Back to Journals » Nature and Science of Sleep » Volume 7
Treatment paradigms for cataplexy in narcolepsy: past, present, and future
Received 10 July 2015
Accepted for publication 19 October 2015
Published 11 December 2015 Volume 2015:7 Pages 159—169
DOI https://doi.org/10.2147/NSS.S92140
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Steven Shea
Todd J Swick1–4
1Department of Neurology, University of Texas School of Medicine-Houston, Houston; 2The Sleep Center at North Cypress Medical Center, Cypress; 3Apnix Sleep Diagnostics, Houston; 4Neurology and Sleep Medicine Consultants, Houston, TX, USA
Abstract: Cataplexy is defined as episodes of sudden loss of voluntary muscle tone triggered by emotions generally lasting <2 minutes. Cataplexy is most commonly associated with and considered pathognomonic for narcolepsy, a sleep disorder affecting ~0.05% of the general population. Knowledge of the pathophysiology of cataplexy has advanced through study of canine, murine, and human models. It is now generally considered that loss of signaling by hypothalamic hypocretin/orexin-producing neurons plays a key role in the development of cataplexy. Although the cause of hypocretin/orexin neuron loss in narcolepsy with cataplexy is unknown, an autoimmune etiology is widely hypothesized. Despite these advances, a literature review shows that treatment of cataplexy remains limited. Multiple classes of antidepressants have been commonly used off-label for cataplexy in narcolepsy and are suggested for this use in expert consensus guidelines based on traditional practice, case reports, and small trials. However, systematic research evidence supporting antidepressants for cataplexy is lacking. The single pharmacotherapy indicated for cataplexy and the guideline-recommended first-line treatment in Europe and the US is sodium oxybate, the sodium salt of gamma-hydroxybutyrate. Clinical trial evidence of its efficacy and safety in cataplexy is robust, and it is hypothesized that its therapeutic effects may occur through gamma-aminobutyric acid receptor type B-mediated effects at noradrenergic, dopaminergic, and thalamocortical neurons. Additional possible mechanisms for cataplexy therapy suggested by preliminary research include antagonism of the histamine H3 autoreceptor with pitolisant and intravenous immunoglobulin therapy for amelioration of the presumed autoimmune-mediated hypocretin/orexin cell loss. Further research and development of therapeutic approaches to cataplexy are needed.
Keywords: cataplexy, narcolepsy, treatment, sodium oxybate, antidepressants, emerging therapies
Cataplexy: definition and characteristics
Cataplexy is defined as episodes of sudden, transient loss of voluntary muscle tone (usually bilateral, but case reports have identified unilateral cases1) triggered by strong emotions. While laughter is the most typical trigger, other triggers include happiness, elation, fright, anger, startle, stress and, less frequently, pain and orgasm, although episodes may also occur spontaneously.2–4 Episodes are typically brief, generally lasting <2 minutes, followed by rapid return of normal muscle tone/function, and range from mild or barely noticeable to severe, with complete postural collapse. While any or all voluntary muscles can be affected (with the exception of the diaphragm and the extraocular muscles of the eye), patients remain conscious and continue to breathe and to move their eyes.3 The most common manifestations are neck weakness, causing head drop; partial or complete ptosis; facial weakness with sagging of the jaw with or without dysarthria; and trembling or buckling of the knees.2,5,6 Positive motor symptoms such as muscle twitching or small jerks of the face or limbs also occur, sometimes contributing to misdiagnosis.2,5 Patients typically sense the onset of an episode, allowing them to sit or brace themselves before its occurrence, thus reducing the risk of injury, although in one survey, up to half of patients reported some kind of injury from their cataplexy.2 Duration of a cataplectic attack generally lasts from a few seconds to several minutes. However, more commonly after the abrupt discontinuation of antidepressant medication (tricyclic antidepressants [TCAs], serotonin reuptake inhibitors [SSRIs], or serotonin-norepinephrine reuptake inhibitor [SNRI]), attacks of cataplexy typically are more frequent and/or more intense (rebound phenomena) and can last up to several hours, at which time they are designated as status cataplecticus.7,8 Frequency of episodes is variable, ranging from <1 episode per year to several per day, but most patients have several episodes per week.2,5,6,9
Narcolepsy with cataplexy is a sleep disorder that is traditionally characterized by a symptom pentad that includes, in addition to cataplexy, excessive daytime sleepiness (EDS), sleep paralysis, hypnagogic hallucinations, and disrupted nighttime sleep.10 Narcolepsy with cataplexy is estimated to affect 0.03% to 0.05% of the general population,11 and onset occurs typically in childhood or adolescence.6,12 Although EDS is present in all patients with narcolepsy and is often the initial presenting symptom, cataplexy occurs in ~70% of patients;3,13 cataplexy is considered pathognomonic for narcolepsy and after sleepiness is the primary behavioral marker.
The third edition of the International Classification of Sleep Disorders (ICSD-3) classifies narcolepsy as either type 1 or type 2, with the presence of cataplexy incorporated into the definition of type 1.13 Type 1 narcolepsy is formally defined in the ICSD-3 as EDS that persists for ≥3 months with “positive” electrophysiological sleep studies that includes the finding of an average sleep-onset latency ≤8 minutes on the MSLT following a nocturnal polysomnogram that was negative for any comorbid sleep disorders, and two or more sleep-onset rapid eye movement (REM) periods on the MSLT (one of these sleep-onset REM periods may come from the preceding nocturnal polysomnogram) with clear historic evidence of cataplexy or low or absent levels of hypocretin/orexin in cerebrospinal fluid (CSF) along with the “positive” sleep studies.13 Hypocretin-1 and hypocretin-2, also called orexin A and orexin B, respectively, are peptide neurotransmitters that facilitate wakefulness and enhance arousal mechanisms as well as stabilizing REM and non-REM sleep states.14,15 Type 2 narcolepsy, or narcolepsy without cataplexy, is defined as EDS persisting for ≥3 months, a positive result on the polysomnogram/Multiple Sleep Latency Test (MSLT), and normal or mildly decreased CSF hypocretin/orexin levels; hypocretin-1 is more stable than hypocretin-2 and thus is the peptide that is measured in CSF as a neurochemical marker of narcolepsy.13
Although cataplexy is often absent as an early symptom of narcolepsy in children, retrospective data suggest that children with narcolepsy who initially present without cataplexy will likely develop cataplexy within 1 year of EDS onset.16–18 Additionally, the initial presenting cataplexy phenotype in children can differ from the presentation commonly seen in adults.16,19,20 Relative to the exclusive loss of voluntary muscle tone in adults, early manifestations of cataplexy in children may variously include both hypotonia and abnormal movements, with prominent facial involvement (often referred to as “cataplectic facies”), including partial ptosis, masked-like facies with jaw slackening, facial grimaces, and perioral and tongue movements/protrusion all with or without dysarthria.16,19,20 Furthermore, chorea-like or dystonic movements, gait disturbances, and complete falls can occur spontaneously without apparent emotional triggers.16,19,20
Cataplexy or cataplexy-like symptoms have been observed in other conditions including Niemann–Pick type C disease, Prader–Willi syndrome, Coffin–Lowry syndrome, Moebius syndrome, Norrie disease, and Wilson’s disease.21–28 Although some of these conditions may also be accompanied by sleep disorders and hypocretin/orexin deficiency,29–31 they are characterized by distinct features including other specific neurological deficits or mental/cognitive changes, or both, such that there is little overlap in diagnosis with narcolepsy with cataplexy.
