")
Back to Journals » Cancer Management and Research » Volume 15
Treatment at Relapse for Synovial Sarcoma of Children, Adolescents and Young Adults: From the State of Art to Future Clinical Perspectives
Authors Ferrari A , Berlanga P, Gatz SA, Schoot RA, van Noesel MM, Hovsepyan S, Chiaravalli S, Bergamaschi L, Minard-Colin V, Corradini N, Alaggio R, Gasparini P, Brennan B, Casanova M, Pasquali S, Orbach D
Received 23 June 2023
Accepted for publication 20 October 2023
Published 27 October 2023 Volume 2023:15 Pages 1183—1196
DOI https://doi.org/10.2147/CMAR.S404371
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Antonella D'Anneo
Andrea Ferrari,1 Pablo Berlanga,2 Susanne Andrea Gatz,3 Reineke A Schoot,4 Max M van Noesel,4,5 Shushan Hovsepyan,6 Stefano Chiaravalli,1 Luca Bergamaschi,1 Veronique Minard-Colin,2 Nadege Corradini,7 Rita Alaggio,8 Patrizia Gasparini,9 Bernadette Brennan,10 Michela Casanova,1 Sandro Pasquali,11,12 Daniel Orbach13
1Pediatric Oncology Unit, Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, Italy; 2Department of Pediatric and Adolescent Oncology, Gustave Roussy Cancer Campus, Université Paris-Saclay, Villejuif, France; 3Cancer Research UK Clinical Trials Unit, Institute of Cancer and Genomic Sciences, University of Birmingham, Birmingham, UK; 4Princess Máxima Center for Pediatric Oncology, Utrecht, the Netherlands; 5Division Imaging & Cancer, University Medical Center Utrecht, Utrecht, the Netherlands; 6Pediatric Cancer and Blood Disorders Center of Armenia, Yerevan, Armenia; 7Department of Pediatric Hematology and Oncology-IHOPe, Léon Bérard Center, Lyon, France; 8Pathology Department, Ospedale Pediatrico Bambino Gesù IRCCS, Roma, Italy; 9Tumor Genomics Unit, Department of Research, Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, Italy; 10Pediatric Oncology, Royal Manchester Children’s Hospital, Manchester, UK; 11Molecular Pharmacology Unit, Department of Applied Research and Technological Development, Molecular Pharmacology Unit, Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, Italy; 12Sarcoma Service, Department of Surgery, Department of Surgery, Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, Italy; 13SIREDO Oncology Center(Care, Innovation and Research for Children, Adolescents and Young Adults with Cancer), Institut Curie, PSL University, Paris, France
Correspondence: Andrea Ferrari, Pediatric Oncology Unit, Fondazione IRCCS Istituto Nazionale dei Tumori, Via G. Venezian, Milano, MI, 1 20133, Italy, Tel +39 02 23902588, Fax +39 02 23902648, Email [email protected]
Abstract: While the overall prognosis is generally quite satisfactory in children, adolescents and young adults with localised synovial sarcoma at first diagnosis, the outcome remains poor for patients after relapse. Conversely to the front-line standardised treatment options, patients with relapse generally have an individualised approach and to date, there is still a lack of consensus regarding standard treatment approaches. Studies on relapsed synovial sarcoma were able to identify some prognostic variables that influence post-relapse survival, in order to plan risk-adapted salvage protocols. Treatment proposals must consider previous first-line treatments, potential toxicities, and the possibility of achieving an adequate local treatment by new surgery and/or re-irradiation. Effective second-line drug therapies are urgently needed. Notably, experimental treatments such as adoptive engineered TCR-T cell immunotherapy seem promising in adults and are currently under validation also in paediatric patients.
Keywords: synovial sarcoma, relapse, second line therapy, surgery, new agents, TCR-T cell therapy, children
Introduction
Synovial sarcoma (SS) is one of the most common types of non-rhabdomyosarcoma soft tissue sarcoma (NRSTS) presenting in children, adolescents and young adults. It accounts for approximately 8 −10% of all soft tissue sarcomas with an annual incidence of 0.8 per million in children and 1.4 per million in adults.1,2 In the general population, the median age at diagnosis is around 30 years and more than 30% of cases are seen mainly in adolescents and adults under the age of 20 years3 The molecular hallmark of SS is the specific chromosomal translocation t(X;18)(p11.2;q11.2), found in more than 90% of cases.
Clinical presentation of SS is mainly a painless and gradually growing soft tissue mass with a predilection for the extremities (66%), followed by the trunk, and head and neck.4 Some patients may however have a slowly evolutive, quite indolent small peri-articular lesion. At the time of diagnosis, less than 10% of patients present with metastatic disease, most commonly in the lungs, and nodal spread is quasi-inexistent.5,6 The prognostic variables that can strongly affect survival are the completeness of tumour removal, the size and site of the disease, and the presence of metastases.7
Historically, the management of paediatric SS stems from the knowledge gained from the protocols designed for rhabdomyosarcoma. In the first decade of the new millennium, however, paediatric cooperative groups developed multimodal risk-adapted trials tailored specifically to NRSTS, ie the European pediatric Soft tissue sarcoma Study Group (EpSSG NRSTS 2005) study8 and the COG (Children’s Oncology Group) ARST0332 study.9 SS patients (aged under 21 years for EpSSG and under 30 years for COG) were included in these two clinical trials that were very similar in terms of rationale, patient stratification and treatment paradigms.10 The two studies were based on a risk-stratified approach that was developed considering previous paediatric experience but also experience coming from adults.
Histology and Molecular Biology
Histologically, SS can be subcategorised into monophasic (spindle cell), biphasic (epithelioid and spindle cells), and poorly differentiated. Regardless of the histological subtype, SS is considered a high-grade tumour with local invasiveness and the potential to metastasize.
As far as we know, SS histological and molecular features in children do not differ from their adult counterparts.11 Biphasic SS shows spindle cells with minimal amounts of cytoplasm and uniform pale nuclei in fascicles that blend with variable amounts of epithelioid cells arranged in nests, cords or glands. In monophasic SS, only a spindle cell component (spindle SS), or, very rarely, only epithelioid cells with gland formation (monophasic epithelial SS) are seen. Poorly differentiated SS is densely cellular, with a high mitotic rate, necrosis and cellular pleomorphism.11
SS shows a t(X;18)(p11.2;q11.2) translocation leading to the juxtaposition of the SYT gene (on chromosome 18) to one of the three SSX genes (SSX1, SSX2 or SSX4) on chromosome X, in a mutually exclusive manner. SYT::SSX1 is the most common fusion (two-thirds of SS),12 while SS18L1::SSX1 fusion arising from a t(X:20) and SS18::SSX4 fusions are rarely found.12 SS18::SSX2 fusion is significantly more frequent in females and is often associated with spindle cell monophasic morphology,13,14 and immunohistochemistry is a helpful diagnostic tool. The antibody detecting SS18::SSX fusion protein is highly specific and sensitive for the diagnosis of SS.15 SS expresses cytokeratins, especially cytokeratins 7 and 19, epithelial membrane antigen, vimentin, CD99 and bcl2. Cytokeratins and EMA are frequently negative in poorly differentiated SS. The SMARCB1/INI1/BAF47 protein is frequently lost or reduced due to an interaction of the SS18-SSX fusion protein with the SWI/SNF chromatin-remodelling complex leading to ejection and subsequent degradation of INI116 (see below).
In this regard, SS18 is involved in the transcriptional activation and interacts with other molecules involved in epigenetic regulation, such as Sin3A and p300. Conversely, SSXs are a group of repressors that interact with polycomb group proteins (PcG).17–19 The SS18-SXX fusion proteins interfere with the functional antagonism between the chromatin remodelling the BRG1/BRM-associated factor (BAF) complex, also called the mammalian SWI/SNF complex, and the Polycomb repressive complexes (PRCs) that ultimately lead to an epigenetic reprogramming resulting in pro-tumorigenic events.20 The deregulation of the antagonism between BAF and PRC complexes has been recognised as a relevant mechanism in many cancer types, as BAF counteracts PRC by exploiting a rapid ATP-dependent eviction that results in the formation of accessible chromatin.21
Kadoch and Crabtree performed several pioneering studies that clarified the pathobiology of SS. The SS18-SSX fusion protein competes with the wild-type SS18 resulting in a dysfunctional BAF complex that lacks the tumour suppressor properties, due to the absence of the suppressor factor BAF47 (hSNF5/SMARCB1/INI1). The BAF complex binds the SOX2 locus and impairs normal repression activity, leading to the transcriptional activation of SOX2; SOX2 is required for tumour proliferation in this sarcoma.16 BAF complexes that include SS18-SSX are characterised by more robust activation of PRC2 and PRC1 compared to wild-type BAF and this is responsible for the open chromatin and the expression of the oncogene SOX2.21 The investigations in the role of BRD9, another subunit of the BAF complex, provided an innovative therapeutic option for SS.22 BRD9 is a subunit of the non-canonical BAF (ncBAF), one of the three BAF complexes together with canonical BAF (cBAF) and PBAF. Specifically BRD9 is a synthetic lethal target for SS as its depletion reduces proliferation of SS cells.23
In addition, consistent with other tumours with loss of BAF47/INI1 expression, SS proved to be dependent on the enhancer of zeste homolog 2 (EZH2) that is the enzymatic subunit of the PRC2. EZH2 catalyses the trimethylation of histone H3 at Lysine 27, leading to the silencing of differentiation effectors and to the preservation of the ability of self-renewal in the tumour cell. SS is characterised by an overexpression of both EZH2, due to the alteration in the dynamic balance of the BAF-PRC interaction generated by the SS18-SSX fusion proteins, and the consequently increased production of the H3K27me3 motif and repression of tumour suppressors16,24 Consistently, the expression of EZH2 correlates with tumour aggressiveness and stage in SS, supporting the role of this protein for disease progression,25–27 but also as a potential therapeutic target.
