")
Back to Journals » International Journal of Nanomedicine » Volume 18
Treatment of Ulcerative Colitis by Cationic Liposome Delivered NLRP3 siRNA
Authors Huang J, Dai M, He M, Bu W, Cao L, Jing J, Cao R , Zhang H , Men K
Received 6 April 2023
Accepted for publication 9 August 2023
Published 16 August 2023 Volume 2023:18 Pages 4647—4662
DOI https://doi.org/10.2147/IJN.S413149
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Anderson Oliveira Lobo
Jing Huang,1,* Mengmeng Dai,1,* Mingxia He,1,* Weicheng Bu,1 Liwen Cao,1 Jing Jing,1 Run Cao,1 Hailong Zhang,1 Ke Men2
1Joint National Laboratory for Antibody Drug Engineering, The First Affiliated Hospital, Henan University, Kaifeng, Henan Province, 475004, People’s Republic of China; 2State Key Laboratory of Biotherapy and Cancer Center, West China Hospital of Sichuan University, Chengdu, Sichuan Province, 610044, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Hailong Zhang, Joint National Laboratory for Antibody Drug Engineering, The First Affiliated Hospital, Henan University, Kaifeng, Henan Province, 475004, People’s Republic of China, Email [email protected] Ke Men, State Key Laboratory of Biotherapy and Cancer Center, West China Hospital of Sichuan University, Chengdu, Sichuan Province, 610044, People’s Republic of China, Email [email protected]
Purpose: The abnormal activation of NLRP3 inflammasome is related to the occurrence and development of ulcerative colitis (UC). However, the ideal drug and delivery system remain important factors limiting the targeting of NLRP3 inflammasome in UC therapy. Gene therapy by delivering siRNA is effective in treating various diseases. Therefore, delivering siNLRP3 using an ideal vector for UC treatment is necessary.
Materials and Methods: Nanoparticles delivering siNLRP3 were developed based on cationic liposome (CLP/siNLRP3). Their ability to inhibit NLRP3 inflammasome activation was monitored using Western blot (WB) and Enzyme-linked Immunosorbent Assay (ELISA). The ASC oligomerization in LPS-primed peritoneal macrophages (PMs) was detected by WB and immunofluorescence. Moreover, we assessed the role of CLP/siNLRP3 on dextran sodium sulfate (DSS)-induced UC by examining NLRP3 levels, pro-inflammatory cytokines expression, and disease-associated index (DAI). Flow cytometry (FCM) was used to detect the contents of macrophages and T cells. Finally, we assessed the safety of CLP/siNLRP3.
Results: The prepared CLP was spherical, with a small particle size (94 nm) and low permeability. The CLP could efficiently protect siNLRP3 from degradation and then deliver siNLRP3 into PMs, inhibiting NLRP3 inflammasome activation. Also, the CLP/siNLRP3 could inhibit the secretion of mature IL-1β and IL-18 from PMs, thereby achieving a favorable anti-inflammation effect. In vivo, CLP/siNLRP3 could effectively alleviate intestinal injury in UC mice, which was attributed to down-regulating levels of IL-1β and IL-18, inhibiting infiltration of macrophages and other immune cells, and the polarization of M1 macrophages. Finally, pathological testing of tissue sections and blood biochemical tests showed no significant toxic effects of CLP/siNLRP3.
Conclusion: We introduced a prospective approach for the efficient delivery of siRNA in vitro and in vivo with high safety and stability, which was found to have great potential in treating NLRP3-driven diseases in an RNA-silencing manner.
Keywords: cationic liposome, NLRP3, siRNA, macrophage, ulcerative colitis
Introduction
Since the 20th century, tThe incidence of ulcerative colitis (UC) has been increasing over time, negatively affecting peoples’ health.1,2 UC is a multifactorial induced inflammatory bowel disease (IBD) characterized by chronic recurrent and remitting mucosal inflammation.3,4 It originates in the rectum and then extends to the proximal segments of the colon.5,6 If not timely and adequately treated, it can lead to persistent intestinal damage and an increased risk of hospitalization, surgery, and even colon cancer.7–9 Considering no radical treatment is available, patients with UC must take life-long medications.10 Therefore, it is an urgent problem for patients and society to find ideal targets and therapeutic strategies for UC based on mechanism studies.
The pathogenesis of UC is complex. Studies have suggested that the susceptibility of UC to intestinal mucosal damage results from a disturbed innate immune response.11,12 Therefore, the modulation of the innate immune response in the intestine may be crucial for treating UC. The NOD-like receptor (NLR) family, pyrin domain-containing protein 3 (NLRP3) inflammasome is an important contributor to macrophage activation and immune response, composed of the core molecule NLRP3, the adapter apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), and the effector molecule pro-caspase-1.13–15 When sensitized to damage-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs), NLRP3 inflammasome can be activated, promoting the activation of pro-caspase-1, which in turn leads to the release of mature interleukin-1β (IL-1β) and interleukin-18 (IL-18).16,17 Numerous studies have shown that NLRP3 inflammasome is an important regulator of intestinal homeostasis, and the abnormal activation of NLRP3 inflammasome is involved in the development of UC.11,12 Elevated levels of NLRP3 have been detected in both UC patients and mouse models.18–20 Further studies have shown that genetic polymorphisms in NLPR3 genes were associated with disease progression.21,22 Meanwhile, inhibition of NLRP3 inflammasome activation or knockdown of NLRP3 expression can down-regulate pro-inflammatory cytokines production and attenuate UC induced by dextran sulfate sodium salt (DSS).20,23–26 However, no drugs currently target NLRP3 inflammasome for treating UC in the clinical setting, which may be related to the diversity of the intestinal environment and the lack of an excellent drug delivery system.
In addition to current therapeutic options, including small-molecule drugs and various biological agents, gene therapy has been increasingly important in treating UC as another potential therapeutic approach.27,28 Small interfering RNA (siRNA) has been identified as a tremendous potential silencing tool capable of regulating gene expression at the messenger RNA (mRNA) level.29–32 Currently, several siRNA drugs are in clinical trials for treating cancer, viral diseases, neurological disorders, etc.33–36 Therefore, applying siRNA agents to UC gene therapy has a broad prospect. Herein, we suggested siRNA targeting of the NLRP3 gene as an effective strategy to ameliorate the occurrence and progression of UC. So far, relatively few studies reported on using siRNA to treat UC, which may be due to the lack of an efficient system.37–40 However, considering high instability and easy degradation by nucleases, achieving tissue-targeted loading and cellular uptake of free siRNA is difficult.41,42 Therefore, there is an urgent need to develop a safe and effective siRNA delivery system targeting the NLRP3 gene.
Both viral and non-viral vector systems can deliver RNA efficiently.10,43 However, the application of viral vector systems is limited by their high immunogenicity, risk of integration into chromosomes, and poor reproducibility.44 In contrast, non-viral vector systems, such as biodegradable polymeric nanoparticles (NPs), lipid NPs, stable nucleic acid-lipid particles (SNALP), lipoplex, ginger-derived NPs, and chitosan NPs, are increasingly favored by researchers.10,43,45–48 Cationic liposomes (CLP) composed of (2,3-Dioleoyloxy-propyl)-trimethylammonium-chloride (DOTAP) and cholesterol have been reported to have lower cytotoxicity and higher delivery efficiency.36,49 It was hypothesized that the CLP could efficiently deliver siRNA to macrophages in vitro and in vivo. Therefore, in this study, we assessed the effect of CLP loaded with NLRP3 siRNA (siNLRP3) on NLRP3-related inflammatory cascades in intraperitoneally derived macrophages and DSS-induced ulcerative colitis.
Materials and Methods
Animals
Male C57BL/6N mice (6–8 weeks) were purchased from Vital River Laboratory Animal Technology Co., Ltd (Zhejiang, China). All animal experiments have been approved by the Biomedical Research Ethics Committee of Henan University (HUSOM2020-028). Moreover, all animal care and use were conducted following the principles of Laboratory Animal - Guidelines for ethical review of animal welfare (GB/T 35892-2018).
Cells
Primary peritoneal macrophages (PMs) were obtained from C57BL/6N mice intraperitoneally injected with 2 mL of 3% fluid thioglycollate medium (Merck, Germany) three days before cell collection. PMs were cultured in 3.5 mm dishes (Nunc, ThermoFisher Scientific, USA) or 6-well plates (Nunc, ThermoFisher Scientific, USA) with RPMI 1640 medium (Hyclone, Cytiva, USA) containing 10% fetal bovine serum (FBS) (Gibco, ThermoFisher Scientific, USA) and 1% penicillin-streptomycin Solution (Cytiva, USA). After 3 hours, the medium was replaced by a fresh RPMI 1640 medium (Hyclone, Cytiva, USA), after which the cells were cultured overnight. The medium was changed to opti-MEM (Gibco, ThermoFisher Scientific, USA) for 1 hour. Cells were then transfected with CLP/siRNA for 48 hours. After that, the cells were primed with ultrapure lipopolysaccharide (LPS) (AdipoGen, Switzerland) for 2 hours and subsequently stimulated with nigericin (20 μM) (AdipoGen, Switzerland) for 45 min. Finally, cell extracts and precipitated supernatants were analyzed by Western blot (WB).
Preparation of CLP NPs
Briefly, 1 mol of DOTAP (Sigma-Aldrich, Merck, Germany) and 1 mol of cholesterol (Solarbio LIFE SCIENCES, China) were dissolved in chloroform (Sigma-Aldrich, Merck, Germany), subjected to rotary evaporation for 30 min, and then a lipid membrane was formed. The lipid membrane was rehydrated with 5% glucose solution (Sigma-Aldrich, Merck, Germany) and redispersed under sonication to form CLP NPs solution.36
Characterization of CLP/siRNA NPs
The loading capacity of CLP NPs on siRNA was determined using an agarose gel retardation assay. CLP/siRNA mixtures with different mass ratios were electrophoresed on a 1% agarose gel. For a gradient set, 0.5 μg siRNA (HANBIO Technologies, China) was mixed with different amounts of CLP NPs. The gel was stained with GelRed (YEASEN, China) before solidification and visualized using a Gel imaging system (Molecular Imager Gel DocTM XR+, Bio-Rad, USA).
