Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 16
Translation Animal Models of Diabetic Kidney Disease: Biochemical and Histological Phenotypes, Advantages and Limitations
Authors Luo W, Tang S, Xiao X, Luo S, Yang Z, Huang W, Tang S ![]()
Received 11 February 2023
Accepted for publication 29 April 2023
Published 5 May 2023 Volume 2023:16 Pages 1297—1321
DOI https://doi.org/10.2147/DMSO.S408170
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Gian Paolo Fadini
Wenting Luo,1 Shiyun Tang,2 Xiang Xiao,1 Simin Luo,1 Zixuan Yang,1 Wei Huang,3 Songqi Tang1
1School of Traditional Chinese Medicine, Hainan Medical University, Haikou, Hainan Province, People’s Republic of China; 2Hospital of Chengdu University of Traditional Chinese Medicine, Chengdu, Sichuan Province, People’s Republic of China; 3School of Basic Medical Sciences, Chengdu University of Traditional Chinese Medicine, Chengdu, Sichuan Province, People’s Republic of China
Correspondence: Wei Huang, School of Basic Medical Sciences, Chengdu University of Traditional Chinese Medicine, Chengdu, Sichuan Province, 611137, People’s Republic of China, Email [email protected] Songqi Tang, School of Traditional Chinese Medicine, Hainan Medical University, Haikou, Hainan Province, 571199, People’s Republic of China, Tel +86 18689713500, Email [email protected]
Abstract: Animal models play a crucial role in studying the pathogenesis of diseases, developing new drugs, identifying disease risk markers, and improving means of prevention and treatment. However, modeling diabetic kidney disease (DKD) has posed a challenge for scientists. Although numerous models have been successfully developed, none of them can encompass all the key characteristics of human DKD. It is essential to choose the appropriate model according to the research needs, as different models develop different phenotypes and have their limitations. This paper provides a comprehensive overview of biochemical and histological phenotypes, modeling mechanisms, advantages and limitations of DKD animal models, in order to update relevant model information and provide insights and references for generating or selecting the appropriate animal models to fit different experimental needs.
Keywords: modeling, advanced diabetic kidney disease, diabetic complications, hypertension, atherosclerosis, non-proteinuric diabetic kidney disease
Introduction
Diabetic kidney disease (DKD) is a common microvascular complication of diabetes mellitus (DM) that is clinically characterized by a gradual decline in renal function, with or without proteinuria, due to prolonged hyperglycemia. There are no substantial differences between patients with type 1 DM (T1D) and those with type 2 DM (T2D) in the basic pathophysiological mechanisms leading to nephropathy.1 The early glomerular pathologic changes in DKD are principally mesangial matrix expansion, glomerular basement membranes (GBM) thickening and compensatory glomerular hypertrophy causing ultrafiltration, which subsequently progresses to diffuse and/or nodular glomerulosclerosis with decreased glomerular filtration rate (GFR) and arterial hyalinosis. In addition, tubulointerstitial injury is a consistent feature of DKD, manifested by interstitial inflammation, tubular basement membrane thickening, tubular atrophy, and interstitial fibrosis. Chronic tubulointerstitial injury may be more severe than diabetic glomerulopathy in patients with T2D-DKD because of the more heterogeneous lesions often associated with atherosclerosis.1
DKD can be found in 25–40% of diabetes and is one of the leading causes of end-stage renal disease (ESRD).1 Additionally, its presence poses a significant cardiovascular risk and greatly increases cardiovascular mortality in patients.2–4 As a prevalent chronic disease, DM already affects the health of more than 350 million people worldwide. Research has indicated that the number of diabetes is expected to reach 600 million by 2035, by which time more than one in ten adults will have DM,5 suggesting that more people may suffer from DKD. Although a combination of glycemic control, anti-hypertension, lipid regulation, and lifestyle interventions has been clinically proven to be effective in slowing down the progression of DKD,6 this disease remains incurable or irreversible.
