Back to Journals » Neuropsychiatric Disease and Treatment » Volume 12
Treadmill exercise promotes neuroprotection against cerebral ischemia–reperfusion injury via downregulation of pro-inflammatory mediators
Authors Zhang Y, Cao RY ![]() , Jia X, Li Q, Qiao L, Yan G, Yang J
, Jia X, Li Q, Qiao L, Yan G, Yang J
Received 7 September 2016
Accepted for publication 16 November 2016
Published 12 December 2016 Volume 2016:12 Pages 3161—3173
DOI https://doi.org/10.2147/NDT.S121779
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Wai Kwong Tang
Ying Zhang,1,* Richard Y Cao,2,* Xinling Jia,3,* Qing Li,1 Lei Qiao,1 Guofeng Yan,4 Jian Yang1
1Department of Rehabilitation, 2Laboratory of Immunology, Shanghai Xuhui Central Hospital, Shanghai Clinical Research Center, Chinese Academy of Sciences, 3School of Life sciences, Shanghai University, 4School of Medicine, Shanghai Jiao Tong University, Shanghai, People’s Republic of China
*These authors contributed equally to this work
Background: Stroke is one of the major causes of morbidity and mortality worldwide, which is associated with serious physical deficits that affect daily living and quality of life and produces immense public health and economic burdens. Both clinical and experimental data suggest that early physical training after ischemic brain injury may reduce the extent of motor dysfunction. However, the exact mechanisms have not been fully elucidated. The aim of this study was to investigate the effects of aerobic exercise on neuroprotection and understand the underlying mechanisms.
Materials and methods: Middle cerebral artery occlusion (MCAO) was conducted to establish a rat model of cerebral ischemia–reperfusion injury to mimic ischemic stroke. Experimental animals were divided into the following three groups: sham (n=34), MCAO (n=39), and MCAO plus treadmill exercise (n=28). The effects of aerobic exercise intervention on ischemic brain injury were evaluated using functional scoring, histological analysis, and Bio-Plex Protein Assays.
Results: Early aerobic exercise intervention was found to improve motor function, prevent death of neuronal cells, and suppress the activation of microglial cells and astrocytes. Furthermore, it was observed that aerobic exercise downregulated the expression of the cytokine interleukin-1β and the chemokine monocyte chemotactic protein-1 after transient MCAO in experimental rats.
Conclusion: This study demonstrates that treadmill exercise rehabilitation promotes neuroprotection against cerebral ischemia–reperfusion injury via the downregulation of pro-inflammatory mediators.
Keywords: rehabilitation, cytokine, chemokine, stroke, rat model
Introduction
Stroke is one of the major causes of morbidity and mortality worldwide,1 which is associated with serious physical deficits, including motor disability, cognitive dysfunction, and learning problems that affect the quality of life.2 Thus, stroke produces immense health and economic burdens globally.3 Unfortunately, it is very difficult to track and determine the exact onset of brain injuries. After stroke, neuroprotection by means of surgery to open up narrowing arteries in the brain, medication to break down the clot, and rehabilitation to recover lost function are critically important. Pharmaceutical drugs that scavenge reactive oxygen species, inhibit programmed cell death, or inhibit excitatory neurotransmitters may reduce tissue injury caused by ischemia. But the clinical trial of NXY-059, a free-radical-trapping agent, failed to show neuroprotective effects in patients with acute ischemic stroke.4 Dietary supplement of omega-3 fatty acid was believed to prevent vascular events. Unfortunately, the results of a meta-analysis of clinical trials did not confirm the neuroprotective role of omega-3 fatty acid in stroke.5 However, a growing number of studies have shown that exercise is an effective rehabilitation program for improving clinical outcomes of stroke.6,7 Beneficial effects of exercise in stroke patients are also supported by studies in animal models through attenuating pro-inflammatory reactions or inhibiting neuron apoptosis.8,9
It has been reported that leukocyte infiltration caused by ischemia–reperfusion plays an important role in the development of cerebral ischemia–reperfusion injury.10 During reperfusion, activated leukocytes release cytokines to produce an inflammatory cascade,11 resulting in the deterioration of the salvageable penumbra. Cytokine is a general name given to small nonstructural proteins, and other names include lymphokine, monokine, chemokine, and interleukins (ILs). Those promoting inflammation are called pro-inflammatory cytokines (for example, IL-1, interferon-γ [IFN-γ], and tumor necrosis factor-α [TNF-α]), which are regulated by M1 macrophages/type 1 T helper (Th1) cells under infection and/or inflammation condition, whereas other cytokines suppressing inflammation are called anti-inflammatory cytokines (for example, IL-4, IL-13, and tumor growth factor-β), which are regulated by M2 macrophages/type 2 T helper (Th2) cells during the phase of tissue repair.12,13 Postischemic inflammation with pro-inflammatory cytokine expression is an essential step in the progression of brain ischemia–reperfusion injury.14 The release of inflammatory factors such as ILs and free radicals in response to tissue damage may in turn cause the release of more free radicals. Such reactive species may also act indirectly in redox signaling to turn on apoptosis.15 Moreover, it has been reported that apoptosis is closely related to inflammation,16 which plays a crucial role in the pathological process of ischemic brain injury. Research data have shown that the occlusive brain responds to ischemic injury with an acute and prolonged inflammatory process, characterized by rapid production of pro-inflammatory mediators such as IL-1β,17 TNF-α,18 and monocyte chemotactic protein-1 (MCP-1)19 and activation of resident cells such as microglia/macrophages in the ischemic brain tissue.20 Resident microglial cells are activated within minutes of ischemia onset and produce an excess of pro-inflammatory mediators, including IL-1β and TNF-α, which exacerbate tissue damage.21,22 Moreover, it seems that the interaction between the pro-inflammatory mediators IL-1β, TNF-α, and MCP-1 and microglia/macrophages in ischemic brain tissues forms a vicious circle to exacerbate brain injury after ischemic stroke.
Recent reports have demonstrated that physical exercise affects behavior and neuroinflammation through elevation of anti-inflammatory cytokines and reduction in pro-inflammatory cytokines.23 Kang et al24 reported that treadmill exercise revoked the increase in high-fat diet-induced pro-inflammatory cytokines TNF-α and IL-1β in the obese rat. While Gomes et al25 found that aerobic exercise could increase anti-inflammatory cytokine IL-10 in the brains of aged rats. Moreover, the previous study has shown that early appropriate treadmill exercise promotes brain repair process in a mouse intracerebral hemorrhage model.26 But the mechanisms of how exercise promotes neuroprotection are unclear. Therefore, a comprehensive study to examine the effects of aerobic exercise on motor function, activation of neuronal and glial cells, and expression of signal molecules such as IL-1β, TNF-α, and MCP-1 in both the brain tissue and peripheral blood was conducted in a rat model of transient middle cerebral artery occlusion (MCAO) to elucidate how aerobic exercise could promote neuroprotection after ischemic stroke. The reason for choosing IL-1β, TNF-α, and MCP-1 was that these pro-inflammatory mediators interact with microglia/macrophages in ischemic brain tissues to form a vicious circle to exacerbate tissue damage.
