Back to Journals » Cancer Management and Research » Volume 10
Trichostatin A promotes GLI1 degradation and P21 expression in multiple myeloma cells
Authors Geng Y, Liu J, Xie Y, Jiang H, Zuo K, Li T, Liu Z
Received 5 March 2018
Accepted for publication 16 May 2018
Published 28 August 2018 Volume 2018:10 Pages 2905—2914
DOI https://doi.org/10.2147/CMAR.S167330
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Antonella D'Anneo
Yan Geng,1,* Jing Liu,2,* Ying Xie,2 Hongmei Jiang,2 Kai Zuo,3 Tao Li,4 Zhiqiang Liu2
1Department of Clinical Laboratory, Shanxi Provincial People’s Hospital, Taiyuan, Shanxi, 030012 China; 2Department of Physiology and Pathophysiology, Tianjin Medical University, Heping, Tianjin, 300070 China; 3Department of Infectious Disease, Binzhou People’s Hospital, Binzhou, Shandong, 264000 China; 4Department of Immunology, School of Medicine, Hunan Normal University, Changsha, Hunan, 410013, China
*These authors contributed equally to this work
Background: Histone deacetylase inhibitors are promising drugs for the future application in cancer therapy. Trichostatin A (TSA), a histone deacetylase inhibitor, exhibits effective antitumor effects in various cancers. However, the effects and underlying mechanisms of TSA on multiple myeloma (MM) are not fully investigated.
Methods: In the present study, RPMI8226 and MM.1S cells treated with TSA were used for cell proliferation, cell cycle, and survival examinations, then the localization and post transcriptional modification of GLI1 protein as well as the target gene P21 were analyzed using immunofluorescence, immunoprecipitation, western blots and qPCR, respectively.
Results: TSA exerted a time and dose-dependent cytotoxicity on MM cell lines, and suppressed the proliferation of MM cells and induced an upregulation of p21 protein accompanied by a decreased expression of cyclin D1. TSA treatment led to a downregulation of GLI1, and the nuclear accumulation of GLI1 was also inhibited. As a result of hedgehog inhibition, the expression of MYC and SURVIVIN was greatly weakened after TSA treatment. Furthermore, TSA accelerated GLI1 degradation in a proteasome-dependent manner. Additionally, p21 induction also contributed to GLI1 downregulation via reducing the transcription of GLI in mRNA level. Rescue experiments verified that exogenous expression of GLI1 alleviated MM cell apoptosis induced by TSA.
Conclusion: These results indicated that TSA represses MM cell growth and induces cell apoptosis. The inhibition of hedgehog signaling is an important mechanism accounting for the cytotoxic effects of TSA.
Keywords: histone deacetylase, trichostatin A, multiple myeloma, hedgehog signaling
Introduction
Histone deacetylase (HDAC) inhibitors are a class of compounds that regulate acetylation states of histone proteins and other non-histone proteins by inhibiting the activity of HDAC.1 HDACs are a group of enzymes capable of removing acetyl groups from histone and other proteins, which are divided into four major classes: HDAC1, 2, 3, and 8 comprise class I HDACs locating in the nucleus, HDAC4, 5, 6, 7, 9, and 10 constitute class II HDACs including locating in both cytoplasm and nucleus, SIRT1, 2, 3, 4, and 5 are members of class III HDACs, and HDAC11 is identified as class IV HDAC.2–4
Acetylation and deacetylation modifications of histones trigger dynamic shifts between transcriptionally active and suppressed states.5 Deacetylation on H3 and H4 histones lysine leads to chromatin de-condensation and consequently influences gene transcription including upregulation of several anti-oncogenes and DNA repair genes.6 Deacetylation of a non-histone protein can enhance or decrease the active status of this substrate, such as transport and localization, stability, and protein interactions. A wide subset of HDAC substrates have been identified, including p53, STAT3, β-catenin, estrogen receptor, NANOG, MYC, hypoxia-inducible factor 1 (HIF1), and NF-kB. For instance, HDAC1/2 can cooperate with NANOG to establish and maintain the pluripotent status of stem cells, while inhibiting the activation of transcriptional factor SP1.7,8 Once exposed to HDAC inhibitors, HDAC1 protein is released from the SP1, thereby SP1 becomes active and increases p21 expression.7
Aberrant expression of HDACs have been widely found in tumors and play important roles in carcinogenesis.2 HDACs seem to be required for the maintenance of a set of key genes associated with survival and growth of cancer cells but not of normal ones. Thus, HDACs have emerged as novel therapeutic targets for cancer chemotherapy. HDAC inhibitors have entered clinical trials in both solid tumors and hematologic malignancies.2 HDAC inhibitors can effectively induce cancer cell cycle arrest, differentiation or cell death, and inhibit angiogenesis and invasion.1 Trichostatin A (TSA), a known class I and II HDAC inhibitor, has been demonstrated to exert powerful antitumor effects. It is reported that TSA strongly blocks cell growth and induces cell cycle arrest, and subsequently induces cell apoptosis in multiple myeloma (MM) cells.9 TSA can sensitize TNF-related apoptosis-inducing factor (TRAIL)-resistant cells by decreasing the antiapoptotic BCL2 proteins.10 A study showed the genome-wide transcriptional response to TSA treatment includes downregulation of several MM proliferation-associated factors.11
However, the effect of TSA on hedgehog signaling in MM has not been well characterized. In this study, we found that TSA caused cell cycle arrest in MM cells. TSA treatment decreased the expression of GLI1, a critical executor of hedgehog signaling. As a consequence, the expression of genes lying downstream of hedgehog pathway was downregulated. Meanwhile, we found that TSA treatment inhibited the nuclear localization and expression of GLI1 protein. TSA accelerated its degradation via promoting GLI acetylation. On the other hand, p21 induced by TSA could independently repress the transcription of GLI1. Given the important role of hedgehog signaling in carcinogenesis, our results imply that the HDAC inhibitor TSA can block cancer progression via repressing hedgehog signaling.
Materials and methods
Cell lines and culture
Myeloma cell lines 8226 and MM.1S were purchased from ATCC (American Type Culture Collection, Manassas, VA, USA). MM cells were cultured in RPMI-1640 medium supplemented with 10% of fetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA), 100 U/mL of penicillin, and 100 mg/mL of streptomycin. The cells were grown at 37°C in a humidified incubator with 5% CO2.
Compliance with ethical standards
This study contains no experimental procedures performed on human participants by any of the authors.
Reagents
TSA and cycloheximide were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). MG132 was purchased from Selleck Chemicals (Houston, TX, USA). TSA was dissolved in dimethyl sulfoxide (DMSO) as a 5 mM stock solution and stored at –20°C.
Cell counting Kit-8 (CCK-8) assay
MM cells (5×103) on 96-well plates were treated with TSA as indicated. About 10 mL of CCK-8 solution (Dojindo, Kumamoto, Japan) was added to each well, and the plates were incubated at 37°C for an additional 4 h. Absorbance was then measured at 450 nm as reference with a Microplate Reader 550 (Bio-Rad Laboratories, Richmond, CA, USA). The following equation was used: cell viability (%) = OD value of treatment group/OD value of control group ×100%.