A high expression of cancer testis antigens (CTAs) has been observed in SS, including Melanoma-Associated antigen 4 (MAGE-A4), New York Esophageal Squamous Cell Carcinoma 1 (NY-ESO-1), and Preferentially Expressed Antigen in Melanoma (PRAME).28–33 Whilst the mechanism remains unclear, the SS18-SSX fusion proteins are expected to mediate this epigenetic dysregulation. Importantly, the expression of CTA in SS is exceptionally high and homogeneous, a condition that is rarely observed in cancer with few exceptions, such as myxoid/round cell liposarcoma. In addition, the expression of CTA in SS is considered a dynamic rather than static process, as a higher expression at protein level can be detected in patients with advanced SS. NY-ESO-1 and MAGE-A4 represent the two most characterised CTA in SS leading to recent clinical immunotherapy trials testing therapeutic strategies against these targets.28–33
SS has been generally considered a “cold” tumour together with other sarcomas driven by a specific molecular event, such as those harbouring a translocation and characterised by low mutational burden. SS has less T-cell infiltration compared to sarcomas characterised by a complex karyotype such as undifferentiated pleomorphic sarcomas and leiomyosarcomas.34 The T-cell infiltrate in SS is also less oligoclonal compared to these tumours. However, the tumour microenvironment in SS is enriched with PD-L1 and PD-1, which has been reported particularly represented in SS,35 and CD8 positive cells particularly at the tumour edge and in the event of distant metastasis.36 Interestingly, T cells co-express PD-1 and CD8 at the tumour edge and are located closely to tumour PD-L1 positive cells within the tumour. In addition, PD-1 positive cells when detected at the tumour edge were associated with a shorter time to disease progression, suggesting a possible exhausted phenotype of CD8 positive T cells expressing PD-1.36
Preclinical findings that investigated the interplay between SS and tumour immune infiltrate applying an integrative approach that combines in-depth tumour sequencing (ie, single-cell RNA sequencing and spatial profiling) with genetic and pharmacological perturbations revealed a malignant subpopulation characterised by immune-deprived niches that were also associated with low patient survival.37 Remarkably, the SS18-SSX fusion protein controlled this malignant cell state through cytokines derived from macrophages and T cells. The combination of HDAC and CDK4/CDK6 inhibitors enhanced the immunogenicity of SS cells in tumour models and induced both T cell reactivity and SS cell death mediated by T cells.37
First Line Therapy
The overall upfront treatment strategy for paediatric patients with SS was developed from numerous retrospective studies from European and American groups. These studies have defined prognostic risk factors on which the current stratification is based, ie the extent of disease at diagnosis and therefore quality of surgical margins (defined in paediatric studies by the International Rhabdomyosarcoma Study [IRS] surgical stage), size (with a 5 cm cut-off to define the risk group), and tumour site (axial tumours have a worse prognosis than tumours occurring in the limbs).7,38–41 Based on the broad experience in adult studies with the ifosfamide–doxorubicin combination,42 this same regimen was introduced in the latest paediatric protocols. However, due to the rarity of this disease, there are still no prospective comparative first-line studies in children with SS to confirm the best chemotherapy combination.
In paediatrics, as in adults, studies have shown the importance of management in expert centres.43,44
Patients with tumours below 5 cm, microscopically completely resected at diagnosis, may be considered at low risk and can be treated with a “surgery-alone” approach,45 thus avoiding the use of adjuvant chemotherapy and radiotherapy and limiting morbidity and mortality.46,47
Chemotherapy and radiotherapy have a role in SS with more advanced presentation. SS is considered to have “intermediate” sensitivity to chemotherapy, definitely less than observed in rhabdomyosarcoma, but generally higher than reported for other adult sarcomas. The response rate has been variously reported in the 45–60% range.7,39,48
Figure 1 represents the risk-adapted treatment for localised SS at diagnosis, developed in the EpSSG NRSTS 2005 study. The first EpSSG report on SS included 138 patients <21 years with initially localised SS treated from August 2005 to August 2012: 5-year event-free survival (EFS) and overall survival (OS) were 80.7% and 90.7%, respectively. Tumour progression or relapse occurred in 27/138 patients after a median time to progression of 22.0 months (1.9–78.8 months): 2 had local progression, 10 relapsed locally, 1 experienced combined local and metastatic relapse, 2 had regional lymph node spread, and 12 had an isolated metastatic recurrence (lung metastases) as the first tumour event.10 Survival rates in the EpSSG series were better than previous paediatric studies7,38,40 and better than those reported in adult series.10,49–53
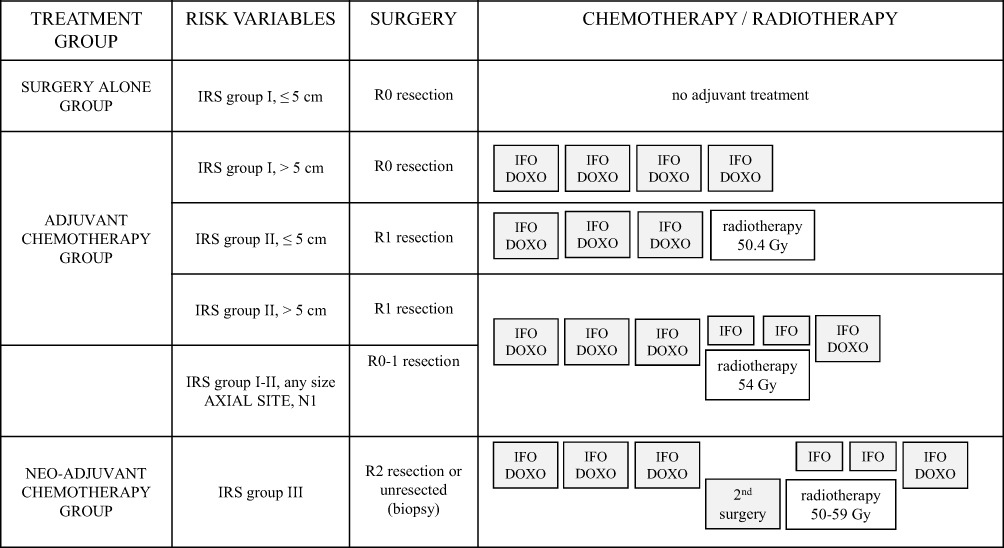 |
Figure 1 Risk-adapted treatment for synovial sarcoma in the European pediatric soft tissue sarcoma study group (EpSSG). Abbreviations: Intergroup Rhabdomyosarcoma study (IRS) grouping system: IRS I, initial complete resection (R0 resection); IRS II, grossly resected tumors with suspected microscopic residual disease (R1); IRS III, macroscopic residual disease (R2), or biopsy only (unresected disease); IFO-DOXO, chemotherapy with ifosfamide and doxorubicin; IFO, chemotherapy with ifosfamide. |
In paediatric studies, chemotherapy is mainly indicated, in neoadjuvant or adjuvant settings, for tumours larger than 5 cm, not completely resectable or metastatic, and/or located on the axial site (head and neck, chest, abdomen). The indications for adjuvant radiotherapy are also based on the quality of the margins, the size and the initial site of the tumour.
Adult oncologists recognise that SS may be quite different from other adult STS, particularly in light of its higher chemosensitivity. In day-to-day clinical practice, many adult oncologists generally recommend chemotherapy for synovial sarcoma patients, not only in cases of advance/unresected disease, but also as an adjuvant treatment after surgery in case of large, high grade deep-seated tumours.54,55
International collaborations associating paediatric and adult medical oncologists are essential for promoting age-related molecular profiling studies in SS, especially in this tumour crossing paediatric and adult ages. Biological studies have suggested that differences in the genomic instability between adult and paediatric SS might offer a biological explanation for the reported differences in outcome (ie fewer metastatic relapses in children). Complexity index in sarcoma (“CINSARC”) is a 67-gene signature related to chromosome integrity and genomic complexity that has shown high prognostic value in adult sarcoma (and in SS, in particular). This biologic signature as well as the genomic index (another measure of genomic complexity) may be potentially used to stratify patients to receive or not receive chemotherapy. Chromosomal instability was reported frequently in adult SS and rarely in paediatric cases.27,56,57
Limited data exist on the clinical behaviour of paediatric patients with SS with distant metastases at onset. The outcome is often dismal, and a standard treatment is still lacking. A recent EpSSG study on 61 patients <21 years old with metastatic NRSTS treated from 2008 to 2016 on two prospective EpSSG protocols reported a 3‐year EFS and OS of 15.4% and 34.9%, respectively. Six SS cases were included in the study.58
Relapsed Synovial Sarcoma
While the overall prognosis is generally quite satisfactory in children and adolescents with localised synovial sarcoma at diagnosis, the outcome remains poor for patients with initial metastatic disease or after relapse.