Materials and methods
Ischemic brain injury model induction
Pathogen-free 8-week-old male Sprague Dawley rats weighing 250–300 g (Shanghai Laboratory Animal Center [SLAC], certificate number: 2007000555791) were housed under controlled temperature at 23°C in a 12 h light/dark cycle with free access to standard chow food (SLAC, Shanghai, People’s Republic of China) and water. The model of rat cerebral ischemia–reperfusion injury was induced by transient MCAO according to Longa’s methods.27 In brief, 76 rats were anesthetized with 10% chloral hydrate and placed on a heated pad to maintain body temperature. Blood flow of the right middle cerebral artery was blocked with an intraluminal 4–0 surgical suture (Ethicon, Brooklyn, NY, USA) on the common carotid artery for 120 min followed by reperfusion. After the surgery, nine rats were excluded because of either no apparent deficits or unconsciousness. A total of 67 rats with successful MCAO were selected for the study and randomly divided into MCAO group (MCAO, n=39) and MCAO with treadmill exercise group (exercise, n=28). A total of 34 rats in the sham group underwent an identical process without cerebral arterial occlusion (Table 1). All the experimental procedures were carried out according to the National Institutes of Health (NIH) guidelines for the care and use of laboratory animals. This study was conducted in the animal care facility of Shanghai Jiao Tong University School of Medicine and approved by the animal ethics committee of Shanghai Jiao Tong University (Ethical code: 2013102). All efforts were made to minimize the number of animals used and their suffering.
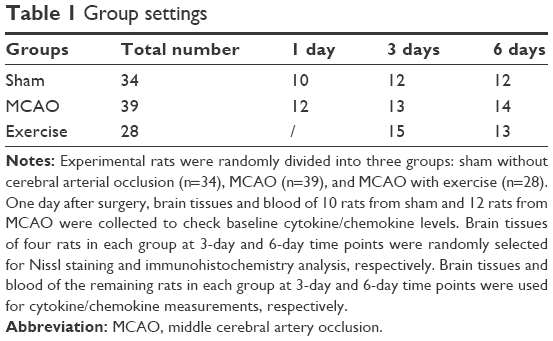 | Table 1 Group settings |
Exercise intervention
Treadmill exercise was started 3 days before the MCAO procedure for adaptability. To avoid impact on neurogenesis, exercise preconditioning procedure was only conducted at a speed of 1 m/min for less than a minute each day for three consecutive days to let each rat get familiar with the treadmill environment. Then, a 30 min intensive aerobic exercise was conducted 1 day after MCAO for rehabilitation in the exercise group. Rats were forced to run on an electric treadmill at the beginning intensity of 6 m/min for 10 min and then 12 m/min for 20 min each day for 3-day and 6-day periods, respectively. Rats in the MCAO non-exercise group were kept relatively static on the treadmill at the same time. Group settings and exercise duration are shown in Table 1. A set of 10 rats in the sham group and 12 rats in the MCAO group were used to check cytokine/chemokine changes 1 day after surgery as baseline control.
Motor function evaluation
Animal behavior was evaluated by foot-fault test at different time points based on modified Bederson scoring methods:28 0, no deficit; 1, forelimb flexion; 2, forelimb flexion plus decreased resistance to lateral push; 3, unidirectional circling; 4, longitudinal spinning or seizure activity; 5, no movement (Figure 1). Evaluation was performed by a laboratory assistant blinded to the experimental design. The average score of each experimental rat from three trials was used for statistical analysis.
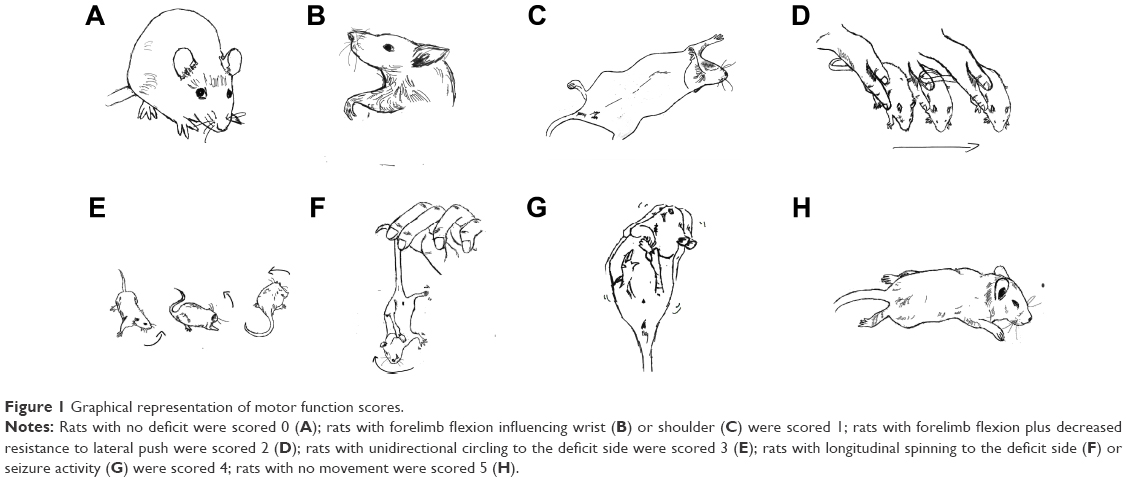 | Figure 1 Graphical representation of motor function scores. |
Blood collection and tissue preparation
Blood was collected from all 101 experimental rats before tissue preparation, and extracted serums were stored at −80°C for later cytokine measurement. Brain tissue preparation was performed as described earlier.26 Briefly, after 3-day and 6-day exercise treatment, four rats in each group at 3-day and 6-day time points, respectively, were anesthetized with 10% chloral hydrate and transcardially perfused with ice-cold phosphate-buffered saline and then with 4% paraformaldehyde. Brains were rapidly removed and fixed overnight in 4% paraformaldehyde at 4°C and transferred into a 30% sucrose solution the next day, then stored at −20°C before cryostatic sectioning. Brain tissues were sectioned coronally at 25 μm thickness and stored at −20°C for later Nissl staining and immunohistochemistry analysis. Brains from the remaining rats in each group at 3-day and 6-day time points were wrapped in foil and immediately placed in liquid nitrogen and then stored at −80°C for later cytokine measurement.
Nissl staining and immunohistochemistry analysis
Nissl staining was conducted to assess neuronal survival in the cerebral cortex and caudatum. Frozen brain sections were dried overnight and then immersed in 0.1% cresyl violet at 37°C for 2 h until the desired depth of staining was achieved. After rinsing with distilled water, sections were dehydrated in a graded series of 70%–100% ethanol, submerged in xylene, and then mounted with neutral balsam. Examined with a light microscope, neurons with round and palely stained nuclei were considered as surviving cells, whereas shrunken cells with pyknotic nuclei were considered as dying cells.
Immunohistochemistry staining was performed as described earlier.26 In brief, frozen brain sections were blocked in 1% bovine serum albumin (BSA) for 1 h, followed by overnight incubation with primary antibodies: mouse anti-NeuN for neuronal cells (Chemicon, Temecula, CA, USA), goat anti-Iba1 for microglial cells (Abcam, Cambridge, UK), mouse anti-glial fibrillary acidic protein (GFAP) for astrocytes (Sigma-Aldrich, St Louis, MO, USA) in 1% BSA at 4°C. Brain sections were incubated with appropriate biotinylated secondary antibodies for 2 h at room temperature followed by the complex of avidin–biotin horseradish peroxidase (HRP) and 3,3′-diaminobenzidine (DAB) HRP substrate detection (Invitrogen, Carlsbad, CA, USA). Five discontinued sections were analyzed from each specimen. Quantification of positively stained cells was conducted using Image-Pro Plus software version 5.1 (Media Cybernetics, Silver Spring, MD, USA). The total number of positive cells was counted in ischemic penumbra of cerebral cortex and caudatum with the lesion under high magnification (40×). For each section, five nonoverlapping visual fields in ischemic penumbra of cerebral cortex and caudatum with the lesion were chosen at random for statistical analysis. Cell counting was performed by a laboratory assistant blinded to the experimental design. Four rats were used in each group at 3-day and 6-day time points, respectively.