Flow cytometry analysis
MM cells were incubated in 60 mm plates and then treated with TSA at various concentrations. After incubation at 37°C for the indicated time, cells were washed twice with PBS. To assess cell cycle progression by flow cytometry, MM cells were suspended in 100 mL of PBS, and 200 mL of 95% ethanol was added. After incubation for 1 h, cells were resuspended in sodium citrate buffer together with RNase for another 30 min. Cellular DNA was then stained with 250 mL propidium iodide (PI) for 30 min at room temperature. Red fluorescence emitted from the PI–DNA complex was analyzed at 488 nm/600 nm (excitation/emission wave length) using an FACS Calibur instrument (BD Biosciences, San Jose, CA, USA). Apoptosis assay was then carried out using the Apoptosis Detection Kit (Keygen Biotech, Nanjing, China) according to the manufacturer’s instructions. A total of 1×105 cells were stained with 5 mL Annexin V-fluorescein isothiocyanate (FITC) and 1 mL of PI in dark. The cells were analyzed by FACS Calibur instrument. The data of flow cytometry were analyzed with CellQuest 3.0 software (BD Biosciences). Cells that were stained negatively with Annexin V and PI were considered viable cells. Early apoptotic cells were positive for Annexin V and negative for PI, and late apoptotic cells were positive for Annexin V and PI.
Transfection and luciferase assay
Plasmid encoding p21 gene is obtained from Addgene; the pEGFP-GLI1 and pBSU6 siRNA GLI1 expressing plasmids were generous gifts from Dr Fritz Aberger (Department of Molecular Biology, University of Salzburg). Plasmids of pCMV-p300 and 8×GLI-BS luciferase reporter were prepared as described previously.12,13 p21 siRNA (sc-29427) and control siRNA were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). For transient transfection of MM cells, the Neon transfection system (Thermo Fisher Scientific) was used according to the manufacturer’s protocols. For luciferase assay, MM cells were transfected with 0.9 mg of 8×GLI-BS Luc plasmid and 0.1 mg of pRL-TK Renila plasmid (Promega, Madison, WI, USA). After transfection, cells were treated with 5 mM TSA for 24 h, and the cell lysate was used to detect luciferase activity in a Dual-Luciferase Reporter assay system (Promega) according to the manufacturer’s protocol.
Western blot
Cells after TSA treatment were collected, washed twice with cold PBS, and suspended in lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10 mM EDTA, 0.5% sodium deoxycholate, 1% NP-40, 1 mM sodium ovanadate, 10 mg/mL aprotinin, 1 mM phenylmethanesulfonyl fluoride, and 10 mg/mL leupeptin) for 20 min on ice. The lysates were centrifuged, and the supernatants were used as whole cell extracts. The protein concentration was determined using the BCA protein assay kit (Beyotime, Shanghai, China). Cell lysate (30 mg) was separated by electrophoresis on SDS-PAGE gel and transferred to polyvinylidene fluoride membranes. Membranes were blocked with 5% non-fat milk for 1 h and incubated with the primary antibodies at 4°C overnight, and then incubated with horseradish peroxidase-conjugated secondary antibodies. Bands were visualized using an enhanced chemiluminescence system (EMD Millipore, Billerica, MA, USA). The primary antibodies for GLI1, p21, cyclin D1, histone H3, and actin were all obtained from Santa Cruz Biotechnology (Dallas, TX, USA), and the antihuman PARP antibody was purchased from Cell Signaling Technology (Danvers, MA, USA).
Immunoprecipitation and ubiquitination assay
Myeloma cells were appropriately treated with TSA and lysed with RIPA buffer. Clarified lysates were precleared with Protein G-Sepharose (Pierce, Rockford, IL, USA), and then immunoprecipitated with anti-GLI1 absorbed to protein G-Sepharose. Proteins were fractionated by SDS-polyacrylamide gel electrophoresis and transferred to PVDF membranes and blotted with anti-acetyl lysine (K) antibody or anti-Ubiquitin antibody.
Nuclear extraction
Nuclear proteins were extracted and isolated from MM cells using the Nuclear and Cytoplasmic Extraction Kit protocol (ThermoFisher Scientific, Waltham, MA, USA). The cells were lysed in cytoplasm extraction reagent and spun at 14,000 ×g to extract the nuclear material. Proteins from the nuclear material were then extracted by adding nuclear extraction reagent to the nuclei and spun at 14,000 ×g. Protein concentrations of the nuclear extracts were measured using Bio-Rad Bradford protein quantification assay.
RNA extraction and real-time PCR
Total RNA was isolated using Trizol (Life Technologies, South San Francisco, CA, USA) according to the manufacturer’s instructions. Total RNA (2 μg) was reverse transcribed using the Super Script II reverse transcriptase (Life Technologies). Quantitative real-time PCR was carried out in a 7500 real-time PCR system (Thermo Fisher Scientific). Gene-specific oligonucleotide primers included the following: GAPDH, forward 5′-AATCCCATCACCATCTTCCA-3′ and reverse 5′-TGGACTCCACGACGTACTCA-3′; c-MYC, forward 5′-ctaccctctcaacgacagca-3′ and reverse 5′-tcttgttcctcctcagagtcg-3′; and SURVIVIN, forward 5′-AAGAACTGGCCCTTCTTGGA-3′ and reverse 5′-CAACCGGACGAATGCTTTT-3′. The expression of each gene was normalized to GAPDH and calculated using the 2-DDCt method.
Immunofluorescence staining
MM cells were fixed in 4% formaldehyde for 10 min. For fluorescence staining, the samples were treated in 0.5% (V/V) Triton X-100 for 15 min and blocked with 10% BSA for 30 min at 37°C. Then, cells were incubated overnight at 4°C with anti-GLI1 antibody, followed by incubation with tetramethylrhodamine (TRITC) -conjugated goat anti-mouse IgG (1:200) for 30 min at room temperature and nucleus counterstaining with DAPI. Imaging was performed using a fluorescence microscope (model IX71; Olympus, Tokyo, Japan).
Statistics
Data are presented as mean ± SD. Statistical analysis was performed using SPSS 11.5 software (SPSS Inc., Chicago, IL, USA). Statistical significance was determined using a two-tailed Student’s t-test. In all experiments, P < 0.05 was considered statistically significant.