Pathophysiology of narcolepsy with cataplexy
Role of hypocretin/orexin in narcolepsy
Located in the posterolateral hypothalamus, hypocretin/orexin neurons project to almost all brain areas and play critical roles in regulation of the sleep/wake cycle, as well as metabolism, feeding, reward, mobility, and autonomic tone (Figure 1).32–36 Hypocretin/orexin neurons innervate noradrenergic, dopaminergic, serotonergic, histaminergic, and cholinergic brain regions and directly excite these neurons, as well as regulate release of glutamate and other neurotransmitters (Figure 2A and B).34,37–39 Studies of neuronal sleep regulation in murine and other animal models have shown that hypocretin/orexin neurons discharge during wakening, promoting wakefulness and the return of postural muscle tone, and virtually cease discharge during sleep; their activity appears to suppress cortical deactivation and muscle atonia.40,41
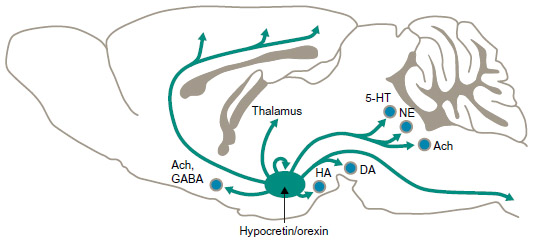 | Figure 1 Hypocretin/orexin neuron innervation. |
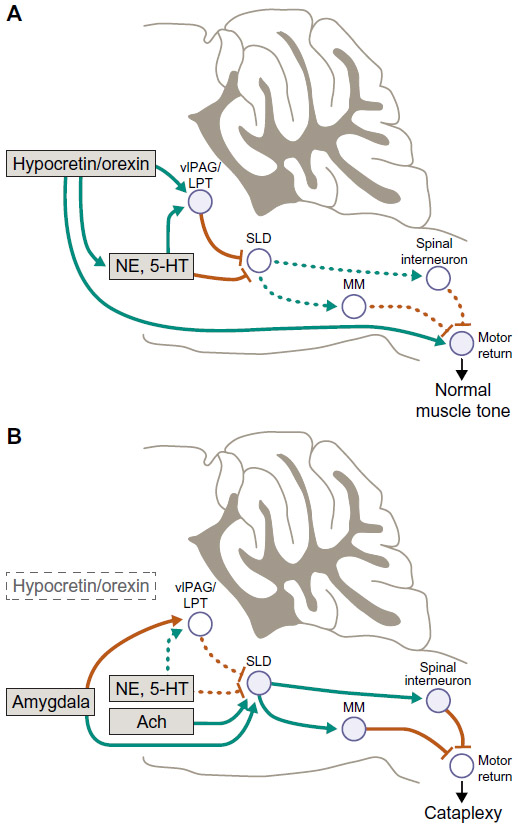 | Figure 2 Atonia pathways triggering cataplexy. |
The discovery that loss of signaling by these hypothalamic neuropeptides is the presumptive cause of narcolepsy in humans, dogs, and mice has been a major advance in our understanding of the regulation of sleep and wakefulness along with the control of REM-on and REM-off states.37,42–44 While the underlying pathologic changes resulting in this loss of signaling are different among the models, genetic mutation of the gene encoding the hypocretin-2 protein in dogs,42 knockout of genes encoding hypocretin receptors in mouse models,43 and selective loss of hypocretin neurons likely due to an autoimmune response in humans,44 the manifestations resulting from the loss of hypocretin signaling support this loss as the pathophysiologic basis of narcolepsy.
Narcolepsy with cataplexy is typically associated with the loss of 90% or more of hypocretin/orexin-producing neurons with low or undetectable CSF hypocretin-1/orexin A levels.34 A CSF hypocretin/orexin level below 110 pg/mL is considered diagnostic for type 1 narcolepsy,13 but a standard test for hypocretin/orexin is not available for general use in the clinical setting. Most patients with narcolepsy without cataplexy have CSF hypocretin/orexin levels in the normal range, although a reduced hypocretin/orexin concentration in this subgroup of patients is associated with worse narcolepsy symptoms than in patients with higher levels.17 Mouse models suggest that cataplexy occurs when loss of hypocretin/orexin-producing neurons is ~95%, whereas early narcolepsy traits such as sleep fragmentation/disturbance and reduced wakefulness were observed even at low levels of neuronal destruction.36,45 Such a relationship was further supported by a case study suggesting that cataplexy may be on a narcolepsy spectrum that results from a decremental loss of hypocretin/orexin.46
Although the etiopathogenesis of the destruction of these hypocretin/orexin-producing neurons has not been fully elucidated, it appears to result from a process selective for these neurons rather than generalized neuronal degeneration.37,47 A widely held hypothesis is that autoimmune processes contribute to the pathogenic mechanisms.47,48 Support for autoimmune mechanisms also comes from the observed association of narcolepsy with specific genotypes, including the human leukocyte antigen HLA-DQB1*06:02, which is found in ~95% of patients with what is currently classified as type 1 narcolepsy, 41% of patients with type 2, and only 18%–35% of the general population.49 An observed association between T-cell receptor variants and loss of hypocretin/orexin-producing neurons further supports the autoimmune hypothesis,50,51 as do the reports of narcolepsy onset associated with seasonal Streptococcus infections, H1N1 influenza, and H1N1 vaccination in individuals with genetic predisposition to induction of autoimmune events. While researchers have yet to definitively determine the specific autoimmune mechanisms involved, a nucleoprotein that is present in both the H1N1 virus and the Pandemrix H1N1 vaccine has been identified that cross-reacts with the hypocretin receptor 2.52 The additional finding of a possible immune response to the hypocretin-2 receptor after the Pandemrix H1N1vaccine in DQB1*0602-positive narcoleptic patients supports the autoimmune hypothesis.52
Neurological pathways of cataplexy
Investigations of the neurophysiology of cataplexy have included studies in humans and animal models (ie, dogs and mice; Table 1). The “REM sleep disassociation hypothesis” suggests that cataplexy and sleep paralysis are dysregulated manifestations, or intrusions into the waking state, of the skeletal muscle motor inhibition that normally occurs during REM sleep to prevent the acting out of dreams, while diaphragmatic breathing and extraocular muscles remain functional.9 Indeed, studies in dogs and humans have suggested that brainstem circuitry is similar in both REM sleep and cataplexy episodes.53,54 However, this does not fully address the triggering of cataplexy by emotional stimulation, and this mechanism has also been an area of investigation and source of alternative hypotheses.55–58
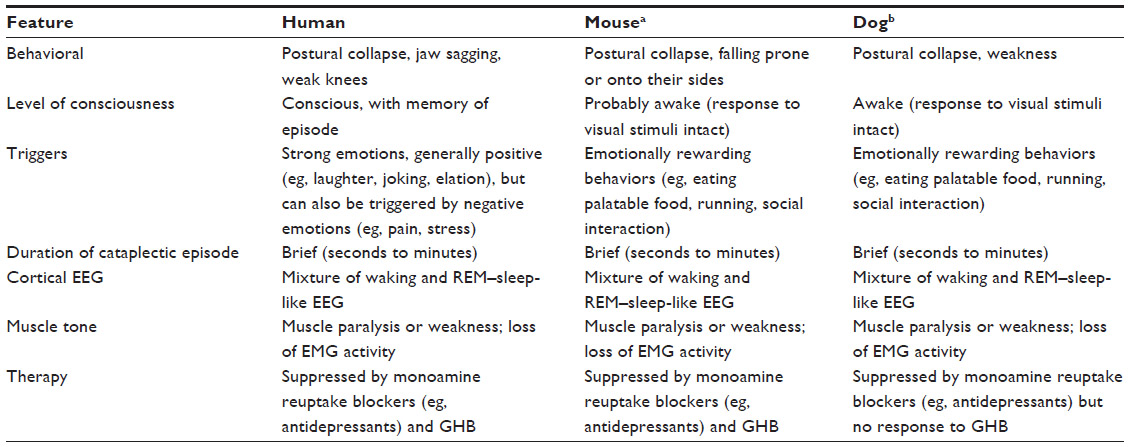 | Table 1 Cataplexy across species |