Conversely to the front-line standardised treatment options, patients with relapse generally have an individualised approach and to date, there is a lack of consensus about standard treatment approaches for relapsing patients, in adults and in children. Approximately 25–32% of patients experience recurrence with a median time to relapse of 24 months.4 International studies have reported a narrow “salvage gap”, defined as the difference between EFS and OS: this means that the chances of further treatments curing patients who progress or relapse are decidedly low.2,4,59
Three retrospective studies have been published on patients with relapsed SS after initial localised disease by national cooperative paediatric oncology groups in the last decade, aiming to investigate the pattern of relapse, the outcome, and the factors associated with survival.4,59,60 The main findings of these studies are summarised in Table 1: relapse was local in 50–73% of the cases and metastatic in 27–66%, with median time to relapse of 20–30 months. The 5-year post-relapse OS ranged from 30% to 46%.2,4,59 Type of recurrence influenced post-relapse survival in all the series, with patients with local recurrence having a better outcome than those with metastases. The likelihood of secondary remission also depended on a good response to second-line chemotherapy and/or the feasibility of complete surgical resection (at both local and distant sites). The Italian series demonstrated that patients who had tumour response to second-line chemotherapy had a statistically significant better survival rate, as well as those who were able to receive secondary surgery.2 The finding that patients who succeeded in undergoing complete surgery had a better outcome was confirmed by the French experience.4 Therefore, complete surgical resection of a local relapse is required to achieve the best outcomes. Aggressive surgical approach may be justified: understanding the potential complications of surgery and the impact on function and quality of life remains a key aspect, but since surgical resection is of critical value, mutilating surgery (eg amputation in localised limb recurrences) might be even considered in selected cases.
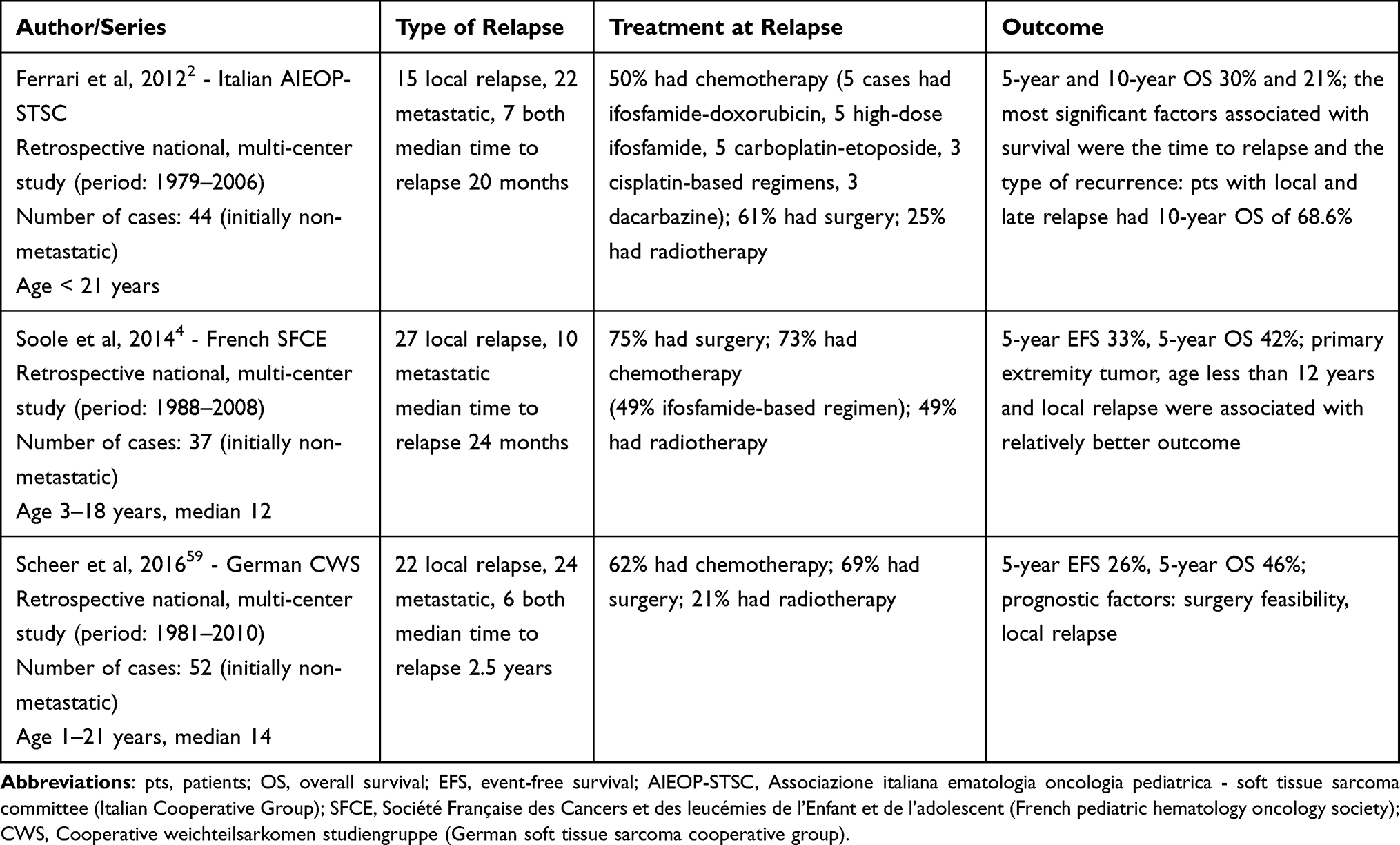 |
Table 1 Published Studies on Relapsed Synovial Sarcoma in Pediatric Age |
Surgery of metastatic lesions may also have a role,4 as suggested by adult experience: aggressive focal therapy of metastases (ie surgery, thermal ablation, stereotactic-guided radiotherapy) is recommended, when feasible, in relapsing adult sarcomas.59
The main prognostic variables can be used to estimate post-relapse survival, for the purposes of planning risk-adapted salvage protocols. In the Italian experience, for example, the time and type of relapse and the possibility to achieve secondary remission were combined to develop a prediction tool for estimating the chance of salvage and orienting decisions regarding second-line therapy, ie to identify those patients who should be treated with standard therapy, those who need aggressive surgery and those – usually the majority – whose outcome is so poor to suggest discussing experimental therapies at once. In fact, in the subset of patients without any risk factor (only 7 out of 41 cases analysed for this purpose), 5-year OS was 85.7%; when at least one factor was present (early relapse or metastatic, secondary remission not reached), 5-year OS drastically decreased to 18.7%.2
Treatment Strategy for Relapsed Synovial Sarcoma
There is no uniform standardised approach to treat young patients with relapsed SS. Multimodal treatment has to be tailored to each patient depending on their previous upfront treatment, type of recurrence, time to relapse and patients/parent’s opinion.
The paediatric sarcoma community is currently aware that any effort should be made to include relapsed SS patients in prospective clinical trials.
In general, there is a consensus that patients with exclusive local relapse should promptly receive microscopically complete tumour resection, whenever possible. Local treatments (surgery and radiotherapy) remain a key stone of the therapeutic approach. Systemic treatment should be considered, however, and probably used in all cases, in neo-adjuvant and/or adjuvant setting.
In case of metastatic relapse, systemic therapy used in neo-adjuvant setting should have a major role, with local treatment in case of concomitant local relapse and treatment on metastases (surgery/radiotherapy/thermal ablation) depending on site, number and size of metastatic lesions. In particular, surgery of resectable lung metastases without extrapulmonary disease should be recommended.
Conventional Chemotherapy
There is no consensus on the best systemic treatment for SS patients who relapse.
The ifosfamide-doxorubicin chemotherapy (currently considered the standard front-line chemotherapy in SS) should certainly be considered the best option for relapsed patients that did not previously receive this combination upfront.