Results
TSA induces growth arrest and apoptosis in MM cells
In order to assess the effects of TSA on MM cell, cell viability was tested in RPMI8226 and MM.1S cell lines by CCK-8 assay. As shown in Figure 1A, TSA showed a dose-dependent cytotoxic effect on MM cells. The viability of MM cells was significantly repressed by TSA at concentrations over 1 mM. After TSA treatment at the dose of 5 uM for 48 h, the relative cell viability declined to 53.15% and 38.44% in RPMI8226 cells and MM.1S cell, respectively. Similar effects could be observed in MM.1S cells. Furthermore, we observed that TSA treatment downregulated the expression of Cyclin D1, one of the cyclins driving the G1/S phase transition of cell mitosis, while enhanced the amount of p21, an important cyclin-dependent kinase inhibitor (Figure 1B). The decline of cyclin D1 together with an increase in p21 protein undoubtedly triggered cell growth arrest. TSA treatment alone arrested RPMI8226 and MM.1S cells at the G0/1 phase and decreased the cell proportion in S phase of the cell cycle (Figure 1C). Subsequently, double-staining of Annexin V-FITC and PI followed by flow cytometry were used to determine the effect of TSA on cell apoptosis. As depicted in Figure 1D, the treatment with 5 mM TSA for 48 h initiated cell apoptosis moderately. Taken together, these data indicated that TSA can exert inhibitory effects on the proliferation and survival of MM cells in a concentration-dependent manner.
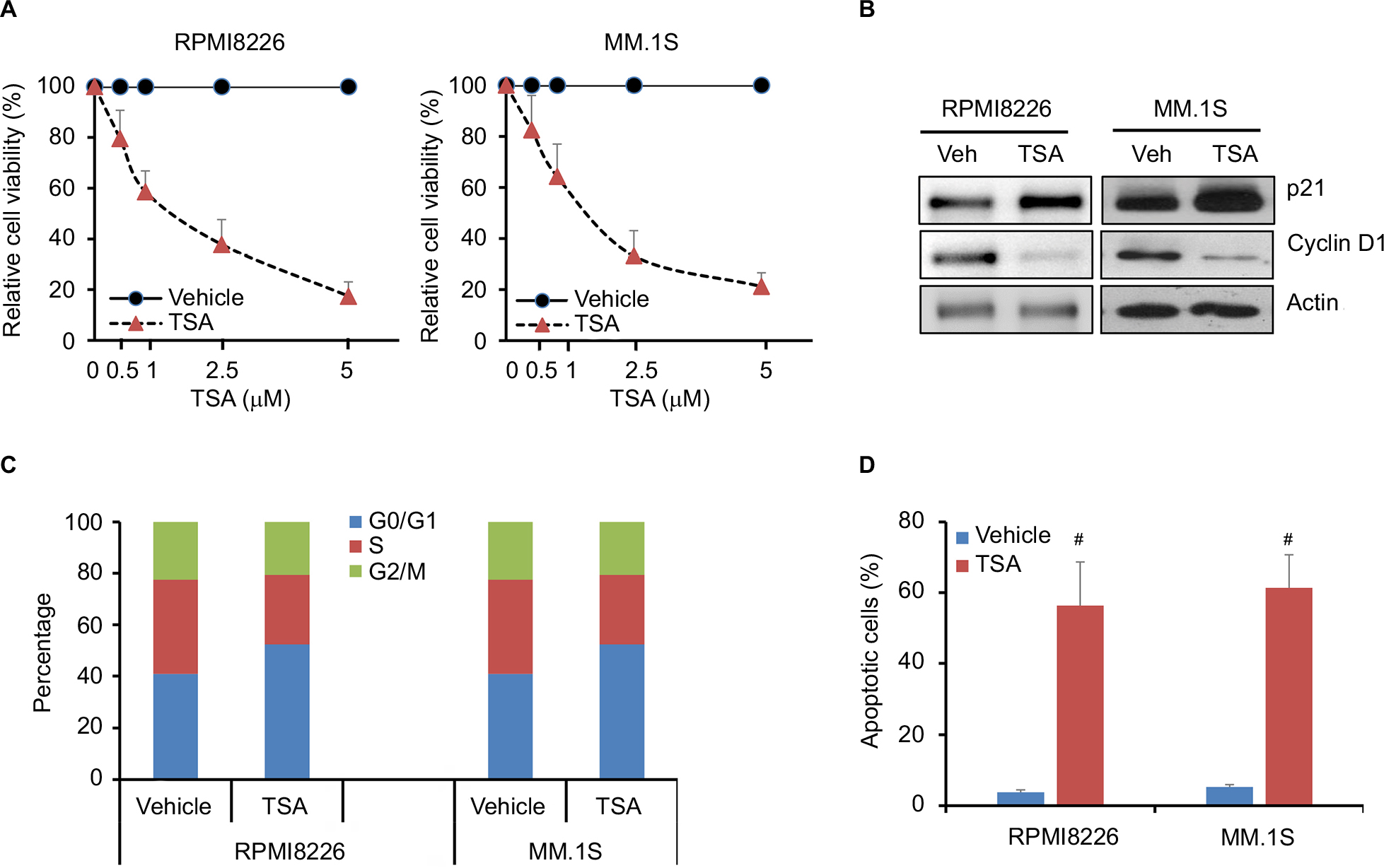 | Figure 1 TSA reduces cell viability of MM cells. Notes: (A) Relative cell viability of RPMI8226 and MM.1S cells treated with TSA at indicated concentrations for 48 h using CCK-8 assay. Results shown are the mean ± SD of three independent experiments. Control cells were treated with DMSO in parallel in each experiment. (B) The protein expression of cyclin D1 and p21 after treatment with 5 μM TSA for 48 h was assessed by Western blot. Actin was used as loading control. Representative images are from at least three independent experiments. (C) RPMI8226 and MM.1S cells were cultured in the presence of 5 μM TSA for 24 h. Cells were stained with PI and subjected to cell cycle analysis by flow cytometry. The statistical analysis of cell percentage of cell cycle distribution is presented. (D) Shown are cell apoptosis rates of RPMI8226 and MM.1S cells treated with 5 μM TSA for 48 h measured by FACS-based Annexin V-FITC/PI double staining. Data are statistical analysis of three similar experiments. #, p<0.001, TSA treatment vs. Vehicle. Abbreviations: TSA, trichostatin A; CCK-8, Cell Counting Kit-8; DMSO, dimethyl sulfoxide; PI, propidium iodide; FACS, fluorescence-activated cell sorter; FITC, fluorescein isothiocyanate; MM, multiple myeloma; Veh, vehicle. |
TSA inhibits GLI1 and its downstream genes
Subsequently, we investigated the mechanisms underlying the cytotoxic effect of TSA. As Cyclin D1 is a well-known downstream gene of hedgehog signaling, its expression was greatly repressed by TSA. We speculated that hedgehog signaling might be a target affected by TSA. Thus, we further examined the effect of TSA on hedgehog signaling. As shown in Figure 2A, TSA exhibited a time- and dose-dependent inhibition on GLI1 protein level, denoting a repression on hedgehog signaling. Additionally, the transcripts of GLI1 were significantly decreased upon the treatment with high dosages of TSA (Figure 2B). In parallel, the luciferase activity of a reporter under the control of eight tandem copies of GLI-binding site declined after exposure to TSA (Figure 2C).