CCK-8 Assay
Raw264.7 (National Collection of Authenticated Cell Cultures, China) and PMs were seeded in 96-well plates (Nunc, ThermoFisher Scientific, USA) at a density of 2×104/well. The next morning, different concentrations of CLP or polyethyleneimine (PEI) (Polysciences, USA) were incubated with the cells for 48 hours, followed by incubation with 10 μL CCK-8 solution (Beyotime Biotechnology, China) for 4 hours. Next, the absorbance value at 450 nm was measured for each solution well. Meanwhile, the culture medium and unstimulated cells were set as blank and negative controls, respectively.
The Transfection of CLP/siRNA in vitro
The optimal quality ratio was verified by transfection of primary macrophages. The siRNA used for transfection was carboxyfluorescein (FAM)-labeled negative control siRNA (siRNAFAM), which was synthesized by HANBIO Technologies (China). PMs were plated overnight in 6-well plates, after which the medium was changed to opti-MEM (Gibco, ThermoFisher Scientific, USA) the following morning. Then, 0.5 μg siRNAFAM was mixed with different concentrations of CLP NPs at 1:0.5, 1:1, 1:2, 1:5 and 1:10 (siRNAFAM: CLP) mass ratios for 5 min at room temperature; CLP NPs were used as negative control. Immediately afterward, the mixture was added to the PMs culture solution. The transfection efficiency was detected by flow cytometry (FCM) (FACS Calibor, BD, USA). Subsequently, we compared the siRNA delivery efficiency of CLP, PEI, Lipofectamine 2000 (Lipo2000, Invitrogen, ThermoFisher Scientific, USA), Lipofectamine 3000 (Lipo3000, Invitrogen, ThermoFisher Scientific, USA), and RNAiMAX (Invitrogen, ThermoFisher Scientific, USA). The mass ratios were 10:1 (CLP:siRNA), 5:1 (RNAiMAX:siRNA), 3:1 (Lipo3000:siRNA), 2.5:1 (Lipo2000:siRNA), and 2:1 (PEI:siRNA), respectively. Meanwhile, transfection reagents without encapsulated siRNA served as the corresponding negative controls. The transfection efficiency was also detected by FCM.
Finally, PMs were transfected with CLP/siNLRP3 complex, and CLP NPs coated with irrelevant siRNA (CLP/NC) was used as a negative control. Meanwhile, the mass ratio of CLP to siNLRP3 was 10:1. Real-Time Fluorescence Quantitative Polymerase Chain Reaction (RT-PCR) and WB were used to detect the level of NLRP3 in PMs.
Internalization Study of the CLP/siRNA Complex
PMs were plated in 6-well plates or 24-well plates and pretreated with different inhibitors for 30 min at 37°C, including amiloride (1.5 mM, Selleckchem, USA), chlorpromazine hydrochloride (CPZ, 15 μg/mL, Selleckchem, USA), genistein (150 μM, Selleckchem, USA), and methyl-β-cyclodextrin (M-β-CD, 12mM, Sigma-Aldrich, Merck, Germany). Amiloride, chlorpromazine hydrochloride (CPZ), genistein, and methyl-β-cyclodextrin (M-β-CD) were the inhibitors of micropinocytosis, clathrin-mediated endocytosis, caveolin-mediated endocytosis, and lipid raft-mediated endocytosis, respectively. Subsequently, PMs were transfected with CLP/siRNA (10:1, w/w) for 48 hours. The transfection efficiency was simultaneously detected by FCM and immunofluorescence (IF). For FCM, PMs were digested with Trypsin-EDTA solution (Servicebio, China) at 37°C for 10 min, and then cells were collected to detect the fluorescence intensity of FAM bound to siNLRP3. In addition, PMs cell membranes were stained with Dil (10 mg/mL, Beyotime, China), and nuclei were stained with Hoechst (1 mg/mL, Solarbio LIFE SCIENCES, China). Fluorescence intensity was detected by confocal microscopy (NIKON-A1R/STORM, NIKON, Japan).
RT-PCR
After 48 hours of CLP/siRNA transfection, total RNA was extracted from PMs using an RNA extraction Kit (Fastagen, China). In parallel, total RNA from colonic tissue was extracted using Trizol (Takara, China) as previously described.50 The RNA was then reverse-transcribed into cDNA using the PrimeScript™ RT reagent kit (Takara, China). The siRNA-targeted NLRP3 (forward: GGUCUCUUCUCAAGUCUAATT and reverse: UUAGACUUGAGAAGA GACCTT) was purchased from HANBIO Technologies (China). The following PCR primers were used: mouse NLRP3 (forward: CAGCCAGAGTGGAATGACACG and reverse: TG AGGTCCACACTCTCACCT), mouse IL-1β (forward: ACTACAGGCTCCGAGATG AAC and reverse: TGGGTGTGCCGTCTTTCATTA), and mouse GAPDH (forward: ACCCAGAAGACTGTGGATGG and reverse: ACACATTGGGGGTAGGAACA) (Sangon Biotech, China). RT-PCR was performed with PowerUp™ SYBR™ Green Master Mix (Thermo Fisher Scientific, USA).
ASC Oligomerization
ASC oligomerization was performed as previously described.51 Firstly, PMs were plated overnight in 3.5 mm plates and transfected with CLP/siRNA for 48 hours the following morning. Secondly, cells were primed with LPS (200ng/mL) for 2 hours and subsequently stimulated with nigericin (20μM) for 45 min. They were subsequently washed in ice-cold phosphate buffer solution (PBS) (Servicebio, China) and then lysed with 300 μL of NP-40 lysis buffer (Beyotime Biotechnology, China) on ice for 30 min. Then, 30 μL of lysate was taken as an input sample, and the rest of the lysate was centrifuged at 400g for 10 min at 4 °C. The pellets were washed twice with ice-cold PBS and then resuspended with 500 μL of PBS. Subsequently, the resuspended pellets were incubated with 4 mM disuccinimydylsuberate (ThermoFisher Scientific, USA) for 30 min at room temperature with rotation. After centrifugation at 400 g for 10 min at 4 °C, the cross-linked pellets were collected and resuspended in 1 × sodium dodecyl sulfate (SDS) sample buffer. Finally, the samples were boiled and then subjected to WB analysis.
Immunofluorescence (IF)
PMs were transfected with CLP/siRNA as described above. After 48 hours, PMs were washed twice with cooled PBS and then fixed in paraformaldehyde (Beyotime Biotechnology, China) for 30 min at room temperature. Subsequently, PMs were blocked with 5% bovine serum albumin (BSA) (Beyotime Biotechnology, China) for 2 hours and incubated overnight at 4°C with anti-ASC antibody (Cell Signaling Technology, USA). The next day, cells were incubated with Alexa FluorTM 594-conjugated goat anti-rabbit IgG (H+L) (Invitrogen, ThermoFisher Scientific, USA) for 1 hour, followed by staining with DAPI (Solarbio LIFE SCIENCES, China) for 10 min. Finally, fluorescence intensity and density were detected by confocal fluorescence microscopy.
Macrophage content in frozen sections of colonic tissue was also detected by IF, as described in previous research.52 The primary antibody was rabbit anti-mouse F4/80 antibody (Abcam, England), and the secondary antibody was Alexa FluorTM 594-conjugated goat anti-rabbit IgG (H+L) (Invitrogen, ThermoFisher Scientific, USA).
DSS-Induced Acute UC
DSS (36–50kDa, MP Biomedicals, USA)-induced UC in mice was established according to the previous article.20,53 Mice were injected intraperitoneally with CLP/siNLRP3 or CLP/NC on day 1, day 3, and day 5, respectively. Disease activity index (DAI) was monitored and recorded daily, which included body weight, fecal morphology, and blood stool status.20 On day 8, mice were sacrificed, and peripheral blood, mesenteric lymph nodes (mLNs), and spleen were collected for FCM. Colonic tissue was collected for RT-PCR, WB, hematoxylin, and eosin (HE) (Solarbio LIFE SCIENCES, China) staining, and IF.
In addition, 4% paraformaldehyde-fixed colonic tissue was treated with 30% sucrose (Beyotime Biotechnology, China) at 4°C overnight. Subsequently, colonic tissue was embedded in optimal cutting temperature compound (OCT) (ThermoFisher Scientific, USA) and cut into slices (7 μm thick), which were then stained with HE. Histopathology scores of the tissue sections were calculated as previously described.54
FCM Analysis of Macrophages and T Cells
On day 8, the number and type of macrophages and T cells in peripheral blood, mLNs, and spleen were detected by FCM. The spleen and mLNs were ground on the 70 μm filter (Corning Incorporated-Life Sciences, USA), and the filter was washed with PBS to obtain single-cell suspensions. The erythrocytes in peripheral blood and single-cell suspensions were lysed by red blood cell lysis buffer (RCLB) (Beyotime Biotechnology, China). After washing with PBS, the cells were incubated with the corresponding antibodies, including APC-conjugated anti-CD4 antibody (BD, USA), PE-conjugated anti-CD8 antibody (BD, USA), or FITC-conjugated anti-CD3 antibody (BD, USA), BB515-conjugated anti-CD11b antibody (BD, USA), PerCP-Cyanine5.5-conjugated anti-F4/80 antibody (eBioscience, USA). Among these, anti-CD3, anti-CD4, and anti-CD8 antibodies were used to detect total T cells and T cell subclasses. Meanwhile, anti-CD11b and anti-F4/80 antibodies were used to detect total macrophages. For macrophage subtype detection, the cells were fixed and permeated by BD Cytofix/CytopermTM Fixation/Permeabilization Kit (BD, USA) for 20 min after incubation with antibodies against membrane surface antigens. Then, cells were incubated with PE-conjugated anti-iNOS antibody (eBioscience, USA) and APC-conjugated anti-CD206 antibody (eBioscience, USA) overnight at 4°C. The following morning, the fluorescence intensity in the cells was detected by FCM.