Cataplectic atonia is caused by inhibition of skeletal motor neuron activity and absence of deep tendon reflexes and the loss of the monosynaptic Hoffman reflex,56 which results from increased inhibitory and reduced excitatory signaling of motor neurons in the brain and spinal cord.34,59,60 Notably, loss of Hoffman reflex activity is common to cataplexy, laughter, and REM sleep.56 Neurochemically, cataplexy is triggered by cholinergic activation and deactivation of monoaminergic systems primarily in the brainstem, especially those of adrenergic pathways, which may be caused by an imbalance of monoamines and acetylcholine.55,59 This inhibitory mechanism is characterized by intense activation of gamma-aminobutyric acid (GABA)–releasing neurons in the medial medulla and central nucleus of the amygdala, which, in turn, inhibits noradrenergic neurons that maintain waking muscle tone such as those in the ventrolateral periaqueductal gray, lateral pontine tegmentum, locus coeruleus, and dorsal raphe.9,58,61 This action turns off release of noradrenaline to motor neurons (both alpha-motor neurons and spinal interneurons), leading to their reduced activity with emergence of cataplectic atonia.9,34 It has also been suggested that the emotionally-induced cataplectic atonia may result from glutamatergic excitatory activation of neurons in the sublaterodorsal tegmental nucleus, which generally regulate muscle atonia during REM sleep;62 in normal individuals, that is, those with normal hypocretin levels, this excitation during waking would be blocked by a hypocretin-mediated GABAergic effect. Other brain regions involved in the neural circuitry of cataplexy may also include basal forebrain, hypothalamus, and limbic structures, although the precise roles of these regions are yet to be fully elucidated.34,57,58,63
Cataplexy treatment
Clinical management of cataplexy is limited by etiological uncertainty as well as by challenges in the clinical identification and diagnosis of the underlying narcolepsy; diagnosis is often delayed by many years after initial symptom onset.64 In particular, there are no standard measures for identifying or assessing cataplexy, increasing the challenges not only of its identification but also of evaluation during treatment. Thus, the recognition of cataplexy has primarily been based on clinical interview and patient self-reports. With evaluation of cataplexy during clinical trials or in the clinical setting relying on patient recall and/or diaries, the manner of assessment often varies and may not always include a full description of frequency and severity. Although self-administered cataplexy questionnaires have been developed for eliciting information on occurrence,65,66 they may be limited by a high burden of administration (one of the validated questionnaires consists of 51 questions65) and/or use only as a screening tool since their sensitivity to treatment effects has not been demonstrated. However, despite these challenges, the pharmacological management of cataplexy has a long history and a potentially promising future.
Pharmacological management: historical overview
Multiple classes of pharmacological agents have been used for treatment of narcolepsy, including stimulants, antidepressants, and hypnosedatives. The majority of these agents have not been rigorously tested for either safety or efficacy in cataplexy. Use of amphetamines was among the earliest approaches to narcolepsy treatment; they were first used for this purpose in 1935 based on their strong wake-promoting effects.67 Although these drugs have consistently been used and are generally considered effective for reducing EDS, their lack of efficacy for cataplexy was recognized early during their clinical use.68 Methylphenidate, with a similar mechanism of action and somewhat better safety profile compared with the amphetamines, has been widely used for narcolepsy since the 1950s,69 but also like the amphetamines, there have been no demonstrable effects vis-à-vis improvement of cataplexy.
TCAs such as clomipramine, imipramine, desipramine, and protriptyline have been used as a therapeutic approach for narcolepsy for many years, and their efficacy for cataplexy was reported as early as 1960.70,71 Since then, evidence from case reports and small open-label studies has further supported their beneficial effects for improving cataplexy.72–77 It is important to note that these drugs have never been evaluated in larger and more formal randomized, placebo-controlled clinical trials. Although the mechanism of action of these drugs for cataplexy remains unknown, it has been thought that it may be related to their augmentation of noradrenergic signaling and/or the relative suppression of REM sleep.78
Current therapeutic approaches
Treatment recommendations for cataplexy are included as part of the overall guidelines for the treatment of narcolepsy issued by the American Academy of Sleep Medicine79 and European Federation of Neurological Societies.80 Both of these guidelines recommend several drugs as being of potential benefit for the treatment of cataplexy, but only sodium oxybate is recommended as a first-line treatment for cataplexy based on high levels of evidence obtained from randomized controlled clinical trials. Sodium oxybate is indicated for the treatment of cataplexy associated with narcolepsy81 and is currently the only medication with such an indication. Additionally, both guidelines recommend sodium oxybate as a first-line therapy for treatment of EDS in narcolepsy,79,80 for which it is indicated,81 and the American Academy of Sleep Medicine guidelines also recommend sodium oxybate as a standard treatment for disrupted sleep, although it is not approved by the US Food and Drug Administration or the European Medicines Agency for use for the treatment of this symptom. Both guidelines further suggest that sodium oxybate be considered for hypnagogic hallucinations and sleep paralysis;79,80 as is the case for disrupted nighttime sleep, it is not specifically indicated for these symptoms, and the level of evidence is lower than for cataplexy and EDS.
As mentioned in the guidelines, the evidentiary basis for therapeutic alternatives to sodium oxybate for the treatment of cataplexy is limited. Suggested alternatives include TCAs, particularly clomipramine, and newer antidepressants such as SSRIs, the SNRI venlafaxine, and norepinephrine reuptake inhibitor reboxetine (not available in the US).79,80 The monoamine oxidase type B inhibitor selegiline is also noted for its efficacy in cataplexy, although both guidelines warn of limitations due to its safety profile including the potential for drug and food interactions.79,80 The European guidelines identify TCAs as the most effective of the alternatives to sodium oxybate for cataplexy, whereas the American guidelines do not state any general preferences among the second-line drug classes.