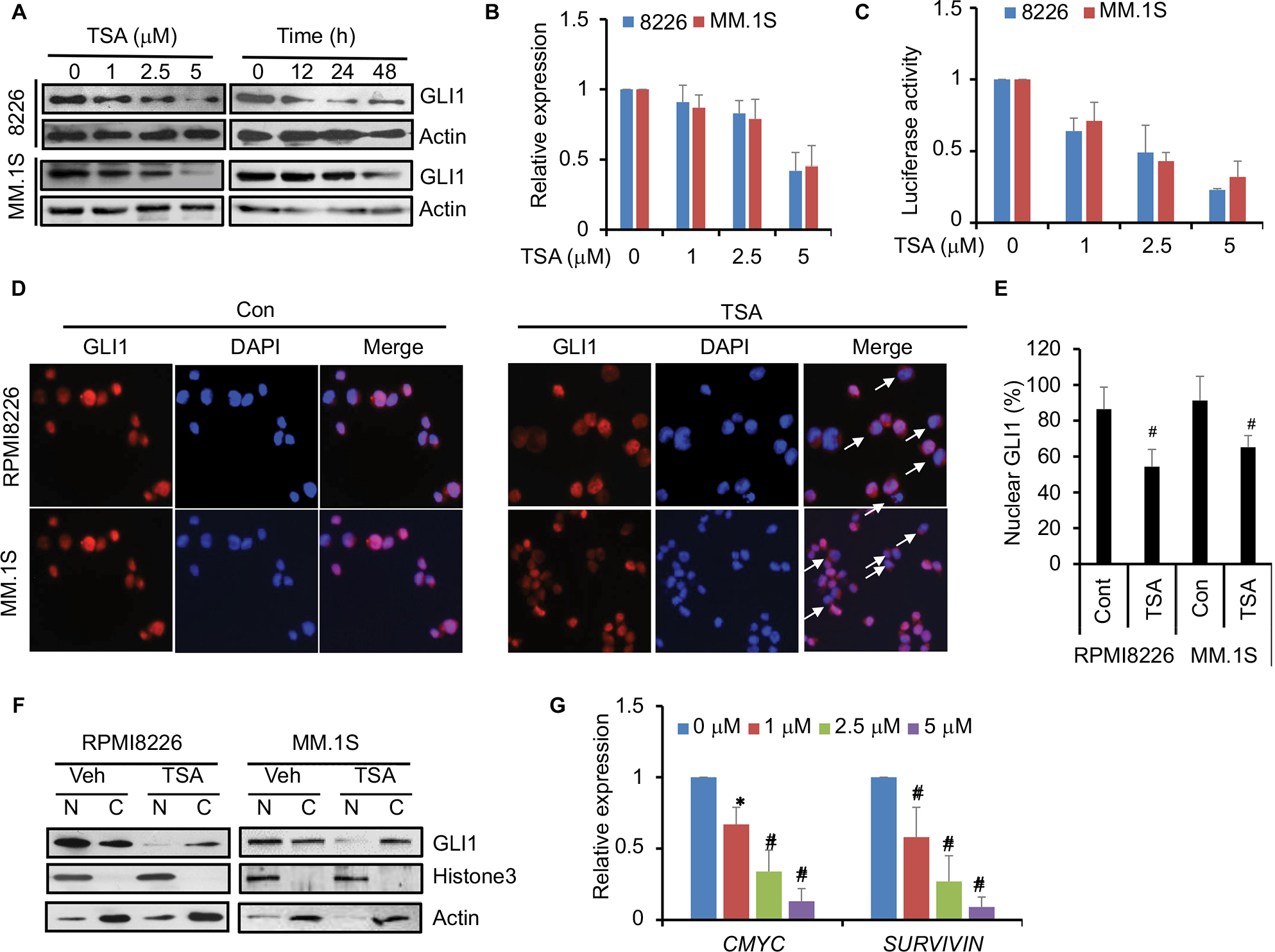 | Figure 2 TSA reduces GLI1 and downstream genes of hedgehog signaling. Notes: (A) The time- and dose-dependent downregulation of GLI1 after TSA treatment. Representative images from at least three independent experiments are shown. (B) The effect of TSA treatment for 48 h on GLI1 expression in mRNA level. (C) Luciferase assay showed the activity of the 8×GLI luciferase reporter responsive to increasing dosages of TSA. (D) Immunofluorescence showed the subcellular localization of GLI1 with or without TSA treatment. Magnification: 200×. Representative images are taken over 10–50 fields of view. Arrows, cells with GLI1 trans-localized into the cytoplasm. (E) Statistical analysis of GLI1 localization in the nucleus of RPMI8226 and MM.1S cells treated with TSA for 24 h. (F) Representative images of Western blot analysis are shown to detect the subcellular distribution of GLI1. Histone H3 was used as an internal control for nuclear, and actin was used as an internal control for cytoplasm. (G) mRNA levels of c-MYC and SURVIVIN were detected by real-time PCR in MM cells treated with 5 μM TSA for 48 h. The median expression value of control group is normalized to 1. Data represent mean ± SD from three independent experiments (#P<0.01, *P<0.05). Abbreviations: TSA, trichostatin A; Cont, control; Veh, vehicle; N, nuclear; C, cytoplasm. |
Furthermore, we investigated the nuclear expression of GLI1 in the presence or absence of TSA, which more reliably reflects hedgehog activation. As shown in Figure 2D, GLI1 protein was mainly localized in the nucleus in the absence of TSA, suggesting a spontaneous activation of hedgehog signaling in MM cells. However, the appearance of TSA altered the cellular distribution of GLI1. Moreover, a significantly increasing proportion of GLI1 protein was shuttled into the cytoplasm after TSA treatment (Figure 2D and E). Western blot analysis also showed that TSA reduced the nuclear accumulation of GLI1 (Figure 2F). These results further validated that TSA treatment caused hedgehog inhibition, as marked by decreased expression and nuclear accumulation of GLI1.
As a consequence of GLI inhibition, two downstream genes of hedgehog signaling, c-MYC and SURVIVIN, were significantly downregulated by TSA treatment (Figure 2G). These results strongly supported the fact that TSA treatment leads to the inhibition of hedgehog signaling pathway.
TSA promotes the degradation of GLI1 via acetylation
GLI1 protein localized in the cytoplasm is more susceptible to degradation than that in the nucleus. Next, we measured the half-life of GLI1 protein after TSA addition. In the presence of cycloheximide (to block de novo protein synthesis), cells were subjected for immunublot assay to detect the degradation rate of GLI1. As shown in Figure 3A, TSA treatment significantly accelerated the degradation of GLI1 in RPMI8226 and MM.1S cell lines. Meanwhile, MG132, a proteasome inhibitor, was capable of rescuing the degradation of GLI1 (Figure 3B). Thus, these results indicated that TSA treatment caused an enhanced degradation of GLI1 in a proteasome-dependent manner, thereby inhibiting hedgehog signaling. Furthermore, immunoprecipitated GLI1 in TSA treatment group exhibited a hyperacetylated status (Figure 3C). Obviously, TSA can increase the acetylation of GLI1 via inhibiting HDACs. Hyperacetylated GLI1 is perhaps more sensitive to degradation via a proteasome-dependent manner. Further experiments corroborated that TSA treatment greatly enhanced GLI1 ubiquitination compared with vehicle control (Figure 3D). Taken together, TSA can remarkably affect GLI1 stabilization via acetylation.
