Back to Journals » Infection and Drug Resistance » Volume 16
Treatment Options for Epstein-Barr Virus-Related Disorders of the Central Nervous System
Authors Andersen O, Ernberg I ![]() , Hedström AK
, Hedström AK ![]()
Received 5 March 2023
Accepted for publication 28 June 2023
Published 13 July 2023 Volume 2023:16 Pages 4599—4620
DOI https://doi.org/10.2147/IDR.S375624
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Suresh Antony
Oluf Andersen,1 Ingemar Ernberg,2 Anna Karin Hedström3
1Department of Clinical Neuroscience, Institute of Neuroscience and Physiology, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden; 2Department of Microbiology, Tumor and Cell Biology, Biomedicum Q8C, Karolinska Institutet, Stockholm, 171 77, Sweden; 3Department of Clinical Neuroscience, Karolinska Institutet, Stockholm, Sweden
Correspondence: Anna Karin Hedström, Department of Clinical Neuroscience, Karolinska Institutet, Stockholm, 171 76, Sweden, Tel +46762736426, Email [email protected]
Abstract: Epstein-Barr virus (EBV), a causative agent for several types of lymphomas and mucosal cancers, is a human lymphotropic herpesvirus with the capacity to establish lifelong latent infection. More than 90% of the human population worldwide is infected. The primary infection is usually asymptomatic in childhood, whereas infectious mononucleosis (IM) is common when the infection occurs in adolescence. Primary EBV infection, with or without IM, or reactivation of latent infection in immunocompromised individuals have been associated with a wide range of neurologic conditions, such as encephalitis, meningitis, acute disseminated encephalomyelitis, and cerebellitis. EBV is also involved in malignant lymphomas in the brain. An increasing number of reports on EBV-related disorders of the central nervous system (CNS) including the convincing association with multiple sclerosis (MS) have put in focus EBV-related conditions beyond its established link to malignancies. In this review, we present the clinical manifestations of EBV-related CNS-disorders, put them in the context of known EBV biology and focus on available treatment options and future therapeutic approaches.
Keywords: Epstein-Barr virus, infectious mononucleosis, central nervous system disease, multiple sclerosis, treatment, vaccination
Introduction
Epstein-Barr virus (EBV), discovered in 1964 by Epstein et al, in cultured lymphoid cells from Burkitt’s lymphoma, is a member of the 1 subfamily of human herpes viruses.1,2 This subfamily is characterized by its tropism for lymphoid cells and the capacity to induce cell proliferation in vivo, resulting in transient or chronic lymphoproliferative disorders. EBV infects only humans in nature, but close viral relatives are found in Old World Higher Primates. Antibodies to EBV have been detected in several primate species, but it is considered that this is due to the presence of cross-reacting antibodies against their own species-specific variant of EBV.3
EBV is one of the most common, widespread human viruses, infecting over 90% of the adult population. Primary infection acquired in childhood is usually asymptomatic, whereas infectious mononucleosis (IM) is common when the infection is delayed until adolescence or young adulthood.4 After acute infection with or without IM, latent EBV infection will usually persist throughout life.5 The virus has co-evolved with humans over millions of years which has resulted in a balance between viral persistence and immune control. Yet, the global burden of disease due to EBV infection is significant.2 An association between EBV infection and several lymphoid and epithelial malignancies, lymphoproliferative and inflammatory conditions has long been known. Although EBV latently persists in resting, predominantly memory B lymphocytes, the virus has the capacity to infect numerous cell types such as epithelial cells, T cells, natural killer cells (NK), macrophages, astrocytes, as well as neurons, however with much lower efficiency.2,6,7 EBV can infect neural tissue directly or indirectly via the vasculature and infected B lymphocytes, and EBV-associated CNS infections can manifest in a variety of neurological symptoms. In addition to the acute neurologic complications that may occur in the context of primary infection or EBV reactivation, latent EBV infection also seems to contribute to the pathogenesis of various chronic CNS disorders. EBV has a profound influence on the immune system, and recent studies have suggested a causal role of EBV in multiple sclerosis (MS), a chronic immune-mediated disease of the central nervous system (CNS).8 In this review, we present clinical manifestations of CNS disorders associated with acute and latent EBV-infection with focus on available treatment options for these disorders and the potential of new therapeutic approaches.
Epstein-Barr Virus
Etiology and Epidemiology
The proportion of the population showing signs of previous EBV infection increases with age. In Sweden, 50% are seropositive at the age of 5, 60–70% at the age of 25, and 90–95% at the age of 25.9 EBV infects via mucous membranes, particularly in the oral cavity and pharynx, penetrates the epithelium and establishes a persistent, lifelong latent infection of memory B lymphocytes. Lytic infection with virus production is common and usually asymptomatic, occurs in the mucous membranes of the mouth-nasal cavities and EBV is then intermittently shed into saliva. Although viral transmission through salivary contact is the most common route of infection, EBV can also be transmitted via other mucous tissues.10–12
Structure of EBV
The EBV genome is a large double-stranded, linear 172 kb DNA molecule, wrapped on a toroid-shaped protein core, and enclosed by an icosahedral capsid that is surrounded by a tegument, containing both viral mRNAs and viral tegument proteins which promote the initiation of infection and release of infectious particles. The tegument is surrounded by a lipid envelope derived from host cell membranes containing the viral attachment and entry proteins.
The genome has 538 bp terminal direct repeats at the 3′ and 5′ ends and internal 3kb direct repeats close to the 5′ end, which divide the EBV genome into short (12 kb) and long (134kb) unique sequences.12 EBV DNA codes for 85 genes, twelve of which are used to control the latent infection, while the others code for enzymes and structural proteins that are expressed during the virus’s lytic, productive cycle. In addition, the virus encodes 44 microRNAs.13
Two major EBV types have been detected in humans. Type A and type B (or I and II) genomes are closely similar, except for genes that encode EBNA2, EBNA3, EBNA4 and EBNA6, and their introns. Both isolates are worldwide; type A is most common in Europe, North America, and Asia, while type B is more frequently found in Africans. Type A is often found in the healthy population, while type B is more often detected in immunosuppressed individuals (due to post-transplant treatment or HIV-infection) and EBV-associated lymphomas in these patients.12,14
Virology and Molecular Biology
Glycoprotein gp350/220 is a viral knob protein expressed late during the lytic virus cycle and inserts into the outer viral envelope.15 The molecular weight is 350 kDa with a splice variant at 220 kDa. It serves as the main viral receptor for binding to host cells. Infection of human B lymphocytes is mediated through the interaction of gp350/220 with the complement receptor CR2 (CD21) on B lymphocytes, followed by interaction of viral glycoprotein gp42 with major histocompatibility complex (MHC) class II molecules.15,16 Gp42 is also a structural viral protein expressed late in the virus production cycle and gets inserted into the outer envelope.15 It is in fact part of a viral three-part glycoprotein complex of gHgL gp42. The two-part complex of gHgL mediates epithelial cell membrane fusion. Viruses lacking the gp42 portion can bind human B lymphocytes but are unable to infect.
CD35, or complement receptor 1 (CR1) is an additional receptor for gp350/220 binding which can provide an alternative route for entry of EBV into CD21-negative cells, including immature B cells.17 CD35 is downregulated by EBV infection.
To infect epithelial cells, which normally do not express the complement receptors, viral protein BMRF-2 binds to cellular β1 integrins. Then, viral protein gH/gL interacts with cellular αvβ6/αvβ8 integrins. This triggers fusion of the viral envelope with the epithelial cell membrane, allowing EBV to enter the epithelial cell.15 After virus entry, the EBV genome is transported to the nucleus where virus replication is initiated.
In latent infection, EBV DNA is localized to the nucleus of the host cells in circular episomal form. The circularized virus genome is found 12–16 hours post-infection in blood lymphocytes infected in vitro. It precedes or coincides with the earliest detected virus gene expression.12 Although the episomal form is the most common, the EBV genome can also persist by integrating into chromosomes of the host. However, episomal DNA is likely to be necessary for lytic cycle EBV DNA replication, since viral production has not been observed in cells that contain only integrated EBV DNA.
EBV genes are functionally defined by their expression in lytic or latent infection. Approximately 75 viral proteins are expressed in lytic infection. During viral replication, these proteins play a fundamental role in regulating viral gene expression, viral DNA replication, viral structural protein synthesis, virus particle assembly and modulation of the host immune response.
EBV Latency Programs
Latently infected B lymphocytes enter one of three main latency programs (latency III, latency II, and latency I/0; Table 1; Figure 1) where the expression of viral proteins and viral RNAs become more restricted with each latency type. Six Epstein-Barr virus nuclear proteins (EBNA1-6, alternatively EBNA1, EBNA2, EBNA3a, EBNA3b, EBNA3c, EBNA-LP) and three latent membrane proteins (LMP1, LMP2A, and LMP2B) are expressed in different combinations in the three latency programs.18–20 The only viral protein expressed in all latency programs is EBNA1.21 Two EBV-encoded small non-coding RNAs (EBER1 and EBER2) and a majority of the 44 known viral microRNAs are also expressed in all three latency types of infected cells (Table 1).13,22
 |
Table 1 Latent and Lytic Cycle Proteins |
 |
Figure 1 EBV infectious cycle in vivo. Notes: Overview of current view on the EBV-infectious cycle in vivo, primary EBV-infection and latency. Free virus from another host (which may be asymptomatic) carrying EBV and secreting it to saliva may be transferred, eg, by kissing. B lymphocytes are infected with oropharyngeal mucosal cells as intermediates. Latent EBV infection is established in a fraction of memory B cells, possible after some amplification by a few divisions of the EBV-infected B cells. Memory B cells carrying EBV can deliver virus by cell–cell contact or by activation of the lytic cycle to the oropharyngeal mucosa, which supports virus replication and release to saliva. Both humoral and cellular immunity are established during the primary infection. Drawing: Ingemar Ernberg. |
Upon EBV infection of B lymphocytes in vitro, the resulting transformed B lymphocytes proliferate continuously, with a pattern of gene expression referred to as latency III. These B lymphocytes express all nine latent proteins, the six EBNAs and three LMPs.12,23 The latency III program is expressed only in B lymphocytes and is active during the acute phase of primary infection before the EBV-specific T cell response develops, during reactivation among patients with impaired immune function, eg, immunosuppressed stem cell transplanted patients but also, most likely, as a means of intermittently refilling the pool of latently infected B cells. Thus, the latency III program is active in the infected B lymphocytes of IM patients (Figure 1).
The infected cells enter germinal centres of the lymph nodes and spleen and subsequently switch into the latency II program, which promote survival and differentiation into memory B cells.12,18 EBV latency II cells express EBNA 1, LMP1, LMP2A and LMP2b and are also detected in the classical Hodgkin lymphoma, nasopharyngeal carcinoma and nasal NK/T lymphoma, when EBV positive.23
When memory B cells enter the peripheral circulation, the EBV gene expression becomes more restricted. In latency I, only one or two virally encoded proteins, EBNA1 and occasionally LMP2A, are expressed. Cells expressing latency I pattern do not proliferate and their phenotype corresponds to non-activated B cells (Figure 1). The asymptomatic EBV persistence in healthy carriers, where no EBV proteins are produced, is referred to as latency 0. By limiting the viral gene expression during latency, EBV evades immune surveillance.