The etiology and pathogenesis of DKD are complex and not well understood, which is a major obstacle to developing clinical control methods. Suitable animal models can provide a basis for clinical treatment and important clues for studying the etiology, pathogenesis, and pathophysiological changes.7 There are various animal models of DKD available; however, no single model can represent all the features of human DKD. Therefore, it is crucial to select a suitable animal model based on research needs and purposes. There are three classical categories of animal models of DKD used in laboratories: induced models (chemical, dietary, and surgical induction), spontaneous models and genetic models. DKD, especially T2D-DKD, is not clinically independent and is often accompanied by complications such as hyperlipidaemia, hypertension, and atherosclerosis. In addition, the increasing number of patients with non-proteinuric DKD has raised concerns. Traditional DKD models cannot meet all the needs of studies due to their non-significant comorbid phenotypes. Scientists have been working on further development of new animal models to better mimic human DKD. In recent years, several new translation models, such as models of DKD with stable hypertension, DKD with atherosclerosis, and non-proteinuric DKD, have emerged, offering more possibilities for DKD research. Based on the signs of human DKD and the optimal characteristics proposed by the Animal Models of Diabetic Complications Consortium (AMDCC, http://www.amdcc.org), which are expected in a DKD model, this paper reviews translation animal models in terms of metabolic indicators, proteinuria (whether increased >10-fold), early/advanced glomerular pathology, tubulointerstitial pathology, and GFR (whether decreased >50%), as well as their modeling mechanisms, advantages and limitations. The purpose is to update the model information and provide important insights and references for generating or selecting reasonable animal models to fit different experimental needs.
Animal Models of T1D-DKD
Streptozotocin (STZ) Induced Animal Models of T1D-DKD
Using chemical agents – STZ or Alloxan is one of the most popular means to establish animal models of DKD. Compared to Alloxan, STZ is used more frequently in the laboratory because of its higher potency, less tissue toxicity, and stronger model stability.8
Model Mechanism
STZ is a 2-deoxy-2(3-(methyl-3-nitrosoureido)-aminoglucose synthesized by the Streptomyces chromogene. It is readily transported into pancreatic β-cells via GLUT-2. On the one hand, as an N-acetylglucosamine (GlcNAc) analogue, STZ selectively inhibits the activity of O-linked N-acetylglucosamine hydrolase (O-GlcNAcase, OGA), leading to irreversible O-glycosylation of intracellular proteins.9 On the other hand, the generation of NO and reactive oxygen species (ROS) during intracellular metabolism of STZ, as well as its strong alkylation properties, can induce DNA fragmentation and apoptosis of β-cells, ultimately leading to an absolute deficiency of insulin.10
Species and Strains
Pancreatic β-cells of rats and mice are more sensitive to the cytotoxicity of STZ than those of rabbits, dogs and pigs.11,12 Besides, the short generation time and low breeding cost make rodents the most commonly used in STZ-induced DKD experiments. For mouse strains, C57BL/6 (B6), DBA/2, CD1, 129/Sv and KK/HIJ are the most popular, among them, DBA/2, KK/HIJ and CD1 background are sensitive to STZ induction, while B6 are relatively resistant to the development of nephropathy.13,14 Even so, B6 mice are still widely used because of the advantage of easy genetic manipulation. For rat strains, the most frequently used are Sprague-Dawley (SD) rats and Wistar rats. In recent years, there has been some progress in STZ-induced DM and related complications in model organisms such as zebrafish and drosophila. High reproduction rate, ease of genetic manipulation and imaging deep tissues in vivo are their significant advantages. These models are most likely to be used for genetic or drug screening, however, the findings need to be replicated in mammals because their primitive kidney cells are functionally very different from those of humans.15
Phenotypes