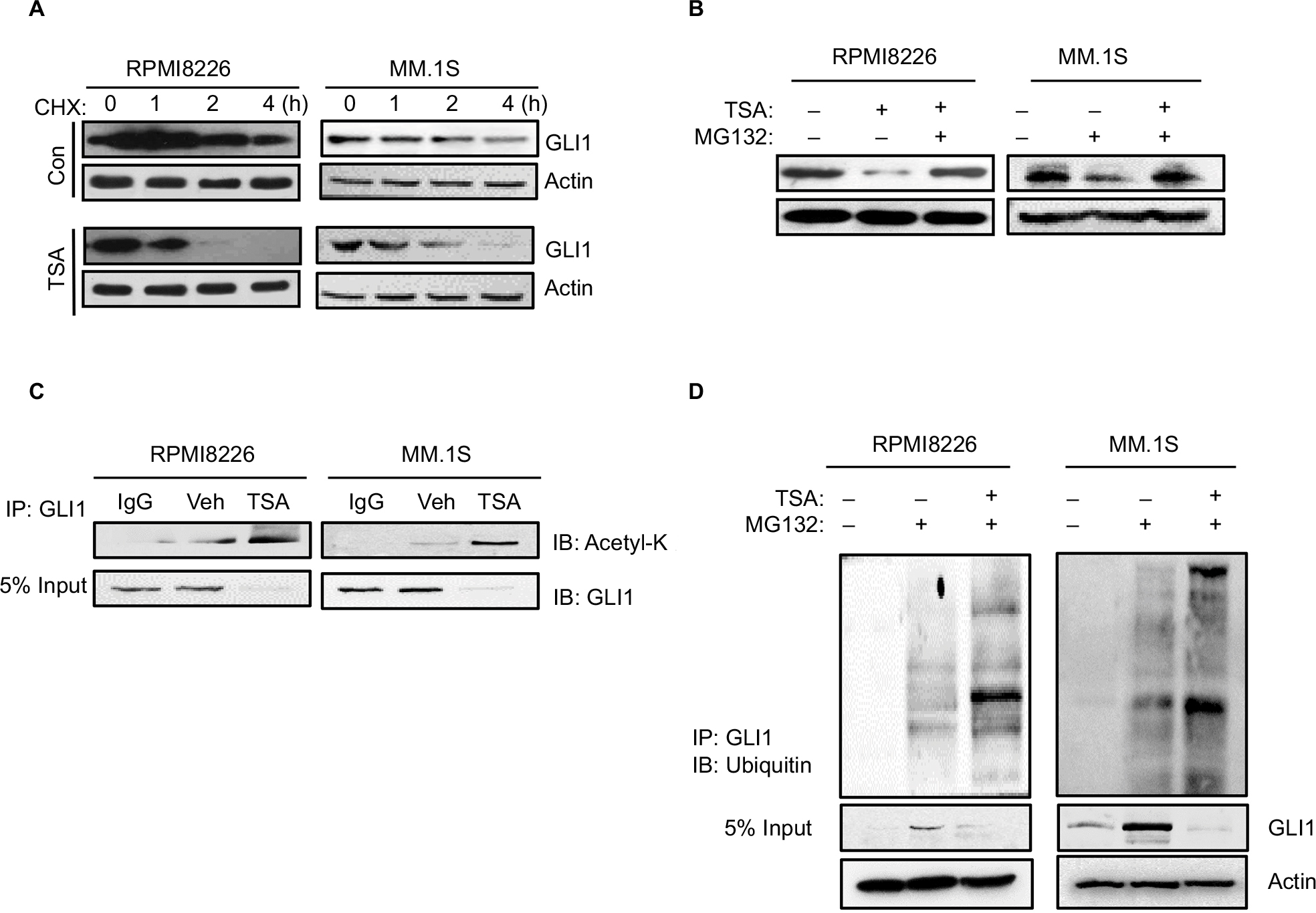 | Figure 3 TSA enhances the degradation of GLI1. Notes: (A) GLI1 protein degradation in RPMI8226 and MM.1S cells treated with 5 μM TSA for 24 h and then 20 μM cycloheximide for up to 4 h. (B) Accumulation of GLI1 protein in RPMI8226 and MM.1S cells treated with 5 μM TSA and 10 μM MG132 for 48 h. (C) Immunoprecipitation assay showed the acetylation status of GLI1 in TSA-treated RPMI8226 and MM.1S cells. (D) Ubiquitination status of GLI1 in TSA-treated RPMI8226 and MM.1S cells were detected with immunoprecipitation. Representative images are from at least three independent experiments. Abbreviations: TSA, trichostatin A; CHX, cycloheximide; Con, control; Veh, vehicle; IB, immunoblot; IP: Immunoprecipitation. |
TSA inhibits GLI1 transcription via p21 induction
TSA can trigger GLI1 degradation via blocking HDAC activity. However, the mechanisms underlying TSA-induced GLI1 downregulation in mRNA level remained obscure, as TSA treatment also induced p21 upregulation, leading to cell cycle arrest. We speculated that TSA-mediated acetylation and/or p21 induction might account for repressed GLI1 transcription. We further investigated the effects of transfection with an acetylase p300 and p21. As shown in Figure 4A, p21 overexpression greatly decreased GLI1 transcription. By comparison, p300 displayed a weak inhibition on GLI1 expression. Transfection with an increasing amount of p21 plasmid caused a gradual downregulation of GLI1 protein (Figure 4B). In contrast, p21 overexpression seemed to unaffect GLI1 stabilization (Figure 4C). To further validate the effects of p21 on GLI1 expression, we performed siRNA-mediated knockdown and overexpression experiments of p21 gene in RPMI8226 and MM.1S cell lines. As shown in Figure 4D, p21 knockdown attenuated TSA-induced GLI1 inhibition to a certain extent. On the contrary, p21 overexpression directly enhanced TSA-mediated repression on GLI1 expression (Figure 4E). Therefore, we concluded that p21 induction can also reduce the transcription of GLI at mRNA level. Collectively, TSA treatment can promote GLI1 degradation via acetylation and repress GLI transcription via p21 induction.