Latent and Lytic Cycle Proteins
EBNA1 is a DNA binding protein, expressed in all three main EBV latency programs.21 It contains a glycine-alanine repetitive sequence of variable length that inhibits its processing through the proteasomes and the subsequent MHC class I association of the derived peptides, impeding its recognition by CD8+ cytotoxic T cell.24,25 This results in a dramatically extended half-life of EBNA1 to more than two weeks and may contribute to its presence in resting B cells without de novo synthesis. EBNA1 plays an important role in latent DNA replication as well as in EBV genome maintenance and segregation into daughter cells.21
EBNA2 is the most important marker to distinguish between the EBV type-A (1) and type-B (2) strains which show 57% homology.12,26 Together with EBNA5, EBNA2 is the first viral protein expressed after B cell primary infection.27 It is a specific transcription activator of cellular and viral genes, such as the B lymphocytes activation marker CD23 and the EBV receptor CD21.23,28 EBNA2 plays an essential role in the immortalization of B cells due to its transactivation of viral promoters (LMP1-2, Cp) and a variety of cellular genes associated with B cell activation and growth. EBNA2 is also required for maintaining the EBV-driven proliferation of B cells.23
The EBNA3 family (EBNA3, EBNA4 and EBNA6, or EBNA3a, EBNA3b and EBNA3c) has similar effects on cellular gene expression as EBNA2. These EBNAs generate highly immunogenic peptides that can associate with MHC class I molecules. Some peptide-HLA class I combinations can induce CD8+ T cell mediated rejection in immunocompetent hosts.28,29
Latent membrane protein 1 (LMP1) is essential for the immortalization of B lymphocytes. It confers a survival advantage on EBV-infected B cells by protecting them from apoptosis. This is largely due to the LMP1-induced upregulation of the anti-apoptotic protein B cell lymphoma 2 (BCL-2) and the block of p53 mediated apoptosis by the latter.23
LMP1 almost completely mimics the function of CD40 mediated signaling and interacts with several proteins of the tumor necrosis factor receptor (TNFR) signaling pathway through its C-terminal activation region. Through its interference with several major signaling pathways in B cells and epithelial cells, LMP1 mediates deregulation of several hundreds of human cellular proteins. LMP1 also induces the expression of adhesion molecules such as intracellular adhesion molecule-1 and lymphocyte function-associated antigen-1, as well as MHC Class I and II.30
The LMP2 contributes to the maintenance of viral latency by preventing normal B cell receptor (BCR) activation, which would otherwise initiate viral lytic replication, while sustaining survival pathways in latently infected B cells.23,31 LMP2a acts as a surrogate BCR in vivo by providing constitutive signalling required for B cell development and survival. Together with LMP1, LMP2a is likely to provide the necessary survival signals for EBV-infected B lymphocytes to differentiate into latently infected, resting memory cells.
EBERs are noncoding RNAs, expressed in abundance in almost all EBV-infected cells, including latency 0/1. They bind the interferon-inducible, double-stranded RNA-activated protein kinase receptor (PKR) and inhibit its antiviral functions, which could be of importance for viral persistence.32
A family of EBV transcripts termed the BARF (BamH1 restriction fragment A right forward transcripts or BARTs) family has been identified in CD19+, but not in CD23+ B cells from peripheral blood of healthy individuals. Expression of the BARTs in peripheral blood B cells suggests that the proteins encoded by these transcripts are likely to be important for maintenance of in vivo latency.12,33
To date, EBV has been found to encode about 44 miRNAs that directly target cellular or viral mRNAs. EBV has evolved to exploit its own miRNAs to modulate the expression of a variety of genes related to viral latency, cell proliferation, apoptosis, and cell transformation. More importantly, EBV miRNAs play powerful roles in subverting host immune responses.13,32 However, the role of EBV-encoded miRNAs in immune regulation remains to be studied in detail. In-depth exploration of EBV miRNAs will be of great importance for further elucidation of the interaction between EBV and host immune system and provide novel insights into how EBV succeeds as a viral pathogen.
ZEBRA (Zta or BZLF1 protein) and Rta are two immediate early proteins instrumental for initiation of the EBV lytical cycle. When latent infection of EBV-infected cell is interrupted and enters the lytic cycle, the first detectable change is expression of the ZEBRA protein. This, together with Rta, transactivates viral and certain cellular promoters.34 Expression of ZEBRA is sufficient to trigger the entire downstream lytic replication cascade.
EBV Pathogenesis and Host Immunity
Serology
Primary infection with EBV induces an EBV-specific antibody response. Acute EBV infection is supported by the detection of heterophile antibodies (Monospot Test) directed against autoantigens and possibly also to food components, which also can be seen in other acute infections, in autoimmune disease and cancer. Heterophile antibodies are present only in acute EBV infection and usually become undetectable after a few weeks.4
EBV-specific antibody profiles are needed for staging the EBV infection. Most patients have IgM-antibodies against the viral capsid (VCA) at the time of clinical onset. These are then replaced by VCA IgG which persists for life and is the preferable test to verify a previous EBV infection. Early antigen (EA) antibodies can be detected in about 70% of patients with IM and remain detectable for 3–6 months. Antibodies against EBNA appear later in the disease course and persist for life. Healthy EBV carriers typically show IgG-antibody titers against VCA and EBNA (Figure 2).12,35,36 An increase in these titers, or appearance of anti-EA antibodies, signals an EBV-host imbalance or disease.
 |
Figure 2 EBV infection and immunity in IM. Notes: Longitudinal course of humoral and cellular anti-EBV immunity following primary EBV-infection manifesting as IM, as seen in lymphoid tissue and mirrored in peripheral blood. The infection usually starts as a lytic infection by saliva-carried virus of epithelial cells in nasopharyngeal epithelial linings. Virus spreads to mucosa-associated lymphoid tissues (MALT) where B cells become infected and enter proliferation (latency III program). These cells are highly immunogenic, inducing a strong cellular immune reaction involving both CD4+ and CD8+ T cells. Activated CD8+ T cells kill latency III cells using the conventional MHC Class I-restricted peptide-based recognition to curb the proliferation. The peptides are derived from EBNA 2–6, LMP1 and LMP2, but initially also from proteins expressed during the EBV lytic cycle. CD4+ T cells are primed and activated via stimulus from MHC Class II-peptide signaling from infected B cells and regulate the humoral immune response regarding T cell dependent antibody production. The CD4+ cells have been shown to terminate the proliferation of latency III cells by other immunoregulatory mechanisms than cell-killing, such as cytokine production. Some EBV antigens, such as EBNA1 with its large repetitive regions, seem to partly evoke a T cell independent response. There are also other types of immune reactions which have been demonstrated in vitro that may play a role in the curbing of the acute infection: neutralizing antibodies, complement-dependent cell-killing antibodies, antibody dependent cellular cytotoxicity involving NK-cells and activated T cells, opsonization by macrophages, as well as early acute responses involving interferons and cytokines. These are most likely involved in reducing the early lytic infection. Drawing: Ingemar Ernberg. |
Primary EBV Infection and Infectious Mononucleosis (IM)
The immune response to EBV is influenced by age, immunocompetence, and host genetics.37,38 Primary infection of infants and young children with EBV is often asymptomatic or results in nonspecific findings such as upper respiratory tract symptoms, pharyngitis, or fever without lymphadenopathy. The incubation period of IM is 3050 days.39,40 There is a prodrome of malaise and fever, followed by the clinical illness of IM. This may present with sore throat, fever, headache, stiff neck, anorexia, abdominal discomfort, pharyngitis, exudative tonsillitis, palatal petechiae, generalised tender lymphadenopathy, splenomegaly (50%), hepatomegaly (25%) and skin rash (50%).
In IM, the host does not seem to be able to control the primary EBV infection (Figure 2). The establishment of controlled virus latency in B lymphocytes is only achieved after a strong primary activation of the immune system. There is a massive expansion of CD8+ T cells, many of which are directed against EBV proteins from the early stages of the lytic cycle.41–43 The cytotoxic CD4+ T cell response contribute to the symptoms of acute IM by massive cytokine production, including Interleukin-7 and TNF-α. Proliferating B lymphoblasts may constitute up to 20% of all peripheral B cells and give rise to a polyclonal antibody production as well as a broad and partly non-specific activation of CD4+ and CD8+ cells. This leads to further activation of B cells, also EBV-negative, which produce antibodies with many specificities, typically also against several viral and host proteins. A condition that can be characterized as an acute autoimmune syndrome develops with massive activation of T cells and cytokine surge, which leads to severe enlargement of lymphoid tissues such as lymph nodes, spleen, and liver due to infiltration and proliferation of B and T cells and inflammation. Within 3–4 weeks the symptoms usually subside, while remaining fatigue is in some cases long-lasting, up to 3–6 months.
The latent infection is dependent on the HLA genotype of the host. Genetic differences in the HLA class I locus have been associated with both the outcome of primary EBV infection and the viral persistence.37,38 Epitopes derived from the latently expressed proteins such as EBNA2 and EBNA3 are recognized by the most prevalent HLA genotypes.
HLA-restricted cytotoxic T cells (CTLs) are generated that are directed against both lytic and latent antigens. While most of the CTLs are CD8+ cells, CD4+ CTLs are also present. Initially, CTLs are predominantly directed against lytic antigens; with time the frequency of CTLs to lytic antigens declines and CTLs to latent antigens increase. CTLs directed against lytic antigens are predominantly CD45RA+, CD45RO-, while CTLs directed against latent antigens are usually CD45RA- –, CD45RO+ +.44 Typically, a dynamic equilibrium arises between latently infected B cells and EBV-specific CTLs. Although the number of infected memory B cells vary 100-fold between individuals it remains rather stable throughout life reflecting a balance between virus-driven B cell proliferation and immune response. Most of the persistent EBV-infected cells in the peripheral blood of healthy carriers are resting B cells with a latency 0/I pattern: expression of LMP2A and EBNA1, as well as the two non-translated RNAs (EBER1 and EBER2).
In rare cases, the individual fails to control the EBV infection, and a chronic persistent condition or fulminant life-threatening forms may develop. Sporadic cases occur, while others are associated with X-linked lymphoproliferative syndrome.45 The patient suffers from fatigue, elevated inflammatory parameters, recurrent febrile episodes, lymphadenopathy, and hepatosplenomegaly. A strong infiltration of both EBV-positive B lymphoblasts and activated T cells can be demonstrated in lymphoid organs, but also in other organs such as the liver, lung, and CNS. Being rare, this condition is difficult to diagnose. A characteristic of chronic symptomatic primary EBV infection is a decline of EBNA1 antibodies which can be used in screening for risk of chronic disease.
EBV-Mediated CNS Pathology
Although relatively rare, EBV infection may cause an astonishing spectrum of neurologic complications. Neurologic disorders related to productive infection include meningitis, encephalitis, cerebellitis, Guillain-Barré syndrome, acute disseminated encephalomyelitis, and peripheral neuropathies, whereas latent infection increases the risk of malignant lymphomas in the brain and may contribute to autoimmune and neurodegenerative disorders.
The neuroinvasive capacity of EBV is elusive. However, recent studies investigating the neurotropic potential of the virus indicate that EBV has the capacity to infect glial cells and neurons as well as endothelial cells of the blood-brain barrier.46–48 There are several potential mechanisms by which EBV may induce CNS pathology. Apart from direct lytic EBV infection of neurons, damage to neural tissue may also arise by infection of endothelial cells of the neurovasculature with release of viral proteins, inflammatory cytokines, and free radicals from infected cells as demonstrated in EBV infected human cell cultures.48 Rare B cells that migrate to the brain and enter lytic virus infection may also lead to inflammation and damage of local neurons that may be reversible depending on the extent of the neural damage. Furthermore, interplay between the CNS infiltrating B cells and the T cell response against them will likely cause local bystander damage.49
The EBV infection may also activate T cells that are cross-reactive with CNS antigens, subsequently inducing an autoimmune response.50 Molecular mimicry could be involved in disorders related to both productive and latent infection. Furthermore, EBV is highly efficient in manipulating the host cell cycle at various stages51 and it has been suggested that EBV-mediated cell-cycle dysregulation could initiate neurodegenerative pathology. The presence of core cell-cycle proteins in terminally differentiated neurons could potentially induce abnormal cell-cycle re-entry upon stress, resulting in neural apoptosis. An abnormal cell cycle re-entry in glial cells leads to activation and proliferation of these cells, with glial scar formation and production of inflammatory cytokines.52 However, although several studies have shown that dysregulation of the cell-cycle plays an important role in neurodegenerative disorders, an association between EBV and neurodegenerative pathology remains elusive.