Male rodents are more often chosen than females as the latter are comparatively insensitive to the toxic effects of STZ, which may be attributed to sexual dimorphism and estrogen.16 In STZ induced mouse models, male B6 mice show a significant increase in blood glucose with mild proteinuria 2 weeks after diabetes induction, but their renal lesions are not significant, with only mild mesangial expansion and GBM thickening.17–19 STZ-129/Sv mice are characterized by the ability to develop hypertension, but do not perform as well as STZ-B6 mice in terms of hyperglycemia and proteinuria.13,17 STZ-DBA/2 mice are an excellent model of T1D-DKD with higher glucose and proteinuria levels and more severe renal lesions, for example, increased mesangial matrix accumulation, podocyte apoptosis, and leukocyte infiltration compared with STZ-B6 mice; moreover, glomerulosclerosis and arterial hyalinization can be observed in these mice.17,20 STZ-KK/HIJ mice have similar but less severe renal pathological alterations as STZ-DBA/2 mice.14 STZ-CD1 mice are advantageous in studying diabetic renal fibrosis; they display renal fibrosis associated with the epithelial–mesenchymal transition (EMT) and abnormal glycolysis at ~4 months after diabetes induction and significant lesions at ~6 months;21–23 these mice die because of diabetic complications by 6–7 months after the first STZ injection.13 For STZ induced rat models, renal lesions are mild in both SD strain and Wistar strain, although elevated blood glucose and increased urinary protein excretion are present.24–28
Advantages and Limitations
STZ-induced wild-type (WT) models have the advantage of low cost and simple experimental procedures.29 STZ destroys islet β-cells causing absolute insulin deficiency, which is similar to the pathogenesis of T1D in humans. However, these models have some non-negligible limitations: (i) renal lesions are mostly mild in most strains of mice and rats, with few advanced pathological changes such as nodular and/or diffuse glomerulosclerosis and decreased GFR; therefore, STZ induction is often combined with other approaches, such as dietary interventions and surgery, or the use of genetic animals to accelerate the disease processes to obtain the phenotypes desired, which was described in other sections of this review; (ii) at high doses or systemic administration, STZ exhibits non-specific cytotoxic to the kidney, especially tubular cells, and subsequently causes acute kidney injury, which makes it difficult to explain whether the kidney lesions are attributable to hyperglycemia or to the direct toxic effects of STZ.30 So, it is recommended that any morphological and functional assessment of experimental animals should be started at least 2–3 weeks after STZ administration.31,32
Uninephrectomy (UNx) + STZ Induced Animal Models of T1D-DKD
Model Mechanism
The addition of UNx to STZ is an intensive means of inducing T1D-DKD. Different from STZ that gradually develops renal ultrafiltration by inducing hyperglycemia, UNx directly alters renal hemodynamics causing an increase in GFR in the residual kidney. Ultrafiltration is an early manifestation of chronic kidney disease, and it can induce mechanical stretch to cause mesangial cell injury, further leading to glomerular hypertrophy and ECM production.33 Excessive activation of the renin–angiotensin system (RAS) after UNx can increase blood pressure, as well as the production of pro-fibroblastic cytokines and ROS,33–37 all of which contribute to progressive kidney injury and accelerate the development of nephropathy. For strains resistant to DKD development, UNx + STZ is the best approach to induce T1D-DKD.38
Species and Strain
B6 mice, SD rats, Wistar rats, spontaneous hypertension rats (SHR).
Phenotypes
Hyperglycemia, albuminuria, and early glomerular pathological changes such as glomerular hypertrophy, GBM thickening, mesangial matrix expansion and mild glomerulosclerosis can be observed in UNx + STZ treated mice and rats.38–43 Furthermore, tubular atrophy, brush border loss, tubular dilatation and tubulointerstitial fibrosis develop in B6 mice at 12th week after the treatment of UNx and multiple low-dose STZ.38 These tubular damages are considered to be caused by glucose toxicity, while they are not evident or present under chemical induction alone.
Advantages and Limitation
In general, UNx + STZ induced models take shorter time to develop more severe kidney disease than those controls with STZ induced alone. However, Wada et al noted that in STZ-sensitive CD1 strain, UNx played a very limited role in accelerating the progression of renal lesions.39 In addition, generating this type of models demands specialized technical knowledge and complete surgical conditions, as well as reasonable antibiotic and analgesic treatments, to avoid the risk of surgical-related deaths.