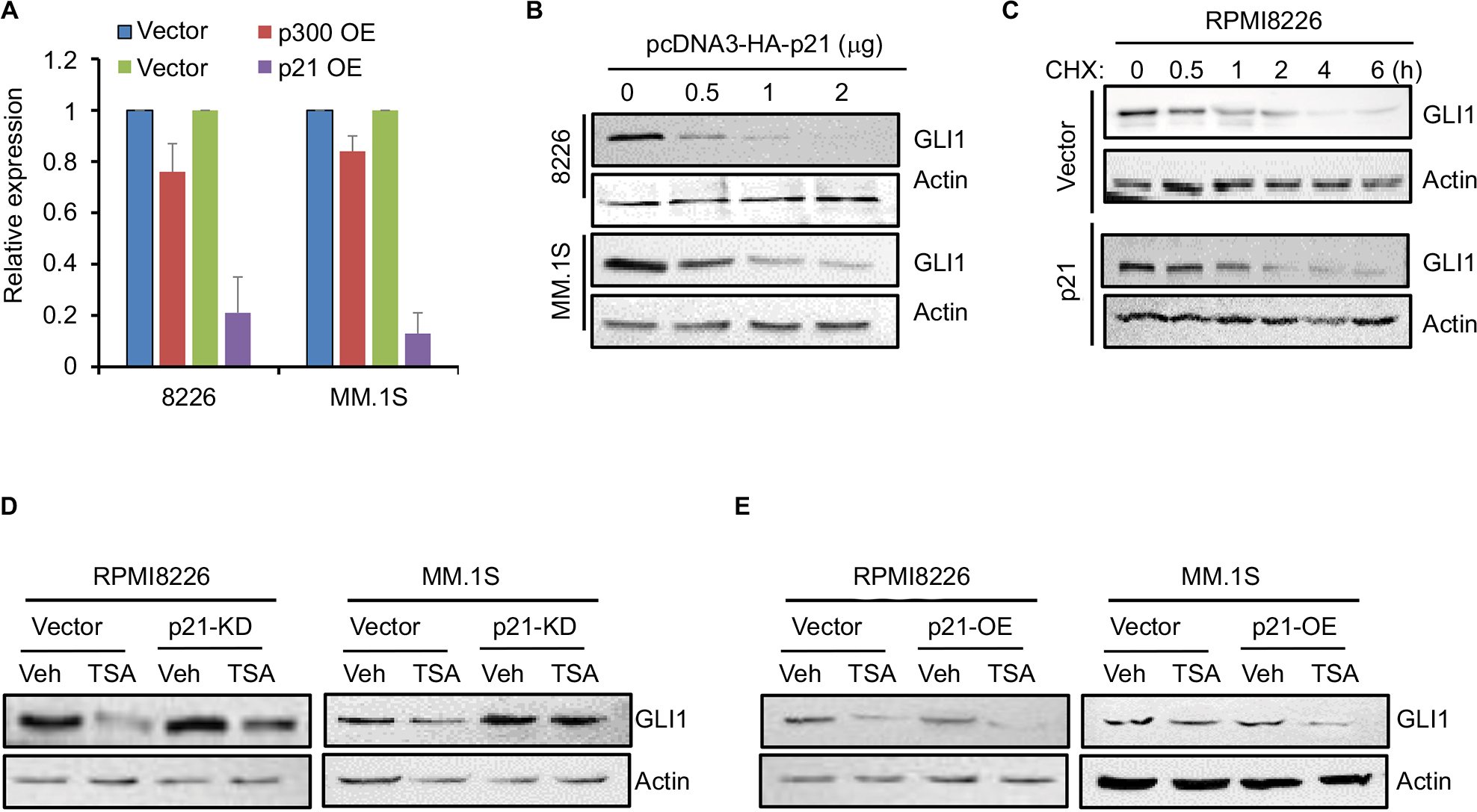 | Figure 4 p21 induced by TSA represses the transcription of GLI1. Notes: (A) The mRNA expression of GLI1 was detected by real-time PCR in MM cells transfected with p300 or p21 expressing (OE) plasmids compared with the vector control (vector). (B) GLI1 protein level in MM cells transfected with an increasing amount of p21 plasmid. (C) Half-life of GLI1 protein in RPMI8226 and MM.1S cells transfected with p21 or control plasmids was detected by Western blot. (D) RPMI8226 and MM.1S cells were transiently transfected with p21-specific or control siRNAs. Twenty-four hours post-transfection, cells were subjected for TSA treatment for another 24 h, and then, GLI1 protein was evaluated by Western blot. (E) Overexpression of p21 in RPMI8226 and MM.1S cells enhanced TSA-induced GLI1 repression. Abbreviations: TSA, trichostatin A; Veh, vehicle; MM, multiple myeloma. |
GLI1 overexpression alleviates TSA-induced apoptosis in MM cells
Subsequently, we estimated the susceptibility of GLI1-overexpressing MM cells to TSA treatment. As shown in Figure 5A, GLI1 expression was successfully elevated in RPMI8226 and MM.1S cells transfected with GLI1-expressing plasmid. When these MM cells were exposed to 5 mM TSA for 48 h, the amount of cleaved PARP-1, a marker of cell apoptosis, was significantly decreased in GLI1-overexpressing cells compared with their parental cells transfected with vector control (Figure 5B). In parallel, GLI1 overexpression greatly reduced the apoptotic rate in both the RPMI8226 and MM.1S cells treated with TSA compared with vector controls, when analyzed using flow cytometry assay (Figure 5C and D). These results indicated that GLI1-overexpresssing MM cells were more resistant to the cytotoxic effects of TSA, and validated that TSA-mediated GLI1 downregulation is indispensable for the cytotoxicity of TSA.
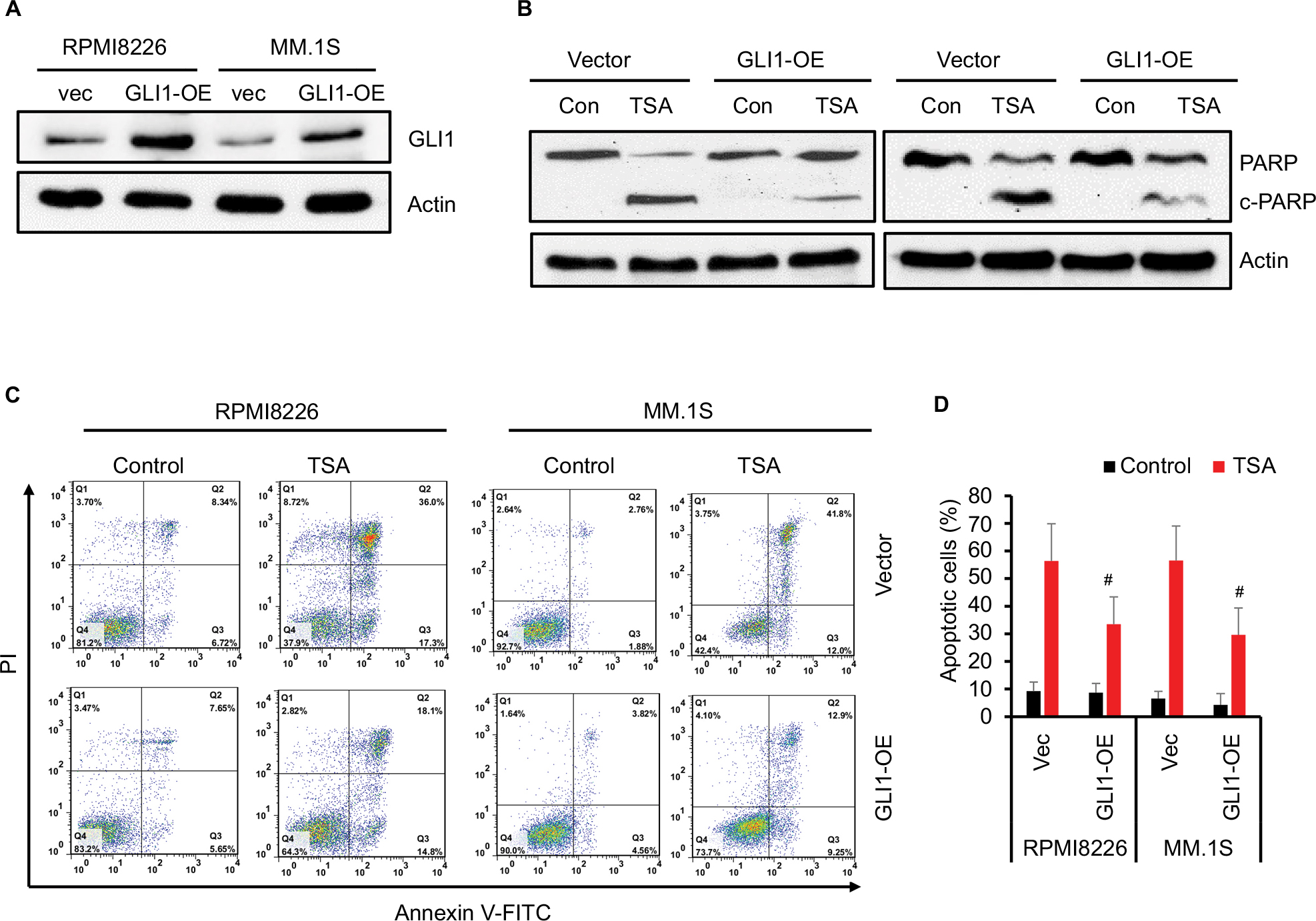 | Figure 5 GLI1 overexpression rescues TSA-induced MM cell apoptosis. Notes: (A) Western blot assay confirmed the GLI1 protein in MM cells transfected with GLI1 expressing (OE) plasmid. (B) TSA treatment induced cleavage of PARP-1 as indicative of apoptosis in the GLI1 overexpressing MM cells compared with the vector controls. (C) Representative flow cytometry analysis of MM cells apoptosis treated with TSA for 48 h using Annexin V-FITC/PI staining. (D) Statistical analysis of three similar experiments for the MM cell apoptosis (#P<0.01). Abbreviations: TSA, trichostatin A; PARP, poly(ADP-ribose) polymerase; PI, propidium iodide; FITC, fluorescein isothiocyanate; vec, vector; Con, control; MM, multiple myeloma. |
Discussion
TSA, a HDAC inhibitor, is a prospective candidate chemotherapeutic agent and is valid to antagonize the progression of numerous cancers, such as breast cancer, ovarian cancer, lung cancer, colorectal cancer, hepatocellular carcinoma, and other cancers. In this study, we evaluated the effects of HDAC inhibitor TSA on cell proliferation and apoptosis in myeloma cell lines RPMI8226 and MM.1S. Our results revealed that TSA dramatically inhibited hedgehog signaling via downregulating GLI1 expression.