Neurologic Disorders Associated with Lytic EBV Infection
EBV Meningitis and Encephalitis
The most common neurologic complication of IM is aseptic meningitis defined by headache, fever, and signs of meningeal irritation, usually occurring in the context of other common manifestations of IM.53 EBV meningitis is almost always self-limiting, and the pathogenesis has therefore not been extensively studied.
EBV encephalitis may be a consequence of direct viral invasion of the brain parenchyma or may occur due to immune-mediated mechanisms. As with other forms of viral encephalitis, clinical symptoms include headache, fever, muscle weakness, photophobia, epileptic seizures, and altered mental status. However, the prognosis is generally good in the pediatric clientele and disabling sequela are uncommon.54,55
The diagnosis is based on EBV DNA in cerebrospinal fluid (CSF) and serum by PCR. The CSF of patients with EBV meningitis/encephalitis typically shows pleocytosis, slightly increased protein levels, and atypical lymphocytes.56 Brain magnetic resonance imaging (MRI) findings and EEG results in EBV meningitis/encephalitis are usually non-specific.57
EBV encephalitis often occurs with other manifestations of IM but may also occur in the absence of systemic findings, making it difficult to diagnose.58 The presence of EBV DNA in the CNS indicates direct viral invasion and aids in the differential diagnosis against acute disseminated encephalomyelitis (ADEM).59 EBV may also be reactivated secondary to other CNS infections, and an increased number of EBV copies may be unspecific, resulting from any pronounced inflammatory pleocytosis including chronically EBV infected memory B cells. There is no consensus on the minimal number of copies in the CSF required for a diagnosis of EBV encephalitis. The window of PCR positivity is narrow, and a diagnosis of EBV encephalitis was often applied despite negative EBV-PCR in the CSF.57 Nevertheless, the number of copies in the CSF was clearly higher in CNS EBV lymphoma than in EBV encephalitis, in turn higher than in EBV-related ADEM, providing some practical aid in differential diagnosis.56
EBV has also been implicated in Rasmussen’s encephalitis (RE), a rare pediatric epileptic disease characterized by progressive hemisphere atrophy and rapid decline in cognitive function. EBV has been detected in RE lesion areas accompanied by a high frequency of CD8+ T cells,60,61 suggesting a T cell mediated response against virus-infected neurons.
Acute Disseminated Encephalomyelitis
ADEM is an uncommon immune-mediated demyelinating disorder of the CNS that has been associated with several viral infections, including post-IM and EBV reactivation. Viral prodromal symptoms often precede an abrupt onset with multifocal neurological deficits that rapidly deteriorate over several days.62,63 The disease is typically monophasic but recurrent forms of ADEM have been reported in case-series, typically in the context of persisting myelin oligodendrocyte glycoprotein (MOG) antibodies.64 ADEM usually affects children and adolescents and may be difficult to distinguish from a first clinical event of MS. However, MRI usually shows a specific pattern of widespread lesions of similar age.65
The induction of MOG-specific autoantibodies following primary EBV infection has been detected following IM and is often found in ADEM.66 The immune-mediated process of EBV-associated ADEM may possibly involve molecular mimicry between EBV antigens and MOG, where antigen-presenting cells induce a T cell response against a viral antigen that also results in an autoimmune response.66,67 However, while MOG-specific antibodies appear to decline to undetectable levels following monophasic ADEM, the anti-MOG-production has been reported to remain elevated in multiphasic forms of ADEM.68 Most children and adolescents with monophasic ADEM eventually recover.62,63 However, the risk of subsequently developing MS is substantially increased.69
Cerebellar Ataxia
Acute cerebellar ataxia is a rare complication of EBV infection, characterized by cerebellar swelling and cerebellar dysfunction. Clinical symptoms include headache, fever, cerebellar signs, and altered consciousness. The condition mainly affects children and young adults.70 The disease course is usually monophasic, and most patients eventually recover completely.71,72
Acute cerebellar ataxia usually presents in the context of IM, but the condition has also been described as the only feature of EBV infection.73 In post-EBV acute cerebellar ataxia, serum IgM autoantibodies have been identified which decreased with clinical improvement and thus suggests an autoimmune nature of the disease,74 possibly as a result of molecular mimicry between EBV proteins and cerebellar tissue. However, viral nucleic acids in the CSF have been detected in some affected patients,71 suggesting that viral infection of brain tissue may also contribute.
Neurologic Disorders Associated with Latent EBV Infection
Primary CNS Lymphoma
Primary CNS lymphoma (PCNSL) is a highly aggressive tumor of B cell origin that accounts for 1% of all lymphomas and 3–5% of primary brain tumors.75 The tumor is confined to the brain, soft meninges, spinal cord, and eyes. Biopsies show angiocentric growth pattern with infiltration of the cerebral parenchyma. EBV-associated PCNSLs are mainly observed in patients who are immunocompromised. The incidence has risen over the last 20 years, particularly among the elderly.76,77
Clinically, PCNSL presents with subacute, progressive mental status changes and with a variable combination of focal weakness, seizures, and headaches, depending on the location of the tumor. The tumors are not typically amenable to surgical resection due to widespread and diffusely infiltrative tumor growth and the clinical outcomes are poor.78,79 The neoplastic cells express type III latency molecules such as LMP-1. Expression of several chemokines, inflammatory cytokines and adhesion molecules are involved in the migration of malignant B cells into the CNS and for their angiocentric positioning.80,81
A genetic variant of LMP1 may be a pathogenic factor for EBV-positive PCNSLs in older immunocompetent patients, since the altered LMP-1 product is less immunogenic for EBV-specific cytotoxic T cells. The pathogenesis of PCNSL in immunocompetent hosts has been suggested to involve decreased specific immunity to EBV due to impaired cytotoxic T cell activity related to aging.82
Multiple Sclerosis
Multiple sclerosis (MS) is a chronic immune-mediated disease of the central nervous system characterized by inflammation, demyelination, and axonal degeneration. For northern Europeans, the lifetime risk of MS is 1:400. The disease typically starts as a relapsing remitting disease (RR MS) characterized by bouts of disease activity between periods of remission. The symptoms vary considerably depending on the location of the lesions within the CNS. Ultimately, the disease usually converts into secondary progressive MS (SPMS) with gradually increasing disability. In a low proportion of people affected by MS, the disease is progressive from onset without preceding clinically observable relapses (primary progressive MS; PPMS).83
The risk of MS is increased after IM, related to the post-childhood acquisition of EBV infection, and patients with MS have higher levels of EBV-specific antibodies than control populations.84 In a seronegative group remaining from a cohort of young adults in the US military, the risk of MS was increased 32-fold after infection with EBV. The study demonstrated that EBV infection preceded the development of MS and that serum levels of neurofilament light chain, a biomarker that reflects neuroaxonal degeneration, increased only after seroconversion.8 Several studies have observed a significant increase in EBNA1 antibody titers many years prior to MS onset.85,86 The consistent findings that EBV infection and elevation of specific EBNA1 antibody titers precede MS onset suggest that EBV is a necessary, but not sufficient, factor in MS development.
EBV-infected B cells and plasma cells have been detected in chronic MS lesions and in areas of demyelination87,88 the B cell infiltration has been correlated with the severity of the disease progression.89,90 In patients with MS, serum EBNA1 antibodies have been correlated with disease-associated oligoclonal Ig-bands in CSF.91 Furthermore, higher frequency of CD4+ T cells specific to EBNA1 has been observed in MS patients,92,93 where a subset of the T cells cross-reacted with CNS-autoantigens.93
Genetic susceptibility to MS is mainly located within the human leukocyte antigen (HLA) complex, where the HLA class II allele DRB1*15:01 exerts the single strongest effect.94 Increased levels of EBNA1 antibodies following EBV infection in DRB1*15:01 carriers implicate a poor class II-mediated immune control of the infection. The exact mechanisms by which EBV predisposes individuals to MS remain unclear but possible mechanisms may involve deficient immune control of EBV infection, molecular mimicry, and bystander damage.95 In the context of DRB1*15:01, it is possible that EBV infection could activate virus-specific T and B cells that are cross-reactive with CNS antigens, and subsequently initiate the onset of MS. EBV may also contribute to the progressive phase of MS. A severe disease progression has been associated with B cell infiltration and follicle formation within CNS where some studies,89,90 but not all96 have detected EBV-infected B cells. If insufficiently controlled EBV-infected B cells accumulate in the CNS or the meninges, they may contribute to CNS-compartmentalized inflammation.