Overall, the currently available treatments for cataplexy act symptomatically, and there is no evidence suggesting that they target the hypocretin/orexin system (Table 2).82 Sodium oxybate, which is the sodium salt of gamma-hydroxybutyrate, was observed to induce REM sleep followed by an increase in slow-wave non-REM sleep and was subsequently investigated for the treatment of narcolepsy on the hypothesis that improvement of nocturnal sleep would reduce EDS and possibly other narcolepsy symptoms, including cataplexy.83,84 Although the mechanism of action of sodium oxybate is unknown, it is hypothesized that its therapeutic effects on cataplexy and EDS are mediated through GABA type B (GABAB) activity, potentially impacting noradrenergic and dopaminergic neuronal function as well as that of thalamocortical neurons.81,85 However, a clinical comparison with the prototypical GABAB agonist baclofen (a racemic mixture of R- and S-isomers of baclofen) showed that while sodium oxybate reduced EDS and cataplexy, baclofen had no effect on these narcolepsy symptoms. These results suggest that the efficacy of sodium oxybate may derive from mechanisms more complex than direct GABAB agonism.86,87
 | Table 2 Commonly used anticataplectic medications and their pharmacological properties |
The mechanism of action of the antidepressants in narcolepsy with cataplexy is generally related to their ability to inhibit reuptake of monoamines,9,87,88 with a high correlation between receptor affinity and potency of antidepressants for their effects on cataplexy.89–91 The TCAs are nonspecific monoamine reuptake inhibitors with effects that promote the availability and activity of serotonin, noradrenaline and, for some agents, dopamine. The TCAs most commonly used in narcolepsy – clomipramine, imipramine, desipramine, and protriptyline – exert, in addition to monoamine reuptake inhibition, anticholinergic actions that may contribute to their effect on cataplexy but may also cause important side effects such as constipation, dry mouth, blurred vision, restless legs, and alterations in cardiac conduction.9,87 While inhibition of serotonin reuptake by SSRIs reduces cataplexy, very selective SSRIs such as escitalopram or fluoxetine are usually not as effective for cataplexy as venlafaxine, an SNRI that is used mainly because it has demonstrated a short onset of action.87 However, abrupt withdrawal of venlafaxine may precipitate rebound cataplexy.8 The monoamine oxidase type B inhibitor selegiline increases availability of the monoamine neurotransmitter dopamine, but its use is limited by potential side effects, as also noted in the narcolepsy treatment guidelines.89,90
As suggested by the high level of evidence used to support its recommendation as a first-line therapy, sodium oxybate has demonstrated efficacy for the treatment of cataplexy and other narcolepsy symptoms in multiple randomized controlled clinical trials.92–94 In particular, the median percent decrease in the number of weekly cataplexy attacks was ~70% after 4 weeks of treatment with 9 g per night, and no rebound cataplexy was observed in patients who had their sodium oxybate abruptly discontinued.93,94 Long-term studies showed that these effects on cataplexy were maintained for 12 months and up to 4 years.95,96 Two systematic reviews and meta-analyses of sodium oxybate provided further support for the robustness of its efficacy for reducing cataplexy episodes, with the greatest effects observed at the highest dose of 9 g per night.97,98 These meta-analyses also reported that sodium oxybate was generally well tolerated, with most adverse events of mild-to-moderate severity. Furthermore, a retrospective analysis of sodium oxybate in a pediatric population suggested that in children and adolescents aged 4 through 16 years, treatment significantly reduced the median frequency of cataplexy episodes from 38 per week to <1 per week (n=14; P<0.001) and reduced cataplexy severity relative to baseline (P<0.001; sodium oxybate is not indicated for use in patients <18 years old).99 It should also be noted that sodium oxybate has the potential for serious adverse effects (eg, central nervous system depression, abuse/misuse, respiratory depression and sleep-disordered breathing, depression and suicidality, other behavioral/psychiatric adverse reactions, and parasomnias)81 and when used at higher than therapeutic doses, whether accidentally or intentionally, it can lead to acute or fatal intoxication.100
In contrast to sodium oxybate, randomized controlled trials evaluating antidepressants for narcolepsy are sparse. A 2008 Cochrane Database System Review of the effects of antidepressant drugs on narcolepsy symptoms, including cataplexy, found only three crossover and two parallel trials of “unclear” methodological quality, resulting in the conclusion that there is “scarce evidence” that antidepressants have a positive effect on cataplexy.101 Although there have not been any additional, more formal evaluations of these drugs for cataplexy, or indeed for narcolepsy in general, TCAs continue to be used off-label for cataplexy on the basis of historical precedent, anecdotal evidence from case reports, and guideline suggestions of their potential efficacy. Off-label use of other antidepressants (SSRIs and SNRIs) for cataplexy is similarly based primarily on case reports102–106 and their listing in guidelines.79,80 The well-recognized side effects associated with antidepressants must also be considered, including anticholinergic effects of TCAs, as noted above, sexual dysfunction, and exacerbation of other sleep disorders caused by most antidepressants of all classes.106–108 Although onset of action in cataplexy is rapid, rebound cataplexy, which can be dramatic and rapid, has been observed to occur with cessation of these treatments, occurring as early as the day following treatment interruption.7,8,109,110
Ongoing clinical cataplexy treatment research
Histamine H3-receptor antagonism using direct receptor antagonists or inverse agonists
In addition to the catecholamines, tuberomammillary histaminergic neurons play a crucial role in maintenance of wakefulness but remain active during cataplexy, helping to preserve consciousness.34,111 Indeed, these neurons appear to be increased in narcolepsy, perhaps as a compensatory response to hypocretin/orexin loss and the resulting deficit in excitatory adrenergic drive.112 Pitolisant is an inverse agonist of the histamine H3 autoreceptor, which theoretically reduces H3 inhibitory activity below basal rates and thereby functions more effectively than H3 antagonists to activate histaminergic neuronal activity in the brain and promote wakefulness. While it reduced EDS in patients with narcolepsy in a small clinical trial,113 a post hoc analysis of another clinical trial reported that pitolisant for 8 weeks resulted in statistically significant reductions in the rate of cataplexy from baseline compared with placebo (P<0.05) and was not “noninferior” relative to modafinil in terms of improvement in daytime sleepiness.114 Pitolisant was also shown to slightly improve cataplexy severity and frequency in a case series of four teenagers with narcolepsy.115 The mechanism by which pitolisant may have positive effects on cataplexy is not clear since histaminergic activity is maintained during cataplexy and likely contributes to maintenance of wakefulness during these events.111 Nevertheless, based on the preliminary results, a Phase III, randomized, controlled trial was initiated to specifically evaluate the effects of pitolisant on cataplexy as a primary outcome compared with placebo (ClinicalTrials.gov identifier NCT01800045).
Hypocretin/orexin replacement therapy
Compensating for hypocretin/orexin deficiency through the use of hypocretin/orexin peptide supplementation or cell replacement therapies may provide a rational approach to narcolepsy with cataplexy therapy.82 Potential techniques include delivery of hypocretin/orexin peptides via intranasal, intravenous, intracisternal, or intracerebroventricular modes; use of prodrugs or agonists; or by genetic engineering or cell replacement techniques.82 While these techniques are still in their early stages of development, the few available human studies have shown potential benefits for sleep and wakefulness, but the effects on cataplexy were not evaluated.116,117 However, in a canine model of narcolepsy, repeated systemic administration of hypocretin-1/orexin-A consolidated waking and sleeping periods and abolished cataplexy completely for periods of ≥3 days.118 Further evaluation of these techniques may confirm the benefits of this therapeutic approach and may also provide insight into the underlying mechanisms of narcolepsy and cataplexy.