Cytokine measurements
Proteins were extracted from lysed and homogenized brain tissues at each time point in each group (sham group: n=10 for 1-day, n=8 for 3-day, and n=8 for 6-day time points; MCAO group: n=12 for 1-day, n=9 for 3-day, and n=10 for 6-day time points; exercise group: n=11 for 3-day and n=9 for 6-day time points). Protein concentrations were determined by the Bradford Protein Assay (Bio-Rad Laboratories, Hercules, CA, USA) with BSA as the standard for equal loading. Ischemic brain tissues and blood serums were measured for IL-1β, TNF-α, and MCP-1 via Bio-Plex Assays (Bio-Rad Laboratories) according to the manufacturer’s instructions.
Statistical analysis
SPSS software version 19.0 (IBM, Armonk, NY, USA) was used for data management and statistical analysis. The comparison of neurological deficit score was performed by Mann–Whitney–Wilcoxon test. All other data were analyzed by unpaired t-test. A P-value of <0.05 was considered significant.
Results
Exercise improved motor function
The effects of exercise on the neurological behavior of rats after ischemic brain injury were evaluated based on a modified Bederson scoring system as given in Table 2. This scoring system was proportional to the degree of nerve injury. Exercise treatment was started 1 day after surgery and lasted for 3-day (n=15) and 6-day (n=13) periods, while the rats in the MCAO group were kept static in the same environment (3 days, n=13; 6 days, n=14). All 34 rats in the sham group were scored 0 for no deficit, which are not presented in this article. The neurological deficit scores did not show statistical difference in the MCAO group in comparison with baseline parameters 3 days (2.6±0.8 vs 2.5±1.0, P>0.05) and 6 days post surgery (2.7±0.8 vs 2.4±0.9, P>0.05), respectively. The neurological deficit scores of the baseline did not show statistical difference between the MCAO group and exercise group either (2.5±1.0 vs 2.3±1.0, P>0.05 and 2.4±0.9 vs 2.4±1.0, P>0.05). Behavioral scores were significantly reduced after 3-day (1.7±1.1 vs 2.6±0.8, P<0.05) and 6-day (1.5±0.9 vs 2.7±0.8, P<0.01) exercises in comparison with the MCAO groups (Figure 2). Thus, the results of the evaluation of the motor function indicate that therapeutic exercise can significantly prevent behavioral dysfunction after ischemic brain injury.
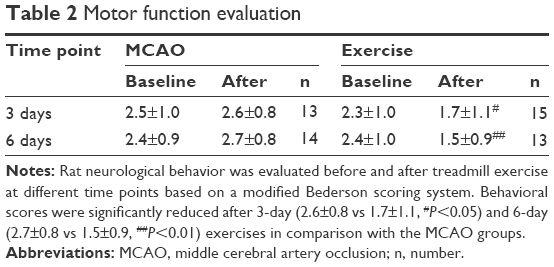 | Table 2 Motor function evaluation |
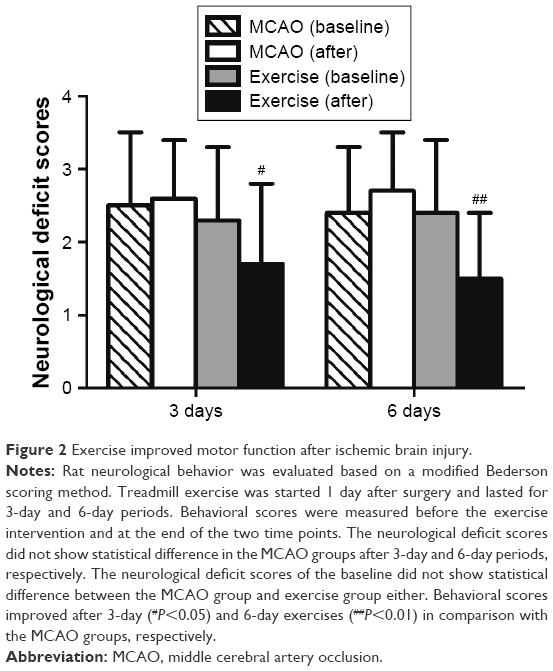 | Figure 2 Exercise improved motor function after ischemic brain injury. |
Exercise prevented death of neuronal cells
To determine the effects of exercise treatment on neuronal survival, Nissl staining was conducted on brain sections in penumbra of both cerebral cortex and caudatum. Healthy neurons with round and palely stained nuclei were found in the sham group in both cerebral cortex and caudatum (Figure 3A[a, d] and B[a, d]). In the MCAO group, neurons were relatively smaller with nuclear pyknosis clearly visible against the darkly stained cytoplasm 3 days after surgery (Figure 3A[b] and B[b]), and healthy neurons were further reduced with more shrunken cells with pyknotic nuclei 6 days after surgery (Figure 3A[e] and B[e]). In the exercise treatment group, relatively more healthy neurons and less nuclear pyknosis were observed in comparison with the MCAO group after 3-day and 6-day exercises, respectively (Figure 3A[c, f] and B[c, f]). The results of Nissl staining demonstrate that exercise prevents death of neuronal cells induced by ischemic brain injury.