Treatment Rationale and Options
Sentinel Event: Infectious Mononucleosis. Treatment and Prophylaxis
Although IM is usually a self-limiting disease, therapeutic and preventive trials are warranted due to its immediate and late complications: persistent fatigue occurs in 9–22% of post-IM patients after 6 months;97,98 the risk of CNS disorders complicating IM as described in the previous section of this review was reported to be 1–18%,;99,100 and beyond the 2- to 3-fold increased risk of MS there is increased risk of other autoimmune disorders. Moreover, IM is associated with EBV-positive Hodgkin lymphoma.101
Antiviral agents tested against IM were mainly acyclovir and its prodrug valacyclovir. Acyclovir is an acyclic guanosine derivative. The viral kinase for phosphorylation is necessary for activation of Acyclovir, so it accumulates virtually only in infected cells undergoing lytic productive infection. It competes for the viral DNA polymerase, expressed only during the lytic cycle, with the activated nucleotide deoxy-guanosine-triphosphate, resulting in chain termination after incorporation into the viral DNA. In spite of the clear rationale, the first trials of acyclovir in IM showed no impact on the clinical course although virus shedding transiently ceased during treatment.102 Authors speculated whether the obstacle was the long incubation time of IM, about 4 weeks, or the low concentrations of acyclovir in the oropharynx. The lack of effect clearly indicates that at the time of clinically overt disease the latently infected B lymphocytes drive the overreactive immune response, while there is little impact of the lytic virus cycle linked to earlier steps of the primary infection.103 However, corticosteroids were not beneficial either.104 A Cochrane review of seven randomized controlled studies of acyclovir and its derivatives confirmed that viral shedding was reduced while on antiviral treatment, although this effect was not sustained when the treatment was stopped. There was a mean reduction in physician assessed time to clinical recovery of five days and a mean reduction in duration of lymphadenopathy of nine days in the treatment group. However, due to wide confidence intervals the Cochrane authors concluded that the therapeutic effect of these antiviral agents in IM was unproven.105 We are not aware of any study using intravenous acyclovir as for herpes encephalitis. Gancyclovir is an acyclic guanosine analog effective in cytomegalovirus (CMV) infections. Similar to acyclovir, its initial phosphorylation is catalyzed by a virus-specified kinase. In a Chinese multicenter study on hospitalized pediatric patients with IM, neither acyclovir nor ganciclovir demonstrated therapeutic effect.106
Vaccination against EBV is still in its early stages after more than 25 years of trials. Due to the life-long infection with mutual immunogenetic linkage between virus and host, vaccination with live attenuated EBV is obviously contraindicated. In order to obtain prophylaxis against EBV-associated disorders, there is a rationale for vaccination inducing neutralizing antibodies in infancy before maternal antibodies wane. It is unknown whether persistent immunity can be achieved and repeat boosters may be required to avoid a later lytic infection. An alternative rationale is to produce neutralizing anti-EBV immunity in seronegative teenagers without eliciting IM. A theoretical objection would be that the EBV seropositive state implicates a manifold higher risk of MS compared with the seronegative state. However, epidemiological studies indicate that IM itself is associated with increased risk of MS and the teenage population will, with few exceptions, be seropositive two or three decades later. Then the rationale is 1) Prevention of IM itself, 2) Prevention of several immediate complications as reviewed here, as well as 3) Lymphomas and other EBV-related malignant disorders. An essential spin-off is the possible prevention of MS (and other autoimmune disorders) which would confirm EBV as a prerequisite for MS. Experimental trials of vaccines based on EBV-like virus in primates showed that it was possible to expand EBV-specific T cell lines against new EBV epitopes,107,108 recently reviewed in Cui et al.109
The first clinical trial of an EBV vaccine was reported in 1995. It used gp340 (gp350) expressed in vaccinia virus.110 Producing neutralizing antibodies against gp350 is still the mainstay of prophylactic EBV vaccines. The virus attaches to and enters the host cell by the lytic proteins gp350 and the three-part glycoprotein complex gp42, gH, and gB, as described above. Immunization combining these antigens experimentally was more effective than immunization based on gp350 alone.111 Since 2002, a number of human Phase 1 and 2 studies that were performed in children and adolescents showed promising effects, as reviewed in.112 Inoculation with recombinant gp350 before kidney transplantation induced neutralizing antibodies, however only transiently.113 In a randomized double-blind study of 181 EBV-seronegative healthy young volunteers, vaccination based on gp350-induced seroconversion in 98.7% of vaccinated participants who remained seropositive for gp350 for more than 18 months. The vaccination prevented the occurrence of IM with mean efficacy rate 78%. No safety problems were reported.114 A tetramer of the gp350 was developed as a future vaccine; experimentally, it showed increased specific immunogenicity compared with the monomer without inducing production of polyvalent antibodies.115 CTLs are probably more important for the defence against EBV, and the HLA restricted epitopes for CTL differ from those for antibodies, as described above. Rapid recovery from IM symptoms was coincident with broad T cell reactivity to multiple CD8 T+ cell epitopes within lytic and latent antigens, whereas a narrowly focused response was associated with protracted illness.116 A Phase I trial of an HLA restricted CD8+ T cell vaccine against a peptide in EBNA3 showed that 4/4 individuals had seroconverted asymptomatically without IM after a follow-up of two to twelve years.117 The T cell-based strategy was pursued in MS prophylaxis (below). Recently, an mRNA vaccine tested in mice based on the late lytic EBV glycoproteins gp350, gB, gH/gL and gp42 produced higher neutralization activity compared to human sera.109,112 The Moderna company currently conducts a phase 1 study of a uridine-modified mRNA vaccine based on these four glycoproteins in mainly EBV-seronegative young adults. The primary outcome parameter is a robust antigenic response against the vaccine proteins with the ultimate goal of preventing IM.118 There is a rationale for other targets such as EBNA2 and LMP1, which are important for EBV immortalization and viral persistence, as discussed above. Most vaccine trials evaluating the T cell response were based on latent-phase proteins such as EBNA and LMP,112 while there is a lack of trials aiming at induction of a T cell response against lytic glycoproteins. EBERs may be relevant for inhibition of chronic infection as it is expressed in latency 0. A further candidate is the immediate early ZEBRA protein which alone is sufficient to trigger the entire downstream lytic replication cascade. However, no IM vaccine is currently in clinical use.
EBV is capable of initiating a wide spectrum of disease processes. A pragmatic division is onset with or without IM, as this will facilitate our ultimate discussion on prophylaxis. Nevertheless, many clinical entities like encephalitis seem to behave equally with or without preceding IM. Moreover, this feature is age-dependent, as the primary EBV infection is much less likely to present as IM in the youngest paediatric clientele. In the following sections, we report on the two main categories of therapy, antiviral (mainly acyclovir) or immunomodulatory (i.v. methylprednisolone, dexamethasone or i.v. gamma globulin) which were used depending on diagnosis and individual findings in the CSF. In Tables 2–4, we present case-reports with convincing acute EBV infection based on elevated VCA IgM or PCR in serum. The tables include information on PCR findings in the CSF and the intervention used in individual cases as well as a note on the outcome. Table 5 summarizes current treatment options in CNS disorders associated with EBV.
 |
Table 2 Selection of Case Reports on CNS Disorders Associated with Mononucleosis Due to Primary EBV-Infection |
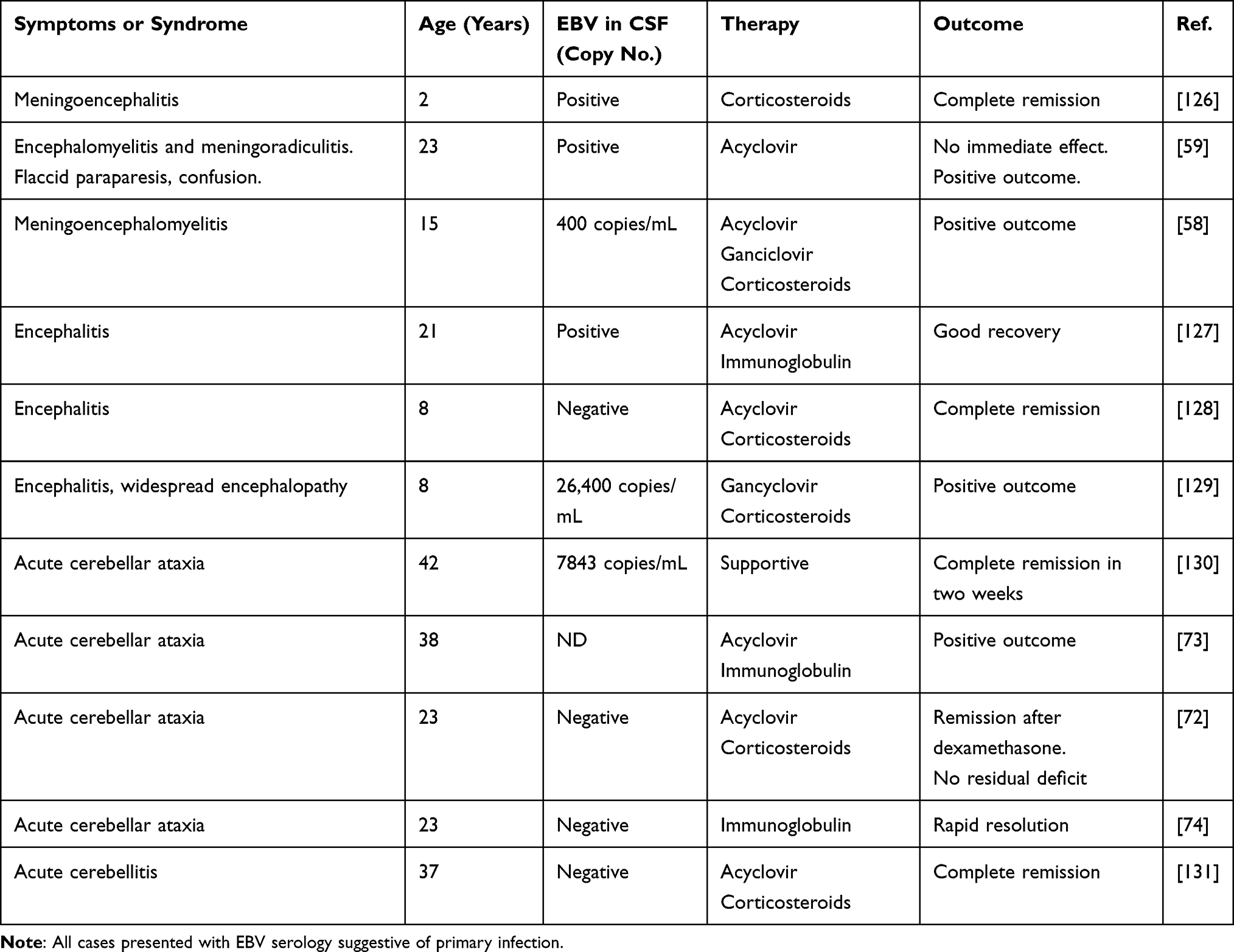 |
Table 3 Selection of Case Reports on CNS Disorders Associated with Primary EBV-Infection Without IM Manifestation |
 |
Table 4 Selection of Case Reports on CNS Disorders Associated with Reactivated EBV-Infection |
 |
Table 5 CNS Disorders Associated with EBV, Possible Pathogenesis and Treatment Options |
Treatment of EBV Meningitis or Encephalitis Following IM
Therapy of EBV encephalitis was reported in casuistics or small case series (Table 2). A case of IM associated encephalitis with typical proliferation of cytotoxic T cells not only in blood but also in the CSF was treated by parenteral acyclovir and dexamethasone.49 Although most cases are of mild or moderate severity, the spectrum is wide. A case of severe encephalitis with decerebrate posturing involving basal ganglia with onset 4 days after IM, with positive EBV-PCR in the CSF and absence of onconeural or synapse antibodies was treated with corticosteroids and i.v. acyclovir. The outcome was positive which the authors did not attribute to any of the treatment modalities.137 EBV meningoencephalitis is apparently never multiphasic (a course reported in herpes type 2 conditions),138 so secondary prophylaxis is not an issue.
Apparently, the documentation of therapy of EBV-induced meningitis and encephalitis in case reports or small case series is limited to Evidence grade IV. Randomized controlled trials are still lacking in the wide spectrum of neurological disorders following primary EBV infection. This highlights the importance of continuing the development of prophylaxis against primary EBV infection.
Treatment of ADEM Following IM
ADEM may be difficult to separate from post-IM encephalitis but is typically characterized by large white matter inflammatory demyelinating lesions demonstrated by T2-weighted MRI and absence of the EBV genome in the CSF. Furthermore, well-defined serum anti-MOG-antibodies are frequently detected.64 Encephalopathy with MRI showing multifocal lesions and significant number of EBV copies in the CSF may represent EBV encephalitis complicated with an ADEM-like immunological reaction, and when treated by both corticosteroids and ganciclovir a favourable course cannot be attributed to a specific pharmacological category. In a multicentre study, 44% (11/25) of children with MOG antibody positive disease (MOGAD) were EBV positive in serum compared with 95% (445/468) of MS pediatric patients.122 The combination of IM with MOGAD is unusual, however it does occur. In an illustrative case report, MOG antibody positive ADEM followed serologically verified IM with absence of the EBV genome in the CSF. The patient improved quickly after pulses of methylprednisolone followed by betamethasone tapering139 (Table 2).
Treatment of Monophasic Cerebellar Ataxia Following IM
The cerebellum is a relatively frequent target for autoimmune attacks.140 Paraneoplastic antibodies such as anti-Yo are often targeting the Purkinje cells. In a Western blot and ELISA study on post-EBV cerebellitis, approximately one-third of the patients had autoantibodies against triosphosphate isomerase (TPI), a ubiquitous protein,74 while few of the controls and notably none of their MS controls showed TPI-antibodies. Intravenous gamma globulin and plasmapheresis have been used in the treatment of cerebellar ataxia complicating EBV primary infection, although this condition is usually benign and self-limiting.141 Treatment with corticosteroids was used in a few cases, with doubtful results73 (Table 2).