Western Blot (WB)
PMs or colonic tissues were lysed with Radio Immunoprecipitation Assay (RIPA) lysis buffer (Beyotime Biotechnology, China) at 4°C for 30 min, and then the lysates were centrifuged at 12,000 g for 10 min. Then, the quantified supernatant was mixed with the loading buffer and subsequently subjected to immunoblot analysis.55 The primary antibodies were mouse anti-mouse GAPDH antibody (Abclonal, China), rabbit anti-mouse pro-caspase-1 antibody (Abcam, England), rabbit anti-mouse pro-IL-1β antibody (Abcam, England), rabbit anti-mouse NLRP3 antibody (Abcam, England), rabbit anti-mouse cleaved-caspase-1 antibody (Cell Signaling Technology, USA), rabbit anti-mouse cleaved-IL-1β antibody (Cell Signaling Technology, USA), and rabbit anti-mouse ASC antibody (Cell Signaling Technology, USA). In addition, the corresponding secondary antibodies included HRP-conjugated goat anti-mouse IgG antibody (Abclonal, China) and HRP-conjugated goat anti-rabbit IgG antibody (Abclonal, China).
ELISA
Supernatants of cell culture or tissue lysate were used to detect the concentrations of IL-1β, IL-18, and tumor necrosis factor-α (TNF-α) according to the manufacturer’s instructions. The kits were purchased from Invitrogen (Invitrogen, ThermoFisher Scientific, USA).
Statistical Analysis
Data were presented as mean ± standard error of the mean (SEM). The experiments were conducted as technical triplicates, and three independent experiments were conducted per analysis. Statistical analysis was performed by two trailing tests or one-way analysis of variance (ANOVA) using Prism 8.0 software (GraphPad Software, La Jolla, USA). P ≤ 0.05 was considered statistically significant.
Results
Preparation and Characterization of the CLP/siRNA Complex
Using DOTAP and cholesterol, CLP cationic liposomes were constructed to deliver siRNA through electronic interaction, forming CLP/siRNA complexes (Figure 1A). The diameter of prepared CLP was 94.76 nm; Zeta potential was +52.42 mV (Figure 1B and C). Meanwhile, CLP exhibited a circular morphology in transmission electron micrographs (Figure 1D). CLP was a self-assembled cationic nanoparticle that could bind siRNA. Based on this, we assayed the ability of CLP to bind siRNA by gel retardation assay. When the mass ratio of CLP:siRNA was 10:1, almost no free siRNA was observed, suggesting that siRNA was completely adsorbed by CLP (Figure 1E). This mass ratio was used in all subsequent assays if not otherwise stated. To assess the ability of CLP nanoparticles to protect siRNA from degradation, CLP/siRNA was incubated with RNase A for different hours. As shown in Figure 1F, CLP/siRNA remained intact after 4 hours, while free siRNA was rapidly degraded after being treated with RNase A (Figure 1F). These results suggest that the prepared CLP nanoparticles might be an ideal strategy for siRNA delivery.
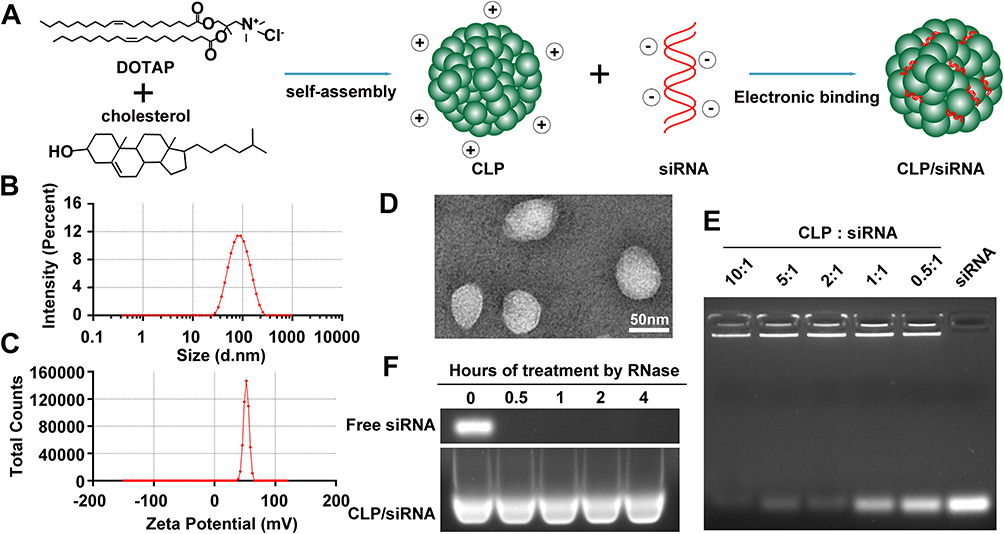 |
Figure 1 Preparation and characterization of the CLP/siRNA complex. (A) Preparation strategy. (B) Size distribution of CLP detected by particle sizer. (C) Zeta potential of CLP. (D) Transmission electron microscopy image of CLP. (E) Gel retardation assay of the CLP/siRNA complex. (F) siRNA degradation assay by agarose gel electrophoresis. |
CLP Can Efficiently Deliver siRNA in vitro Through Clathrin-Mediated Endocytosis
To evaluate the ability of CLP to deliver siRNA into PMs, we treated PMs with different transfection reagents mixed with siRNAFAM, representing siRNA conjugated to carboxyfluorescein (FAM). As shown in Figure 2A, CLP showed stronger transfection capacity than other transfection reagents, such as RNAiMAX, PEI, Lipo2000 and Lipo3000. Meanwhile, the transfection efficiency of CLP showed a positive correlation with the ratio of CLP to siRNAFAM, where the mass of siRNAFAM was fixed. When the mass ratio was 10:1, the transfection efficiency was 68.6%. When the the mass ratio was 5:1, the transfection efficiency decreased to 52.7% (Figure 2B). We further detected the capacity of siRNA delivery of RNAiMAX, PEI, Lipo2000, and Lipo3000, which were used as conventional nucleic acid delivery vectors. Data showed that CLP had greater siRNA transfection efficiency compared to other vectors in PMs (Figure 2C).
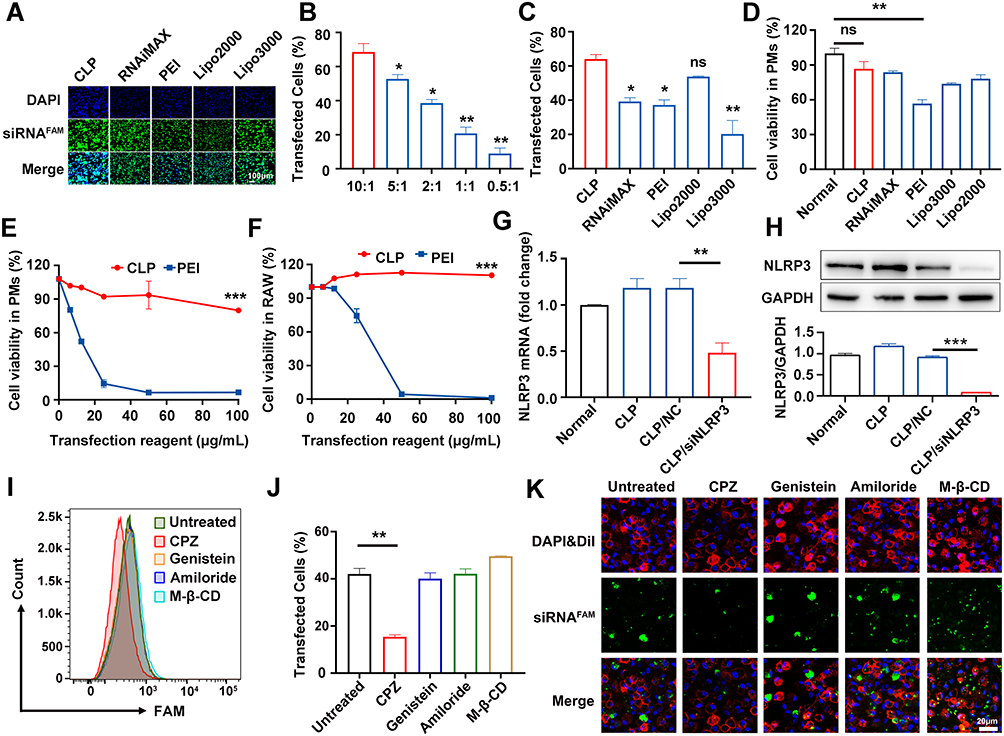 |
Figure 2 CLP could efficiently deliver siRNA in vitro through clathrin-mediated endocytosis. (A) Laser confocal scanning microscope images of PMs cells transfected different vectors coated with siRNAFAM (green channel) and stained cell nuclei with DAPI (blue channel). (B) FCM for the transfection efficiency of CLP/siRNAFAM complex for different mass ratios. (C) FCM for the transfection efficiency of different vectors delivering siRNAFAM for 48 hours. (D) CCK-8 analysis of the cell viability of PMs treated with different vectors for 48 hours. (E and F) CCK-8 analysis of the cell viability of PMs (E) and RAW264.7 (F) transfected with different concentrations of CLP and PEI. (G and H) RT-PCR (G) and WB (H) for NLRP3 levels in PMs transfected for 48 hours with CLP/siNLRP3 or CLP/NC and then stimulated with LPS. (I–K) FCM (I and J) and Laser confocal scanning microscope images (K) for PMs cells treated with cellular uptake inhibitor before transfection with CLP/ siRNAFAM. Data were mean± SEM. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. |
To assess the safety of CLP, we detected the cell viability of macrophages transfected with CLP and other transfection reagents. Our results showed that except for PEI (a “gold standard” nucleic acid delivery vector), reagents were not significantly cytotoxic. Meanwhile, CLP showed the highest cell viability among all the reagents without significant toxicity (Figure 2D). Furthermore, we compared the effects of different concentrations of CLP and PEI on cell viability. The results showed that PEI exhibited remarked cytotoxicity in PMs with an IC50 < 10 μg/mL (Figure 2E). However, CLP still showed no significant cytotoxicity, even at 100 μg/mL concentrations. We also observed similar results in RAW264.7 cells.