Genetic Animal Models of T1D-DKD
Genetic animals are popular in DKD research because they often have more advanced nephropathy manifestations than traditional induced models. Islet-related genetic changes can generate T1D-DKD models, such as Akita mice and OVE26 mice.
Akita Mouse Lines
Model Mechanism
Akita mice carry an autosomal dominant spontaneous point mutation (Ins2C96Y) in the pre-insulin gene (Ins2). This mutation leads to improper folding of insulin protein, resulting in proteotoxicity to pancreatic β-cells, which causes dysfunction and cell death.44
Mouse Strains
B6, DBA/2, 129/SvEv, KK/Ta, Balb/c.
Phenotypes
Akita mice can develop significant and persistent hyperglycemia at 4 week-old, with some degree of elevated blood pressure, then spontaneously progress to DKD.45 However, the severity of renal lesions is highly dependent on the mouse strain. The original line, B6-Ins2+/C96Y, exhibits moderate albuminuria (~2.8-fold increase over B6-WT) and mesangial expansion. Attempts were made to cross over the variant to strains more susceptible to nephropathy (eg, DBA/2, 129/SvEv and KK/Ta), and new Akita mouse lines have been successfully developed. In contrast to B6-Ins2+/C96Y, DBA/2-Ins2+/C96Y mice and 129/SvEv-Ins2+/C96Y mice show more pronounced albuminuria (3-fold and 5-fold relative change, respectively), but no exacerbation in renal pathology.44 Besides, male KK/Ta-Ins2+/C96Y mice show progressive nephrotic changes, with a marked increase in urinary albumin excretion started at ~5 weeks old and a more than 600-fold increase in urinary microalbumin/creatinine (ACR) compared to non-diabetic controls after 15 weeks old; an increase in GFR occurred at ~10 weeks old, presenting a glomerular hyperfiltration state, then a decline started at ~20 weeks old, a sign of disease progression to end-stage renal failure;46 their renal pathology reveals moderate mesangial dilatation, irregular thickening of GBM, glomerulosclerosis, small arteriolar hyalinosis and focal tubulointerstitial fibrosis.47 On the other hand, Yu et al found that induction of two insulin 2 allele mutations (Ins2C96Y/C96Y), ie, Akita knockout (KO), in Balb/c background allowed 80% of mice to survive for more than 6 months, unlike the other backgrounds in which most of the homozygotes died perinatally; moreover, Balb/c-Ins2C96Y/C96Y mice likewise develop more severe DKD than the original line, such as decreased GFR, a sustained increase in ACR (3-fold more than non-diabetic controls); the renal pathology is seen as mesangial dilatation, glomerulosclerosis and GBM thickening.48
Advantages and Limitations
These mice are advantageous in reflecting progressive nephropathology. Unfortunately, none of the available Akita mouse lines show the characteristic changes of advanced DKD with diffuse and/or nodular glomerulosclerosis and a significant decrease in GFR (~50%).
OVE26 Mice
Model Mechanism
Overexpression (OE) of calcineurin in pancreatic β-cells under the regulation of the rat insulin 2 promoter causes cell damage and inadequate insulin secretion in OVE26 mice.49
Mouse Strain
FVB.