Treatment of Encephalitis Following EBV Primary Infection Without Systemic Findings, and EBV Reactivation
A proportion of patients with EBV-related encephalitis do not present with preceding IM, notably in pediatric clienteles. In a series of 21 children, median age 13, with convincing EBV diagnosis based on serology or PCR, only one had typical symptoms of IM.58 Reactivation is suggested by temporal relation to immune deficiency state due to HIV infection or post-transplantation drug-induced immune deficiency, as well as by a constellation of EBV antibodies with high anti-EBNA1 and low anti-VCA IgM.142 Six of ten encephalitis patients received antiviral therapy, acyclovir or gancyclovir. They improved clinically, regardless of underlying conditions, use of EBV-specific antiviral therapy, or EBV load. The use of antivirals did not influence the outcome, although a rapid decrease in the CSF EBV load in two patients whose CSF was tested after 7 and 14 days of treatment, respectively, suggested that the drugs inhibited the viral replication56 (Table 3 and Table 4).
Treatment of CNS Disorders Characterized by Specific EBV Latency Stages
Current treatment of PCNSL associated with latency III does not include antiviral drugs,79 but consists of conventional lymphoma therapy with chemotherapy and consolidation with whole-brain radiotherapy or high-dose chemotherapy supported by autologous stem cell transplantation. High-dose methotrexate is the mainstay, recently in combination with Bruton’s tyrosine kinase (BTK) inhibitors, new monoclonal antibodies, or checkpoint blockers, as reviewed.75 Efforts are ongoing to identify EBV lytic and latent peptide HLA restricted specific T cell expansion for cellular therapy. Further novel therapies were reviewed.143 Proposed therapies include apart from the BTK inhibitor ibrutinib, third party EBV-specific T cells144 and CAR-T cell therapy. Furthermore, EBV-encoded miRNAs are abundantly expressed in EBV-associated tumors, and they are implicated in tumor development by targeting viral or cellular genes. Controlling the activities of EBV-encoded miRNAs in EBV-positive tumors may represent a promising approach to restoring host anti-tumor immunity, which provides a novel therapeutic strategy for the treatment of EBV-associated tumors.145
PCNSL appears to be more common in immunocompromised individuals and among those with autoimmune disorders requiring immunosuppression.146 It can be challenging to differentiate PCNSL from occasional large hemispheric demyelinating lesions in MS and the disorders may also co-occur, particularly in MS patients treated with fingolimod or natalizumab. In this context, autologous hematopoietic stem cell transplant, apart from conventional lymphoma therapy, may be an option.
Treatment of CNS Disorders Putatively Associated with EBV Latency 0/1
Multiple Sclerosis
According to contemporary opinion on the involvement of EBV in MS pathogenesis, prophylaxis against MS will partly be identical to prophylaxis against the IM and the caveats discussed above. The ultimate proof of EBV involvement in MS pathogenesis would be if EBV prophylaxis turn out to be efficacious in preventing MS. Stringent follow-up would be needed for several years at a sufficient scale (eg, in national registers) to determine such preventive effects. Selecting an MS prodrome rather than manifest MS for outcome might shorten the observation time.
More than a dozen immunomodulatory treatments are available in relapsing remitting MS, and two agents are now approved for progressive MS although with moderate effects, probably limited to cases with inflammatory lesions.147 However, the prospect of possible antiviral therapy is interesting. Previous small studies of anti-viral treatment in MS included a 2-year randomized study of high-dose Acyclovir and further trials of Acyclovir and Valacyclovir.148–150 While none show statistically significant effects on disease activity, intriguing trends were observed. Neither Acyclovir nor Valacyclovir affect EBV infection in latency 0–1; in fact, there are no currently available drugs with an effect on latent EBV infection. However, neither available anti-viral drugs nor neutralizing antibodies against glycoproteins can induce clearance of latently EBV infected memory B cells, whereas T cell immunity is essential for controlling the infection in latency.151 After EBV-specific CD8+ T cell lines originating from patients with progressive MS were produced by in vitro stimulation with LMP and EBNA1, the patients were treated by adoptive transfer of these cells. In a two-year follow-up of 10 patients, four participants showed clinical improvement.152
B cell depleting therapies are highly efficacious in relapsing remitting MS. Recently, a study integrated in the Swedish MS registry demonstrated that rituximab was superior to fumarate in reducing the frequency of relapses.153 It is unknown whether the efficacy of anti-CD20 therapy partly depends on its capacity to eliminate CD20+ B cells, including CD20+ memory B cells harbouring the EBV. To the best of our knowledge, there are no studies comparing the relationship between anti-CD20 clinical efficacy, CD20+ B cell concentration and EBV genome count. Exploration of such a mutual relationship should be an immediate priority.
The Challenges of Vaccination to EBV
If MS is associated with a high degree of - and in some respects aberrant – immunoreactivity (as in IM polyspecific immunoreactivity),154 a similar challenge as discussed for IM above would be to obtain a sufficient neutralizing response without eliciting the acute IM autoimmune response. However, for a wide range of EBV associated malignancies, the object is to completely avoid the primary infection, implicating prophylaxis at an early age (1–2 years), maybe necessitating repeat boosters to avoid a later lytic infection. A survey was performed of recent and ongoing phase 1 and 2 trials of prophylactic vaccines for IM and MS, and large studies were proposed, scheduled from power calculations.155
Treatment of CNS Disorders Associated with Possible Co-Infection with EBV and HHV-6
There are indications that human herpes virus (HHV)-6A, together with EBV, may be involved in MS. While virtually all MS patients experienced a preceding EBV infection, only a smaller proportion (approximately 40%) had a preceding HHV-6A infection according to their serological state.156 In a histological study on Rasmussens’s encephalitis, 17 vs 15 of 30 brain specimens contained EBV vs HHV-6 (not differentiated between A and B) antigens.61 No antiviral drug has been approved or systematically investigated for treatment of HHV-6 infection, however HHV-6 fusion inhibitors with a rhodamine structure were tested in human cells.157
Integrated Rationale for the Spectrum of EBV Infection Sequalae
- The intensity of the primary EBV infection is dependent on either the infectious dosage or the individual propensity for a forceful cellular, mainly CD8+ response.
- Several types of immediate complications after EBV primary infection, with or without IM, were apparently treated successfully with antiviral or immunosuppressive agents, however with no randomized trials available.
- The long-term consequences of the primary infection, particularly when severe, are probably dependent on either a larger burden of or less stable EBV in latency 0-I or a pathological setting of the humoral and cellular anti-EBV immunoreactivity.
- Anti-EBV therapy in MS was essentially negative, suggesting that MS ultimately develops from pathological processes based in latent infection, if EBV is a prerequisite for the disease.
- Therefore, the single most promising action would be prevention of the primary EBV infection. The rationale is prevention of IM, implicating prevention of its established immediate complications discussed in this review as well as several EBV-dependent neoplastic disorders. Prophylaxis against MS may be an important spin-off, with the further asset of obtaining strong evidence of a causal relationship between EBV and MS.
- To obtain an effective EBV prevention, the target could be known viral receptors eliciting neutralizing antibodies, EBV proteins essential for the lytic infection, as well as EBV proteins important for cellular immunity.
- Vaccine trials against IM should be implemented in a robust administrative context using nation-wide hospital-based or disease-specific registers, enabling long-term follow-up with reasonable coverage. Aiming at MS prevention, specific risk groups such as MS trait or radiologically isolated syndrome could be selected.
The present review demonstrates that many of the disorders in the large spectrum of complications to EBV infection are of autoimmune nature. IM is associated with polyspecific antiviral and heterophile immunoreactivity and several of its immediate complications are due to autoimmune reactions towards CNS antigens, although EBV persistence was often revealed in the CSF. Late complications may be due to deficient cellular immune control. In MS, there is a polyspecific antiviral immunoreactivity in the CSF similar to that seen in IM, while several lymphomas and mucosal carcinomas are dependent on the persistence of the EBV-genome. A common countermeasure against the EBV primary infection and its spectrum of complications would be prophylaxis against the primary infection. The rationale for IM vaccination is stronger, aiming at its immediate and malignant complications, while the rationale for IM vaccination as MS prophylaxis is pending confirmation of an EBV-MS causal relationship. However, MS prophylaxis could be an important spin-off from IM vaccination, aiming at immediate and malignant complications.
Author Contributions
All authors made a significant contribution to the work reported, including study design, execution, and acquisition of data. The authors contributed equally to writing and revising the manuscript, and agreed to the journal to which the article was submitted. They reviewed and agreed on all versions of the article before submission and accepted the final version for publication and agree to take responsibility and be accountable for the contents of the article.
Disclosure
The authors have nothing to declare.
References
1. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet. 1964;15:702–703. doi:10.1016/S0140-6736(64)91524-7
2. Young LS, Yap LF, Murray PG. Epstein-Barr virus: more than 50 years old and still providing surprises. Nat Rev Cancer. 2016;16(12):789–802. doi:10.1038/nrc.2016.92
3. Gerber P, Kalter SS, Schidlovsky G, Peterson WD Jr, Daniel MD. Biologic and antigenic characteristics of Epstein-Barr virus-related Herpesviruses of chimpanzees and baboons. Int J Cancer. 1977;20(3):448–459. doi:10.1002/ijc.2910200318
4. Dunmire SK, Hogquist KA, Balfour HH. Infectious mononucleosis. Curr Top Microbiol Immunol. 2015;390(Pt 1):211–240. doi:10.1007/978-3-319-22822-8_9
5. Thorley-Lawson DA. EBV persistence–introducing the virus. Curr Top Microbiol Immunol. 2015;390(Pt 1):151–209. doi:10.1007/978-3-319-22822-8_8
6. Menet A, Speth C, Larcher C, et al. Epstein-Barr virus infection of human astrocyte cell lines. J Virol. 1999;73(9):7722–7733. doi:10.1128/JVI.73.9.7722-7733.1999
7. Jones K, Rivera C, Sgadari C, et al. Infection of human endothelial cells with Epstein-Barr virus. J Exp Med. 1995;182(5):1213–1221. doi:10.1084/jem.182.5.1213
8. Bjornevik K, Cortese M, Healy BC, et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science. 2022;375(6578):296–301. doi:10.1126/science.abj8222
9. Demissie A, Svmyr A. Age distribution of antibodies to EB virus in Swedish females as studied by indirect immunofluorescence on Burkitt cells. Acta Pathol Microbiol Scand. 1969;75(3):457–465.
10. Sixbey JW, Nedrud JG, Raab-Traub N, Hanes RA, Pagano JS. Epstein- Barr virus replication in oropharyngeal epithelial cells. N Engl J Med. 1984;310(19):1225–1230. doi:10.1056/NEJM198405103101905
11. Sixbey JW, Lemon SM, Pagano JS. A second site for Epstein-Barr virus shedding: the uterine cervix. Lancet. 1986;328(8516):1122–1124. doi:10.1016/S0140-6736(86)90531-3
12. Young LS, Arrand JR, Murray PG. EBV gene expression and regulation. In: Arvin A, Campadelli-Fiume G, Mocarski E, editors. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge: Cambridge University Press; 2007:Chapter 27.