Other chemotherapy options that showed some activity in patients with relapsed SS (in limited retrospective experiences) are:
a) High-dose ifosfamide. In a retrospective monocentric study of 17 patients, 4 patients achieved a partial response with high-dose ifosfamide (defined as doses ≥12–14 g/m²) given as a rechallenge, ie after a previous ifosfamide-doxorubicin regimen (when ifosfamide was given at the dose of 9 g/m²).61 High-dose ifosfamide can be administered as a prolonged continuous infusion, i.e.1 g/m²/day on 14 consecutive days every 3–4 weeks by elastomeric pump infusion. This regimen has demonstrated a favourable toxicity profile and allows outpatient administration with obvious advantages in terms of the patient’s quality of life, as well as the related economic costs.62
b) Trabectedin. A retrospective multicentre study reported 9 partial responses (plus 2 minor responses) out of 61 adult patients with metastatic SS, for an overall response rate of 15%, a tumour control rate of 50%, a median progression-free survival of 3 months, with 23% of patients free from progression at 6 months.60 Trabectedin is approved for the treatment of adult patients with advanced soft-tissue sarcoma, after failure of anthracyclines and ifosfamide, or who are unsuited to receive these agents, based on the efficacy observed in liposarcoma and leiomyosarcoma patients. It is not approved in paediatric patients and can be given only on compassionate basis or off-label.
Antitumour activity with other chemotherapeutic regimens such as gemcitabine/docetaxel seems to be much lower, with only 1 partial response out of 21 treated patients treated,63 and is therefore not recommended. Another retrospective study, assessing adult patients ≥24 years of age with refractory STS, showed that dacarbazine as a second-line treatment achieved no complete responses and a median PFS of 2 months.64
Multi-Targeted Tyrosine Kinase Inhibitors and Other Targeted Therapies
Comprehensive molecular profiling with high-throughput technologies (large panel NGS, WES and RNAseq) has failed to identify game-changing actionable gene alterations in SS, as is the case for most fusion-positive sarcomas.65,66
Multi-targeted tyrosine kinase inhibitors (MTKI) with anti-angiogenic activities have been tested in patients with relapsed SS with progression-free survival (PFS) improvement compared to placebo in adults, but limited objective responses as a single agent and gave no clear improvement on OS.67,68 Pazopanib is currently approved in the USA and Europe for the treatment of adult patients with specific subtypes of advanced soft-tissue sarcoma who have received prior chemotherapy for metastatic disease or have progressed within 12 months after adjuvant therapy. This approval is based on the results of the PALETTE Phase III study, with a median PFS of 17.9 weeks for pazopanib compared with 4.1 weeks for the placebo (p = 0.005) in patients with SS (but with no improvement in OS).67 Pazopanib is not approved in children and adolescents (below 18 years); in the absence of active clinical trials, administration may only be possible through compassionate or off-label use.
In the REGOSARC phase III trial, the median PFS with regorafenib was 5.6 months versus 1.0 with placebo, although only 1 patient had a partial response out of the 13 patients treated with regorafenib.68 Other MTKIs such as anlotinib have shown similar limited activity.49,50 Experience on MTKI in children/adolescents with SS is scarce, mainly limited to case reports.69
In order to increase efficacy, the combination of an MTKI with chemotherapy may be interesting. The COG ARST1321 trial (conducted between 2014 and 2018 by the COG in cooperation with adult oncology cooperative groups) investigated the value of the addition of pazopanib to preoperative ifosfamide-doxorubicin chemoradiotherapy in an upfront setting, in children/adults with unresected intermediate-grade/high-grade NRSTS. The study included 85 eligible patients, and SS was the most common histotype (42 cases; n = 22 chemo + pazopanib and n = 20 chemotherapy only). At a planned interim analysis, the trial showed significant improvement in pathological response rate (the primary endpoint) with the addition of pazopanib (over all patients), and enrolment was stopped.70 Outcome data, however, did not confirm the benefit of pazopanib: 3-year EFS was 52.5% (95% CI: 34.8%–70.2%) for Regimen A (ifosfamide-doxorubicin plus pazopanib) and 50.6% (32%–69.2%) for Regimen B (standard therapy) (p = 0.8677), while 3-year OS was 75.7% (59.7%–91.7%) for Regimen A and 65.4% (48.1%–82.7%) for Regimen B (p = 0.19).71 Subset numbers for SS are too small to draw any conclusions on the potential benefit of pazopanib added to chemotherapy in SS patients.
In a relapse setting, the combination of the monoclonal anti-vascular endothelial growth factor (VEGF) antibody ramucirumab with gemcitabine/docetaxel has been tested in patients with relapsed/refractory SS (NCT04145700), but results are not yet available. Previously, the combination of another monoclonal anti-VEGF antibody, bevacizumab, to standard upfront chemotherapy at diagnosis failed to improve the outcome in patients with all types of metastatic NRSTS enrolled in the EpSSG Bernie study (the study included 2 SS cases out of 49 NRSTS).72
EZH2 inhibitors, such as tazemetostat, have been tested in different tumour types with INI1 (SMARCB1/BAF47) loss. EZH2 is the core component of the polycomb complex interacting with the BAF complex and is overactive in SS. In SS, the INI loss is induced due to ejection of INI1 out of the BAF complex followed by degradation. In contrast to the genomic loss/mRNA expression loss, in other INI1 deficient tumours (ie malignant rhabdoid tumours or epithelioid sarcomas), the INI1 loss is due to the genomic loss/mRNA expression loss of SMARCB1. No objective responses were observed in the 33 SS patients included in the Phase II Tazemetostat trial with a 4-month PFS of 15%.73 These findings were presented in abstract form, and correlative studies to investigate possible predictive factors have not yet been reported. Therefore, this drug cannot be recommended outside a prospective clinical trial.
Immune Check-Point Inhibitors
Immune check-point inhibitors have shown encouraging activity in some specific sarcoma histotypes. Indeed, atezolizumab has recently been approved by the FDA for the treatment of adult and paediatric patients (aged 2 years and older) with unresectable or metastatic alveolar soft part sarcoma. However, anti-PD-1/anti-PD-L1 antibodies have limited activity in patients with relapsed SS. Only 1 partial response has been reported out of 10 patients treated with pembrolizumab in the SAR028 trial,74 and no objective response to atezolizumab with a 6-month PFS of 9.5%.75
Despite these disappointing findings, epigenetic regulation of transcriptional programmes, occurring at different levels, has been reported to influence the immune landscape and ultimately the response to immunotherapy in different solid tumours, including soft tissue sarcomas.76 A tight crosstalk between activation of the oncogenic programme and immune-deprivation in specific tumour regions has been recently described in SS. Treatment of SS cells with epigenetic drugs represses the oncogenic programme and increases tumour cell immunogenicity. Importantly, restoring functional T cells and M1 polarised macrophages suppresses the oncogenic programme, thus indicating a bi-directional synergism between immunotherapy and epigenetic drugs.37
Emerging Targeted and Cellular Therapies
BRD9 is a non-BET bromodomain protein and a subunit of the ncBAF complex that represents a synthetic lethal target for SS following evidence that its depletion reduces proliferation of SS cells both in vitro and in vivo.22 Indeed, following the generation of a BRD9 chemical degrader which bridges the BRD9 bromodomain and the E3 ubiquitin ligase,77 several inhibitors of BRD9 have been developed.78–80 Two BRD9 inhibitors – CFT8634 and FHD-609 – are currently being investigated in early phase clinical trials. CFT8634 is an oral heterobifunctional degrader, which bridges BRD9 with the cereblon (CRBN) E3 ubiquitin ligase, leading to BRD9 ubiquitination and proteasomal degradation.81 This drug is under investigation in a Phase I/II study that is enrolling patients (from 12 years of age) with locally advanced or metastatic SMARCB1-perturbed cancers, including SS and SMARCB1-negative tumours (NCT05355753). FHD-609 is similar to CFT8634 (it bridges BRD9 with CRBN E3 ubiquitin ligase) but administered intravenously.82 It is undergoing a Phase I trial in patients over 16 years with the same tumours (NCT04965753).
Adoptive immunotherapy with T-cell therapy targeting cancer testis antigens (CTAs) represents a promising new approach for relapsing SS patients.28 The reprogramming of T lymphocytes by a transgenic T-Cell Receptor (TCR) is possible by gene transfer of α and β chains of a TCR specific to a tumour antigen of interest.
In recent years, interesting results have been reported with TCR-T cells targeting cancer testis antigens (CTAs) such as NY-ESO-1, MAGE-A4, and PRAME. CTAs are overexpressed in many cancers, whereas they are not expressed in healthy tissues, except for the testis, thus limiting the risks of potential side effects when targeting these proteins.
The NY-ESO-1 specific TCR-T cells (letetresgene autoleucel; lete-cel; GSK3377794) recognise the peptides presented by HLA-A*02. NY-ESO-1, a hydrophobic cancer-testis antigen, is expressed by 70–80% of SS.29,30,83,84 Promising results were reported with genetically modified T-cells transduced with NY-ESO-1 T-cell receptor.31,85
In particular, the Phase 1 results with lete-cel have been reported.29,86,87 Patients with refractory or relapsed SS were divided into 4 cohorts based on NY-ESO-1 tumour expression and received lete-cel after lymphodepletion. Fifty patients were enrolled and 45 received lete-cel infusion. Response rates ranged from 20% to 50% depending on the cohorts, with one complete response and 14 partial responses. Responses were more pronounced for patients with high expression of NY-ESO-1 and intense lymphodepletion. Adverse events (40% of patients) were mainly haematological. Phase II is ongoing (NCT03462316) from 14 years old age.