Acetylation of histone and other essential cellular proteins is an important modification to regulate cellular function. Aberrant acetylation of histone and other proteins along with overexpressed HDACs are often observed in the development of disparate types of cancer. Thus, HDAC inhibitors are emerging as promising anticancer agents, particularly in the combination with other chemotherapeutic drugs and/or radiotherapy. The resultant inhibition of HDACs causes pleiotropic effects. HDAC inhibitors loosen chromatin structure to make transcription factor binding sites more accessible, thereby altering gene expression profile. Generally, HDAC inhibitors allow expression of suppressed genes and also downregulate oncogenes. HDAC inhibitors upregulate cell cycle gene p21 and block the cyclin D1/CDK4 complex with ensuing cell cycle arrest and differentiation. HDAC inhibition also promotes apoptosis via inducing elements of the apoptotic pathway such as Fas, TNF-α, as well as the TRAIL receptor; it is also well known that HDAC inhibitors affect the activity of some signaling pathways such as Wnt, ERK, PI3K/Akt, and PKC.14 HDAC inhibitors can also affect cancer angiogenesis and interfere with the metastatic process. The antiangiogenic effects of HDAC inhibitors attribute to downregulation of pro-angiogenic genes such as vascular endothelial growth factor (VEGF).2–4 In this study, our results revealed that hedgehog signaling is also a target for HDAC inhibitors.
Another mechanism of HDAC inhibition affecting cellular function is modulation of proteasomal degradation. Acetylation of p53 induced by HDAC inhibitors decreases the formation of a complex p53/Mdm2 E3 ligase and lowers p53 degradation by the proteasome, thereby cancelling p53-mediated growth arrest and apoptosis.15 On the other hand, HDAC inhibitors increase acetylation of the pro-angiogenic transcription factor HIF-1α, enhancing its degradation.16 In addition, the occurrence of HDAC inhibitors can alter the expression of several proteasomal enzymes, including Ubc8 E2 ubiquitin conjugase and RLIM subunit of the SCF E3 family of the ubiquitin ligase.17 The present study provided evidences that HDAC inhibition can modulate GLI1 degradation.
Notably, mechanisms of anticancer effects of HDAC inhibitors have not been concluded an agreement. The effects of HDAC inhibitors may be different among varying cancers. It depends on the genotype and context of cancer, as well as on some other factors. For instance, HDAC inhibitors have not induced cell cycle inhibition in some neuroblastoma cell lines such as SH-SY5Y and SK-N-BE.18 With regard to MM, hedgehog signaling is frequently active and plays important roles in cancer development.10,14,15 Our results showed that GLI1 normally accumulates in the nucleus, implicating a consecutive activation of hedgehog signaling in MM cells. It lays a foundation for HDAC inhibitor to affect cell proliferation and apoptosis via modulating hedgehog signaling. Meanwhile, GLI1-overexpresssing MM cells were resistant to TSA treatment compared with control cells. Thus, it seems that TSA-mediated GLI1 downregulation is indispensable for the cytotoxicity of TSA.