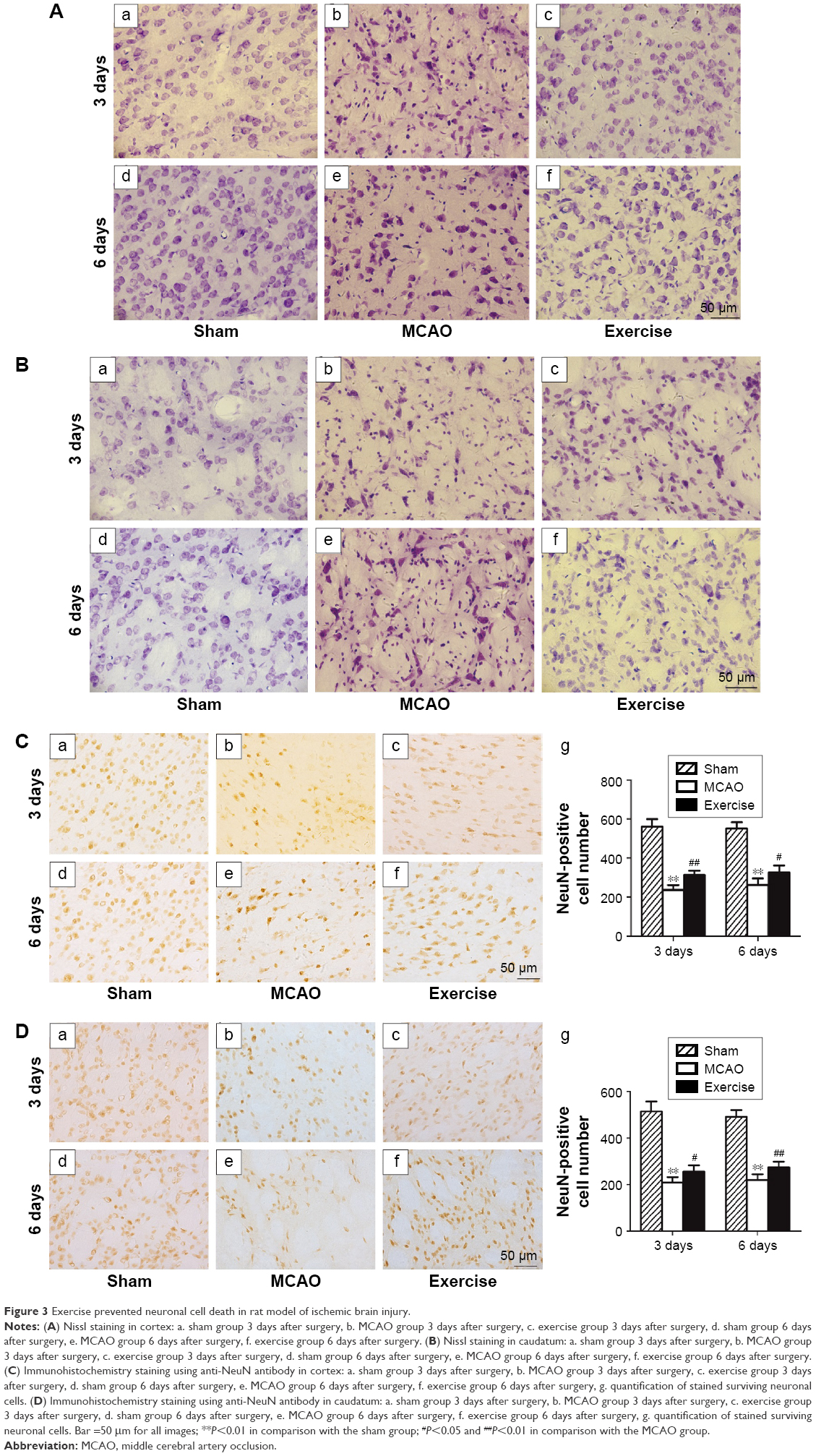 | Figure 3 Exercise prevented neuronal cell death in rat model of ischemic brain injury. |
To verify the abovementioned observation, anti-NeuN antibody was used in the immunohistochemistry experiment to determine the number of surviving neuronal cells. In agreement with the results of Nissl staining, quantified data revealed that the MCAO reduced the number of NeuN-positive cells in comparison with sham in both cerebral cortex and caudatum at 3-day (P<0.01 in both cerebral cortex and caudatum) and 6-day time points (P<0.01 in both cerebral cortex and caudatum), which were partially reversed by the exercise treatment in both cerebral cortex and caudatum at 3-day (P<0.01 in cerebral cortex and P<0.05 in caudatum) and 6-day time points (P<0.05 in cerebral cortex and P<0.01 in caudatum), respectively (Figure 3C and D). These data further demonstrate that exercise treatment is effective in rescuing neuronal cell death after ischemic brain injury.
Exercise inhibited the activation of microglial cells and astrocytes
To evaluate the activity of microglial cells, Iba1 antibody was used by immunohistochemistry staining in ischemic penumbra of cerebral cortex (Figure 4A) and caudatum (Figure 4B) after 3 days and 6 days of surgery. In the sham group, normal microglial cells were dark brown with small cell bodies and multiple slender branched protuberances (Figure 4A[a, d] and B[a, d]). In the MCAO group, microglial cells were evidently activated after 3 days (P<0.01 in both cerebral cortex and caudatum) and 6 days (P<0.01 in both cerebral cortex and caudatum) of surgery with an increase in cell density, and many of them appeared hypertrophic and amoeboid in shape with statistical difference in comparison with the sham group (Figure 4A[b, e] and B[b, e]). A declining trend in the activation of microglial cells was observed in the exercise treatment group in comparison with the MCAO group. The morphology of the microglial cells appeared to revert back to a state comparable with the sham group after 3 days and 6 days of exercise (Figure 4A[c, f] and B[c, f]). Quantitative analysis confirmed that the number of microglial cells in the exercise treatment group was significantly reduced in comparison with the MCAO group after 3 days (P<0.01) and 6 days of exercise (P<0.01) in cerebral cortex (Figure 4A[g]); in caudatum, the number of microglial cells in the exercise treatment group was also reduced with statistical difference (P<0.01 for both 3 days and 6 days of exercise; Figure 4B[g]).
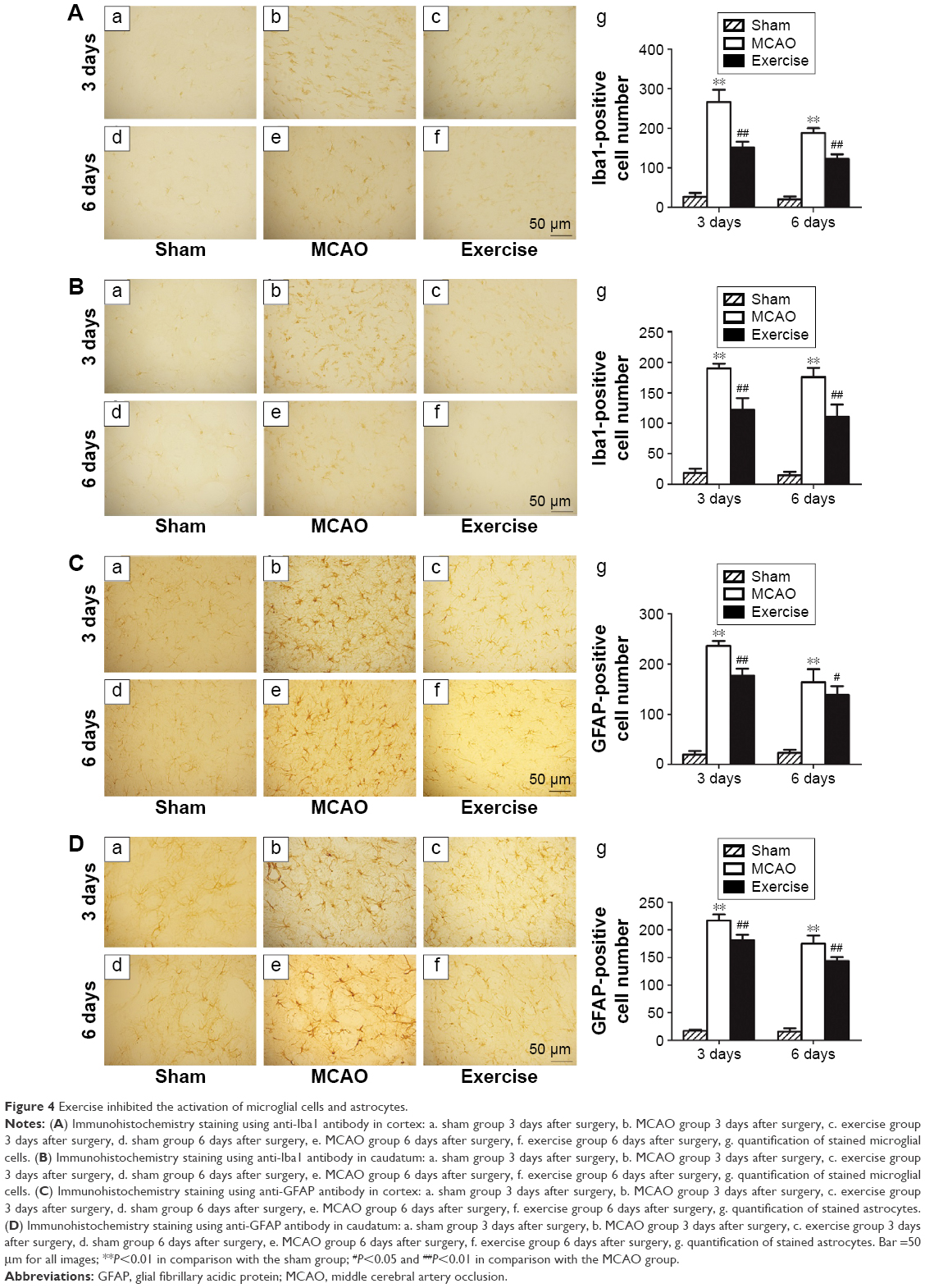 | Figure 4 Exercise inhibited the activation of microglial cells and astrocytes. |