Furthermore, PMs were transfected with CLP/siNLRP3 for 48 hours, followed by 2 hours of stimulation with LPS. RT-PCR and WB revealed that the expression of NLRP3 was significantly decreased in PMs transfected with CLP/siNLRP3 (Figure 2G and H). These results indicated that CLP has a high transfection efficiency in macrophages, while its safety profile is excellent.
Cells take up NPs via four endocytosis pathways, including micropinocytosis, clathrin-mediated endocytosis, caveolin-mediated endocytosis, and lipid raft-mediated endocytosis. To determine which endocytosis pathways were responsible for siRNA delivery by CLP, we treated PMs with the corresponding inhibitors and then transfected them with CLP/siRNAFAM. FCM and immunofluorescence analysis showed that micropinocytosis (amiloride), caveolin-mediated endocytosis (genistein), and lipid raft-mediated endocytosis (M-β-CD) were not involved, while CPZ, an inhibitor of clathrin-mediated endocytosis could strikingly inhibit CLP-mediated siRNA delivery (Figure 2I–K). These results suggest that the CLP/siRNA complex can efficiently and safely deliver siRNA through clathrin-mediated endocytosis.
CLP/siNLRP3 Can Efficiently Inhibit the Activation of NLRP3 Inflammasome in vitro
The activation of NLRP3 inflammasome is dependent on the assembly of inflammasome complexes containing NLRP3, ASC, and pro-caspase-1. To confirm the function of CLP/siNLRP3 in the activation of NLRP3 inflammasome, PMs transfected with CLP/siNLRP3 were stimulated by LPS and nigericin. As shown in Figure 3A, the maturation of pro-caspase-1 and pro-IL-1β was strikingly suppressed. Consistent with these data, ELISA analysis showed reduced secretion of matured IL-1β and IL-18 (Figure 3B and C). However, CLP/siNLRP3 did not affect the production of TNF-α (Figure 3D).
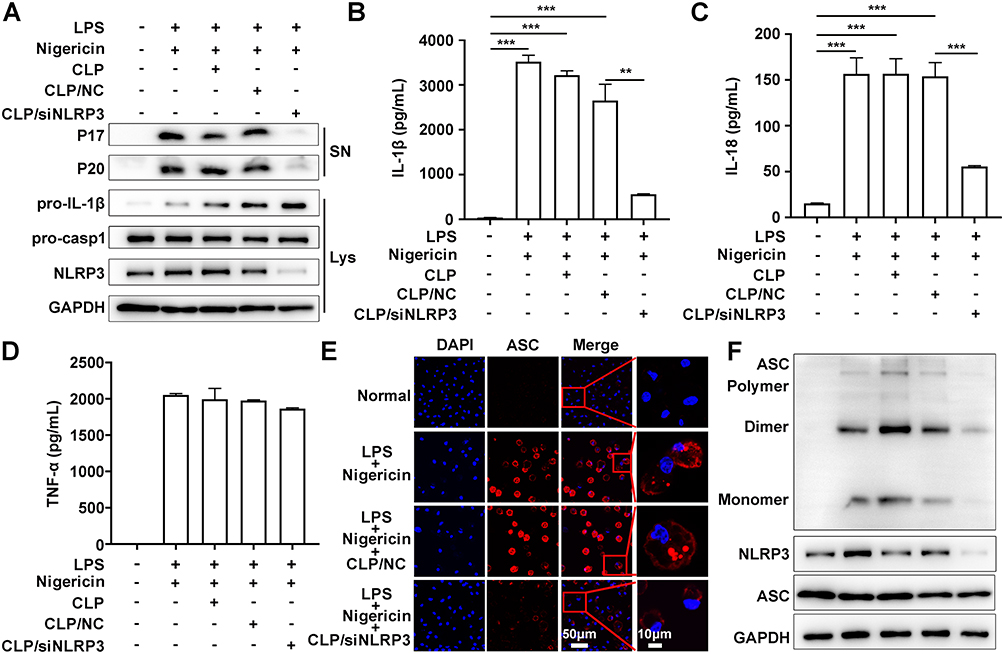 |
Figure 3 CLP/siNLRP3 could efficiently inhibit the activation of NLRP3 inflammasome in vitro. PMs were transfected with CLP/siNLRP3 for 48 hours and then stimulated with LPS and nigericin. Subsequently, the culture supernatants (SN) and lysates (Lys) were subjected to the following operations. (A) WB analysis of matured IL-1β (P17) and cleaved caspase-1 (P20) in SN, and pro-IL-1β and pro-caspase-1 in Lys. (B–D) ELISA of IL-1β (B), IL-18 (C), and TNF-α (D) in SN. (E) Immunofluorescence of ASC specks in PMs. The red channel represented ASC, and the blue channel represented DAPI. (F) WB of ASC oligomerization in Lys of PMs. Data were mean± SEM. **P ≤ 0.01, ***P ≤ 0.001. |
ASC oligomerization was vital for the assembly and subsequent activation of NLRP3 inflammasome. Immunofluorescence analysis also showed that ASC speck was remarkedly inhibited in PMs treated with CLP/siNLRP3 (Figure 3E). We also found that CLP/siNLRP3 could notably down-regulate the levels of ASC monomers, dimers, and oligomers, which meant that ASC oligomerization was disturbed (Figure 3F).
Altogether, these results indicate that CLP/siNLRP3 can effectively inhibit the activation of NLRP3 inflammasome by knocking down the expression of NLRP3.
CLP/siNLRP3 Effectively Alleviates DSS-Induced UC in Mice
As CLP/siNLRP3 could inhibit NLRP3 inflammasome activation in vitro, we evaluated the anti-inflammatory effects of CLP/siRNA in vivo. First, to assess the transfection efficiency of CLP/siRNAFAM, mice were injected intraperitoneally with the complex. We found that the presence of FAM was detectable in colonic tissue 6 hours after the injection of CLP/siRNAFAM, and the expression efficiency was higher after 24 hours (Supplementary Figure 1). Then, an ulcerative colitis model was established by feeding mice with 3% DSS, which exhibited symptoms such as weight loss, diarrhea, and rectal bleeding (Figure 4A–C). CLP/siNLRP3 could effectively improve the above symptoms in a siNLRP3 dose-dependent manner (Figure 4B and C). In addition, as the disease progressed, the colon became shorter, and CLP/siNLRP3 significantly improved the length of the colon (Figure 4D and E).
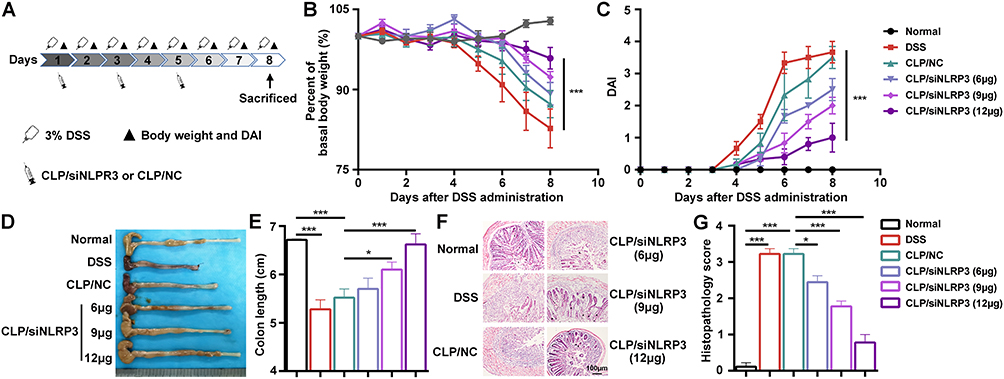 |
Figure 4 CLP/siNLRP3 could effectively alleviate DSS-induced UC in mice. (A) The schematic of the dosing regimen of CLP/siNLRP3 and DSS-induced UC. (B and C) Body weight loss (B) and DAI (C) in mice with DSS-induced UC. (D and E) Photographs of the colon (D) and its length statistics (E) in mice with DSS-induced UC. (F and G) HE staining (F) and histopathology score (G) of frozen sections of the colon in mice with DSS-induced UC (n=6 per group). Data were mean± SEM. *P ≤ 0.05, ***P ≤ 0.001. |
HE staining was used to further determine the anti-inflammatory effect of CLP/siNLRP3, ie, to assess the pathological changes in colonic tissue. As shown in Figure 4F and G, CLP/siNLRP3 could significantly blunt the pathological damages, including inflammatory cell infiltration, goblet cell disappearance, loss of crypt, mucosal injury, and necrosis, which were obviously visible in colonic tissue sections of mice with UC. These results demonstrated that CLP/siNLRP3 can efficiently deliver siNLRP3 in vivo and exert excellent anti-inflammatory effects.
CLP/siNLRP3 Remarkedly Inhibited the Activation of NLRP3 Inflammasome in vivo
To investigate whether the anti-inflammatory effects of CLP/siNLRP3 correlated with the inhibition of NLRP3 inflammasome activation, we detected the mRNA levels of IL-1β and NLRP3 with RT-PCR. The results showed that CLP/siNLRP3 could strikingly down-regulate IL-1β and NLRP3 mRNAs in vivo (Figure 5A and B). Meanwhile, the secretion of mature IL-1β was also significantly inhibited in a dose-dependent manner (Figure 5C). In addition, we found that CLP/siNLRP3 down-regulated TNF-α levels (Figure 5D). This phenomenon may be related to complex immune regulatory networks in mice. Consistent with these data, WB analysis also showed reduced protein levels of mature IL-1β and cleaved pro-caspase-1 (Figure 5E), implying the inhibitory effect of CLP/siNLRP3 on pro-caspase-1 maturation and NLRP3 inflammasome activation.