13. Skalsky RL, Cullen BR. EBV Noncoding RNAs. Curr Top Microbiol Immunol. 2015;391:181–217. doi:10.1007/978-3-319-22834-1_6
14. Zimber U, Adldinger HK, Lenoir GM, et al. Geographical prevalence of two types of Epstein-Barr virus. Virology. 1986;154(1):56–66. doi:10.1016/0042-6822(86)90429-0
15. Shannon-Lowe C, Rowe M. Epstein Barr virus entry; kissing and conjugation. Curr Opin Virol. 2013;4:78–84. doi:10.1016/j.coviro.2013.12.001
16. Haan KM, Longnecker R. Coreceptor restriction within the HLA-DQ locus for Epstein-Barr virus infection. Proc Natl Acad Sci U S A. 2000;97(16):9252–9257. doi:10.1073/pnas.160171697
17. Ogembo JG, Kannan L, Ghiran I, et al. Human complement receptor type 1/CD35 is an Epstein-Barr Virus receptor. Cell Rep. 2013;3(2):371–385. doi:10.1016/j.celrep.2013.01.023
18. Tierney RJ, Steven N, Young LS, Rickinson AB. Epstein-Barr virus latency in blood mononuclear cells: analysis of viral gene transcription during primary infection and in the carrier state. J Virol. 1994;68(11):7374–7385. doi:10.1128/jvi.68.11.7374-7385.1994
19. Kanda T. EBV-ENCODED LATENT GENES. Adv Exp Med Biol. 2018;1045:377–394.
20. Chen F, Zou JZ, Di Renzo L, et al. A subpopulation of normal B cells latently infected with Epstein-Barr virus resembles Burkitt lymphoma cells in expressing EBNA-1 but not EBNA-2 or LMP1. J Virol. 1995;69(6):3752–3758. doi:10.1128/jvi.69.6.3752-3758.1995
21. Leight ER, Sugden B. EBNA-1: a protein pivotal to latent infection by Epstein-Barr virus. Rev Med Virol. 2000;10(2):83–100. doi:10.1002/(SICI)1099-1654(200003/04)10:2<83::AID-RMV262>3.0.CO;2-T
22. Howe JG, Steitz JA. Localization of Epstein-Barr virus-encoded small RNAs by in situ hybridization. Proc Natl Acad Sci U S A. 1986;83(23):9006–9010. doi:10.1073/pnas.83.23.9006
23. Klein G, Ernberg I. Effects on apoptosis, cell cycle and transformation, and comparative aspects of EBV with other known DNA tumor viruses. In: Arvin A, Campadelli-Fiume G, Mocarski E, et al., editors. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge: Cambridge University Press; 2007:Chapter 29.
24. Levitskaya J, Coram M, Levitsky V, et al. Inhibition of antigen processing by the internal repeat region of the Epstein-Barr virus nuclear antigen-1. Nature. 1995;375(6533):685–688. doi:10.1038/375685a0
25. Ressing ME, van Gent M, Gram AM, Hooykaas MJ, Piersma SJ, Wiertz EJ. Immune evasion by Epstein-Barr virus. Curr Top Microbiol Immunol. 2015;391:355–381. doi:10.1007/978-3-319-22834-1_12
26. Hennessy K, Kieff E. A second nuclear protein is encoded by Epstein-Barr virus in latent infection. Science. 1985;227(4691):1238–1240. doi:10.1126/science.2983420
27. Rickinson AB, Young LS, Rowe M. Influence of the Epstein-Barr virus nuclear antigen EBNA 2 on the growth phenotype of virus-transformed B cells. J Virol. 1987;61(5):1310–1317. doi:10.1128/jvi.61.5.1310-1317.1987
28. Ling PD, Hsieh JJ, Ruf IK, Rawlins DR, Hayward SD. EBNA-2 upregulation of Epstein-Barr virus latency promoters and the cellular CD23 promoter utilizes a common targeting intermediate, CBF1. J Virol. 1994;68(9):5375–5383. doi:10.1128/jvi.68.9.5375-5383.1994
29. Styles CT, Paschos K, White RE, Farrell PJ. The cooperative functions of the EBNA3 proteins are central to EBV persistence and latency. Pathogens. 2018;7(1):31. doi:10.3390/pathogens7010031
30. Lo AK, Dawson CW, Lung HL, Wong KL, Young LS. The role of EBV-encoded LMP1 in the NPC tumor microenvironment: from function to therapy. Front Oncol. 2021;11:640207. doi:10.3389/fonc.2021.640207
31. Longnecker R. Epstein-Barr virus latency: LMP2, a regulator or means for Epstein- Barr virus persistence? Adv Cancer Res. 2000;79:175–200.
32. Nanbo A, Takada K. The role of Epstein-Barr virus-encoded small RNAs (EBERs) in oncogenesis. Rev Med Virol. 2002;12(5):321–326. doi:10.1002/rmv.363
33. Sadler RH, Raab-Traub N. Structural analyses of the Epstein-Barr virus BamHI A transcripts. J Virol. 1995;69(2):1132–1141. doi:10.1128/jvi.69.2.1132-1141.1995
34. McKenzie J, El-Guindy A. Epstein-Barr virus lytic cycle reactivation. Curr Top Microbiol Immunol. 2015;391:237–261. doi:10.1007/978-3-319-22834-1_8
35. Evans AS, Niederman JC, McCollum RW. Seroepidemiologic studies of infectious mononucleosis with EB virus. N Engl J Med. 1968;279(21):1121–1127. doi:10.1056/NEJM196811212792101
36. Niller HH, Bauer G. Epstein-Barr Virus: clinical diagnostics. Methods Mol Biol. 2017;1532:33–55.
37. McAulay KA, Higgins CD, Macsween KF, et al. HLA class I polymorphisms are associated with development of infectious mononucleosis upon primary EBV infection. J Clin Invest. 2007;117:3042–3048. doi:10.1172/JCI32377
38. Agostini S, Mancuso R, Guerini FR, et al. HLA alleles modulate EBV viral load in multiple sclerosis. J Transl Med. 2018;16:80. doi:10.1186/s12967-018-1450-6
39. Svedmyr E, Ernberg I, Seeley J, et al. Virologic, immunologic, and clinical observations on a patient during the incubation, acute, and convalescent phases of infectious mononucleosis. Clin Immunol Immunopathol. 1984;30(3):437–450. doi:10.1016/0090-1229(84)90029-1
40. Rickinson AB, Fox CP. Epstein-Barr virus and infectious mononucleosis: what students can teach us. J Infect Dis. 2013;207(1):6–8. doi:10.1093/infdis/jis647
41. Lee SP, Thomas WA, Murray RJ, et al. HLA A2.1-restricted cytotoxic T cells recognizing a range of Epstein-Barr virus isolates through a defined epitope in latent membrane protein LMP2. J Virol. 1993;67(12):7428–7435. doi:10.1128/jvi.67.12.7428-7435.1993
42. Svedmyr E, Jondal M. Cytotoxic effector cells specific for B cell lines transformed by Epstein-Barr virus are present in patients with infectious mononucleosis. Proc Natl Acad Sci U S A. 1975;72(4):1622–1626. doi:10.1073/pnas.72.4.1622
43. Callan MF, Tan L, Annels N, et al. Direct visualization of antigen- specific CD8+ T cells during the primary immune response to Epstein-Barr virus in vivo. J Exp Med. 1998;187(9):1395–1402. doi:10.1084/jem.187.9.1395
44. Hislop AD, Annels NE, Gudgeon NH, Leese AM, Rickinson AB. Epitope-specific evolution of human CD8+ T cell responses from primary to persistent phases of Epstein-Barr virus infection. J Exp Med. 2002;195(7):893–905. doi:10.1084/jem.20011692
45. Fujiwara S, Nakamura H. Chronic active Epstein–Barr virus infection: is it immunodeficiency, malignancy, or both? Cancers. 2020;12(11):3202. doi:10.3390/cancers12113202
46. Jakhmola S, Jha HC. Glial cell response to Epstein-Barr virus infection: a plausible contribution to virus-associated inflammatory reactions in the brain. Virology. 2021;559:182–195. doi:10.1016/j.virol.2021.04.005
47. Jha HC, Mehta D, Lu J, et al. Gammaherpesvirus infection of human neuronal cells. mBio. 2015;6(6):e01844–e01915. doi:10.1128/mBio.01844-15
48. Casiraghi C, Dorovini-Zis K, Horwitz M. Epstein-Barr virus infection of human brain microvessel endothelial cells: a novel role in multiple sclerosis. J Neuroimmunol. 2011;230(1–2):173–177. doi:10.1016/j.jneuroim.2010.08.003
49. Lehrnbecher T, Chittka B, Nanan R, et al. Activated T lymphocytes in the cerebrospinal fluid of a patient with Epstein-Barr virus-associated meningoencephalitis. Pediatr Infect Dis J. 1996;15(7):631–633. doi:10.1097/00006454-199607000-00016
50. Jakhmola S, Sk MF, Chatterjee A, et al. A plausible contributor to multiple sclerosis; presentation of antigenic myelin protein epitopes by major histocompatibility complexes. Comput Biol Med. 2022;148:105856. doi:10.1016/j.compbiomed.2022.105856
51. Fan Y, Sanyal S, Bruzzone R. Breaking bad: how viruses subvert the cell cycle. Front Cell Infect Microbiol. 2018;8:396. doi:10.3389/fcimb.2018.00396
52. Tiwari D, Mittal N, Chandra Jha H. Unraveling the links between neurodegeneration and Epstein-Barr virus-mediated cell cycle dysregulation. Curr Res Neurobiol. 2022;3:100046. doi:10.1016/j.crneur.2022.100046
53. Grillo E, da Silva RJM, Barbato Filho JH. Epstein-Barr virus acute encephalomyelitis in a 13-year-old boy. Eur J Paediatr Neurol. 2008;12(5):417–420. doi:10.1016/j.ejpn.2007.10.016
54. Dyachenko P, Smiianova O, Kurhanskaya V, Oleshko A, Dyachenko A. Epstein-Barr virus-associated encephalitis in a case-series of more than 40 patients. Wiad Lek. 2018;71(6):1224–1230.
55. Cheng H, Chen D, Peng X, et al. Clinical characteristics of Epstein–Barr virus infection in the pediatric nervous system. BMC Infect Dis. 2020;20(1):886. doi:10.1186/s12879-020-05623-1
56. Weinberg A, Li S, Palmer M, Tyler KL. Quantitative CSF PCR in Epstein-Barr virus infections of the central nervous system. Ann Neurol. 2002;52(5):543–548. doi:10.1002/ana.10321
57. Abul-Kasim K, Palm L, Maly P, Sundgren PC. The neuroanatomic localization of Epstein-Barr virus encephalitis may be a predictive factor for its clinical outcome: a case report and review of 100 cases in 28 reports. J Child Neurol. 2009;24(6):720–726. doi:10.1177/0883073808327842
58. Doja A, Bitnun A, Jones ELF, et al. Pediatric Epstein-Barr virus—associated encephalitis: 10-year review. J Child Neurol. 2006;21(5):384–391. doi:10.1177/08830738060210051101
59. Tselis A, Duman R, Storch GA, Lisak RP. Epstein-Barr virus encephalomyelitis diagnosed by polymerase chain reaction: detection of the genome in the CSF. Neurology. 1997;48(5):1351–1355. doi:10.1212/WNL.48.5.1351
60. Bauer J, Bien CG, Lassmann H. Rasmussenʼs encephalitis: a role for autoimmune cytotoxic T lymphocytes. Curr Opin Neurol. 2002;15(2):197–200. doi:10.1097/00019052-200204000-00012
61. Liu D, Wang X, Wang Y, et al. Detection of EBV and HHV6 in the brain tissue of patients with Rasmussen’s encephalitis. Virol Sin. 2018;33(5):402–409. doi:10.1007/s12250-018-0063-9
62. Leake JAD, Albani S, Kao AS, et al. Acute disseminated encephalomyelitis in childhood: epidemiologic, clinical and laboratory features. Pediatr Infect Dis J. 2004;23(8):756–764. doi:10.1097/01.inf.0000133048.75452.dd
63. Shen J, Lin D, Jiang T, Gao F, Jiang K. Clinical characteristics and associated factors of pediatric acute disseminated encephalomyelitis patients with MOG antibodies: a retrospective study in Hangzhou, China. BMC Neurol. 2022;22(1):418. doi:10.1186/s12883-022-02963-0
64. Hennes E-M, Baumann M, Schanda K, et al. Prognostic relevance of MOG antibodies in children with an acquired demyelinating syndrome. Neurology. 2017;89(9):900–908. doi:10.1212/WNL.0000000000004312
65. Prineas J, McDonald WI, Franklin R. Demyelinating diseases. In: Graham D, Lantos P, editors. Greenfield’s Neuropathology.