Another approach involved a lentiviral vector (LV305) targeting dendritic cells to prime NY-ESO-1-specific T-cells: the CMB305 therapeutic vaccine induces NY-ESO-1-specific T-cell responses in SS.83,88
MAGE-A4 specific TCR-T cells (afami-cel; adaptimmune) are also being developed in patients with relapsed or refractory SS (and some other solid tumours) with two conditions: 1) HLA-A*02 positive (~ 45% of 6167 patients in the screening trial NCT0263685533 (5)), and 2) with 30% or more of the tumour cells expressing MAGE-A4 (>2+) on immunohistochemistry (67% of HLA-eligible patients with SS met study requirements for MAGE-A4 expression).33 The preliminary results of the phase II trial were presented at the ASCO meeting in 2021.32 Recently, at the 2022 CTOS Conference,84 the final analysis of SPEARHEAD-1 clinical trial was presented. It showed that the overall response rate of afami-cel was 38.6% (24.36–54.50) for 44 patients with SS. Responses were durable with a median duration of 50.3 weeks (range: 11.7–122 weeks). Toxicities include cytokine release syndrome (> Gr 3 in one patient only) and reversible hematologic toxicities, in line with previous findings indicating an acceptable safety profile. Responses were observed across all evaluated subpopulations with trends for higher response rates observed in female patients and those who had higher MAGE-A4 expression, had lower disease burden at baseline, or needed bridging therapy. Exploratory analyses (6 and 7) showed that afami-cel infiltrate tumours have an interferon-γ-driven mechanism of action and trigger adaptive immune responses. Phase II is ongoing in SS from 10 years of age (NCT04044768). Future studies are developing (some under way) combining an anti-PD-1 in MAGE-A4 positive tumours to increase the persistence of TCR-T cells or developing HLA-independent TCR-T cells. Based on these encouraging early results of engineered TCR-T cell therapies, we encourage clinicians to consider this therapeutic option, and propose early screening for HLA typing and CTA tumour expression in case of relapse/refractory SS.
Finally, SS is potentially a suitable candidate for PRAME-specific TCR-gene therapy.89–91
The SS18-SSX fusion protein has further been shown to induce synthetic lethality in the presence of ATR inhibition in various cell line in vitro models including SS cell lines, and this effect could be enhanced with the addition of PARP inhibitors. CCNE1 expression was identified as a key biomarker to relate this sensitivity in the presence of the fusion protein.92 The presence of an SS18-SSX fusion is a preselection biomarker for enrolment in the replication/transcription stress cohort of the arm N (ATR inhibitor ceralasertib and PARP inhibitor Olaparib) in the European Proof-of-Concept Therapeutic Stratification Trial of Molecular Anomalies in Relapsed or Refractory Tumors (ESMART) platform trial (NCT02813135).93
Conclusions
The treatment of young patients with relapsed SS is currently challenging. A standardised approach is still lacking, and patient outcomes remain poor, although promising new therapies seem to be on the horizon.
Figure 2 describes a possible flow diagram with different therapeutic options, as discussed in this review. In addition to considering previous therapies and the clinical prognostic factors influencing post-relapse survival to estimate the chance of salvage with second-line treatment, it is important to consider two aspects:
- To refer the patient to a highly specialised high-volume referral centre, to optimise the chance of being enrolled in available clinical trials.
- To consider the option to perform a biopsy on the relapsed tumour to obtain deep molecular profiling. This might have the potential to provide molecularly guided suggestions on target therapies (that might be administered mainly within commercial or academic early clinical trials). Worthy of note, different molecular profiling/precision medicine programs on relapsed or refractory paediatric solid malignancies have been conducted and/or are ongoing in Europe, ie MAPPYACTS,66 iTHER,94 INFORM95 and Stratified Medicine Paediatrics (SMPaeds)/NHS England.96
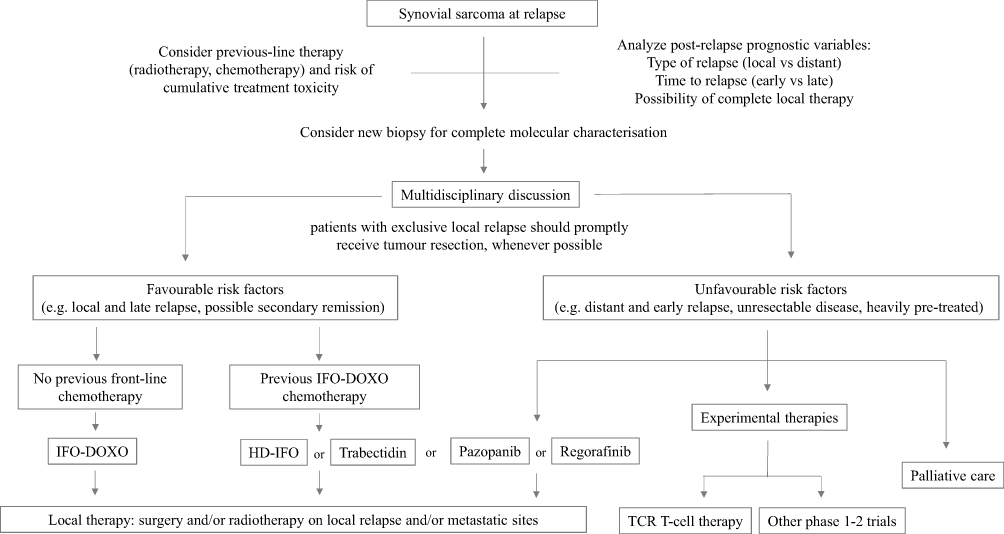 |
Figure 2 Possible flow diagram for synovial sarcoma at relapse. Abbreviations: IFO-DOXO, chemotherapy with ifosfamide and doxorubicin; HD-IFO, chemotherapy with high-dose ifosfamide; TCR-T cell, adoptive immunotherapy with T-Cell Receptor T cell. |
In patients with favourable features, chemotherapy might be the first therapeutic approach. Ifosfamide-chemotherapy is the treatment of choice for patients who did not receive it as first-line. In patients who already had ifosfamide-doxorubicin, possible options might be high-dose ifosfamide, trabectedin or pazopanib (even though the access to trabectedin or pazopanib for patients below 18 years old could be an issue).
Local treatment (surgery and/or radiotherapy) is mandatory. Also, aggressive and mutilating surgery may be considered in cases with local relapse. Aggressive focal therapy of metastases is also recommended, when feasible.
Patients with unfavourable features should be candidates to receive experimental treatment, preferably in the context of clinical trials. The referral of the patients to centres where such trials are open is highly recommended.
New comprehensive approaches are needed, however, to improve the outcome of young patients with SS who relapse. Developing dedicated prospective clinical trials, and involving patients in such studies, is the challenging goal. Tumour rarity is a major barrier for this aim (SS cases are rare, and relapsing cases are even more so), but other issues need to be addressed, requiring cultural and practical changes. Thinking towards a global-scale international cooperation that involves both the paediatric and adult sarcoma community is the first key step. This may require a new working model, new cooperative relations and a new infrastructure. The development of new targeted agents for sarcomas has had less impact on the paediatric population than on adult cases. Joint paediatric/adult clinical research trials that span both paediatric and adult populations should be the ideal goal. More generally, in fact, trials should be based on the molecular target and mechanism of action rather than age. Age eligibility criteria should only exist where there is a biological rationale or safety concerns/evidence, and, for example, the inclusion of adolescents in early adult phase I/II trials should be supported.97,98
New strategies to improve understanding of tumorigenesis and identify new targets/pathways relevant to tumour growth may be – and should be – implemented through such broader networking and collaboration.
Funding
No specific funding sources.