GABAB agonist
Baclofen, a GABAB agonist that has been suggested to improve nighttime sleep in patients with several neurological conditions including narcolepsy, has not demonstrated efficacy for cataplexy or EDS associated with narcolepsy.86 However, a more recent study in a mouse model of narcolepsy showed that R-baclofen, an enantiomer with a three-fold higher affinity for GABAB receptors than the clinically available racemate, had greater efficacy than placebo in reducing cataplexy-like activity and non-REM sleep disturbances.119 Further research is needed to determine if these observations translate into clinical benefits for the treatment of cataplexy.
Immunomodulation
The autoimmune hypothesis of narcolepsy with cataplexy provides a rationale for use of immunomodulation therapy,82 which is exemplified by experimental use of intravenous immunoglobulin therapy, although it has only been evaluated in case studies in adults and children.120–128 While benefits were not consistently demonstrated in all studies, several studies did report that, if administered shortly after disease onset, intravenous immunoglobulin therapy may be effective in reducing narcolepsy symptoms including cataplexy and may have long-term benefits.123–127 These initial results suggest that further, more formal evaluation of intravenous immunoglobulin may be warranted.
In summary, despite advances in our knowledge of narcolepsy, treatment of cataplexy remains challenging. Only one medication, sodium oxybate, has been approved, has a high level of evidence of sustained efficacy for the treatment of cataplexy, and should be considered a first-line therapy. Other drugs, such as antidepressants, some of which have been traditionally used for narcolepsy with cataplexy for more than 50 years, still lack adequate evidence supporting their use, especially for chronic treatment of cataplexy. While formal studies can more clearly evaluate their efficacy and characterize those patients for whom they may be appropriate, they remain important components of treatment and should be considered as second-line therapy. The limited availability of treatment options for cataplexy suggests a need for development and evaluation of new approaches to the management of cataplexy. Ideally, drugs are needed that target cataplexy as well as other narcolepsy symptoms while maintaining an acceptable tolerability profile.
Acknowledgments
E Jay Bienen, PhD, and Larry Deblinger of the Curry Rockefeller Group, LLC, provided medical writing assistance in developing this manuscript. This review was funded by Jazz Pharmaceuticals, Inc.
Disclosure
Dr Swick is an employee of Neurology and Sleep Medicine Consultants; has received consultancy fees and/or honoraria from Jazz Pharmaceuticals, Inc., Vanda Pharmaceuticals, XenoPort Pharmaceuticals, UCB Pharma, Merck, and Aerial BioPharma; has received grant/research funding from Jazz Pharmaceuticals, Inc., Aerial BioPharma, GSK Pharmaceuticals, Otsuka Pharmaceuticals, Teva Pharmaceuticals, Vanda Pharmaceuticals, UCB Pharma, XenoPort Pharmaceuticals, and Merck; and is on the speakers bureaus for Jazz Pharmaceuticals, Inc., XenoPort Pharmaceuticals, Teva Pharmaceuticals, Merck, and UCB Pharma.
References
McCarty DE. A case of narcolepsy with strictly unilateral cataplexy. J Clin Sleep Med. 2010;6(1):75–76. | |
Overeem S, van Nues SJ, van der Zande WL, Donjacour CE, van Mierlo P, Lammers GJ. The clinical features of cataplexy: a questionnaire study in narcolepsy patients with and without hypocretin-1 deficiency. Sleep Med. 2011;12(1):12–18. | |
National Institute of Neurological Disorders and Stroke. Narcolepsy Fact Sheet. NIH publication 13-1637; last updated January 5, 2015. Available from: http://www.ninds.nih.gov/disorders/narcolepsy/detail_narcolepsy.htm#241213201. Accessed August 9, 2015. | |
Poryazova R, Khatami R, Werth E, Bassetti CL. Weak with sex: sexual intercourse as a trigger for cataplexy. J Sex Med. 2009;6(8):2271–2277. | |
Sturzenegger C, Bassetti CL. The clinical spectrum of narcolepsy with cataplexy: a reappraisal. J Sleep Res. 2004;13(4):395–406. | |
Okun ML, Lin L, Pelin Z, Hong S, Mignot E. Clinical aspects of narcolepsy-cataplexy across ethnic groups. Sleep. 2002;25(1):27–35. | |
Martinez-Rodriguez J, Iranzo A, Santamaria J, et al. [Status cataplecticus induced by abrupt withdrawal of clomipramine]. Neurologia. 2002;17(2):113–116. Spanish. | |
Wang J, Greenberg H. Status cataplecticus precipitated by abrupt withdrawal of venlafaxine. J Clin Sleep Med. 2013;9(7):715–716. | |
Dauvilliers Y, Siegel JM, Lopez R, Torontali ZA, Peever JH. Cataplexy – clinical aspects, pathophysiology and management strategy. Nat Rev Neurol. 2014;10(7):386–395. | |
Nishino S. Clinical and neurobiological aspects of narcolepsy. Sleep Med. 2007;8(4):373–399. | |
Longstreth WT Jr, Koepsell TD, Ton TG, Hendrickson AF, van Belle G. The epidemiology of narcolepsy. Sleep. 2007;30(1):13–26. | |
Dauvilliers Y, Montplaisir J, Molinari N, et al. Age at onset of narcolepsy in two large populations of patients in France and Quebec. Neurology. 2001;57(11):2029–2033. | |
American Academy of Sleep Medicine. The International Classification of Sleep Disorders – Third Edition (ICSD-3). Darien, IL: American Academy of Sleep Medicine; 2014. | |
Adamantidis AR, Zhang F, Aravanis AM, Deisseroth K, de Lecea L. Neural substrates of awakening probed with optogenetic control of hypocretin neurons. Nature. 2007;450(7168):420–424. | |
Espana RA, Scammell TE. Sleep neurobiology from a clinical perspective. Sleep. 2011;34(7):845–858. | |
Serra L, Montagna P, Mignot E, Lugaresi E, Plazzi G. Cataplexy features in childhood narcolepsy. Mov Disord. 2008;23(6):858–865. | |
Andlauer O, Moore Ht, Hong SC, et al. Predictors of hypocretin (orexin) deficiency in narcolepsy without cataplexy. Sleep. 2012;35(9):1247–1255. | |
Nevsimalova S, Pisko J, Buskova J, et al. Narcolepsy: clinical differences and association with other sleep disorders in different age groups. J Neurol. 2013;260(3):767–775. | |
