomic cephalalgias (TACs) could manifest as the only initial symptom of ICAD caused by EDS type IV.
Ethics Approval And Consent To Participate
The study was approved by the Ethics Committees of Second Affiliated Hospital of Zhejiang University School of Medicine. Written informed consent was obtained for the publication of this case report and the accompanying images from the patient who participated in this study.
Case Presentation
Here, we present the case of a young EDS type IV patient who manifested TACs as the only initial symptoms. The patient was a 27-year-old man who complained about pain in the right eye for 1 month and right-side headache for 2 weeks. He had a 1-year history of tension headache and several years of hypertension. One month before presentation, the patient experienced pain in his right eye after fatigue. At the beginning, the ophthalmodynia was mild and intermittent, with an attack frequency of three to five times a day and a duration of half an hour for each episode. In addition, the patient exhibited conjunctival injection and ptosis in the ipsilateral eye. No fever, nausea, vomiting, or visual field deficit was reported. There were no reports of recent trauma or chiropractic manipulation of his neck. He sought medical care in the Ophthalmology Department and was diagnosed with conjunctivitis. Levofloxacin was prescribed and his pain was partially alleviated. However, his conjunctival injection and ptosis were not relieved at all. Two weeks later, he experienced lancinating pain in the right side of his head, predominantly in the right frontotemporal and retro-orbital regions. The pain was intermittent, lasting about a few seconds with intermission of hours to a day. He was then admitted to the Emergency Neurology Department. Physical examination revealed conjunctival injection, ptosis, miosis, and enophthalmos in the right eye. He was initially suspected of having trigeminal autonomic cephalalgia. But because of the atypical headache and persistence of conjunctival injection and ptosis, brain computed tomography (CT) and magnetic resonance imaging (MRI) scanning were performed to exclude the secondary forms of TACs, revealing unremarkable findings. Computed tomography angiography (CTA) of intra–extracranial vessels showed ICAD on the right side (Figure 1A–C). A Doppler ultrasound scan of the carotid artery showed stenosis of the right carotid artery and changes in the blood-flow spectrum. The patient was treated with dual antiplatelet therapy with aspirin 100 mg/day and clopidogrel 75 mg/day. One week after antiplatelet treatment, a high-resolution enhanced MRI for cerebral artery plaques was performed, which suggested intramural hematoma but partial internal carotid recanalization compared to the previous CTA (Figure 1D). Dual antiplatelet treatment for 3 months was prescribed for the patient after discharge.
 |
Figure 1 (A–C) Computed tomography angiography of intra–extracranial vessels performed on 12 May 2018: dissection at the passage between the extra- and intracranial part of the right internal carotid artery (white arrows). (D) High-resolution enhanced magnetic resonance imaging for cerebral artery plaques performed on 5 June 2018: a semicircular-shaped enlargement of the carotid wall with a hyperintense signal on the right suggested intramural hematoma but partial internal carotid recanalization (white arrow). |
One month after the antiplatelet treatment, the patient presented with acute onset of right abdominal pain. Laboratory tests included a white blood cell count of 19,700 cells/mm3 (normal range 4,000–10,000 cells/mm3), C-reactive protein 181.5 mg/L (<10 mg/L), erythrocyte sedimentation rate 58.00 mm/h (<20 mm/h), and negative findings for anti-nuclear antibodies, anti-phospholipid antibodies, rheumatoid factor, anti-cardiolipin antibodies, and anti-double-stranded DNA. The findings of ultrasound of the urinary system and appendix were negative. An abdominal enhanced CT scan showed right renal infarction and right renal artery stenosis. An abdominal computed tomography angiogram (CTA) demonstrated dissection and small pseudoaneurysms of the right renal artery and splenic artery, in addition to partial infarction of the right kidney and spleen (Figure 2A). Antiplatelet combined with anticoagulation therapy (aspirin 100 mg/day and dabigatran 110 mg/bid) was prescribed. A suspected diagnosis of systemic vasculitis was made and low-dose glucocorticoid was prescribed. The pain in his right waist and abdomen was gradually relieved. He underwent a second ultrasound of the urinary system, revealing right renal infarction. He was then discharged with a therapeutic regimen of aspirin 100 mg/day, dabigatran 110 mg/bid, and prednisone 40 mg/day.