Disclosure
Dr Pablo Berlanga received grants for Drug supply for a trial from BAYER; an advisor for EUSA Pharma, outside the submitted work. Dr Susanne Andrea Gatz reports sponsorship of clinical trial from AstraZeneca, grants from BAYER, advisory board participation and received consultant fees from EMD Serono/MERCK KGaA, consultant fees for being reviewer on joint French Cancer Institute/AMGEN call from AMGEN, sponsorship of clinical trial from GSK, outside the submitted work. Dr Daniel Orbach reports grants from Bayer Inc., outside the submitted work. The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
1. Sultan I, Rodriguez-Galindo C, Saab R, Yasir S, Casanova M, Ferrari A. Comparing children and adults with synovial sarcoma in the Surveillance, Epidemiology, and End Results program, 1983 to 2005. Cancer. 2009;115(15):3537–3547. doi:10.1002/cncr.24424
2. Ferrari A, Salvo GL, Dall’Igna P, et al. Salvage rates and prognostic factors after relapse in children and adolescents with initially localised synovial sarcoma. Eur J Cancer. 2012;48(18):3448–3455. doi:10.1016/j.ejca.2012.06.017
3. Ferrari A, Sultan I, Huang TT, et al. Soft tissue sarcoma across the age spectrum: a population-based study from the surveillance epidemiology and end results database. Pediatr Blood Cancer. 2011;57(6):943–949. doi:10.1002/pbc.23252
4. Soole F, Maupain C, Defachelles AS, et al. Synovial sarcoma relapses in children and adolescents: prognostic factors, treatment, and outcome. Pediatr Blood Cancer. 2014;61(8):1387–1393. doi:10.1002/pbc.25001
5. Ferrari A, De Salvo GL, Oberlin O, et al. Synovial sarcoma in children and adolescents: a critical reappraisal of staging investigations in relation to the rate of metastatic involvement at diagnosis. Eur J Cancer. 2012;48(9):1370–1375. doi:10.1016/j.ejca.2012.01.013
6. Pappo AS, Fontanesi J, Luo X, et al. Synovial sarcoma in children and adolescents: the St Jude Children’s Research Hospital experience. J Clin Oncol. 1994;12(11):2360–2366. doi:10.1200/JCO.1994.12.11.2360
7. Orbach D, Dowell HM, Rey A, Bouvet N, Kelsey A, Stevens MC. Sparing strategy does not compromise prognosis in pediatric localized synovial sarcoma: experience of the International Society of pediatric oncology, malignant mesenchymal tumors (SIOP-MMT) Working Group. Pediatr Blood Cancer. 2011;57(7):1130–1136. doi:10.1002/pbc.23138
8. Ferrari A, van Noesel MM, Brennan B, et al. Paediatric non-rhabdomyosarcoma soft tissue sarcomas: the prospective NRSTS 2005 study by the European Pediatric Soft Tissue Sarcoma Study Group (EpSSG). Lancet Child Adolesc Health. 2021;5(8):546–558. doi:10.1016/S2352-4642(21)00159-0
9. Spunt SL, Million L, Chi YY, et al. A risk-based treatment strategy for non-rhabdomyosarcoma soft-tissue sarcomas in patients younger than 30 years (ARST0332): a Children’s Oncology Group prospective study. Lancet Oncol. 2020;21(1):145–161. doi:10.1016/S1470-2045(19)30672-2
10. Ferrari A, De Salvo GL, Brennan B, et al. Synovial sarcoma in children and adolescents: the European Pediatric Soft Tissue Sarcoma Study Group prospective trial (EpSSG NRSTS 2005). Ann Oncol. 2015;26(3):567–572. doi:10.1093/annonc/mdu562
11. WHO Classification of Tumours Editorial Board. Soft Tissue and Bone Tumours.
12. Storlazzi CT, Mertens F, Mandahl N, et al. A novel fusion gene, SS18L1/SSX1, in synovial sarcoma. Genes Chromosomes Cancer. 2003;37(2):195–200. doi:10.1002/gcc.10210
13. Ladanyi M, Antonescu CR, Leung DH, et al. Impact of SYT-SSX fusion type on the clinical behavior of synovial sarcoma: a multi-institutional retrospective study of 243 patients. Cancer Res. 2002;62(1):135–140.
14. Jones KB, Barrott JJ, Xie M, et al. The impact of chromosomal translocation locus and fusion oncogene coding sequence in synovial sarcomagenesis. Oncogene. 2016;35(38):5021–5032. doi:10.1038/onc.2016.38
15. Baranov E, McBride MJ, Bellizzi AM, et al. A Novel SS18-SSX Fusion-specific Antibody for the Diagnosis of Synovial Sarcoma. Am J Surgical Pathol. 2020;44(7):922–933. doi:10.1097/PAS.0000000000001447
16. Kadoch C, Crabtree GR. Reversible Disruption of mSWI/SNF (BAF) Complexes by the SS18-SSX Oncogenic Fusion in Synovial Sarcoma. Cell. 2013;153(1):71–85. doi:10.1016/j.cell.2013.02.036
17. Soulez M, Saurin AJ, Freemont PS, Knight JC. SSX and the synovial-sarcoma-specific chimaeric protein SYT-SSX co-localize with the human Polycomb group complex. Oncogene. 1999;18(17):2739–2746. doi:10.1038/sj.onc.1202613
18. Hale R, Sandakly S, Shipley J, Walters Z. Epigenetic Targets in Synovial Sarcoma: a Mini-Review. Front Oncol. 2019;9. doi:10.3389/fonc.2019.01078
19. Alfert A, Moreno N, Kerl K. The BAF complex in development and disease. Epigenetics Chromatin. 2019;12(1):19. doi:10.1186/s13072-019-0264-y
20. El Beaino M, Rassy E, Hadid B, Araujo DM, Pavlidis N, Lin PP. Synovial Sarcoma: a Complex Disease with Multifaceted Signaling and Epigenetic Landscapes. Curr Oncol Rep. 2020;22(12):124. doi:10.1007/s11912-020-00985-w
21. Kadoch C, Williams RT, Calarco JP, et al. Dynamics of BAF–Polycomb complex opposition on heterochromatin in normal and oncogenic states. Nat Genet. 2017;49(2):213–222. doi:10.1038/ng.3734
22. Brien GL, Remillard D, Shi J, et al. Targeted degradation of BRD9 reverses oncogenic gene expression in synovial sarcoma. Elife. 2018;7:56. doi:10.7554/eLife.41305
23. Michel BC, D’Avino AR, Cassel SH, et al. A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat Cell Biol. 2018;20(12):1410–1420. doi:10.1038/s41556-018-0221-1
24. Su L, Sampaio AV, Jones KB, et al. Deconstruction of the SS18-SSX Fusion Oncoprotein Complex: insights into Disease Etiology and Therapeutics. Cancer Cell. 2012;21(3):333–347. doi:10.1016/j.ccr.2012.01.010
25. Karolak M, Tracy I, Shipley J, Walters ZS. Targeting EZH2 for the treatment of soft tissue sarcomas. J Cancer Metastasis Treat. 2021. doi:10.20517/2394-4722.2021.05
26. Shen JK, Cote GM, Gao Y, et al. Targeting EZH2-mediated methylation of H3K27 inhibits proliferation and migration of Synovial Sarcoma in vitro. Sci Rep. 2016;6(1):25239. doi:10.1038/srep25239
27. Lagarde P, Przybyl J, Brulard C, et al. Chromosome Instability Accounts for Reverse Metastatic Outcomes of Pediatric and Adult Synovial Sarcomas. J Clin Oncol. 2013;31(5):608–615. doi:10.1200/JCO.2012.46.0147
28. Mitchell G, Pollack SM, Wagner MJ. Targeting cancer testis antigens in synovial sarcoma. J Immunother Cancer. 2021;9(6):e002072. doi:10.1136/jitc-2020-002072
29. D’Angelo S, Demetri G, Tine B, et al. 298 Final analysis of the phase 1 trial of NY-ESO-1–specific T-cell receptor (TCR) T-cell therapy (letetresgene autoleucel; GSK3377794) in patients with advanced synovial sarcoma (SS). In: regular and young investigator award abstracts. BMJ Publishing Group Ltd. 2020;A182–3.