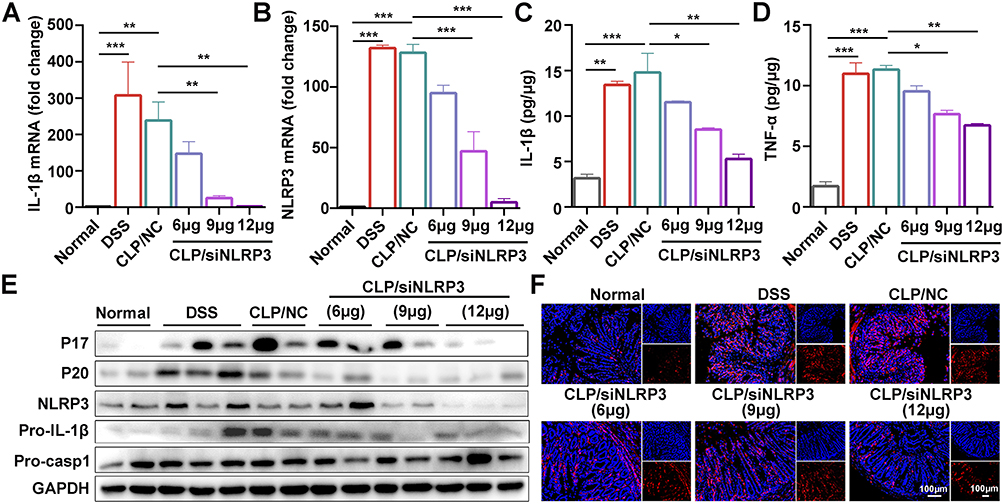 |
Figure 5 CLP/siNLRP3 alleviated DSS-induced UC by inhibiting NLRP3 inflammasome activation. (A and B) The gene levels of pro-IL-1β (A) and NLRP3 (B) in colonic tissue detected by RT-PCR. (C and D) The protein level of mature IL-1β (C) and TNF-α (D) in colonic tissue detected by ELISA. (E) WB analysis of IL-1β, caspase-1, and NLRP3 in colonic tissue. (F) Immunofluorescence detection of F4/80+ macrophages (red) in frozen sections of colonic tissue (n=6 per group), nuclei were stained with DAPI (blue). Data were mean± SEM. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. |
In addition, macrophages have a crucial role in the development of UC. Damaged tissues recruited macrophages to areas of injury via chemokines and DAMPs, further exacerbating the inflammatory response and tissue damage by secreting pro-inflammatory cytokines and chemokines. To investigate whether CLP/siNLRP3 affected macrophage chemotaxis, we detected the number of macrophages in colonic tissue by immunofluorescence. As shown in Figure 5F, CLP/siNLRP3 could effectively down-regulate the number of macrophages in colonic tissue, especially in colonic lamina propria.
These data revealed that the therapeutic effect of CLP/siNLRP3 on UC was mainly dependent on its inhibition of NLRP3 inflammasome activation and macrophage infiltration.
CLP/siNLRP3 Modulates Macrophage Polarization and Down-Regulates CD4+ T Cells Production
As CLP/siNLRP3 could inhibit macrophage infiltration into colonic tissue, we hypothesized that CLP/siNLRP3 could regulate the number of monocytes/macrophages in peripheral blood, mLNs, and spleen. We found that macrophages (CD11b+F4/80+) were increased in mice fed 3% DSS, which was effectively eliminated by CLP/siNLRP3 (Figure 6A and B).
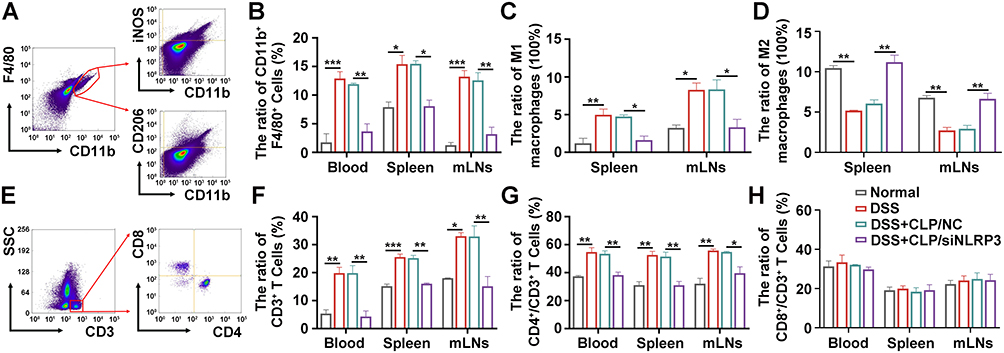 |
Figure 6 CLP/siNLRP3 modulated macrophage polarization and down-regulated CD4+ T cell production. (A) The schematic diagram of macrophage detection by FCM. (B) The percentage of macrophages in peripheral blood, spleen, and mLNs detected by FCM. (C and D) The percentage of M1 (C) and M2 (D) macrophages in the mLNs and spleen detected by FCM. (E) The schematic diagram of T cells and T cell subtypes detection by FCM. (F–H) The percentage of CD3+ (F), CD4+ (G), and CD8+ T cells (H) in peripheral blood, spleen, and mLNs detected by FCM. N=6 per group, data were mean± SEM. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. |
Activated macrophages can polarize into M1, which has pro-inflammatory characteristics, and M2, which exerts anti-inflammatory effects. To investigate the effect of CLP/siNLRP3 on macrophage polarization, we examined the number of M1 (CD11b+F4/80+iNOS+) and M2 (CD11b+F4/80+CD206+) macrophages in spleens and mLNs. Our results showed that M1 macrophages were significantly increased in the spleen and mLNs of the model mice, while CLP/siNLRP3 could down-regulate them to near-normal levels (Figure 6C). However, the changing trend in M2 macrophages was the opposite of M1 macrophages (Figure 6D).
M1 macrophages not only mediated tissue injury by directly secreting pro-inflammatory cytokines but also recruited other immune cells to damaged areas through chemokines, especially CD4+ T cells. Therefore, we detected the number of T cells in peripheral blood, spleen, and mLNs. As shown in Figure 6E and F, CLP/siNLRP3 efficiently reduced the number of CD3+ T cells, which was significantly up-regulated in mice with UC. Furthermore, FCM showed that CD8+ T cells were unaffected, while CD4+ T cell production was remarkedly suppressed (Figure 6G and H).
These results suggest that CLP/siNLRP3 can regulate macrophage polarization, promote M2 production and inhibit M1 production, thereby down-regulating CD4+ T cell content.
CLP/siNLRP3 Had No Significant Side Effects in vivo
Although CLP/siRNA had shown a good safety profile in vitro, it remained to be determined whether it was consistent in vivo. Consequently, we performed pathological analyses of mice tissues, including the heart, liver, spleen, lung, and kidney. HE staining showed that CLP/siNLRP3 treatment did not cause significant tissue damage even at the highest concentration (12μg) (Figure 7A). In addition, there were no significant changes in blood levels of LDH, BUN, ALT, and AST (Figure 7B–E). These data implied that CLP/siNLRP3 also has a favorable safety profile in vivo.
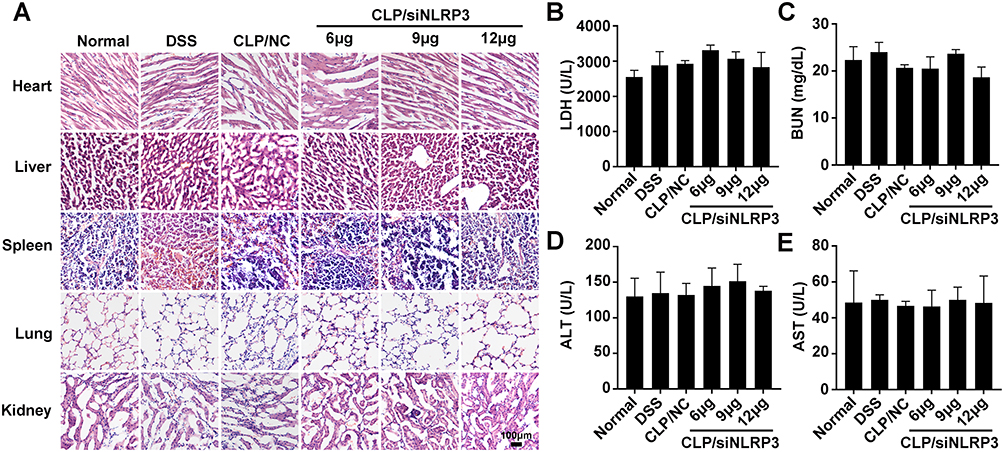 |
Figure 7 CLP/siNLRP3 had no significant side effects in vivo. (A) HE staining of frozen sections of major organs (heart, liver, spleen, lung, and kidney) in mice with DSS-induced UC. (B–E) The levels of LDH (B), BUN (C), ALT (D), and AST (E) were detected with blood biochemistry to assess cardiac (LDH), kidney (BUN) and liver (ALT and AST) function, respectively. N=6 per group, data were mean± SEM. |
Discussion
The activation of NLRP3 inflammasome has been implicated in the pathogenesis of many diseases, including UC.17,56,57 However, there are no clinically effective therapies targeting NLRP3 inflammasome. In this study, we developed a liposome delivery system conjugated with cholesterol nanoparticles to deliver siNLRP3 for the treatment of UC. These results showed that the prepared CLP system efficiently delivers siNLRP3 to primary macrophages and inhibits NLRP3 inflammasome activation and pro-inflammatory cytokines production in vitro and in vivo with a high safety profile. In addition, the CLP/siNLRP3 complex down-regulated the content of M1 macrophages and CD4+ T cells and up-regulated the content of M2 macrophages, significantly improving UC disease. Our studies suggest that CLP is an ideal siRNA delivery system and that the CLP/siNLRP3 complex is a potential candidate for treating NLRP3-related diseases.