Astrocytes in cerebral cortex (Figure 4C) and caudatum (Figure 4D) were analyzed based on the immunohistochemistry staining of GFAP. In the sham group, GFAP immunopositive astrocytes were typically stellar in shape, with small dark staining cell bodies and long fine slender processes (Figure 4C[a, d] and D[a, d]). However, astrocytes in the cerebral cortex and caudatum around ischemic penumbra were activated to a large degree, as indicated by dark blue GFAP staining and had large cell bodies and thick protuberances in the MCAO group with statistical difference in comparison with the sham group (P<0.01 for both 3 days and 6 days after surgery in both cerebral cortex and caudatum; Figure 4C[b, e] and D[b, e]). In the exercise treatment group, the morphological profile of these astrocytes was altered, the cell bodies were reduced, and there were less emitting processes (Figure 4C[c, f] and D[c, f]). Quantitative analysis of astrocytes revealed that the numbers of GFAP-positive cells in the exercise treatment group after 3-day and 6-day exercises were significantly reduced in both cerebral cortex (P<0.01 after 3-day exercise and P<0.05 after 6-day exercise) and caudatum (P<0.01 for both 3-day and 6-day exercises) in comparison with the MCAO group (Figure 4C[g] and D[g]). Thus, it is clear that exercise treatment 24 h after the MCAO procedure can alleviate the activation of microglial cells and astrocytes if it reaches the right amount of exercise intensity.
Time course of pro-inflammatory mediator expression
To determine the time course of pro-inflammatory mediator expression in brain tissues and serums after MCAO, protein levels of IL-1β, TNF-α and MCP-1 after 1, 3, and 6 days of surgery were measured by Bio-Plex Multiplex Cytokine Assays.
In the brain, the level of IL-1β significantly increased 1 day post MCAO in comparison with sham, declined gradually after 3 days and 6 days (Figure 5A[a]). The level of MCP-1 was increased in brain tissue after MCAO and peaked at 3-day time point (Figure 5A[b]). There were no significant changes in the level of TNF-α at different time points in comparison with the sham group (Figure 5A[c]).
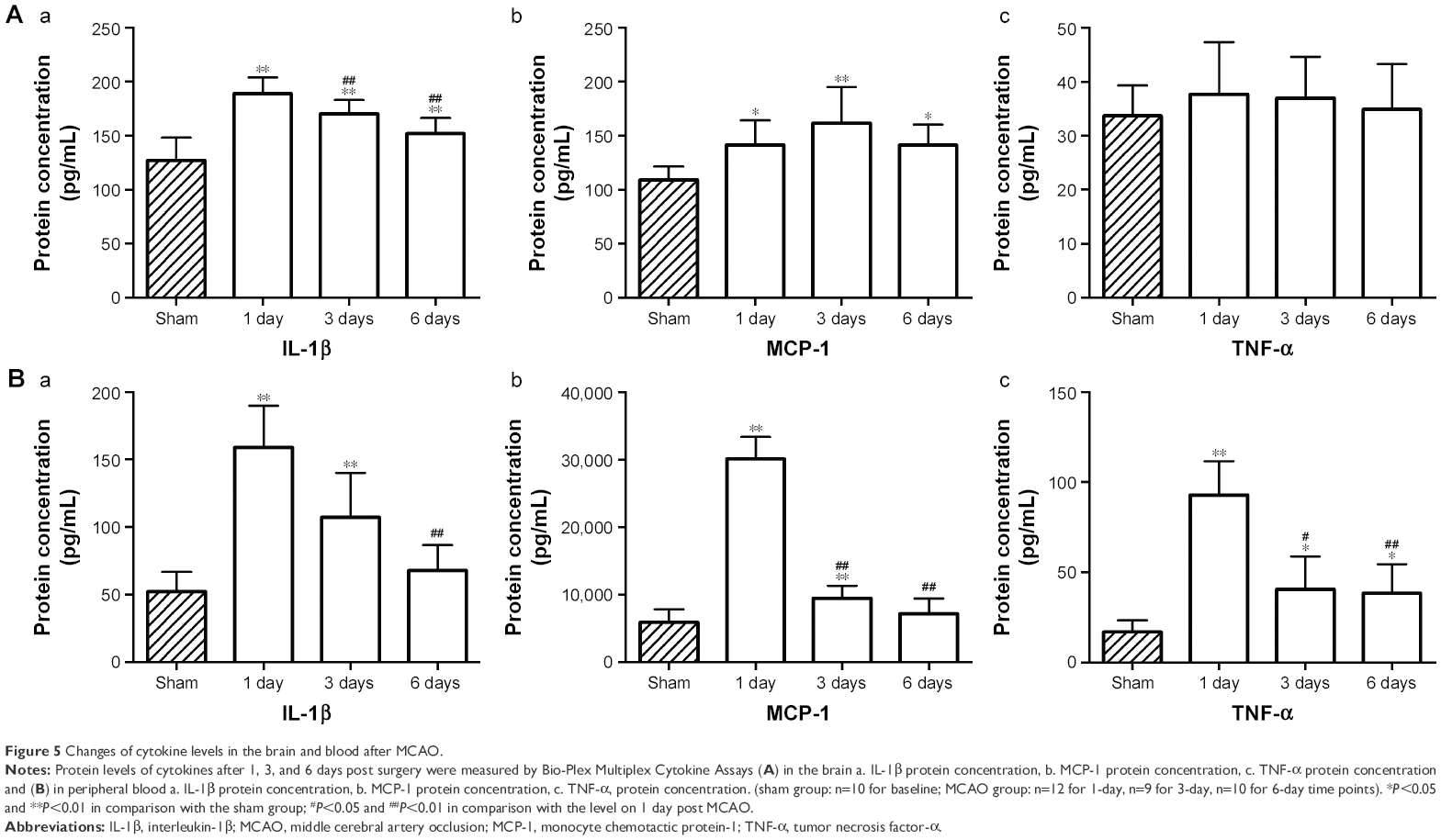 | Figure 5 Changes of cytokine levels in the brain and blood after MCAO. |
In peripheral blood, the expression of IL-1β rapidly increased to a maximum level 1 day after MCAO, declined thereafter and dropped significantly at 6-day time point (Figure 5B[a]), which is consistent with the observation in the brain. Serum levels of MCP-1 and TNF-α increased significantly 1 day post MCAO and progressively decreased at 3-day and 6-day time points (Figure 5B[b, c]). These results imply that exercise intervention should be performed at the peak level of cytokine response, which is 1 day post MCAO.