Phenotypes
These mice develop DM within the first weeks of life and can survive for more than a year without insulin treatment while remaining at near-normal weight.50 They can also develop DKD at an early age (~2 months old) and reproduce many features of late-stage lesions, such as marked renal hypertrophy, severe progressive albuminuria (70–140 times that of controls at 9 months old), marked thickening of GBM (~87% increase), diffuse and nodular mesangial matrix expansion, progressive decrease in GFR (to 83–53% of controls at 9 months old), and the appearance of PAS-positive glomerular nodules similar to Kimmelstiel-Wilson (KW) disease, the most typical lesion of human nodular glomerulosclerosis; additionally, tubular atrophy, interstitial monocytes infiltration and fibrosis can be seen in these mice.50 By performing UNx in 2-month-old OVE26 mice, Zheng et al described late-stage pathologic changes at 10 weeks postoperatively (~4.5-month-old) in the surgical group, comparable to the 8-month-old non-surgical group, implying that surgery greatly shortened the disease process so that the high mortality rate due to aging in older OVE26 mice could be avoided.51 Furthermore, it has been reported that, unlike most DKD models, female OVE26 mice present more severe nephropathy than males, such as more severe albuminuria, greater podocyte loss, additional fibrosis and significantly more inflammatory cell infiltration, although their blood glucose levels may be lower.52 Reduced circulating estradiol and renal estrogen receptors are responsible for this phenomenon.52
Advantages and Limitations
OVE26 mice are considered an accurate model of early-onset T1D with advanced kidney disease and are valuable in studying the mechanisms and treatment of diabetic renal failure. Additionally, they are advantageous in gender studies of DKD and an excellent model to elucidate the potential mechanisms by which females are sensitive to renal injury. Previous studies have shown normal calmodulin expression in their kidney tissue, suggesting that the kidney damage is caused by DM itself, which fits the natural pathogenesis of human T1D-DKD.50 However, some limitations of this model cannot be ignored: (i) transgenesis can only be achieved in FVB strain to obtain the desired characteristics; (ii) extreme and persistent hyperglycemia (≥600 mg/dl) does not correspond to the clinical reality; and (iii) any extent of arterial hyalinosis is not observed.
Animal Models of T2D-DKD
STZ Induced Animal Models of T2D-DKD
Model Mechanism
To induce T2D-DKD, STZ is often used in combination with nicotinamide (NAD), a derivative of vitamin B3, whose powerful antioxidant capacity protects β-cells partially against the toxic damage from STZ and keeps animals from developing absolute insulin deficiency.15 The elevated blood glucose in these models is associated with a reduction in β-cell mass and insulin secretion.
Mouse Strains
Wistar rats, B6 mice.
Phenotypes
Both rat and mouse models with NAD + STZ induction develop moderate levels of hyperglycemia and albuminuria. DKD symptoms typically appear after 30–45 days, for example, mild mesangial expansion, stromal hyperplasia and fibrosis.53–61 Kishore et al described that after 30 days of NAD + STZ induction, serum total cholesterol (TC), triglyceride (TG), low-density lipoproteins (LDL), and very low-density lipoproteins (VLDL) significantly increased in DKD rats.58 However, the findings of lipid disorders in these models are not consistent.
Advantages and Limitations
These models are less time-consuming and less expensive than that created by genetic engineering or high-calorie diets feeding and have been widely used for testing the therapeutic effects of numerous anti-diabetic or renoprotective materials.62 They are non-obese that may provide a unique platform for the study of non-obese DKD. However, these models do not develop insulin resistance and glucose intolerance, which is partly different from the pathogenesis of human T2D-DKD.
Diet Induced Animal Models of T2D-DKD
Model Mechanism
It is widely accepted that T2D is driven by the interaction of genetic factors and poor lifestyle. Unhealthy dietary structure such as high fat (especially referring to high trans fatty acids and low unsaturated fatty acids) and high carbohydrate are important contributors to the development of T2D. Meanwhile, excessive obesity (IBM ≥ 30 kg/m2) is considered the strongest independent risk factor for T2D.63 Obesity induces cellular stress and metabolic syndrome that includes insulin resistance, glucose intolerance, dyslipidemia and hypertension. Oxidative stress and macrophage interaction-mediated cellular inflammation exacerbate damage to renal lipid metabolism, proteins and DNA, subsequently leading to DKD.64 Diet-induced models replicate unhealthy dietary patterns in humans, making it close to the pathogenesis of T2D-DKD. High-calorie diets include various dietary structures, for example, high-fat diet (HFD), high-sugar-high-fat diet (HsuHF), high-sugar diet (HsuD) and high-fat-high-sugar-high-cholesterol diet (HFHsuHC); the first two are the most common among the dietary regimens for inducing T2D-DKD. There is no unified international standard for the exact fat content and composition in HFD. Generally, HFD is considered a diet structure in which 40–60% of total food energy is provided by fat. It should be noted that the fat mainly refers to animal fats and vegetable oils containing ω-6/ω-9 fatty acids, but not fish oils or vegetable glycerols, because the latter two do not cause obesity, metabolic disorders and insulin resistance and even have certain benefits for animals.65,66
Species and Strains
B6 mice, Chinese Bama minipigs.