Non-steroidal anti-inflammatory drugs (mesalazine), glucocorticoids (hydrocortisone), synthetic immunosuppressants (ciclosporin), and biological agents (TNF-α neutralizing antibodies) are currently considered the main clinical drugs to treat UC.58–63 However, the clinical use of these medications is limited due to the risks associated with their long-term use, including metabolic abnormalities, opportunistic infections and other systemic adverse effects.17 In recent years, RNA interference (RNAi)-based therapeutic approaches have attracted considerable attention in treating tumors, brain disorders, and inflammatory bowel disease.64 Due to the structural characteristics (hydrophilicity and negative charge) and instability of siRNA, one of the key issues in using siRNA to treat disease is an effective and safe delivery system.65,66 Thus, more work has been done to address this issue.64,67,68 In particular, lots of novel structural non-viral nanodelivery systems from edible plants have been synthesized for disease treatment, and some candidate systems for cancer are already at the clinical research stage, such as TKM-100201 (siRNA-SNALP complex for the treatment of Ebola virus), siRNA-EphA2-DOPC (siRNA-Neutral liposome for the treatment of advanced cancers), Atu027 (siRNA-Lipoplex complex for advanced solid tumors), and TKM-080301 (siRNA-SNALP complex for the treatment of advanced solid cancers).42,69,70 In addition, some delivery systems have also shown promising results in ulcerative colons. Aouadi et al prepared a hollow porous shell NP encapsulated with Map4k4 siRNA using β-1,3-D-glucans (GeRPs), which can significantly down-regulate pro-inflammatory cytokines and attenuate the inflammatory injury.71 Laroui et al prepared TNF-α siRNA-loaded aminated NPs (ANPs++) using cationic lipid and PEG5K-b-PLGA10K by a double emulsion method, which can effectively accumulate in the injured colons, inhibit TNF-α production, and ameliorate tissue damage.72 Zhang et al applied ginger-derived NPs (GDNPs) to deliver CD98 siRNA and protect mice against UC.45 Huang et al demonstrated galactosylated chitosan (GC)-modified poly (lactic-co-glycolic acid) (PLGA) NPs could effectively deliver TNF-α siRNA and relieved inflammation.73 However, due to the colon’s structural characteristics and intestinal flora, it is still necessary to develop ideal design strategies or vectors for successful siRNA delivery and therapeutic effect.10,66,73 Herein, CLPNPs were constructed and used to deliver siNLRP3 into macrophages, and their anti-inflammatory ability was subsequently evaluated. Our results showed that the prepared CLP NPs possess high siRNA delivery efficiency (up to 66.7%), which is superior to other “gold standard” delivery systems. Meanwhile, CLP NPs also demonstrated excellent delivery efficiency in vivo. More importantly, CLP had demonstrated an excellent safety profile both in vivo and in vitro, which was related to its composition.
Although numerous studies have shown that abnormal activation of the NLRP3 inflammasome positively correlates with the development of UC, inhibiting its activation may alleviate the severity of experimental UC.11,74,75 However, some studies found that NLRP3 inflammasome-related component knockout mice are more sensitive to DSS-induced UC and have more severe tissue damage than normal mice.76–78 In addition, Hirota reported decreased levels of IL-10 and TGF-β and enhanced macrophage and neutrophil infiltration in the colonic tissue of DSS-induced NLRP3−/− mouse UC models.77 Meanwhile, oxazolone-induced UC models using NLRP3−/− mice had lower levels of IL-1β and IL-18. Furthermore, exogenous administration of IL-18 reduced tissue damage and inflammatory symptoms in both DSS and oxazolone-induced UC models.79 However, a previous study by our group showed enhanced activation of NLRP3 inflammasome and increased expression of pro-inflammatory cytokines in UC model mice, with significant immune cell infiltration in the tissues.20 In the present study, we observed the same phenomenon in colonic tissues. Meanwhile, after the nanodelivery system knocked down NLRP3 levels, the mice had lower levels of NLRP3 and inflammatory factors, reduced levels of immune cells, and improved tissue damage. Therefore, we speculate that the bidirectional role of NLRP3 inflammasome may be related to the intestinal flora or animal strain. Although we strongly believe in a facilitative role of NLRP3 inflammasome in the development of UC, robust and reliable animal and human data (primary human macrophages from volunteers and/or UC patients) are still needed to solidify this theory. In addition, UC is predominantly a chronic disease. However, we established an acute UC model in this study. Therefore, it is still necessary to further evaluate the effect and long-term safety of CLP/siNLRP3 in the chronic model, which is one of the limitations of the present study.
As a central regulator of the innate immune system, macrophages have an important role in the development of UC. In addition, macrophages are divided into M1 pro-inflammatory phenotype and M2 anti-inflammatory phenotype.80,81 Under normal conditions, the two phenotypes of macrophages are in a state of mutual equilibrium. Once the intestinal micro-environment is disrupted, the associated DAMP or PAMP induces the polarization of M2 to M1 macrophages, which in turn promotes more pro-inflammatory cells to enter the intestinal mucosa, exacerbating tissue damage.82,83 Studies have shown that the adoptive transfer of M2 macrophages or induction of M1 into M2 macrophages could improve UC.84–86 Moreover, NLRP3 has an important role in macrophage polarization.87,88 Liu et al found that NLRP3 silencing promotes M1 macrophage polarization and up-regulates M1/M2.89 However, more studies have shown that blocking NLRP3 inflammasome or knocking out NLRP3 could promote M2 macrophage polarization.20,87,90–92 In our study, the infiltrate of macrophages and the polarization of M1 phenotype macrophages in the DSS group were up-regulated, while the CLP/siNLRP3 group down-regulated the infiltrate of macrophages and the polarization of M1 phenotype macrophages and then decreased the infiltration of immune cells and ameliorated tissue injury. Immune cells have an important role in the initial development of UC, including dendritic cells, neutrophils, T cells, and macrophages.93 Macrophages are crucial in maintaining UC homeostasis via phagocytic microorganisms and secretion of cytokines to regulate T cell subtypes.93 Our results showed that CLP/siRNA down-regulated the level of T cells and CD4+ T cells. These results demonstrated that the expression of NLRP3 was silenced by high-efficiency delivery siNLRP3 with CLP, followed by the inhibited NLRP3 inflammasome activation and the ameliorated tissue injury.
Conclusion
To the best of our knowledge, there are few studies on targeting NLRP3 inflammasome for gene therapy-related diseases. In our work, several advantages of the CLP nanoparticles, such as high delivery efficiency in vitro and in vivo, low cytotoxicity in cells and high safety in mice, and relatively specific cellular uptake, have been demonstrated. Altogether, our results showed that CLP/siNLRP3 is effective in treating UC and thus may provide a new strategy for treating NLRP3-related diseases.
Abbreviations
UC, ulcerative colitis; CLP, cationic liposome; PMs, peritoneal macrophages; DOTAP, (2,3-Dioleoyloxy-propyl)-trimethylammonium-chloride; PEI, polyethylenimine; DSS, dextran sodium sulfate; DAI, disease-associated index; FCM, flow cytometry; IBD, inflammatory bowel disease; DAMPs, damage-associated molecular patterns; NLRP3, NOD-like receptor family, pyrin domain-containing protein 3; PAMPs, pathogen-associated molecular patterns; siRNA, small interfering RNA; mRNA, messenger RNA; NPs, nanoparticles; SNALP, stable nucleic acid-lipid particles; siNLRP3, NLRP3 siRNA; FBS, fetal bovine serum; RT-PCR, real-time fluorescence quantitative polymerase chain reaction; HE, hematoxylin and eosin.
Acknowledgments
We would like to thank Medsci for providing linguistic assistance while preparing this manuscript.