Exercise downregulated pro-inflammatory mediators
To evaluate the effects of exercise on cytokine/chemokine changes in brain tissues and serums, Bio-Plex Multiplex Cytokine Assays were performed to measure protein levels of IL-1β, MCP-1, and TNF-α at 3-day and 6-day time points among the groups of sham, MCAO, and exercise, respectively.
In brain tissues, IL-1β level in the MCAO group increased significantly post surgery in comparison with the sham group, but reduced significantly after 3-day and 6-day exercise treatment (Figure 6A[a]). TNF-α level did not show significant differences among the sham, MCAO, and exercise groups (Figure 6A[c]). The level of MCP-1 increased significantly in the MCAO group at both 3-day and 6-day time points compared with the sham group, but significantly decreased in the exercise treatment group at 6-day time point, although there were no statistical changes after 3-day exercise (Figure 6A[b]). This implies that 3-day exercise intensity does not reach the threshold to alter MCP-1 expression significantly, but 6-day exercise does.
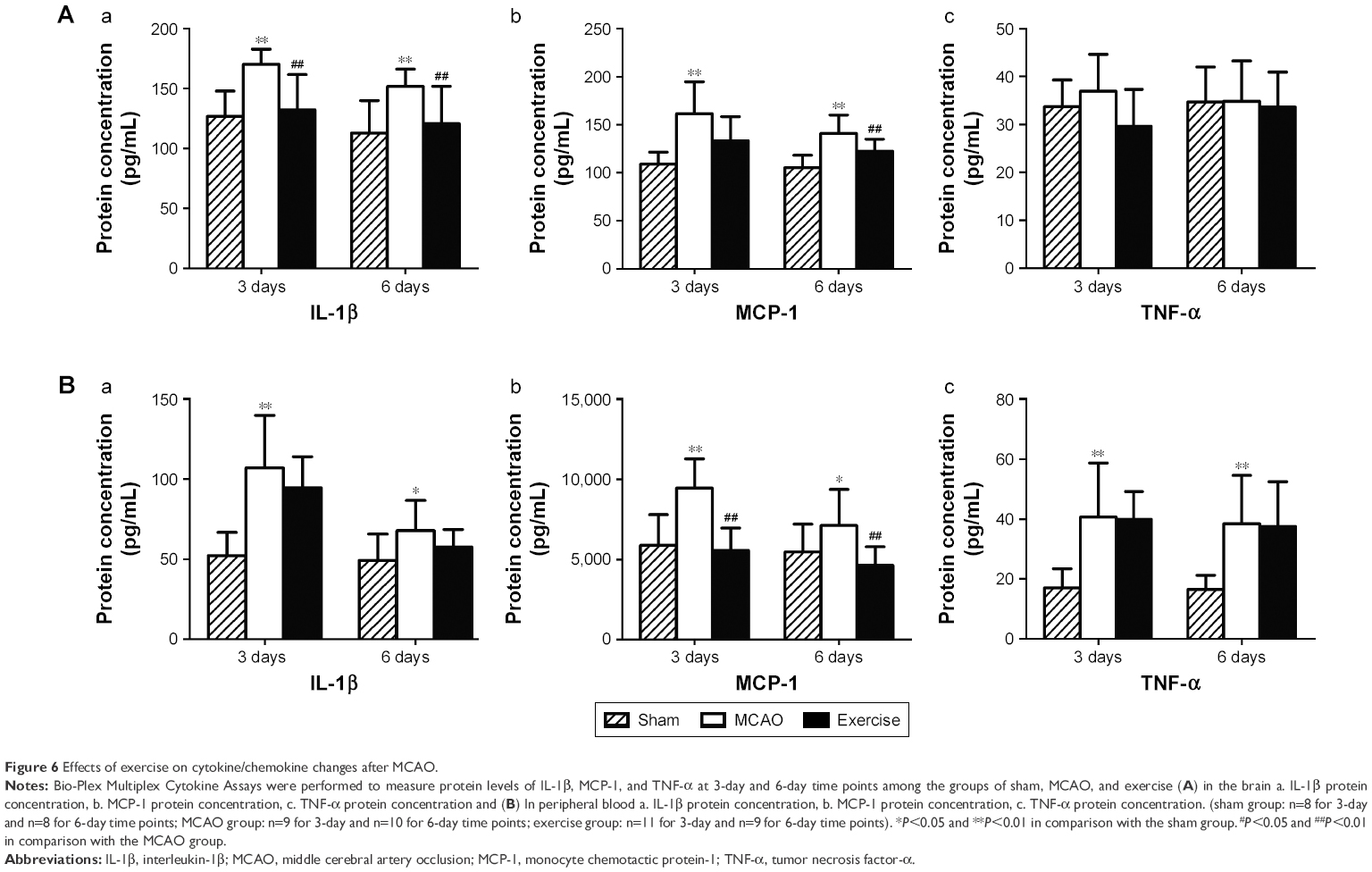 | Figure 6 Effects of exercise on cytokine/chemokine changes after MCAO. |
In blood, IL-1β level in the MCAO group increased significantly post surgery in comparison with the sham group, but exercise treatment did not show significant effects on the level of IL-1β at either 3-day or 6-day time point compared with the MCAO group (Figure 6B[a]). The level of MCP-1 increased after MCAO at both 3-day and 6-day time points, but it significantly decreased after 3-day and 6-day exercise (Figure 6B[b]). MCAO increases TNF-α level at both 3-day and 6-day time points, but exercise did not show significant effects on the level of TNF-α compared with the MCAO group (Figure 6B[c]). The results indicate that exercise intervention attenuates the protein level of MCP-1 significantly, but did not alter TNF-α. Exercise can decrease the protein level of IL-1β, although there is no statistical significance.
Discussion
The current study investigated the effects of aerobic exercise on the score of neurobehavior, survival of neuronal cells, activation of glial cells, and expression of pro-inflammatory mediators after ischemic brain injury in a rat MCAO–reperfusion model. It was found that exercise improved motor function and downregulated the expression of the pro-inflammatory mediators IL-1β and MCP-1.
The MCAO–reperfusion model is widely used in ischemic stroke research, because certain facets of this model resemble human disease acquisition.27,28 For example, in most survived patients, restoration of blood flow achieved by means of thrombolysis or mechanical recanalization may exacerbate the initial ischemic injury through reperfusion and cause disability. Pharmacological approaches to reduce ischemia–reperfusion insult have been tested in this MCAO model.29–31
Recent clinical trials suggest that exercise rehabilitation has the same beneficial effects as pharmacological drugs to improve health-related quality of life not only in patients6,32 with ischemic stroke but also in patients post myocardial infarction.7 Although intensive exercise may exacerbate brain cell damage,33 numerous experimental data have shown that appropriate exercise protects against ischemic brain injury.34,35 Consistent with these findings, the results demonstrated that appropriate early treadmill exercise treatment improved the neurobehavioral scores and increased the number of survival neurons (Figures 2 and 3), although the underlying mechanisms were not completely elucidated. Cotman and Engesser-Cesar36 reported that exercise increased the production of neurotrophic factors, which might promote cell growth and enhance neuronal activity. Zhang et al’s9 research team showed that early exercise protected against cerebral ischemic injury through inhibiting neuron apoptosis. Schimidt et al37 demonstrated that physical exercise attenuated oxidative stress and promoted better neuroprotection to an ischemic event. Ma et al’s38 results indicated that exercise therapy downregulated the overexpression of toll-like receptors after cerebral ischemia. These studies showed the beneficial role of exercise through different mechanisms.