Plazzi G, Pizza F, Palaia V, et al. Complex movement disorders at disease onset in childhood narcolepsy with cataplexy. Brain. 2011; 134 (Pt 12):3480–3492. | |
Pizza F, Franceschini C, Peltola H, et al. Clinical and polysomnographic course of childhood narcolepsy with cataplexy. Brain. 2013;136 (Pt 12):3787–3795. | |
Vanier MT. Niemann-Pick diseases. Handb Clin Neurol. 2013;113:1717–1721. | |
Tobias ES, Tolmie JL, Stephenson JB. Cataplexy in the Prader-Willi syndrome. Arch Dis Child. 2002;87(2):170. | |
Nelson GB, Hahn JS. Stimulus-induced drop episodes in Coffin-Lowry syndrome. Pediatrics. 2003;111(3):e197–e202. | |
Fryssira H, Kountoupi S, Delaunoy JP, Thomaidis L. A female with Coffin-Lowry syndrome and “cataplexy”. Genet Couns. 2002;13(4):405–409. | |
Tyagi A, Harrington H. Cataplexy in association with Moebius syndrome. J Neurol. 2003;250(1):110–111. | |
Vossler DG, Wyler AR, Wilkus RJ, Gardner-Walker G, Vlcek BW. Cataplexy and monoamine oxidase deficiency in Norrie disease. Neurology. 1996;46(5):1258–1261. | |
Portala K, Westermark K, Ekselius L, Broman JE. Sleep in patients with treated Wilson’s disease. A questionnaire study. Nord J Psychiatry. 2002;56(4):291–297. | |
Nevsimalova S, Buskova J, Bruha R, et al. Sleep disorders in Wilson’s disease. Eur J Neurol. 2011;18(1):184–190. | |
Kanbayashi T, Abe M, Fujimoto S, et al. Hypocretin deficiency in Niemann-Pick type C with cataplexy. Neuropediatrics. 2003;34(1):52–53. | |
Oyama K, Takahashi T, Shoji Y, et al. Niemann-Pick disease type C: cataplexy and hypocretin in cerebrospinal fluid. Tohoku J Exp Med. 2006;209(3):263–267. | |
Vankova J, Stepanova I, Jech R, et al. Sleep disturbances and hypocretin deficiency in Niemann-Pick disease type C. Sleep. 2003;26(4):427–430. | |
Peyron C, Tighe DK, van den Pol AN, et al. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 1998;18(23):9996–10015. | |
Cason AM, Smith RJ, Tahsili-Fahadan P, Moorman DE, Sartor GC, Aston-Jones G. Role of orexin/hypocretin in reward-seeking and addiction: implications for obesity. Physiol Behav. 2010;100(5):419–428. | |
Burgess CR, Scammell TE. Narcolepsy: neural mechanisms of sleepiness and cataplexy. J Neurosci. 2012;32(36):12305–12311. | |
Mieda M, Tsujino N, Sakurai T. Differential roles of orexin receptors in the regulation of sleep/wakefulness. Front Endocrinol (Lausanne). 2013;4:57. | |
Inutsuka A, Inui A, Tabuchi S, Tsunematsu T, Lazarus M, Yamanaka A. Concurrent and robust regulation of feeding behaviors and metabolism by orexin neurons. Neuropharmacology. 2014;85:451–460. | |
Peyron C, Faraco J, Rogers W, et al. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med. 2000;6(9):991–997. | |
Peever JH, Lai YY, Siegel JM. Excitatory effects of hypocretin-1 (orexin-A) in the trigeminal motor nucleus are reversed by NMDA antagonism. J Neurophysiol. 2003;89(5):2591–2600. | |
Dauvilliers Y, Jaussent I, Lecendreux M, et al. Cerebrospinal fluid and serum cytokine profiles in narcolepsy with cataplexy: a case-control study. Brain Behav Immun. 2014;260–266. | |
Lee MG, Hassani OK, Jones BE. Discharge of identified orexin/hypocretin neurons across the sleep-waking cycle. J Neurosci. 2005; 25(28):6716–6720. | |
Takahashi K, Lin JS, Sakai K. Neuronal activity of orexin and non-orexin waking-active neurons during wake-sleep states in the mouse. Neuroscience. 2008;153(3):860–870. | |
Lin L, Faraco J, Li R, et al. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98(3):365–376. | |
Chemelli RM, Willie JT, Sinton CM, et al. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98(4):437–451. | |
Nishino S, Ripley B, Overeem S, Lammers GJ, Mignot E. Hypocretin (orexin) deficiency in human narcolepsy. Lancet. 2000;355(9197):39–40. [Letter.] | |
Tabuchi S, Tsunematsu T, Black SW, et al. Conditional ablation of orexin/hypocretin neurons: a new mouse model for the study of narcolepsy and orexin system function. J Neurosci. 2014;34(19):6495–6509. | |
Pizza F, Vandi S, Liguori R, et al. Primary progressive narcolepsy type 1: the other side of the coin. Neurology. 2014;83(23):2189–2190. | |
Nishino S, Okuro M, Kotorii N, et al. Hypocretin/orexin and narcolepsy: new basic and clinical insights. Acta Physiol (Oxf). 2010;198(3):209–222. | |
Partinen M, Kornum BR, Plazzi G, Jennum P, Julkunen I, Vaarala O. Does autoreactivity have a role in narcolepsy? Lancet Neurol. 2014; 13(11):1072–1073. [Letter.] | |
Mignot E, Hayduk R, Black J, Grumet FC, Guilleminault C. HLA DQB1*0602 is associated with cataplexy in 509 narcoleptic patients. Sleep. 1997;20(11):1012–1020. | |
Hallmayer J, Faraco J, Lin L, et al. Narcolepsy is strongly associated with the T-cell receptor alpha locus. Nat Genet. 2009;41(6):708–711. | |
Faraco J, Lin L, Kornum BR, et al. ImmunoChip study implicates antigen presentation to T cells in narcolepsy. PLoS Genet. 2013;9(2):e1003270. | |
Ahmed SS, Volkmuth W, Duca J, et al. Antibodies to influenza nucleoprotein cross-react with human hypocretin receptor 2. Sci Transl Med. 2015;7(294):294ra105. | |
Gulyani S, Wu MF, Nienhuis R, John J, Siegel JM. Cataplexy-related neurons in the amygdala of the narcoleptic dog. Neuroscience. 2002; 112(2):355–365. | |
Hong SB, Tae WS, Joo EY. Cerebral perfusion changes during cataplexy in narcolepsy patients. Neurology. 2006;66(11):1747–1749. | |
Overeem S, Lammers GJ, van Dijk JG. Cataplexy: ‘tonic immobility’ rather than ‘REM-sleep atonia’? Sleep Med. 2002;3(6):471–477. | |
Overeem S, Reijntjes R, Huyser W, Lammers GJ, van Dijk JG. Corticospinal excitability during laughter: implications for cataplexy and the comparison with REM sleep atonia. J Sleep Res. 2004;13(3):257–264. | |
Schwartz S, Ponz A, Poryazova R, et al. Abnormal activity in hypothalamus and amygdala during humour processing in human narcolepsy with cataplexy. Brain. 2008;131(Pt 2):514–522. | |
Burgess CR, Oishi Y, Mochizuki T, Peever JH, Scammell TE. Amygdala lesions reduce cataplexy in orexin knock-out mice. J Neurosci. 2013; 33(23):9734–9742. | |