Among three GLI transcriptional factors, GLI1 functions primarily as a constitutive activator and potentiates the transcriptional output of hedgehog signaling, whereas GLI3 is predominantly a transcriptional repressor; GLI2 acts as a principal activator to trigger the expression of GLI1.19 Activation of hedgehog signaling has profound effects to affect the stability of GLI transcriptional factors. In the absence of hedgehog ligands, the three isoforms of GLI transcriptional factors are readily ubiquitinated by a phosphorylation-dependent E3 ubiquitin ligase b-TrCP. Ubiquitination of GLI proteins requires priming phosphorylations by PKA, CK1, and GSK-3β. After ubiquitination, GLI1 is largely degraded and removed from the cell by the proteasomal system, whereas GLI2 and GLI3 are partially cleaved to form truncated repressor forms. In contrast to the full-length GLI2 as an activator, its truncated form migrates to the nucleus, binds to consensus sequences, and represses transcription of hedgehog target genes. The occurrence of hedgehog ligands ameliorates this cleavage and leads to the accumulation of full-length activator GLI2.14,15
Acetylation also plays an important role in regulating the stability of GLI proteins. As demonstrated by previous studies, GLI1 can be acetylated and HDAC-mediated deacetylation promotes GLI1 transcriptional activation. HDAC1/2 and GLI1 can form a complex and are recruited onto the promoters of GLI-target genes.20 In contrast, the acetyltransferase p300/CBP-associated factor promotes GLI1 ubiquitination and subsequent proteasome-dependent degradation.21 Additionally, β-arrestin1 (Arrb1) is found to repress the transcriptional activity of GLI1, by potentiating its p300-mediated acetylation.22 Thus, it was found that selective inhibition of HDAC1/2 can inhibit hedgehog signaling and decrease tumor growth in preclinical models of SHH medulloblastoma.23
Consistent with this, our results indicated that HDAC inhibition can accelerate the degradation of GLI1 in a proteasome-dependent manner, accompanied by the downregulation of hedgehog downstream genes. Moreover, our results implicated that TSA-induced p21 upregulation contributed to the reduced transcription of GLI1. It is well known that TSA potentiates the transcriptional activity of p53 via acetylation, thereby enhancing the expression of p53 downstream genes such as p21. In this study, we found that overexpressing p21 directly downregulated the mRNA level of GLI1, but silencing p21 did the opposite. Thus, TSA could exert inhibition on GLI1 expression and stabilization via different mechanisms. It is well known that hedgehog inhibition can lead to cell cycle arrest. Seemingly, there exists a reciprocal crosstalk between hedgehog signaling and cell cycle program. In this study, we provided strong evidence exhibiting the effect of p21 overexpression on GLI1 transcription. But, the precise mechanism remained unknown. We speculated that p21 might act as cyclin-dependent kinase inhibitor to inhibit cyclin/CDK complexes and further modify the activities of target transcriptional factors. More investigation is required to uncover the underlying mechanisms.
Conclusion
Overall, our results indicated that the HDAC inhibitor TSA can repress MM cell growth and induce cell apoptosis via abrogation of hedgehog signaling. TSA can exert versatile effects on the hedgehog transcriptional factor GLI1. TSA-mediated acetylation promotes the proteasome-dependent degradation of GLI1, while TSA-induced p21 upregulation impairs the transcription of GLI1.
Acknowledgment
This work was supported by the National Natural Science Foundation of China (81670201, ZL; 81101930, TL) and the Natural Science Foundation of Tianjin (16JCYBJC42600, ZL).
Disclosure
The authors report no conflicts of interest in this work.
References
Suraweera A, O’Byrne KJ, Richard DJ. Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: achieving the full therapeutic potential of HDACi. Front Oncol. 2018;8:92. | ||
Chun P. Histone deacetylase inhibitors in hematological malignancies and solid tumors. Arch Pharm Res. 2015;38:933–949. | ||
Golabek K, Strzelczyk JK, Wiczkowski A, Michalski M. Potential use of histone deacetylase inhibitors in cancer therapy. Contem Oncol. 2015;19: 436–440. | ||
Imai Y, Maru Y, Tanaka J. Action mechanisms of histone deacetylase inhibitors in the treatment of hematological malignancies. Cancer Sci. 2016;107:1543–1549. | ||
Wapenaar H, Dekker FJ. Histone acetyltransferases: challenges in targeting bi-substrate enzymes. Clin Epigenetics. 2016;8:59. | ||
Ginsburg DS, Anlembom TE, Wang J, et al. NuA4 links methylation of histone H3 lysines 4 and 36 to acetylation of histones H4 and H3. J Biol Chem. 2014;289: 32656–32670. | ||
Sowa Y, Orita T, Hiranabe-Minamikawa S, et al. Histone deacetylase inhibitor activates the p21/WAF1/Cip1 gene promoter through the Sp1 sites. Ann N Y Acad Sci. 1999;886:195–199. | ||
Saunders A, Huang X,Fidalgo M, et al. The SIN3A/HDAC corepressor complex functionally cooperates with NANOG to promote pluripotency. Cell Rep. 2017;18:1713–1726. | ||
Moreaux J, Reme T, Leonard W, et al. Gene expression-based prediction of myeloma cell sensitivity to histone deacetylase inhibitors. Br J Cancer. 2013;109:676–685. | ||
Fandy TE, Srivastava RK. Trichostatin A sensitizes TRAIL-resistant myeloma cells by downregulation of the antiapoptotic Bcl-2 proteins. Cancer Chemother Pharmacol. 2006;58:471–477. | ||
Heller, G, Schmidt WM, Ziegler B, et al. Genome-wide transcriptional response to 5-aza-2′-deoxycytidine and trichostatin A in multiple myeloma cells. Cancer Res. 2008;68:44–54. | ||
Li T, Li YM, Jia ZQ, Chen P, Ma KT, Zhou CY. Carboxyl terminus of Nkx2.5 impairs its interaction with p300. J Mol Biol. 2007;370:976–992. | ||
Liu Z, Xu J, He J, et al. A critical role of autocrine sonic hedgehog signaling in human CD138+ myeloma cell survival and drug resistance. Blood. 2014;124:2061–2071. | ||
Eckschlager T, Plch J, Stiborova M, Hrabeta J. Histone deacetylase inhibitors as anticancer drugs. Int J Mol Sci. 2107;18. pii:E1414. | ||
Tang Y, Zhao W, Chen Y, Zhao Y, Gu W. Acetylation is indispensable for p53 activation. Cell. 2008;133:612–626. | ||
Jeong JW, Bae MK, Ahn MY, et al. Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell. 2001;111:709–720. | ||
Kramer OH, Zhu P, Ostendorff M, et al. The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J. 2013;22:3411–3420. | ||
Stockhausen MT, Sjolund J, Manetopoulos C, Axelson H. Effects of the histone deacetylase inhibitor valproic acid on Notch signalling in human neuroblastoma cells. Br J Cancer. 2005;92:751–759. | ||
Ok CY, Singh RR, Vega F. Aberrant activation of the hedgehog signaling pathway in malignant hematological neoplasms. Am J Pathol. 2012;180:2–11. | ||
Canettieri G, Di Marcotullio L, Greco A, et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates hedgehog signalling through Gli acetylation. Nat Cell Biol. 2010;12:132–142. | ||
Mazza D, Infante P, Colichhia V, et al. PCAF ubiquitin ligase activity inhibits hedgehog/Gli1 signaling in p53-dependent response to genotoxic stress. Cell Death Differ. 2013;20;1688–1697. | ||
Miele E, Po A, Begalli F, et al. Beta-arrestin1-mediated acetylation of Gli1 regulates hedgehog/Gli signaling and modulates self-renewal of SHH medulloblastoma cancer stem cells. BMC Cancer. 2017;17:488. | ||
Coni S, Mancuso AB, Di Mango L, et al. Selective targeting of HDAC1/2 elicits anticancer effects through Gli1 acetylation in preclinical models of SHH medulloblastoma. Sci Rep. 2017;7:44079. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.