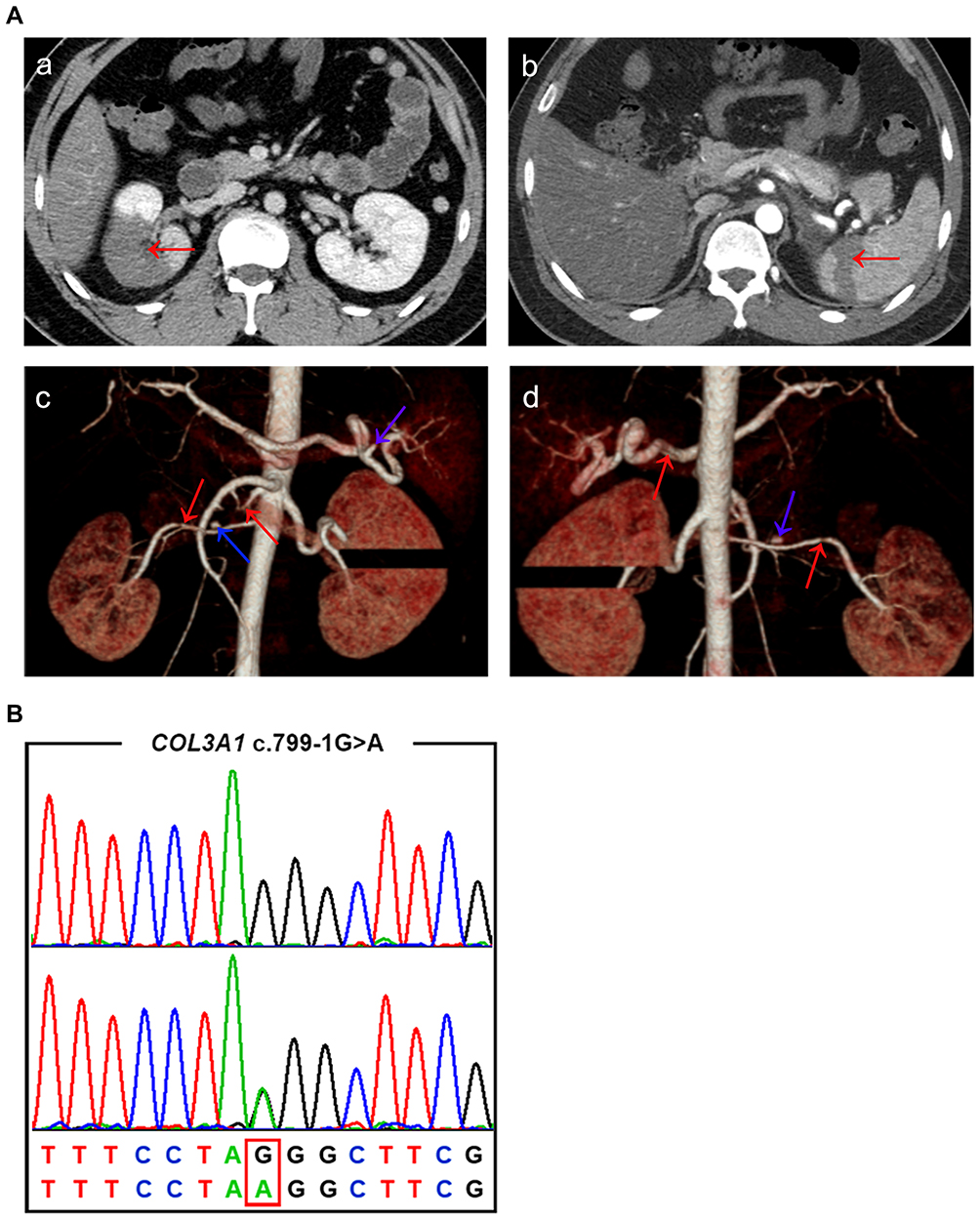 |
Figure 2 (A) (a, b) Abdominal computed tomography angiogram (CTA) performed on 19 July 2018: partial infarction of right kidney (red arrow in a) and spleen (red arrow in b); (c, d) three-dimensional coronal reconstruction from CTA: dissection (red arrows) and small pseudoaneurysms (blue arrows) of right renal artery and splenic artery. (B) The upper chromatogram in each frame represents the reference sequence, and the lower one depicts the mutant sequence (c.799-1G>A within COL3A1) of the patient. |
About 3 months after his right renal and spleen infarction, he underwent a third ultrasound of the urinary system. This implicated that he had recovered from the right renal infarction without abnormal findings. About 6 months after his ICAD, he underwent a Doppler ultrasound scan of the carotid artery, and the findings were normal. At a follow-up visit 11 months after the first discharge, the patient did not complain of any recurrence of painful episodes, although a slight enophthalmos, ptosis, and conjunctival injection in the right eye were still present.