Increasing evidences have demonstrated that inflammation resulted from leukocyte infiltration via reperfusion may play an important role in the pathogenesis of ischemic stroke.11,38 During brain Ischemia–reperfusion injury, pro-inflammatory mediators such as IL-1β, TNF-α, and MCP-1 are produced by microglia/macrophages and astrocytes,39 which are believed to contribute to ischemic stroke.40 Inhibiting the activities of these glial cells and their pro-inflammatory mediators would be an important strategy to protect against brain injury. The current study observed that exercise triggered brain preservation mechanisms to attenuate activities of microglial cells and astrocytes (Figure 4), which were the most sensitive and reliable indicators of brain pathological changes.41,42 Activated microglial cells are also the source of IL-1β and TNF-α production, which in turn activate microglial cells to cause more brain injury.43 IL-1β has been demonstrated to induce MCP-1 expression in human airway smooth muscle cells.44 MCP-1, an adhesion molecule that recruits monocytes, memory T-cells, and dendritic cells to the sites of tissue injury, has been reported to be involved in the neuroinflammatory processes45 and expressed in glial cells during ischemic brain injury.19 Therefore, inhibiting either the activation of glial cells or the expression of pro-inflammatory mediators may protect against brain injury. This hypothesis is in agreement with the results of Bio-Plex Multiplex Cytokine Assays. It was found that exercise reduced the protein levels of IL-1β and MCP-1 not only in the cerebral ischemic penumbra but also in peripheral blood after 3-day and 6-day treadmill running post MCAO, although there was no statistical difference for reduced IL-1β in peripheral blood (Figure 6). Similarly, a French research team examined the influence of physical training (treadmill running) on IL-1β, IL-6, and IL-1ra in rats and found that the training program induced a significant decrease in IL-1β concentration in the brain but not in serum.46 It is implied that IL-1β production is more sensitive to aerobic exercise in the central nerve system (CNS) than in the circulation. Thus, the results indicate that early exercise triggers brain preservation to downregulate pro-inflammatory mediators such as IL-1β and MCP-1, but not TNF-α, for preventing brain damage with systematic effects of MCP-1 on peripheral blood. The possible explanation of the TNF-α neutral role was either that 6-day exercise intensity did not reach the threshold to alter TNF-α expression on the experimental rats or TNF-α was not as sensitive as MCP-1 to be changed rapidly and dramatically. In agreement with the results, a clinical study to evaluate biomarkers for patients at increased risk of stroke did not observe any changes of TNF-α in symptomatic patients with unstable plaques and high-grade carotid artery stenosis compared with asymptomatic patients with stable plaques.47 Furthermore, a very recent investigation on 619 ischemic stroke patients and 612 controls indicated that TNF-α gene polymorphisms may not contribute to the risk of ischemic stroke.48 One study even got opposite results and concluded that exercise-induced TNF-α markedly decreased blood–brain barrier dysfunction in stroke,49 although others still supported the pro-inflammatory role of TNF-α that can exacerbate tissue damage in stroke.18,50 Moreover, TNF-α may have neutral role or contradictory effects in stroke as summarized by Hallenbeck.51
However, little is known about the timing of molecular changes post MCAO. To determine the dynamic molecular changes, inflammatory mediators were also measured in brain tissues and serums at 1-day, 3-day, and 6-day time points after MCAO and found that except brain MCP-1 level peaked at 3-day time point, IL-1β and TNF-α from the brain and blood and MCP-1 from the blood all peaked on day 1 post MCAO compared with the sham surgery group (Figure 5). Thus, the data indicate that early exercise intervention should be performed at the peak level of cytokine/chemokine response on day 1 post MCAO.
There was a limitation in this study. The effects of aerobic exercise on the infarct volume are important, but they are not quantified in the study. This parameter is linked to the poststroke rehabilitation and will significantly improve the current work.
Conclusion
This study demonstrates that appropriate aerobic exercise contributes to neuroprotection against ischemic brain injury by improving motor function, preventing neuronal cell death, and inhibiting microglia and astrocyte activation via downregulation of the pro-inflammatory mediators IL-1β in CNS and MCP-1 in both CNS and circulation. These novel findings indicate that exercise combined with pharmacological intervention to block the inflammatory process through the IL-1β/MCP-1 signaling pathway may prevent the progression of ischemic stroke.
Acknowledgments
This work was funded by Shanghai Disabled Persons’ Federation (ZY201511), Shanghai Key Discipline Construction Grant (ZK2012A40), and National Natural Science Foundation of China (81672260). The authors would like to thank Yanyong Lu for graphical assistance and Ian Martin for proofreading.
Disclosure
The authors report no conflicts of interest in this work.
References
GBD 2013 Mortality and Causes of Death Collaborators. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;385(9963):117–171. | ||
Saxena SK, Ng TP, Koh G, Yong D, Fong NP. Is improvement in impaired cognition and depressive symptoms in post-stroke patients associated with recovery in activities of daily living? Acta Neurol Scand. 2007;115(5):339–346. | ||
Mozaffarian D, Benjamin EJ, Go AS, et al. Heart disease and stroke statistics – 2015 update: a report from the American Heart Association. Circulation. 2015;131(4):434–441. | ||
Koziol JA, Feng AC. On the analysis and interpretation of outcome measures in stroke clinical trials. Stoke. 2006;37(10):2644–2647. | ||
Rizos EC, Ntzani EE, Bika E, Kostapanos MS, Elisaf MS. Association between omega-3 fatty acid supplementation and risk of major cardiovascular disease events: a systematic review and meta-analysis. JAMA. 2012;308(10):1024–1033. | ||
Shimodozono M, Noma T, Nomoto Y, et al. Benefits of a repetitive facilitative exercise program for the upper paretic extremity after subacute stroke: a randomized controlled trial. Neurorehabil Neural Repair. 2013;27(4):296–305. | ||
Peixoto TC, Begot I, Bolzan DW, et al. Early exercise-based rehabilitation improves health-related quality of life and functional capacity after acute myocardial infarction: a randomized controlled trial. Can J Cardiol. 2015;31(3):308–313. | ||
Zhang P, Zhang Q, Pu H, et al. Very early-initiated physical rehabilitation protects against ischemic brain injury. Front Biosci (Elite Ed). 2012;4:2476–2489. | ||
Zhang P, Zhang Y, Zhang J, et al. Early exercise protects against cerebral ischemic injury through inhibiting neuron apoptosis in cortex in rats. Int J Mol Sci. 2013;14(3):6074–6089. | ||