Funding
This research was supported by the National Natural Science Foundation of China (82100897, 81970322), the China Postdoctoral Science Foundation (2021M701061), the Science and Technology Development Plan of Henan Province (212102310245), the Natural Science Foundation of Henan Province (202300410051), and the Medical Science and technology research in Henan Province (SBGJ202102196).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Peyrin-Biroulet L, Sandborn W, Sands BE, et al. Selecting Therapeutic Targets in Inflammatory Bowel Disease (STRIDE): determining therapeutic goals for treat-to-target. Am J Gastroenterol. 2015;110(9):1324–1338. doi:10.1038/ajg.2015.233
2. Bateman RM, Sharpe MD, Jagger JE, et al. 36th international symposium on intensive care and emergency medicine: Brussels, Belgium. 15–18 March 2016. Crit Care. 2016;20(Suppl 2):94. doi:10.1186/s13054-016-1208-6
3. Kappelman MD, Rifas-Shiman SL, Porter CQ, et al. Direct health care costs of Crohn’s disease and ulcerative colitis in US children and adults. Gastroenterology. 2008;135:1907–1913. doi:10.1053/j.gastro.2008.09.012
4. Kappelman MD, Moore KR, Allen JK, et al. Recent trends in the prevalence of Crohn’s disease and ulcerative colitis in a commercially insured US population. Dig Dis Sci. 2013;58(2):519–525. doi:10.1007/s10620-012-2371-5
5. Ng SC, Shi HY, Hamidi N, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2017;390(10114):2769–2778. doi:10.1016/S0140-6736(17)32448-0
6. Ng SC, Bernstein CN, Vatn MH, et al. Geographical variability and environmental risk factors in inflammatory bowel disease. Gut. 2013;62(4):630–649. doi:10.1136/gutjnl-2012-303661
7. Axelrad JE, Lichtiger S, Yajnik V. Inflammatory bowel disease and cancer: the role of inflammation, immunosuppression, and cancer treatment. World J Gastroenterol. 2016;22(20):4794–4801. doi:10.3748/wjg.v22.i20.4794
8. Torres J, Mehandru S, Colombel JF, et al. Crohn’s disease. Lancet. 2017;389:1741–1755. doi:10.1016/S0140-6736(16)31711-1
9. Baumgart DC, Sandborn WJ. Crohn’s disease. Lancet. 2012;380(9853):1590–1605. doi:10.1016/S0140-6736(12)60026-9
10. Alfagih IM, Aldosari BN, AlQuadeib BT, et al. An overview of nano delivery systems for targeting RNA interference-based therapy in ulcerative colitis. Curr Pharm Des. 2021;27(25):2904–2914. doi:10.2174/1381612827666210617120302
11. Lv Q, Xing Y, Liu J, et al. Lonicerin targets EZH2 to alleviate ulcerative colitis by autophagy-mediated NLRP3 inflammasome inactivation. Acta Pharm Sin B. 2021;11(9):2880–2899. doi:10.1016/j.apsb.2021.03.011
12. Shen -H-H, Yang Y-X, Meng X, et al. NLRP3: a promising therapeutic target for autoimmune diseases. Autoimmun Rev. 2018;17(7):694–702. doi:10.1016/j.autrev.2018.01.020
13. Sharma BR, Kanneganti T-D. NLRP3 inflammasome in cancer and metabolic diseases. Nat Immunol. 2021;22(5):550–559. doi:10.1038/s41590-021-00886-5
14. Kelley N, Jeltema D, Duan Y, et al. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. 2019;20(13):3328. doi:10.3390/ijms20133328
15. Sharif H, Wang L, Wang WL, et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature. 2019;570(7761):338–343. doi:10.1038/s41586-019-1295-z
16. Huang Y, Xu W, Zhou R. NLRP3 inflammasome activation and cell death. Cell Mol Immunol. 2021;18(9):2114–2127. doi:10.1038/s41423-021-00740-6
17. Zhen Y, Zhang H. NLRP3 inflammasome and inflammatory bowel disease. Front Immunol. 2019;10:276. doi:10.3389/fimmu.2019.00276
18. Coccia M, Harrison OJ, Schiering C, et al. IL-1β mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4+ Th17 cells. J Exp Med. 2012;209(9):1595–1609. doi:10.1084/jem.20111453
19. Lazaridis L-D, Pistiki A, Giamarellos-Bourboulis EJ, et al. Activation of NLRP3 inflammasome in inflammatory bowel disease: differences between Crohn’s Disease and ulcerative colitis. Dig Dis Sci. 2017;62(9):2348–2356. doi:10.1007/s10620-017-4609-8
20. Cao R, Ma Y, Li S, et al. 1,25(OH)(2) D(3) alleviates DSS-induced ulcerative colitis via inhibiting NLRP3 inflammasome activation. J Leukoc Biol. 2020;108:283–295. doi:10.1002/JLB.3MA0320-406RR
21. Zhang H-X, Wang Z-T, Lu -X-X, et al. NLRP3 gene is associated with ulcerative colitis (UC), but not Crohn’s disease (CD), in Chinese Han population. Inflamm Res. 2014;63(12):979–985. doi:10.1007/s00011-014-0774-9
22. Hanaei S, Sadr M, Rezaei A, et al. Association of NLRP3 single nucleotide polymorphisms with ulcerative colitis: a case-control study. Clin Res Hepatol Gastroenterol. 2018;42(3):269–275. doi:10.1016/j.clinre.2017.09.003
23. Bauer C, Duewell P, Mayer C, et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut. 2010;59(9):1192–1199. doi:10.1136/gut.2009.197822
24. Du X, Chen W, Wang Y, et al. Therapeutic efficacy of carboxyamidotriazole on 2,4,6-trinitrobenzene sulfonic acid-induced colitis model is associated with the inhibition of NLRP3 inflammasome and NF-kappaB activation. Int Immunopharmacol. 2017;45:16–25. doi:10.1016/j.intimp.2017.01.015
25. Zhou W, Liu X, Zhang X, et al. Oroxylin A inhibits colitis by inactivating NLRP3 inflammasome. Oncotarget. 2017;8(35):58903–58917. doi:10.18632/oncotarget.19440
26. Bauer C, Duewell P, Lehr H-A, et al. Protective and aggravating effects of Nlrp3 inflammasome activation in IBD models: influence of genetic and environmental factors. Dig Dis. 2012;30(Suppl 1):82–90. doi:10.1159/000341681
27. Burri E, Maillard MH, Schoepfer AM, et al. Treatment algorithm for mild and moderate-to-severe ulcerative colitis: an update. Digestion. 2020;101(Suppl 1):2–15. doi:10.1159/000504092
28. Vaishnaw AK, Gollob J, Gamba-Vitalo C, et al. A status report on RNAi therapeutics. Silence. 2010;1(1):14. doi:10.1186/1758-907X-1-14
29. Dong Y, Siegwart DJ, Anderson DG. Strategies, design, and chemistry in siRNA delivery systems. Adv Drug Deliv Rev. 2019;144:133–147. doi:10.1016/j.addr.2019.05.004
30. Kim B, Park J-H, Sailor MJ. Rekindling RNAi therapy: materials design requirements for in vivo siRNA delivery. Adv Mater. 2019;31(49):e1903637. doi:10.1002/adma.201903637
31. Saw PE, Song E-W. siRNA therapeutics: a clinical reality. Sci China Life Sci. 2020;63(4):485–500. doi:10.1007/s11427-018-9438-y
32. Hu B, Zhong L, Weng Y, et al. Therapeutic siRNA: state of the art. Signal Transduct Target Ther. 2020;5(1):101. doi:10.1038/s41392-020-0207-x
33. Adams D, Gonzalez-Duarte A, O’Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11–21. doi:10.1056/NEJMoa1716153
34. Lee K, Jang B, Lee Y-R, et al. The cutting-edge technologies of siRNA delivery and their application in clinical trials. Arch Pharm Res. 2018;41(9):867–874. doi:10.1007/s12272-018-1069-4
35. Hu B, Weng Y, Xia X-H, et al. Clinical advances of siRNA therapeutics. J Gene Med. 2019;21(7):e3097. doi:10.1002/jgm.3097
36. Zhang H, Men K, Pan C, et al. Treatment of colon cancer by degradable rrPPC nano-conjugates delivered STAT3 siRNA. Int J Nanomedicine. 2020;15:9875–9890. doi:10.2147/IJN.S277845
37. Song P, Yang C, Thomsen JS, et al. Lipidoid-siRNA nanoparticle-mediated IL-1β gene silencing for systemic arthritis therapy in a mouse model. Mol Ther. 2019;27(8):1424–1435. doi:10.1016/j.ymthe.2019.05.002
38. Duan W, Li H. Combination of NF-kB targeted siRNA and methotrexate in a hybrid nanocarrier towards the effective treatment in rheumatoid arthritis. J Nanobiotechnology. 2018;16(1):58. doi:10.1186/s12951-018-0382-x
39. Luo X, Wang W, Dorkin JR, et al. Poly(glycoamidoamine) brush nanomaterials for systemic siRNA delivery in vivo. Biomater Sci. 2017;5(1):38–40. doi:10.1039/c6bm00683c
40. Yang C, Merlin D. Nanoparticle-mediated drug delivery systems for the treatment of IBD: current perspectives. Int J Nanomedicine. 2019;14:8875–8889. doi:10.2147/IJN.S210315
41. Scherman D, Rousseau A, Bigey P, et al. Genetic pharmacology: progresses in siRNA delivery and therapeutic applications. Gene Ther. 2017;24(3):151–156. doi:10.1038/gt.2017.6
42. Singh A, Trivedi P, Jain NK. Advances in siRNA delivery in cancer therapy. Artif Cells Nanomed Biotechnol. 2018;46(2):274–283. doi:10.1080/21691401.2017.1307210
43. Patil P, Gao YG, Lin L, et al. The development of functional non-viral vectors for gene delivery. Int J Mol Sci. 2019;20(21):5491. doi:10.3390/ijms20215491
44. Ura T, Okuda K, Shimada M. Developments in viral vector-based vaccines. Vaccines. 2014;2(3):624–641. doi:10.3390/vaccines2030624
45. Zhang M, Viennois E, Prasad M, et al. Edible ginger-derived nanoparticles: a novel therapeutic approach for the prevention and treatment of inflammatory bowel disease and colitis-associated cancer. Biomaterials. 2016;101:321–340. doi:10.1016/j.biomaterials.2016.06.018
46. Brooks PJ, Yang NN, Austin CP. Gene therapy: the view from NCATS. Hum Gene Ther. 2016;27(1):7–13. doi:10.1089/hum.2016.29018.pjb
47. Zheng Q, Lin D, Lei L, et al. Engineered non-viral gene vectors for combination cancer therapy: a review. J Biomed Nanotechnol. 2017;13(12):1565–1580. doi:10.1166/jbn.2017.2489
48. Wu P, Chen H, Jin R, et al. Non-viral gene delivery systems for tissue repair and regeneration. J Transl Med. 2018;16(1):29. doi:10.1186/s12967-018-1402-1