His family history revealed that his father and grandmother had hypertension and had suffered intracerebral hemorrhage. However, the definite cause of their intracerebral hemorrhage was not clear.
Considering the recurrent arterial dissection of this young patient and his family history of intracerebral hemorrhage, we performed genetic investigation for the patient. Whole-exome sequencing was conducted, followed by Sanger sequencing. We found a novel splicing variant c.799-1G>A within COL3A1 (Figure 2B). This variant was absent in the ExAC, 1000G, GnomAD, ESP6500, and 200 in-house controls. It was predicted to be deleterious by MutationTaster, CADD, and DANN. The prediction tool Human Splicing Finder revealed that alteration of the WT acceptor site most probably affects splicing and this variant causes the activation of an intronic cryptic acceptor site and potential alteration of splicing. According to the American College of Medical Genetics and Genomics (ACMG) guidelines, this variant should be assigned as a “likely pathogenic” variant (PVS1+PM2). Therefore, this patient was finally diagnosed with Ehlers–Danlos syndrome type IV. However, genetic testing of the proband parents was not performed since they refused to participate.
Discussion And Conclusions
In this study, we presented the case of a young patient with EDS type IV who manifested TACs as the only initial symptom. Further examinations showed that his TACs were caused by right ICAD. In the following month, this patient suffered dissection of the right renal artery and splenic artery, in addition to partial infarction of the right kidney and spleen. Genetic investigations revealed a novel mutation within COL3A1. To our knowledge, TACs have never been previously documented as the initial symptom of ICAD caused by EDS type IV.
TACs can be classified as primary and secondary forms. Secondary forms, such as ICAD, may lead to a severe outcome and even mortality, whereas primary TACs are relatively mild. Thus, differential diagnosis of primary and secondary TACs is significant. In our case, primary TACs were considered initially. But the atypical pain intensity and frequency, in addition to persistent conjunctival injection and ptosis, raised our suspicion of secondary forms. CTA of intra–extracranial vessels was performed, revealing ICAD on the right side, which confirmed our suspicion. In this case, the new attack of TACs occurred for the first time in close temporal relation to ICAD, meeting the criteria of secondary TACs, the pain intensity and frequency of which are usually atypical compared with primary TACs.4–6 To be specific, the symptoms meet the diagnostic criteria of headache or facial or neck pain attributed to cervical carotid or vertebral artery dissection in The International Classification of Headache Disorders, 3rd edition (beta version) (ICHD-3 beta).4 Definite diagnosis made it possible for the correct treatment to be administered, lessening the harm of this event. This suggests that when a patient presents with a new attack of atypical headache similar to TACs, secondary forms of TACs such as ICAD should be taken into account. Different investigations of the vascular system should be combined to rule out the existence of intra–extracranial arterial dissection.4,5
ICAD can be caused by many factors, one of which is vascular EDS.2,3,7 Chronic recurrent headaches may comprise the neurological manifestation of EDS patients, mainly presenting as migraine with aura, migraine without aura, tension headaches, or a combination of migraine and tension headaches.8 These headaches, in general, do not lead to serious consequences. In our case, the patient had a 1-year medical history of tension headache. This may be the presentation of EDS. However, the attack of TACs was triggered by ICAD caused by EDS type IV, which required urgent medical intervention. As some previous studies illustrated, in vascular EDS, arterial dissection usually occurs in young and middle-aged patients, with a preference for medium- and large-sized arteries.3,9,10 Therefore, when ICAD occurs in a young or middle-aged patient without definite causes, vascular EDS should be suspected, especially when the patient has an obvious family history of similar manifestations or has a second arterial dissection, like the patient in our case. Genetic testing for COL3A1 was recommended. An early definite diagnosis of vascular EDS may help a patient to modify their lifestyle, lessen the harms of medical and surgical complications, and influence the management of pregnancy and reproductive counseling.3,7
From a treatment aspect, EDS type IV is a rare but severe genetic disorder with high mortality. It does not seem to respond well to new medical or surgical treatment methods.9,11 There is no consensus on specific treatment for EDS type IV and the interventions mainly focus on symptomatic relief and prevention of complications.7,10,11 The prevention measures include periodic arterial screening, blood pressure monitoring, and avoiding trauma. Invasive imaging techniques are contraindicated because of the risk of vascular injury.1,7
In conclusion, our report expands the genetic spectrum and clinical manifestations of EDS type IV and provides a significant implication for the diagnosis of EDS type IV when the initial symptom manifests as TACs, not the typical presentation of EDS type IV.