Pan J, Konstas A, Bateman B, Ortolano GA, Pile-Spellman J. Reperfusion injury following cerebral ischemia: pathophysiology, MR imaging, and potential therapies. Neuroradiology. 2007;49(2):93–102. | ||
Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol. 2010;87(5):779–789. | ||
Zhang J, An J. Cytokines, inflammation and pain. Int Anesthesiol Clin. 2007;45(2):27–37. | ||
Dinarello CA. Proinflammatory cytokines. Chest. 2000;118(2):503–508. | ||
Shichita T, Sakaguchi R, Suzuki M, Yoshimura A. Post-ischemic inflammation in the brain. Front Immunol. 2012;3(132):1–7. | ||
Circu ML, Aw TY. Reactive oxygen species, cellular redox systems and apoptosis. Free Radic Biol Med. 2010;48(6):749–762. | ||
Srivastava KD, Rom WN, Jagirdar J, Yie TA, Gordon T, Tchou-Wong KM. Crucial role of interleukin-1beta and nitric oxide synthase in silica-induced inflammation and apoptosis in mice. Am J Respir Crit Care Med. 2002;165(4):527–533. | ||
Wang X, Yue TL, Barone FC, White RF, Gagnon RC, Feuerstein GZ. Concomitant cortical expression of TNF-α and IL-1β mRNAs follows early response gene expression in transient focal ischemia. Mol Chem Neuropathol. 1994;23(2–3):103–114. | ||
Liu T, Clark RK, McDonnell PC, et al. Tumor necrosis factor-α expression in ischemic neurons. Stroke. 1994;25(7):1481–1488. | ||
Kim JS, Gautam SC, Chopp M, et al. Expression of monocyte chemoattractant protein-1 and macrophage inflammatory protein-1 after focal cerebral ischemia in the rat. J Neuroimmunol. 1995;56(2):127–134. | ||
Nakajima K, Kohsaka S. Microglia: activation and their significance in the central nervous system. J Biochem. 2001;130(2):169–175. | ||
Rothwell N, Allan S, Toulmond S. The role of interleukin 1 in acute neurodegeneration and stroke: pathophysiological and therapeutic implications. J Clin Invest. 1997;100(11):2648–2652. | ||
Barone FC, Arvin B, White RF, et al. Tumor necrosis factor-α: a mediator of focal ischemic brain injury. Stroke. 1997;28(6):1233–1244. | ||
Svensson M, Lexell J, Deierborg T. Effects of physical exercise on neuroinflammation, neuroplasticity, neurodegeneration, and behavior: what we can learn from animal models in clinical settings. Neurorehabil Neural Repair. 2015;29(6):577–589. | ||
Kang EB, Koo JH, Jang YC, et al. Neuroprotective effects of endurance exercise against high-fat diet-induced hippocampal neuroinflammation. J Neuroendocrinol. 2016;28(5):12385. | ||
Gomes SS, Simoes PS, Mortara RA, et al. Exercise-induced hippocampal anti-inflammatory response in aged rats. J Neuroinflammation. 2013;10(10):61. | ||
Chen J, Qin J, Su Q, Liu Z, Yang J. Treadmill rehabilitation treatment enhanced BDNF-TrkB but not NGF-TrkA signaling in a mouse intracerebral hemorrhage model. Neurosci Lett. 2012;529(1):28–32. | ||
Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniotomy in rats. Stroke. 1989;20(1):84–91. | ||
Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17(3):472–476. | ||
Khan M, Elango C, Ansari MA, Singh I, Singh AK. Caffeic acid phenethyl ester reduces neurovascular inflammation and protects rat brain following transient focal cerebral ischemia. J Neurochem. 2007;102(2):365–377. | ||
Choi IY, Lee JC, Ju C, et al. A3 adenosine receptor agonist reduces brain ischemic injury and inhibits inflammatory cell migration in rats. Am J Pathol. 2011;179(4):2042–2052. | ||
Yaidikar L, Thakur S. Punicalagin attenuated cerebral ischemia-reperfusion insult via inhibition of proinflammatory cytokines, up-regulation of Bcl-2, down-regulation of Bax, and caspase-3. Mol Cell Biochem. 2015;402(1–2):141–148. | ||
Linder SM, Rosenfeldt AB, Bay RC, Sahu K, Wolf SL, Alberts JL. Improving quality of life and depression after stroke through telerehabilitation. Am J Occup Ther. 2015;69(2):1–10. | ||
Scopel D, Fochesatto C, Cimarosti H, et al. Exercise intensity influences cell injury in rat hippocampal slices exposed to oxygen and glucose deprivation. Brain Res Bull. 2006;71(1–3):155–159. | ||
Shan T, Zhang Y, Song T, et al. Early exercise training improves ischemic outcome in rats by cerebral hemodynamics. Brain Res. 2013;1533:114–121. | ||
Lee SU, Kim DY, Park SH, Choi DH, Park HW, Han TR. Mild to moderate early exercise promotes recovery from cerebral ischemia in rats. Can J Neurol Sci. 2009;36(4):443–449. | ||
Cotman CW, Engesser-Cesar C. Exercise enhances and protects brain function. Exerc Sport Sci Rev. 2002;30(2):75–79. | ||
Schimidt HL, Vieira A, Altermann C, et al. Memory deficits and oxidative stress in cerebral ischemia-reperfusion: neuroprotective role of physical exercise and green tea supplementation. Neurobiol Learn Mem. 2014;114:242–250. | ||
Ma Y, He M, Qiang L. Exercise therapy downregulates the overexpression of TLR4, TLR2, MyD88 and NF-kB after cerebral ischemia in rats. Int J Mol Sci. 2013;14(2):3718–3733. | ||
Huang J, Upadhyay UM, Tamargo RJ. Inflammation in stroke and focal cerebral ischemia. Surg Neurol. 2006;66(3):232–245. | ||
Gao S, Mo J, Chen L, et al. Astrocyte GGTI-mediated Rac1 prenylation upregulates NF-κB expression and promotes neuronal apoptosis following hypoxia/ischemia. Neuropharmacology. 2016;103:44–56. | ||
Ling EA, Ng YK, Wu CH, Kaur C. Microglia: its development and role as a neuropathology sensor. Prog Brain Res. 2001;132:61–79. | ||
Mabuchi T, Kitagawa K, Ohtsuki T, et al. Contribution of microglia/macrophages to expansion of infarction and response of oligodendrocytes after focal cerebral ischemia in rats. Stroke. 2000;31(7):1735–1743. | ||
Wu J, Yang S, Xi G, et al. Microglial activation and brain injury after intracerebral hemorrhage. Acta Neurochir Suppl. 2008;105:59–65. | ||
Wuyts WA, Vanaudenaerde BM, Dupont LJ, Demedts MG, Verleden GM. Modulation by cAMP of IL-1beta-induced eotaxin and MCP-1 expression and release in human airway smooth muscle cells. Eur Respir J. 2003;22(2):220–226. | ||
Gerard C, Rollins BJ. Chemokines and disease. Nat Immunol. 2001;2(2):108–115. | ||
Chennaoui M, Drogou C, Gomez-Merino D. Effects of physical training on IL-1beta, IL-6 and IL-1ra concentrations in various brain areas of the rat. Eur Cytokine Netw. 2008;19(1):8–14. | ||
Jaroslav P, Christian R, Stefan O, et al. Evaluation of serum biomarkers for patients at increased risk of stroke. Int J Vasc Med. 2012;2012:906954. | ||
Gu L, We G, Su L, et al. TNF-a (-238G/A and -308G/A) gene polymorphisms may not contribute to the risk of ischemic stroke. Int J Neurosci. 2016;126(3):219–226. | ||
Guo M, Lin V, Davis W, et al. Preischemic induction of TNF-alpha by physical exercise reduces blood-brain barrier dysfunction in stroke. J Cereb Blood Flow Metab. 2008;28(8):1422–1430. | ||
de Lima NM, Ferreira EO, Fernandes MY, et al. Neuroinflammatory response to experimental stroke is inhibited by boldine. Behav Pharmacol. Epub 2016 Oct 19. | ||
Hallenbeck JM. The many faces of tumor necrosis factor in stroke. Nat Med. 2002;8(12):1363–1368. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.