30. Lai JP, Rosenberg AZ, Miettinen MM, Lee CCR. NY-ESO-1 expression in sarcomas. Oncoimmunology. 2012;1(8):1409–1410. doi:10.4161/onci.21059
31. Robbins PF, Kassim SH, Tran TLN, et al. A Pilot Trial Using Lymphocytes Genetically Engineered with an NY-ESO-1–Reactive T-cell Receptor: long-term Follow-up and Correlates with Response. Clin Cancer Res. 2015;21(5):1019–1027. doi:10.1158/1078-0432.CCR-14-2708
32. Hong DS, Van Tine BA, Biswas S, et al. Autologous T cell therapy for MAGE-A4+ solid cancers in HLA-A*02+ patients: a phase 1 trial. Nat Med. 2023;29(1):104–114. doi:10.1038/s41591-022-02128-z
33. Wang T, Navenot JM, Rafail S, et al. Abstract LB001: identifying MAGE-A4-positive tumors for SPEAR T-cell therapies in HLA-A*02-eligible patients. Cancer Res. 2022;82(12_Supplement):LB001–LB001. doi:10.1158/1538-7445.AM2022-LB001
34. Pollack SM, He Q, Yearley JH, et al. T-cell infiltration and clonality correlate with programmed cell death protein 1 and programmed death-ligand 1 expression in patients with soft tissue sarcomas. Cancer. 2017;123(17):3291–3304. doi:10.1002/cncr.30726
35. van Erp AEM, Versleijen-Jonkers YMH, Hillebrandt-Roeffen MHS, et al. Expression and clinical association of programmed cell death-1, programmed death-ligand-1 and CD8+ lymphocytes in primary sarcomas is subtype dependent. Oncotarget. 2017;8(41):71371–71384. doi:10.18632/oncotarget.19071
36. Nowicki TS, Akiyama R, Huang RR, et al. Infiltration of CD8 T Cells and Expression of PD-1 and PD-L1 in Synovial Sarcoma. Cancer Immunol Res. 2017;5(2):118–126. doi:10.1158/2326-6066.CIR-16-0148
37. Jerby-Arnon L, Neftel C, Shore ME, et al. Opposing immune and genetic mechanisms shape oncogenic programs in synovial sarcoma. Nat Med. 2021;27(2):289–300. doi:10.1038/s41591-020-01212-6
38. Ferrari A, Bisogno G, Alaggio R, et al. Synovial sarcoma of children and adolescents: the prognostic role of axial sites. Eur J Cancer. 2008;44(9):1202–1209. doi:10.1016/j.ejca.2008.03.016
39. Okcu MF, Munsell M, Treuner J, et al. Synovial Sarcoma of Childhood and Adolescence: a Multicenter, Multivariate Analysis of Outcome. J Clin Oncol. 2003;21(8):1602–1611. doi:10.1200/JCO.2003.07.008
40. Brennan B, Stevens M, Kelsey A, Stiller CA. Synovial sarcoma in childhood and adolescence: a retrospective series of 77 patients registered by the Children’s Cancer and Leukaemia Group between 1991 and 2006. Pediatr Blood Cancer. 2010;55(1):85–90. doi:10.1002/pbc.22453
41. Stegmaier S, Leuschner I, Poremba C, et al. The prognostic impact of SYT-SSX fusion type and histological grade in pediatric patients with synovial sarcoma treated according to the CWS (Cooperative Weichteilsarkom Studie) trials. Pediatr Blood Cancer. 2017;64(1):89–95. doi:10.1002/pbc.26206
42. Italiano A, Penel N, Robin YM, et al. Neo/adjuvant chemotherapy does not improve outcome in resected primary synovial sarcoma: a study of the French Sarcoma Group. Ann Oncol. 2009;20(3):425–430. doi:10.1093/annonc/mdn678
43. Collignon C, Carton M, Brisse HJ, et al. Soft tissue sarcoma in children, adolescents and young adults: outcomes according to compliance with international initial care guidelines. Eur J Surg Oncol. 2020;46(7):1277–1286. doi:10.1016/j.ejso.2019.11.518
44. de Pinieux G, Karanian M, Le Loarer F, et al. Nationwide incidence of sarcomas and connective tissue tumors of intermediate malignancy over four years using an expert pathology review network. PLoS One. 2021;16(2):e0246958. doi:10.1371/journal.pone.0246958
45. Brecht IB, Ferrari A, Int-Veen C, et al. Grossly-resected synovial sarcoma treated by the German and Italian Pediatric Soft Tissue Sarcoma Cooperative Groups: discussion on the role of adjuvant therapies. Pediatr Blood Cancer. 2006;46(1):11–17. doi:10.1002/pbc.20502
46. Ferrari A, Chi YY, De Salvo GL, et al. Surgery alone is sufficient therapy for children and adolescents with low-risk synovial sarcoma: a joint analysis from the European paediatric soft tissue sarcoma Study Group and the Children’s Oncology Group. Eur J Cancer. 2017;78:1–6. doi:10.1016/j.ejca.2017.03.003
47. Ferrari A, Brennan B, Casanova M, et al. Pediatric Non-Rhabdomyosarcoma Soft Tissue Sarcomas: standard of Care and Treatment Recommendations from the European Paediatric Soft Tissue Sarcoma Study Group (EpSSG). Cancer Manag Res. 2022;14:2885–2902. doi:10.2147/CMAR.S368381
48. Morosi C, Bergamaschi L, Livellara V, et al. Relapsing pediatric non-rhabdomyosarcoma soft tissue sarcomas: the impact of routine imaging surveillance on early detection and post-relapse survival. Eur J Cancer. 2022;175:274–281. doi:10.1016/j.ejca.2022.08.028
49. van der Graaf WTA, Orbach D, Judson IR, Ferrari A. Soft tissue sarcomas in adolescents and young adults: a comparison with their paediatric and adult counterparts. Lancet Oncol. 2017;18(3):e166–75. doi:10.1016/S1470-2045(17)30099-2
50. Ferrari A, Brecht IB, Koscielniak E, et al. The role of adjuvant chemotherapy in children and adolescents with surgically resected, high-risk adult-type soft tissue sarcomas. Pediatr Blood Cancer. 2005;45(2):128–134. doi:10.1002/pbc.20376
51. Brecht IB. Adjuvant chemotherapy for localised resectable soft tissue sarcoma in adults. Cochrane Database Sys Rev. 2000.
52. Gronchi. Short, Full-Dose Adjuvant Chemotherapy in High-Risk Adult Soft Tissue Sarcomas: A Randomized Clinical Trial from the Italian Sarcoma Group and the Spanish Sarcoma Group.
53. Ferrari A, Chiaravalli S, Casanova M, Gasparini P, Corradini N, Orbach D. Considering chemotherapy in synovial sarcoma. Expert Opin Orphan Drugs. 2015;3(10):1111–1124. doi:10.1517/21678707.2015.1076723
54. Eilber FC, Brennan MF, Eilber FR, et al. Chemotherapy Is Associated With Improved Survival in Adult Patients With Primary Extremity Synovial Sarcoma. Ann Surg. 2007;246(1):105–113. doi:10.1097/01.sla.0000262787.88639.2b
55. Canter RJ, Qin LX, Maki RG, Brennan MF, Ladanyi M, Singer S. A Synovial Sarcoma-Specific Preoperative Nomogram Supports a Survival Benefit to Ifosfamide-Based Chemotherapy and Improves Risk Stratification for Patients. Clin Cancer Res. 2008;14(24):8191–8197. doi:10.1158/1078-0432.CCR-08-0843
56. Orbach D, Mosseri V, Pissaloux D, et al. Genomic complexity in pediatric synovial sarcomas (Synobio study): the European pediatric soft tissue sarcoma group (EpSSG) experience. Cancer Med. 2018;7(4):1384–1393. doi:10.1002/cam4.1415
57. Chakiba C, Lagarde P, Pissaloux D, et al. Response to chemotherapy is not related to chromosome instability in synovial sarcoma. Ann Oncol. 2014;25(11):2267–2271. doi:10.1093/annonc/mdu362
58. Ferrari A, Orbach D, Casanova M, et al. Metastatic adult‐type non‐rhabdomyosarcoma soft tissue sarcomas in children and adolescents: a cohort study from the European paediatric Soft tissue sarcoma Study Group. Cancer. 2023;129(16):2542–2552. doi:10.1002/cncr.34814
59. Scheer M, Dantonello T, Hallmen E, et al. Synovial Sarcoma Recurrence in Children and Young Adults. Ann Surg Oncol. 2016;23(S5):618–626. doi:10.1245/s10434-016-5535-2
60. Sanfilippo R, Dileo P, Blay JY, et al. Trabectedin in advanced synovial sarcomas: a multicenter retrospective study from four European institutions and the Italian Rare Cancer Network. Anticancer Drugs. 2015;26(6):678–681. doi:10.1097/CAD.0000000000000228
61. Noujaim J, Constantinidou A, Messiou C, et al. Successful Ifosfamide Rechallenge in Soft-Tissue Sarcoma. Am J Clin Oncol. 2018;41(2):147–151. doi:10.1097/COC.0000000000000243
62. Meazza C, Casanova M, Luksch R, et al. Prolonged 14-day continuous infusion of high-dose ifosfamide with an external portable pump: feasibility and efficacy in refractory pediatric sarcoma. Pediatr Blood Cancer. 2010;55(4):617–620. doi:10.1002/pbc.22596
63. Pender A, Davis EJ, Chauhan D, et al. Poor treatment outcomes with palliative gemcitabine and docetaxel chemotherapy in advanced and metastatic synovial sarcoma. Med Oncol. 2018;35(10):131. doi:10.1007/s12032-018-1193-5
64. Zucali PA, Bertuzzi A, Parra HJS, Campagnoli E, Quagliuolo V, Santoro A. The “old drug” dacarbazine as a second/third line chemotherapy in advanced soft tissue sarcomas. Invest New Drugs. 2008;26(2):175–181. doi:10.1007/s10637-007-9086-z
65. Vlenterie M, Hillebrandt-Roeffen MHS, Flucke UE, et al. Next generation sequencing in synovial sarcoma reveals novel gene mutations. Oncotarget. 2015;6(33):34680–34690. doi:10.18632/oncotarget.5786