Dauvillers Y, Billiard M, Montplaisir J. Clinical aspects and pathophysiology of narcolepsy. Clin Neurophysiol. 2003;114:2000–2017. | |
Peever J. Control of motoneuron function and muscle tone during REM sleep, REM sleep behavior disorder and cataplexy/narcolepsy. Arch Ital Biol. 2011;149(4):454–466. | |
Siegel JM, Nienhuis R, Fahringer HM, et al. Neuronal activity in narcolepsy: identification of cataplexy-related cells in the medial medulla. Science. 1991;252(5010):1315–1318. | |
Luppi PH, Clement O, Sapin E, et al. The neuronal network responsible for paradoxical sleep and its dysfunctions causing narcolepsy and rapid eye movement (REM) behavior disorder. Sleep Med Rev. 2011;15(3):153–163. | |
Nishino S, Tafti M, Reid MS, et al. Muscle atonia is triggered by cholinergic stimulation of the basal forebrain: implication for the pathophysiology of canine narcolepsy. J Neurosci. 1995;15 (7 Pt 1):4806–4814. | |
Thorpy MJ, Krieger AC. Delayed diagnosis of narcolepsy: characterization and impact. Sleep Med. 2014;15:502–507. | |
Anic-Labat S, Guilleminault C, Kraemer HC, Meehan J, Arrigoni J, Mignot E. Validation of a cataplexy questionnaire in 983 sleep-disorders patients. Sleep. 1999;22(1):77–87. | |
Moore WR, Silber MH, Decker PA, et al. Cataplexy Emotional Trigger Questionnaire (CETQ) – a brief patient screen to identify cataplexy in patients with narcolepsy. J Clin Sleep Med. 2007;3(1):37–40. | |
Prinzmetal M, Bloomberg W. The use of benzedrine for the treatment of narcolepsy. JAMA. 1935;105(20):2051–2054. | |
Parkes JD, Fenton GW. Levo(–) amphetamine and dextro(+) amphetamine in the treatment of narcolepsy. J Neurol Neurosurg Psychiatry. 1973;36(6):1076–1081. | |
Daly DD, Yoss RE. The treatment of narcolepsy with methyl phenylpiperidylacetate: a preliminary report. Proc Staff Meet Mayo Clin. 1956;31(23):620–625. | |
Akimoto H, Honda Y, Takahashi Y. Pharmacotherapy in narcolepsy. Dis Nerv Syst. 1960;21:704–706. | |
Hishikawa Y, Ida H, Nakai K, Kaneko Z. Treatment of narcolepsy with imipramine (tofranil) and desmethylimipramine (pertofran). J Neurol Sci. 1966;3(5):453–461. | |
Shapiro WR. Treatment of cataplexy with clomipramine. Trans Am Neurol Assoc. 1974;99:99–102. | |
Guilleminault C, Raynal D, Takahashi S, Carskadon M, Dement W. Evaluation of short-term and long-term treatment of the narcolepsy syndrome with clomipramine hydrochloride. Acta Neurol Scand. 1976;54(1):71–87. | |
Parkes JD, Schachter M. Clomipramine and clonazepam in cataplexy. Lancet. 1979;2(8151):1085–1086. | |
Schachter M, Parkes JD. Fluvoxamine and clomipramine in the treatment of cataplexy. J Neurol Neurosurg Psychiatry. 1980;43(2):171–174. | |
Langdon N, Shindler J, Parkes JD, Bandak S. Fluoxetine in the treatment of cataplexy. Sleep. 1986;9(2):371–373. | |
Chen SY, Clift SJ, Dahlitz MJ, Dunn G, Parkes JD. Treatment in the narcoleptic syndrome: self assessment of the action of dexamphetamine and clomipramine. J Sleep Res. 1995;4(2):113–118. | |
Winokur A, Gary KA, Rodner S, Rae-Red C, Fernando AT, Szuba MP. Depression, sleep physiology, and antidepressant drugs. Depress Anxiety. 2001;14(1):19–28. | |
Morgenthaler TI, Kapur VK, Brown T, et al. Practice parameters for the treatment of narcolepsy and other hypersomnias of central origin. Sleep. 2007;30(12):1705–1711. | |
Billiard M, Bassetti C, Dauvilliers Y, et al. EFNS guidelines on management of narcolepsy. Eur J Neurol. 2006/10 2006;13(10):1035–1048. | |
Xyrem® [sodium oxybate] oral solution US prescribing information. Palo Alto, CA: Jazz Pharmaceuticals, Inc.; April 2014. | |
Mignot E, Nishino S. Emerging therapies in narcolepsy-cataplexy. Sleep. 2005;28(6):754–763. | |
Broughton R, Mamelak M. Effects of nocturnal gamma-hydroxybutyrate on sleep/waking patterns in narcolepsy-cataplexy. Can J Neurol Sci. 1980;7(1):23–31. | |
Mamelak M, Scharf MB, Woods M. Treatment of narcolepsy with gamma-hydroxybutyrate. A review of clinical and sleep laboratory findings. Sleep. 1986;9 (1 Pt 2):285–289. | |
Pardi D, Black J. γ-Hydroxybutyrate/sodium oxybate. Neurobiology, and impact on sleep and wakefulness. CNS Drugs. 2006;20(12):993–1018. | |
Huang YS, Guilleminault C. Narcolepsy: action of two gamma-aminobutyric acid type B agonists, baclofen and sodium oxybate. Pediatr Neurol. 2009;41(1):9–16. | |
Mignot EJ. A practical guide to the therapy of narcolepsy and hypersomnia syndromes. Neurotherapeutics. 2012;9(4):739–752. | |
Gowda CR, Lundt LP. Mechanism of action of narcolepsy medications. CNS Spectr. 2014;19(Suppl 1):25–34. | |
Mignot E, Renaud A, Nishino S, Arrigoni J, Guilleminault C, Dement WC. Canine cataplexy is preferentially controlled by adrenergic mechanisms: evidence using monoamine selective uptake inhibitors and release enhancers. Psychopharmacology (Berl). 1993;113(1):76–82. | |
Nishino S, Arrigoni J, Shelton J, Dement WC, Mignot E. Desmethyl metabolites of serotonergic uptake inhibitors are more potent for suppressing canine cataplexy than their parent compounds. Sleep. 1993;16(8):706–712. | |
Nishino S, Fruhstorfer B, Arrigoni J, Guilleminault C, Dement WC, Mignot E. Further characterization of the alpha-1 receptor subtype involved in the control of cataplexy in canine narcolepsy. J Pharmacol Exp Ther. 1993;264(3):1079–1084. | |
Lammers GJ, Arends J, Declerck AC, Ferrari MD, Schouwink G, Troost J. Gammahydroxybutyrate and narcolepsy: a double-blind placebo-controlled study. Sleep. 1993;16(3):216–220. | |
The US Xyrem Multi-Center Study Group. A randomized, double blind, placebo-controlled multicenter trial comparing the effects of three doses of orally administered sodium oxybate with placebo for the treatment of narcolepsy. Sleep. 2002;25:42–49. | |
Xyrem International Study Group. Further evidence supporting the use of sodium oxybate for the treatment of cataplexy: a double-blind, placebo-controlled study in 228 patients. Sleep Med. 2005;6(5):415–421. | |
US Xyrem Multicenter Study Group. A 12-month, open-label, multicenter extension trial of orally administered sodium oxybate for the treatment of narcolepsy. Sleep. 2003;26(1):31–35. | |