66. Kakalacheva K, Regenass S, Wiesmayr S, et al. Infectious mononucleosis triggers generation of IgG auto-antibodies against native myelin oligodendrocyte glycoprotein. Viruses. 2016;8(2):51. doi:10.3390/v8020051
67. Peschl P, Bradl M, Höftberger R, et al. Myelin oligodendrocyte glycoprotein: deciphering a target in inflammatory demyelinating diseases. Front Immunol. 2017;8:529. doi:10.3389/fimmu.2017.00529
68. Hennes E-M, Baumann M, Lechner C, Rostasy K. MOG spectrum disorders and role of MOG-Antibodies in clinical practice. Neuropediatrics. 2018;49(01):3–11. doi:10.1055/s-0037-1604404
69. Wingerchuk DM. The clinical course of acute disseminated encephalomyelitis. Neurol Res. 2006;28(3):341–347. doi:10.1179/016164106X98251
70. Davis DP, Marino A. Acute cerebellar ataxia in a toddler: case report and literature review. J Emerg Med. 2003;24(3):281–284. doi:10.1016/S0736-4679(02)00746-1
71. Nussinovitch M, Prais D, Volovitz B, Shapiro R, Amir J. Post-infectious acute cerebellar ataxia in children. Clin Pediatr. 2003;42(7):581–584. doi:10.1177/000992280304200702
72. Al-Shokri SD, Karumannil SA, Mohammed SS, Sadek MS. Post-Epstein-Barr virus acute cerebellitis in an adult. Am J Case Rep. 2020;21:e918567. doi:10.12659/AJCR.918567
73. Ali K, Lawthom C. Epstein-Barr virus-associated cerebellar ataxia. BMJ Case Rep. 2013;2013(apr22 1):bcr2013009171. doi:10.1136/bcr-2013-009171
74. Uchibori A, Sakuta M, Kusunoki S, Chiba A. Autoantibodies in postinfectious acute cerebellar ataxia. Neurology. 2005;65(7):1114–1116. doi:10.1212/01.wnl.0000178802.38268.1e
75. Paydas S. Primary central nervous system lymphoma: essential points in diagnosis and management. Med Oncol. 2017;34(4):61. doi:10.1007/s12032-017-0920-7
76. Enblad G, Martinsson G, Baecklund E, et al. Population-based experience on primary central nervous system lymphoma 2000–2012: the incidence is increasing. Acta Oncol. 2017;56(4):599–607. doi:10.1080/0284186X.2016.1270465
77. Kleinschmidt-DeMasters BK, Damek DM, Lillehei KO, Dogan A, Giannini C. Epstein Barr virus-associated primary CNS lymphomas in elderly patients on immunosuppressive medications. J Neuropathol Exp Neurol. 2008;67(11):1103–1111. doi:10.1097/NEN.0b013e31818beaea
78. Bataille B, Delwail V, Menet E, et al. Primary intracerebral malignant lymphoma: report of 248 cases. J Neurosurg. 2000;92(2):261–266. doi:10.3171/jns.2000.92.2.0261
79. Grommes C, DeAngelis LM. Primary CNS lymphoma. J Clin Oncol. 2017;35(21):2410–2418. doi:10.1200/JCO.2017.72.7602
80. Venetz D, Ponzoni M, Schiraldi M, et al. Perivascular expression of CXCL9 and CXCL12 in primary central nervous system lymphoma: t-cell infiltration and positioning of malignant B cells. Int J Cancer. 2010;127(10):2300–2312. doi:10.1002/ijc.25236
81. Rubenstein JL, Fridlyand J, Shen A, et al. Gene expression and angiotropism in primary CNS lymphoma. Blood. 2006;107(9):3716–3723. doi:10.1182/blood-2005-03-0897
82. Sugita Y, Terasaki M, Niino D, et al. Epstein–Barr virus-associated primary central nervous system lymphomas in immunocompetent elderly patients: analysis for latent membrane protein-1 oncogene deletion and EBNA-2 strain typing. J Neuro Oncol. 2010;100(2):271–279. doi:10.1007/s11060-010-0191-z
83. Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National multiple sclerosis society (USA) advisory committee on clinical trials of new agents in multiple sclerosis. Neurology. 1996;46(4):907–911. doi:10.1212/WNL.46.4.907
84. Ascherio A, Munger KL. Epstein–Barr virus infection and multiple sclerosis: a review. J Neuroimmune Pharmacol. 2010;5(3):271–277. doi:10.1007/s11481-010-9201-3
85. Levin LI, Munger K, Rubertone MV, et al. Temporal relationship between elevation of Epstein-Barr virus antibody titers and initial onset of neurological symptoms in multiple sclerosis. JAMA. 2005;293(20):2493–2500. doi:10.1001/jama.293.20.2496
86. Sundström P, Juto P, Wadell G, et al. An altered immune response to Epstein-Barr virus in multiple sclerosis: a prospective study. Neurology. 2004;62(12):2277–2282. doi:10.1212/01.WNL.0000130496.51156.D7
87. Moreno MA, Or-Geva N, Aftab BT, et al. Molecular signature of Epstein-Barr virus infection in MS brain lesions. Neurol Neuroimmunol Neuroinflamm. 2018;5(4):e466. doi:10.1212/NXI.0000000000000466
88. Veroni C, Serafini B, Rosicarelli B, et al. Transcriptional profile and Epstein-Barr virus infection status of laser-cut immune infiltrates from the brain of patients with progressive multiple sclerosis. J Neuroinflammation. 2018;15(1):18. doi:10.1186/s12974-017-1049-5
89. Magliozzi R, Howell O, Vora A, et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain. 2006;130(4):1089–1104. doi:10.1093/brain/awm038
90. Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol. 2004;14(2):164–174. doi:10.1111/j.1750-3639.2004.tb00049.x
91. Nociti V, Frisullo G, Marti A, et al. Epstein-Barr virus antibodies in serum and cerebrospinal fluid from multiple sclerosis, chronic inflammatory demyelinating polyradiculoneuropathy and amyotrophic lateral sclerosis. J Neuroimmunol. 2010;225(1–2):149–152. doi:10.1016/j.jneuroim.2010.04.007
92. Lünemann JD, Edwards N, Muraro PA, et al. Increased frequency and broadened specificity of latent EBV nuclear antigen-1-specific T cells in multiple sclerosis. Brain. 2006;129(6):1493–1506. doi:10.1093/brain/awl067
93. Lynemann JD, Jelcic I, Roberts S, et al. EBNA1-specific T cells from patients with multiple sclerosis cross react with myelin antigens and co-produce IFN-γ and IL-2. J Exp Med. 2008;205(8):1763–1773. doi:10.1084/jem.20072397
94. Sawcer S, Hellenthal G, Pirinen M, et al. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–219.
95. Bjornevik K, Münz C, Cohen JI, Ascherio A. Epstein–Barr virus as a leading cause of multiple sclerosis: mechanisms and implications. Nat Rev Neurol. 2023;19(3):160–171. doi:10.1038/s41582-023-00775-5
96. Peferoen LAN, Lamers F, Lodder LNR, et al. Epstein Barr virus is not a characteristic feature in the central nervous system in established multiple sclerosis. Brain. 2010;133(5):e137. doi:10.1093/brain/awp296
97. Balfour, Jr. HH Jr, Holman CJ, Hokanson KM, et al. A prospective clinical study of Epstein-Barr virus and host interactions during acute infectious mononucleosis. J Infect Dis. 2005;192(9):1505–1512. doi:10.1086/491740
98. Macsween KF, Higgins CD, McAulay KA, et al. Infectious mononucleosis in university students in the United Kingdom: evaluation of the clinical features and consequences of the disease. Clin Infect Dis. 2010;50(5):699–706. doi:10.1086/650456
99. Luzuriaga K, Sullivan JL. Infectious mononucleosis. N Engl J Med. 2010;362(21):1993–2000. doi:10.1056/NEJMcp1001116
100. Martelius T, Lappalainen M, Palomaki M, Anttila VJ. Clinical characteristics of patients with Epstein Barr virus in cerebrospinal fluid. BMC Infect Dis. 2011;11:281. doi:10.1186/1471-2334-11-281
101. Hjalgrim H, Smedby KE, Rostgaard K, et al. Infectious mononucleosis, childhood social environment, and risk of Hodgkin lymphoma. Cancer Res. 2007;67(5):2382–2388. doi:10.1158/0008-5472.CAN-06-3566
102. Andersson J, Sköldenberg B, Henle W, et al. Acyclovir treatment in infectious mononucleosis: a clinical and virological study. Infection. 1987;15(Suppl 1):S14–S20. doi:10.1007/BF01650106
103. Tengvall K, Huang J, Hellström C, et al. Molecular mimicry between anoctamin 2 and Epstein-Barr virus nuclear antigen 1 associates with multiple sclerosis risk. Proc Natl Acad Sci U S A. 2019;116(34):16955–16960. doi:10.1073/pnas.1902623116
104. Tynell E, Aurelius E, Brandell A, et al. Acyclovir and prednisolone treatment of acute infectious mononucleosis: a multicenter, double-blind, placebo-controlled study. J Infect Dis. 1996;174:324–331. doi:10.1093/infdis/174.2.324
105. De Paor M, O’Brien K, Fahey T, Smith SM. Antiviral agents for infectious mononucleosis (glandular fever). Cochrane Database Syst Rev. 2016;12(12):CD011487. doi:10.1002/14651858.CD011487.pub2
106. Hu H, Deng H, Bi J, et al. Clinical characteristics and effectiveness of antiviral agents in hospitalized children with infectious mononucleosis in China: A multicenter retrospective study. Pediatr Investig. 2021;5(3):188–194.
107. Silveira EL, Fogg MH, Leskowitz RM, et al. Therapeutic vaccination against the rhesus lymphocryptovirus EBNA-1 homologue, rhEBNA-1, elicits T cell responses to novel epitopes in rhesus macaques. J Virol. 2013;87(24):13904–13910. doi:10.1128/JVI.01947-13
108. Ruiss R, Jochum S, Wanner G, et al. A virus-like particle-based Epstein-Barr virus vaccine. J Virol. 2011;85(24):13105–13113. doi:10.1128/JVI.05598-11
109. Cui X, Snapper CM. Epstein Barr virus: development of vaccines and immune cell therapy for EBV-associated diseases. Front Immunol. 2021;12:734471. doi:10.3389/fimmu.2021.734471
110. Gu SY, Huang TM, Ruan L, et al. First EBV vaccine trial in humans using recombinant vaccinia virus expressing the major membrane antigen. Dev Biol Stand. 1995;84:171–177.