49. Bhattacharya S, Bajaj A. Fluorescence and thermotropic studies of the interactions of PEI-cholesterol based PEI-chol lipopolymers with dipalmitoyl phosphatidylcholine membranes. Biochim Biophys Acta. 2008;1778(10):2225–2233. doi:10.1016/j.bbamem.2008.05.005
50. Gao C, Yu S, Zhang X, et al. Dual functional Eudragit® S100/L30D-55 and PLGA colon-targeted nanoparticles of iridoid glycoside for improved treatment of induced ulcerative colitis. Int J Nanomedicine. 2021;16:1405–1422. doi:10.2147/IJN.S291090
51. Ren G, Zhang X, Xiao Y, et al. ABRO1 promotes NLRP3 inflammasome activation through regulation of NLRP3 deubiquitination. EMBO J. 2019;38(6). doi:10.15252/embj.2018100376
52. Zhang J, Cui -W-W, Du C, et al. Knockout of DNase1l1l abrogates lens denucleation process and causes cataract in zebrafish. Biochim Biophys Acta Mol Basis Dis. 2020;1866(5):165724. doi:10.1016/j.bbadis.2020.165724
53. Zhu S, Zeng M, Feng G, et al. Platinum nanoparticles as a therapeutic agent against dextran sodium sulfate-induced colitis in mice. Int J Nanomedicine. 2019;14:8361–8378. doi:10.2147/IJN.S210655
54. Zhang H, Ma Y, Cao R, et al. Soluble uric acid induces myocardial damage through activating the NLRP3 inflammasome. J Cell Mol Med. 2020;24(15):8849–8861. doi:10.1111/jcmm.15523
55. Zhang H, Chen X, Zong B, et al. Gypenosides improve diabetic cardiomyopathy by inhibiting ROS-mediated NLRP3 inflammasome activation. J Cell Mol Med. 2018;22:4437–4448. doi:10.1111/jcmm.13743
56. Paik S, Kim JK, Silwal P, et al. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mol Immunol. 2021;18(5):1141–1160. doi:10.1038/s41423-021-00670-3
57. Schwaid AG, Spencer KB. Strategies for targeting the NLRP3 inflammasome in the clinical and preclinical space. J Med Chem. 2021;64(1):101–122. doi:10.1021/acs.jmedchem.0c01307
58. Kucharzik T, Koletzko S, Kannengiesser K, et al. Ulcerative colitis-diagnostic and therapeutic algorithms. Dtsch Arztebl Int. 2020;117(33–34):564–574. doi:10.3238/arztebl.2020.0564
59. Kato S, Ishibashi A, Kani K, et al. Optimized management of ulcerative proctitis: when and how to use mesalazine suppository. Digestion. 2018;97(1):59–63. doi:10.1159/000484224
60. Lamb CA, Kennedy NA, Raine T, et al. British society of gastroenterology consensus guidelines on the management of inflammatory bowel disease in adults. Gut. 2019;68:s1–s106. doi:10.1136/gutjnl-2019-318484
61. Parihar V, Maguire S, Shahin A, et al. Listeria meningitis complicating a patient with ulcerative colitis on concomitant infliximab and hydrocortisone. Ir J Med Sci. 2016;185(4):965–967. doi:10.1007/s11845-015-1355-9
62. Durai D, Hawthorne AB. Review article: how and when to use ciclosporin in ulcerative colitis. Aliment Pharmacol Ther. 2005;22(10):907–916. doi:10.1111/j.1365-2036.2005.02680.x
63. Bhattacharya A, Osterman MT. Biologic therapy for ulcerative colitis. Gastroenterol Clin North Am. 2020;49(4):717–729. doi:10.1016/j.gtc.2020.08.002
64. Setten RL, Rossi JJ, Han SP. The current state and future directions of RNAi-based therapeutics. Nat Rev Drug Discov. 2019;18:421–446. doi:10.1038/s41573-019-0017-4
65. Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nat Rev Drug Discov. 2009;8(2):129–138. doi:10.1038/nrd2742
66. Weng Y, Huang Q, Li C, et al. Improved nucleic acid therapy with advanced nanoscale biotechnology. Mol Ther Nucleic Acids. 2020;19:581–601. doi:10.1016/j.omtn.2019.12.004
67. Shang S, Monfregola L, Caruthers MH. Peptide-substituted oligonucleotide synthesis and non-toxic, passive cell delivery. Signal Transduct Target Ther. 2016;1(1):16019. doi:10.1038/sigtrans.2016.19
68. Oliveira C, Ribeiro AJ, Veiga F, et al. Recent advances in nucleic acid-based delivery: from bench to clinical trials in genetic diseases. J Biomed Nanotechnol. 2016;12(5):841–862. doi:10.1166/jbn.2016.2245
69. Charbe NB, Amnerkar ND, Ramesh B, et al. Small interfering RNA for cancer treatment: overcoming hurdles in delivery. Acta Pharm Sin B. 2020;10:2075–2109. doi:10.1016/j.apsb.2020.10.005
70. Titze-de-Almeida R, David C, Titze-de-Almeida SS. The race of 10 synthetic RNAi-based drugs to the pharmaceutical market. Pharm Res. 2017;34(7):1339–1363. doi:10.1007/s11095-017-2134-2
71. Aouadi M, Tesz GJ, Nicoloro SM, et al. Orally delivered siRNA targeting macrophage Map4k4 suppresses systemic inflammation. Nature. 2009;458(7242):1180–1184. doi:10.1038/nature07774
72. Laroui H, Viennois E, Xiao B, et al. Fab’-bearing siRNA TNFalpha-loaded nanoparticles targeted to colonic macrophages offer an effective therapy for experimental colitis. J Control Release. 2014;186:41–53. doi:10.1016/j.jconrel.2014.04.046
73. Huang Y, Guo J, Gui S. Orally targeted galactosylated chitosan poly(lactic-co-glycolic acid) nanoparticles loaded with TNF-a siRNA provide a novel strategy for the experimental treatment of ulcerative colitis. Eur J Pharm Sci. 2018;125:232–243. doi:10.1016/j.ejps.2018.10.009
74. Tong L, Hao H, Zhang Z, et al. Milk-derived extracellular vesicles alleviate ulcerative colitis by regulating the gut immunity and reshaping the gut microbiota. Theranostics. 2021;11(17):8570–8586. doi:10.7150/thno.62046
75. Cosin-Roger J, Simmen S, Melhem H, et al. Hypoxia ameliorates intestinal inflammation through NLRP3/mTOR downregulation and autophagy activation. Nat Commun. 2017;8:98. doi:10.1038/s41467-017-00213-3
76. Zaki MH, Boyd KL, Vogel P, et al. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010;32(3):379–391. doi:10.1016/j.immuni.2010.03.003
77. Hirota SA, Ng J, Lueng A, et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm Bowel Dis. 2011;17(6):1359–1372. doi:10.1002/ibd.21478
78. Qu S, Fan L, Qi Y, et al. Akkermansia muciniphila alleviates Dextran Sulfate Sodium (DSS)-induced acute colitis by NLRP3 activation. Microbiol Spectr. 2021;9(2):e0073021. doi:10.1128/Spectrum.00730-21
79. Itani S, Watanabe T, Nadatani Y, et al. NLRP3 inflammasome has a protective effect against oxazolone-induced colitis: a possible role in ulcerative colitis. Sci Rep. 2016;6:39075. doi:10.1038/srep39075
80. Funes SC, Rios M, Escobar-Vera J, et al. Implications of macrophage polarization in autoimmunity. Immunology. 2018;154(2):186–195. doi:10.1111/imm.12910
81. Orecchioni M, Ghosheh Y, Pramod AB, et al. Macrophage polarization: different gene signatures in M1(LPS+) vs. classically and M2(LPS–) vs. alternatively activated macrophages. Front Immunol. 2019;10:1084. doi:10.3389/fimmu.2019.01084
82. Wu MM, Wang QM, Huang BY, et al. Dioscin ameliorates murine ulcerative colitis by regulating macrophage polarization. Pharmacol Res. 2021;172:105796. doi:10.1016/j.phrs.2021.105796
83. Yan YX, Shao MJ, Qi Q, et al. Artemisinin analogue SM934 ameliorates DSS-induced mouse ulcerative colitis via suppressing neutrophils and macrophages. Acta Pharmacol Sin. 2018;39(10):1633–1644. doi:10.1038/aps.2017.185
84. Zhu Y, Li X, Chen J, et al. The pentacyclic triterpene lupeol switches M1 macrophages to M2 and ameliorates experimental inflammatory bowel disease. Int Immunopharmacol. 2016;30:74–84. doi:10.1016/j.intimp.2015.11.031
85. Wei -Y-Y, Fan Y-M, Ga Y, et al. Shaoyao decoction attenuates DSS-induced ulcerative colitis, macrophage and NLRP3 inflammasome activation through the MKP1/NF-kappaB pathway. Phytomedicine. 2021;92:153743. doi:10.1016/j.phymed.2021.153743
86. Zhuang H, Lv Q, Zhong C, et al. Tiliroside ameliorates ulcerative colitis by restoring the M1/M2 macrophage balance via the HIF-1alpha/glycolysis pathway. Front Immunol. 2021;12:649463. doi:10.3389/fimmu.2021.649463
87. Zhang J, Liu X, Wan C, et al. NLRP3 inflammasome mediates M1 macrophage polarization and IL-1beta production in inflammatory root resorption. J Clin Periodontol. 2020;47:451–460. doi:10.1111/jcpe.13258
88. Liu T, Wang L, Liang P, et al. USP19 suppresses inflammation and promotes M2-like macrophage polarization by manipulating NLRP3 function via autophagy. Cell Mol Immunol. 2021;18(10):2431–2442. doi:10.1038/s41423-020-00567-7
89. Liu Y, Gao X, Miao Y, et al. NLRP3 regulates macrophage M2 polarization through up-regulation of IL-4 in asthma. Biochem J. 2018;475(12):1995–2008. doi:10.1042/BCJ20180086
90. Ye Y, Jin T, Zhang X, et al. Meisoindigo protects against focal cerebral ischemia-reperfusion injury by inhibiting NLRP3 inflammasome activation and regulating microglia/macrophage polarization via TLR4/NF-kappaB signaling pathway. Front Cell Neurosci. 2019;13:553. doi:10.3389/fncel.2019.00553
91. Wu K, Yuan Y, Yu H, et al. The gut microbial metabolite trimethylamine N-oxide aggravates GVHD by inducing M1 macrophage polarization in mice. Blood. 2020;136(4):501–515. doi:10.1182/blood.2019003990
92. Wang Q, Zhou H, Bu Q, et al. Role of XBP1 in regulating the progression of non-alcoholic steatohepatitis. J Hepatol. 2022;77(2):312–325. doi:10.1016/j.jhep.2022.02.031
93. Neurath MF. Current and emerging therapeutic targets for IBD. Nat Rev Gastroenterol Hepatol. 2017;14(5):269–278. doi:10.1038/nrgastro.2016.208
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.