US Xyrem Multicenter Study Group. Sodium oxybate demonstrates long-term efficacy for the treatment of cataplexy in patients with narcolepsy. Sleep Med. 2004;5(2):119–123. | |
Alshaikh MK, Tricco AC, Tashkandi M, Mamdani M, Straus SE, Bahammam AS. Sodium oxybate for narcolepsy with cataplexy: systematic review and meta-analysis. J Clin Sleep Med. 2012;8(4):451–458. | |
Boscolo-Berto R, Viel G, Montagnese S, Raduazzo DI, Ferrara SD, Dauvilliers Y. Narcolepsy and effectiveness of gamma-hydroxybutyrate (GHB): a systematic review and meta-analysis of randomized controlled trials. Sleep Med Rev. 2012;16(5):431–443. | |
Mansukhani MP, Kotagal S. Sodium oxybate in the treatment of childhood narcolepsy-cataplexy: a retrospective study. Sleep Med. 2012;13:606–610. | |
Corkery JM, Loi B, Claridge H, et al. Gamma hydroxybutyrate (GHB), gamma butyrolactone (GBL) and 1,4-butanediol (1,4-BD; BDO): a literature review with a focus on UK fatalities related to non-medical use. Neurosci Biobehav Rev. 2015;53:52–78. | |
Vignatelli L, D’Alessandro R, Candelise L. Antidepressant drugs for narcolepsy. Cochrane Database Syst Rev. 2008;(1):CD003724. | |
Frey J, Darbonne C. Fluoxetine suppresses human cataplexy: a pilot study. Neurology. 1994;44(4):707–709. | |
Izzi F, Placidi F, Marciani MG, et al. Effective treatment of narcolepsy-cataplexy with duloxetine: a report of three cases. Sleep Med. 2009; 10(1):153–154. [Letter.] | |
Moller LR, Ostergaard JR. Treatment with venlafaxine in six cases of children with narcolepsy and with cataplexy and hypnagogic hallucinations. J Child Adolesc Psychopharmacol. 2009;19(2):197–201. | |
Ratkiewicz M, Splaingard M. Treatment of cataplexy in a three-year-old using venlafaxine. J Clin Sleep Med. 2013;9(12):1341–1342. [Case report.] | |
Sonka K, Kemlink D, Pretl M. Cataplexy treated with escitalopram – clinical experience. Neuro Endocrinol Lett. 2006;27(1–2):174–176. | |
Karacan I. Erectile dysfunction in narcoleptic patients. Sleep. 1986; 9(1 Pt 2):227–231. | |
Ju YE, Larson-Prior L, Duntley S. Changing demographics in REM sleep behavior disorder: possible effect of autoimmunity and antidepressants. Sleep Med. 2011;12(3):278–283. | |
Poryazova R, Siccoli M, Werth E, Bassetti CL. Unusually prolonged rebound cataplexy after withdrawal of fluoxetine. Neurology. 2005; 65(6):967–968. | |
Ristanovic RK, Liang H, Hornfeldt CS, Lai C. Exacerbation of cataplexy following gradual withdrawal of antidepressants: manifestation of probable protracted rebound cataplexy. Sleep Med. 2009;10(4):416–421. | |
John J, Wu MF, Boehmer LN, Siegel JM. Cataplexy-active neurons in the hypothalamus: implications for the role of histamine in sleep and waking behavior. Neuron. 2004;42(4):619–634. | |
Valko PO, Gavrilov YV, Yamamoto M, et al. Increase of histaminergic tuberomammillary neurons in narcolepsy. Ann Neurol. 2013;74(6):794–804. | |
Lin JS, Dauvilliers Y, Arnulf I, et al. An inverse agonist of the histamine H(3) receptor improves wakefulness in narcolepsy: studies in orexin-/- mice and patients. Neurobiol Dis. 2008;30(1):74–83. | |
Dauvilliers Y, Bassetti C, Lammers GJ, et al. Pitolisant versus placebo or modafinil in patients with narcolepsy: a double-blind, randomised trial. Lancet Neurol. 2013;12(11):1068–1075. | |
Inocente C, Arnulf I, Bastuji H, et al. Pitolisant, an inverse agonist of the histamine H3 receptor: an alternative stimulant for narcolepsy-cataplexy in teenagers with refractory sleepiness. Clin Neuropharmacol. 2012;35(2):55–60. | |
Baier PC, Hallschmid M, Seeck-Hirschner M, et al. Effects of intranasal hypocretin-1 (orexin A) on sleep in narcolepsy with cataplexy. Sleep Med. 2011;12(10):941–946. | |
Weinhold SL, Seeck-Hirschner M, Nowak A, Hallschmid M, Goder R, Baier PC. The effect of intranasal orexin-A (hypocretin-1) on sleep, wakefulness and attention in narcolepsy with cataplexy. Behav Brain Res. 2014;262:8–13. | |
John J, Wu MF, Siegel JM. Systemic administration of hypocretin-1 reduces cataplexy and normalizes sleep and waking durations in narcoleptic dogs. Sleep Res Online. 2000;3(1):23–28. | |
Black SW, Morairty SR, Chen TM, et al. GABAB agonism promotes sleep and reduces cataplexy in murine narcolepsy. J Neurosci. 2014;34(19):6485–6494. | |
Fronczek R, Verschuuren J, Lammers GJ. Response to intravenous immunoglobulins and placebo in a patient with narcolepsy with cataplexy. J Neurol. 2007;254(11):1607–1608. | |
Plazzi G, Poli F, Franceschini C, et al. Intravenous high-dose immunoglobulin treatment in recent onset childhood narcolepsy with cataplexy. J Neurol. 2008;255(10):1549–1554. | |
Valko PO, Khatami R, Baumann CR, Bassetti CL. No persistent effect of intravenous immunoglobulins in patients with narcolepsy with cataplexy. J Neurol. 2008;255(12):1900–1903. | |
Lecendreux M, Maret S, Bassetti C, Mouren MC, Tafti M. Clinical efficacy of high-dose intravenous immunoglobulins near the onset of narcolepsy in a 10-year-old boy. J Sleep Res. 2003;12(4):347–348. [Letter.] | |
Dauvilliers Y, Carlander B, Rivier F, Touchon J, Tafti M. Successful management of cataplexy with intravenous immunoglobulins at narcolepsy onset. Ann Neurol. 2004;56(6):905–908. | |
Dauvilliers Y. Follow-up of four narcolepsy patients treated with intravenous immunoglobulins. Ann Neurol. 2006;60(1):153. | |
Dauvilliers Y, Abril B, Mas E, Michel F, Tafti M. Normalization of hypocretin-1 in narcolepsy after intravenous immunoglobulin treatment. Neurology. 2009;73(16):1333–1334. | |
Knudsen S, Mikkelsen JD, Bang B, Gammeltoft S, Jennum PJ. Intravenous immunoglobulin treatment and screening for hypocretin neuron-specific autoantibodies in recent onset childhood narcolepsy with cataplexy. Neuropediatrics. 2010;41(5):217–222. | |
Knudsen S, Biering-Sorensen B, Kornum BR, et al. Early IVIg treatment has no effect on post-H1N1 narcolepsy phenotype or hypocretin deficiency. Neurology. 2012;79(1):102–103. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.