111. Cui X, Cao Z, Chen Q, et al. Rabbits immunized with Epstein-Barr virus Gh/gL or gB recombinant proteins elicit higher serum virus neutralizing activity than Gp350. Vaccine. 2016;34(34):4050–4055. doi:10.1016/j.vaccine.2016.06.021
112. Sun C, Chen XC, Kang YF, Zeng MS. The status and prospects of Epstein-Barr virus prophylactic vaccine development. Front Immunol. 2021;12:677027. doi:10.3389/fimmu.2021.677027
113. Rees L, Tizard EJ, Morgan AJ, et al. A phase I trial of Epstein-Barr virus gp350 vaccine for children with chronic kidney disease awaiting transplantation. Transplantation. 2009;88(8):1025–1029. doi:10.1097/TP.0b013e3181b9d918
114. Sokal EM, Hoppenbrouwers K, Vandermeulen C, et al. Recombinant gp350 vaccine for infectious mononucleosis: a Phase 2, randomized, double-blind, placebo-controlled trial to evaluate the safety, immunogenicity, and efficacy of an Epstein-Barr virus vaccine in healthy young adults. J Infect Dis. 2007;196(12):1749–1753. doi:10.1086/523813
115. Cui X, Cao Z, Sen G, et al. A novel tetrameric gp350 1–470 as a potential Epstein-Barr virus vaccine. Vaccine. 2013;31(30):3039–3045. doi:10.1016/j.vaccine.2013.04.071
116. Bharadwaj M, Burows SR, Burrows JM, et al. Longitudinal dynamics of antigen-specific CD8+ cytotoxic T lymphocytes following primary Epstein-Barr virus infection. Blood. 2001;98:2588–2589. doi:10.1182/blood.V98.8.2588
117. Elliott SL, Suhrbier A, Miles JJ, et al. Phase I trial of a CD8+ T-cell peptide epitope-based vaccine for infectious mononucleosis. J Virol. 2008;82(3):1448–1457. doi:10.1128/JVI.01409-07
118. Internetbased information. Available from: http://clinicaltrials.gov/ct2/show/NCT05164094.
119. Tsuruyama Y, Mori N, Yoshida S, Hayashi T. Epstein-Barr virus-related encephalitis in a young woman: a case-report. J Infect Chemother. 2020;26:741–744. doi:10.1016/j.jiac.2020.02.005
120. Hayton E, Wakerley B, Bowler IC, et al. Successful outcome of Epstein-Barr virus encephalitis managed with bilateral craniectomy, corticosteroids and aciclovir. Pract Neurol. 2012;12:234–237. doi:10.1136/practneurol-2012-000234
121. Hussain RS, Hussain NA. Ataxia and encephalitis in a young adult with EBV mononucleosis: a case report. Case Rep Neurol Med. 2013;2013:516325. doi:10.1155/2013/516325
122. Gaudioso CM, Mar S, Casper TC, et al. MOG and AQP4 antibodies among children with multiple sclerosis and controls. Ann Neurol. 2023;93:271–284. doi:10.1002/ana.26502
123. Murasawa E, Masazumi M, Koichi I, et al. Adult-onset acute disseminated encephalomyelitis with Epstein-Barr virus infection. Case Rep Radiol. 2022;2022:6149501. doi:10.1155/2022/6149501
124. Deeba A, Cardos B, Gorur Y, et al. A rare case of adult acute disseminated encephalomyelitis associated with primary Epstein-Barr virus infection. Eur J Cas Rep Intern Med. 2019;6:001094.
125. Mohsen H, Zeinah GFA, Elsotouhy AH, Mohamed K. Acute disseminated encephalomyelitis following infectious mononucleosis in a toddler. BMJ Case Rep. 2013;2013:bcr2013010048. doi:10.1136/bcr-2013-010048
126. Pangprasertkul S, Sanguansermsri C, Sudjaituk T. Epstein-Barr virus meningoencephalitis in a young immunocompetent child: a case report. Heliyon. 2022;8:e11150. doi:10.1016/j.heliyon.2022.e11150
127. Koning MT, Brik T, Hagenbeek R, et al. A case of fulminant Epstein-Barr virus encephalitis in an immune-competent adult. J Neurovirol. 2019;25(3):422–425. doi:10.1007/s13365-018-0718-1
128. Celik T, Celik U, Tolunay O, et al. Epstein-Barr virus encephalitis with substantia nigra involvement. J Pediatr Neurosci. 2015;10(4):401–403. doi:10.4103/1817-1745.174436
129. Zhang S, Feng J, Shi Y. Transient widespread cortical and splenial lesions in acute encephalitis/encephalopathy associated with primary Epstein-Barr virus infection. Int J Infect Dis. 2016;42:7–10. doi:10.1016/j.ijid.2015.11.009
130. Muscat K, Galea R, Vella M. An adult case of acute EBV cerebellitis. Eur J Case Rep Intern Med. 2017;4(1):000519. doi:10.12890/2016_000519
131. Samkar AV, Poulsen MNF, Beinfait HP, Van Leeuwen RB. Acute cerebellitis in adults: a case report and review of the literature. BMC Res Notes. 2017;10:610. doi:10.1186/s13104-017-2935-8
132. Wang Y, Dong Q, Chen Y, et al. Intracranial Epstein-Barr virus infection appearing as an unusual case of meningitis in an immunocompetent woman: a case report. J Int Med Res. 2020;48:300060520903215. doi:10.1177/0300060520903215
133. Crocchiolo R, Ciccolini J, El-Cheikh J, et al. Successful treatment of post-transplant Epstein-Barr virus-related meningoencephalitis by intravenous rituximab monotherapy. Leuk Lymphoma. 2012;53(10):2063–2065. doi:10.3109/10428194.2012.670232
134. Nakamura J, Yanagida M, Saito K, et al. Epstein-Barr virus encephalitis in a patient with rheumatoid arthritis. Mod Rheumatol Case Rep. 2022;6:160–162. doi:10.1093/mrcr/rxab045
135. Ueda M, Tateishi T, Shigeto H, et al. A case of acute disseminated encephalomyelitis associated with Epstein-Barr virus reactivation during infliximab therapy. Rinsho Shinkeigaku. 2010;50:461–466. doi:10.5692/clinicalneurol.50.461
136. Caucheteux N, Maarouf A, Daelman L, et al. Acute disseminated encephalomyelitis in two renal transplant patients: is there a role for Epstein-Barr virus reactivation? Mult Scler. 2013;19(9):1222–1225. doi:10.1177/1352458513478674
137. Rodrigo-Armenteros P, Kapetanovic-Garcia S, Anton-Mendez L, et al. Akinetic mutism and status epilepticus due to Epstein Barr virus encephalitis. Clin Neurol Neurosurg. 2019;185:105492. doi:10.1016/j.clineuro.2019.105492
138. Görander S, Andersen O, Leiram B, et al. Multiphasic encephalomyelitis in a patient with recurrent herpes simplex type 2 meningitis. Scand J Infec Dis. 2006;38:942. doi:10.1080/00365540600606499
139. Nakamura Y, Nakajima H, Tani H, et al. Anti-MOG antibody-positive ADEM following infectious mononucleosis due to a primary EBV infection: a case report. BMC Neurol. 2017;17(1):76. doi:10.1186/s12883-017-0858-6
140. Hampe CS, Mitoma H. A breakdown of immune tolerance in the cerebellum. Brain Sci. 2022;12(3):328. doi:10.3390/brainsci12030328
141. D’Ambrosio E, Khalighinejad F, Ionete C. Intravenous immunoglobulins in an adult case of post-EBV cerebellitis. BMJ Case Rep. 2020;13(2). doi:10.1136/bcr-2019-231661
142. Sanefuji M, Ohga S, Kira R, et al. Epstein-Barr virus-associated meningoencephalomyelitis: intrathecal reactivation of the virus in an immunocompetent child. J Child Neurol. 2008;23(9):1072–1077. doi:10.1177/0883073808315414
143. Fortin Ensign SP, Gathers D, Wiedmeier JE, Mrugala MM. Central nervous system lymphoma: novel therapies. Curr Treat Options Oncol. 2022;23(1):117–136. doi:10.1007/s11864-021-00921-5
144. Law SC, Hoang T, O’Rourke K, et al. Successful treatment of Epstein-Barr virus-associated primary central nervous system lymphoma due to post-transplantation lymphoproliferative disorder, with ibrutinib and third-party Epstein-Barr virus-specific T cells. Am J Transplant. 2021;21:3465–3471. doi:10.1111/ajt.16628
145. Wang M, Gu B, Chen X, et al. The function and therapeutic potential of Epstein-Barr virus-encoded microRNAs in cancer. Mol Ther Nucleic Acids. 2019;17:657–668. doi:10.1016/j.omtn.2019.07.002
146. Kaulen LD, Karschnia P, Dietrich J, Baehring JM. Autoimmune disease-related CNS lymphoma: systematic review and meta-analysis. J Neurooncol. 2020;149:153–159. doi:10.1007/s11060-020-03583-9
147. Kappos L, Bar-Or A, Cree BAC, et al. Siponimod versus placebo in secondary progressive multiple sclerosis (EXPAND): a double-blind, randomised, Phase 3 study. Lancet. 2018;391(10127):1263–1273. doi:10.1016/S0140-6736(18)30475-6
148. Lycke J, Svennerholm B, Hjelmquist E, et al. Acyclovir treatment of relapsing-remitting multiple sclerosis. A randomized, placebo-controlled, double-blind study. J Neurol. 1996;243(3):214–224. doi:10.1007/BF00868517
149. Bech E, Lycke J, Gadeberg P, et al. A randomized, double-blind, placebo-controlled MRI study of anti-herpes virus therapy in MS. Neurology. 2002;58(1):31–36. doi:10.1212/WNL.58.1.31
150. Friedman JE, Zabriskie JB, Plank C, et al. A randomized clinical trial of valacyclovir in multiple sclerosis. Mult Scler. 2005;11(3):286–295. doi:10.1191/1352458505ms1185oa
151. Pender MP, Csurhes PA, Smith C, et al. Epstein-Barr virus-specific T cell therapy for progressive multiple sclerosis. JCI Insight. 2020;5(20). doi:10.1172/jci.insight.144624
152. Ioannides ZA, Csurhes PA, Douglas NL, et al. Sustained clinical improvement in a subset of patients with progressive multiple sclerosis treated with Epstein-Barr virus-specific T cell therapy. Front Neurol. 2021;12:652811. doi:10.3389/fneur.2021.652811
153. Svenningsson A, Frisell T, Burman J, et al. Safety and efficacy of rituximab versus dimethyl fumarate in patients with relapsing-remitting multiple sclerosis or clinically isolated syndrome in Sweden: a rater-blinded, phase 3- randomisex controlled trial. Lancet Neurol. 2022;21:693–703. doi:10.1016/S1474-4422(22)00209-5
154. Jons D, Persson Berg L, Sundstrom P, et al. Follow-up after infectious mononucleosis in search of serological similarities with presymptomatic multiple sclerosis. Mult Scler Relat Disord. 2021;56. doi:10.1016/j.msard.2021.103288
155. Maple PA, Ascherio A, Cohen JI, et al. The potential for EBV vaccines to prevent multiple sclerosis. Front Neurol. 2022;13:887794. doi:10.3389/fneur.2022.887794
156. Engdahl E, Gustafsson R, Huang J, et al. Increased serological response against human herpesvirus 6A is associated with risk for multiple sclerosis. Front Immunol. 2019;10:2715. doi:10.3389/fimmu.2019.02715
157. Gentili V, Turrin G, Marchetti P, et al. Synthesis and biological evaluation of novel rhodanine-based structures with antiviral activity towards HHV-6 virus. Bioorg Chem. 2022;119:105518. doi:10.1016/j.bioorg.2021.105518
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.