Author Contributions
Mei-Jiao Chen: data acquisition, analysis and interpretation of data, statistical analysis, genomic sequence analysis, drafting the manuscript, ensuring the accuracy or integrity of any part of the work. Shanying Mao: design of the paper, data acquisition, analysis and interpretation of data, critical revision of the manuscript, final approval of the version to be published, ensuring the accuracy or integrity of any part of the work. Hong-Fu Li: study design and conceptualization, data acquisition, analysis and interpretation of data, technical and material support, drafting and critical revision of the manuscript, ensuring the accuracy or integrity of any part of the work. All authors read and approved the final manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Germain DP. Ehlers-Danlos syndrome type IV. Orphanet J Rare Dis. 2007;2:32. doi:10.1186/1750-1172-2-32
2. North KN, Whiteman DA, Pepin MG, Byers PH. Cerebrovascular complications in Ehlers-Danlos syndrome type IV. Ann Neurol. 1995;38:960–964. doi:10.1002/ana.410380620
3. Pepin M, Schwarze U, Superti-Furga A, Byers PH. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med. 2000;342:673–680. doi:10.1056/NEJM200003093421001
4. Headache Classification Committee of the International Headache Society (IHS). The international classification of headache disorders, 3rd edition (beta version). Cephalalgia. 2013;33:629–808. doi:10.1177/0333102413485658
5. Candeloro E, Canavero I, Maurelli M, et al. Carotid dissection mimicking a new attack of cluster headache. J Headache Pain. 2013;14:84. doi:10.1186/1129-2377-14-84
6. Hardmeier M, Gobbi C, Buitrago C, Steck A, Lyrer P, Engelter S. Dissection of the internal carotid artery mimicking episodic cluster headache. J Neurol. 2007;254:253–254. doi:10.1007/s00415-006-0337-2
7. Byers PH, Belmont J, Black J, et al. Diagnosis, natural history, and management in vascular Ehlers-Danlos syndrome. Am J Med Genet C Semin Med Genet. 2017;175:40–47. doi:10.1002/ajmg.c.31553
8. Jacome DE. Headache in Ehlers-Danlos syndrome. Cephalalgia. 1999;19:791–796. doi:10.1046/j.1468-2982.1999.1909791.x
9. Ohshima T, Miyachi S, Isaji T, Matsuo N, Kawaguchi R, Takayasu M. Bilateral vertebral artery dissection and unilateral carotid artery dissection in case of Ehlers-Danlos syndrome type IV. World Neurosurg. 2019;121:83–87. doi:10.1016/j.wneu.2018.09.236
10. Alcorn SR, Sorel MA, Auerbach S, Shaffer K. Ehlers-Danlos syndrome presenting as severe headache in a young adult. Radiol Case Rep. 2008;3:172. doi:10.2484/rcr.v3i2.172
11. Bergqvist D, Bjorck M, Wanhainen A. Treatment of vascular Ehlers-Danlos syndrome: a systematic review. Ann Surg. 2013;258:257–261. doi:10.1097/SLA.0b013e31829c7a59
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.