66. Berlanga P, Pierron G, Lacroix L, et al. The European MAPPYACTS Trial: precision Medicine Program in Pediatric and Adolescent Patients with Recurrent Malignancies. Cancer Discov. 2022;12(5):1266–1281. doi:10.1158/2159-8290.CD-21-1136
67. van der Graaf WT, Blay JY, Chawla SP, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled Phase 3 trial. Lancet. 2012;379(9829):1879–1886. doi:10.1016/S0140-6736(12)60651-5
68. Mir O, Brodowicz T, Italiano A, et al. Safety and efficacy of regorafenib in patients with advanced soft tissue sarcoma (REGOSARC): a randomised, double-blind, placebo-controlled, Phase 2 trial. Lancet Oncol. 2016;17(12):1732–1742. doi:10.1016/S1470-2045(16)30507-1
69. Casanova M, Basso E, Magni C, et al. Response to Pazopanib in Two Pediatric Patients with Pretreated Relapsing Synovial Sarcoma. Tumori J. 2017;103(1):e1–3. doi:10.5301/tj.5000548
70. Weiss AR, Chen YL, Scharschmidt TJ, et al. Pathological response in children and adults with large unresected intermediate-grade or high-grade soft tissue sarcoma receiving preoperative chemoradiotherapy with or without pazopanib (ARST1321): a multicentre, randomised, open-label, phase 2 trial. Lancet Oncol. 2020;21(8):1110–1122. doi:10.1016/S1470-2045(20)30325-9
71. Weiss AR, Chen YL, Scharschmidt T, et al. Outcomes following preoperative chemoradiation ± pazopanib in non-rhabdomyosarcoma soft tissue sarcoma (NRSTS): a report from Children’s Oncology Group (COG) and NRG Oncology. J Clin Oncol. 2022;40(16_suppl):11504. doi:10.1200/JCO.2022.40.16_suppl.11504
72. Ferrari A, Merks JHM, Chisholm JC, et al. Outcomes of metastatic non-rhabdomyosarcoma soft tissue sarcomas (NRSTS) treated within the BERNIE study: a randomised, phase II study evaluating the addition of bevacizumab to chemotherapy. Eur J Cancer. 2020;130:72–80. doi:10.1016/j.ejca.2020.01.029
73. Schoffski P, Agulnik M, Stacchiotti S, et al. Phase 2 multicenter study of the EZH2 inhibitor tazemetostat in adults with synovial sarcoma (NCT02601950). J Clin Oncol. 2017;35(15_suppl):11057. doi:10.1200/JCO.2017.35.15_suppl.11057
74. Tawbi HA, Burgess M, Bolejack V, et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017;18(11):1493–1501. doi:10.1016/S1470-2045(17)30624-1
75. Chawla SP, Van Tine BA, Pollack SM, et al. Phase II Randomized Study of CMB305 and Atezolizumab Compared With Atezolizumab Alone in Soft-Tissue Sarcomas Expressing NY-ESO-1. J Clin Oncol. 2022;40(12):1291–1300. doi:10.1200/JCO.20.03452
76. Tazzari M, Bergamaschi L, De Vita A, et al. Molecular Determinants of Soft Tissue Sarcoma Immunity: targets for Immune Intervention. Int J Mol Sci. 2021;22(14):7518. doi:10.3390/ijms22147518
77. Remillard D, Buckley DL, Paulk J, et al. Degradation of the BAF Complex Factor BRD9 by Heterobifunctional Ligands. Angew Chem Int Ed Engl. 2017;56(21):5738–5743. doi:10.1002/anie.201611281
78. Theodoulou NH, Bamborough P, Bannister AJ, et al. Discovery of I-BRD9, a Selective Cell Active Chemical Probe for Bromodomain Containing Protein 9 Inhibition. J Med Chem. 2016;59(4):1425–1439. doi:10.1021/acs.jmedchem.5b00256
79. Clark PGK, Vieira LCC, Tallant C, et al. LP99: discovery and Synthesis of the First Selective BRD7/9 Bromodomain Inhibitor. Angew Chem Weinheim Bergstr Ger. 2015;127(21):6315–6319. doi:10.1002/ange.201501394
80. Martin LJ, Koegl M, Bader G, et al. Structure-Based Design of an in Vivo Active Selective BRD9 Inhibitor. J Med Chem. 2016;59(10):4462–4475. doi:10.1021/acs.jmedchem.5b01865
81. Jackson KL, Agafonov RV, Carlson MW, et al. Abstract ND09: the discovery and characterization of CFT8634: a potent and selective degrader of BRD9 for the treatment of SMARCB1-perturbed cancers. Cancer Res. 2022;82(12_Supplement):ND09–ND09. doi:10.1158/1538-7445.AM2022-ND09
82. Collins M, Wan AB, Garbitt-Amaral V, Wu HJ, Xu C Preclinical validation of target engagement assay and investigation of mechanistic impacts of FHD-609, a clinical-stage BD9 degrader being developed for the treatment of synovial sarcoma.
83. D’Angelo SP, Melchiori L, Merchant MS, et al. Antitumor Activity Associated with Prolonged Persistence of Adoptively Transferred NY-ESO-1 c259T Cells in Synovial Sarcoma. Cancer Discov. 2018;8(8):944–957. doi:10.1158/2159-8290.CD-17-1417
84. Van Tine BA, Araujo DM, Blay JY. Final Analysis from SPEARHEAD-1 Cohort 1 of Afamitresgene Autoleucel (“Afami-cel” [Formerly ADP-A2M4]) in Advanced Synovial Sarcoma and Myxoid/Round Cell Liposarcoma. 2022.
85. Robbins PF, Morgan RA, Feldman SA, et al. Tumor Regression in Patients With Metastatic Synovial Cell Sarcoma and Melanoma Using Genetically Engineered Lymphocytes Reactive With NY-ESO-1. J Clin Oncol. 2011;29(7):917–924. doi:10.1200/JCO.2010.32.2537
86. D’Angelo SP, Noujaim JC, Thistlethwaite F, et al. IGNYTE-ESO: a master protocol to assess safety and activity of letetresgene autoleucel (lete-cel; GSK3377794) in HLA-A*02+ patients with synovial sarcoma or myxoid/round cell liposarcoma (Substudies 1 and 2). J Clin Oncol. 2021;39(15_suppl):TPS11582–TPS11582. doi:10.1200/JCO.2021.39.15_suppl.TPS11582
87. D’Angelo SP, Druta M, Van Tine BA, et al. Primary efficacy and safety of letetresgene autoleucel (lete-cel; GSK3377794) pilot study in patients with advanced and metastatic myxoid/round cell liposarcoma (MRCLS). J Clin Oncol. 2022;40(16_suppl):11500. doi:10.1200/JCO.2022.40.16_suppl.11500
88. Pollack SM, Lu H, Gnjatic S, et al. First-in-Human Treatment With a Dendritic Cell-targeting Lentiviral Vector-expressing NY-ESO-1, LV305, Induces Deep, Durable Response in Refractory Metastatic Synovial Sarcoma Patient. J Immunother. 2017;40(8):302–306. doi:10.1097/CJI.0000000000000183
89. Luk SJ, van der SDM, Hagedoorn RS, et al. PRAME and HLA Class I expression patterns make synovial sarcoma a suitable target for PRAME specific T-cell receptor gene therapy. Oncoimmunology. 2018;7(12):e1507600. doi:10.1080/2162402X.2018.1507600
90. Wermke M, Tsimberidou AM, Mohamed A, et al. 959 Safety and anti-tumor activity of TCR-engineered autologous, PRAME-directed T cells across multiple advanced solid cancers at low doses – clinical update on the ACTengine® IMA203 trial. J Immunother Cancer. 2021;9(Suppl 2):A1009–A1009. doi:10.1136/jitc-2021-SITC2021.959
91. Roszik J, Wang WL, Livingston JA, et al. Overexpressed PRAME is a potential immunotherapy target in sarcoma subtypes. Clin Sarcoma Res. 2017;7(1):11. doi:10.1186/s13569-017-0077-3
92. Jones SE, Fleuren EDG, Frankum J, et al. ATR Is a Therapeutic Target in Synovial Sarcoma. Cancer Res. 2017;77(24):7014–7026. doi:10.1158/0008-5472.CAN-17-2056
93. Gatz SA, Simón ARS, Archambaud B, et al. Abstract CT019: phase I/II study of the PARP inhibitor olaparib and ATR inhibitor ceralasertib in children with advanced malignancies: arm N of the AcSé-ESMART trial. Cancer Res. 2023;83(8_Supplement):CT019–CT019. doi:10.1158/1538-7445.AM2023-CT019
94. Langenberg KPS, Meister MT, Bakhuizen JJ, et al. Implementation of paediatric precision oncology into clinical practice: the Individualized Therapies for Children with cancer program ‘iTHER. Eur J Cancer. 2022;175:311–325. doi:10.1016/j.ejca.2022.09.001
95. Worst BC, van Tilburg CM, Balasubramanian GP, et al. Next-generation personalised medicine for high-risk paediatric cancer patients – the INFORM pilot study. Eur J Cancer. 2016;65:91–101. doi:10.1016/j.ejca.2016.06.009
96. George SL, Izquierdo E, Campbell J, et al. A tailored molecular profiling programme for children with cancer to identify clinically actionable genetic alterations. Eur J Cancer. 2019;121:224–235. doi:10.1016/j.ejca.2019.07.027
97. Ferrari A, Stark D, Peccatori FA, et al. Adolescents and young adults (AYA) with cancer: a position paper from the AYA Working Group of the European Society for Medical Oncology (ESMO) and the European Society for Paediatric Oncology (SIOPE). ESMO Open. 2021;6(2):100096. doi:10.1016/j.esmoop.2021.100096
98. Gaspar N, Marshall LV, Binner D, et al. Joint adolescent–adult early phase clinical trials to improve access to new drugs for adolescents with cancer: proposals from the multi-stakeholder platform—ACCELERATE. Ann Oncol. 2018;29(3):766–771. doi:10.1093/annonc/mdy002
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.