Phenotypes
Among rodents, B6 mice are more responsive to HFD. Male HFD-fed (≥12 weeks) B6 mice demonstrate metabolic disorders like obesity, hyperglycemia, hyperinsulinemia and dyslipidemia, and develop early nephrotic manifestations such as proteinuria, glomerular hypertrophy, mesangial expansion, thickening of GBM dominated by increased fibronectin and collagen, tubular dilatation and vacuolization, renal lipid deposition and inflammatory infiltration.64,67,68 Prolonged dietary interventions have been attempted to induce more severe nephropathy; unfortunately, even when the HFD-feeding time is increased to 32 weeks, advanced pathological changes are still not detected in B6 mice.69 In contrast, high-calorie diets (HFD or HSuHF) do not induce significant nephropathy in rats, but only mild renal endothelial dysfunction, mesangial expansion and GBM thickening,70 therefore, STZ + HFD or operating on genetic rats (eg, ZDF, ZSF1) would be a better means to generate relevant rat models. Pigs are very promising in DKD research because of their similarity to humans in anatomy, circadian rhythms and metabolism. In addition, omnivorous pigs are more closely resembling humans in terms of diet structure and feeding behavior. Liu et al found that after feeding HSuHF or HFHsuHC (10% lard, 37% sucrose and 2% cholesterol in 51% normal minipig feed) for 5 months, Chinese Bama minipigs developed metabolic syndrome and islet destruction; their kidneys demonstrated ectopic lipid deposition, glomerular hypertrophy, podocyte fusion, basement membrane thickening, mesangial expansion, moderate glomerulosclerosis and tubulointerstitial fibrosis, with production of microalbuminuria.71,72 However, severe hyperglycemia is rarely seen in these minipig models.73–75
Advantages and Limitations
These models simulate the pathogenesis of most human T2D-DKD and also are excellent for studying metabolic disorders, potential mechanisms of insulin resistance, as well as dietary intervention therapies. However, the majority of mice and rats are resistant to the development of obesity, DM and associated nephropathy induced by high-calorie diets alone, so a combination with other means is often required to accelerate disease progression, such as: (i) low-dose STZ to partially destroy β-cells and render islets dysfunctional; (ii) use of genetic animals; and (iii) nephrectomy to reduce kidney mass.
STZ + HFD Induced Animal Models of T2D-DKD
Model Mechanism
Insulin resistance and β-cell dysfunction are two major features of T2D-DKD.63 HFD induces metabolic disorders, while STZ reduces β-cell mass, ultimately leading to T2D-DKD in animals.
Species and Strains
SD rats, Wistar rats, B6 mice.
Phenotypes
Male SD or Wistar rats receive a single low-dose STZ (30–35 mg/kg, i.p.) after being fed HFD or HSuHF for 4–6 weeks, and almost all of these rats develop significant hyperglycemia and proteinuria after induction, with early renal pathology such as glomerular hypertrophy, mesangial expansion, extracellular matrix (ECM) deposition and mild glomerulosclerosis.76–79 In male B6 mice, T2D-DKD can be induced by HFD-feeding with a single low-dose STZ (40 mg/kg, i.p.), or medium-dose STZ (100–120 mg/kg, i.p.), or multiple low-dose STZ (40–50 mg/kg, i.p., for 5 days).80–86 These mice can develop glomerulosclerosis and tubulointerstitial fibrosis, which is not observed in HFD induced alone. However, Glastras et al noted that one dose STZ cannot aggravate the altered renal pathology in B6 mice.87
Advantages and Limitations
The addition of STZ to dietary intervention to reduce β-cell mass can further mimic the features as well as accelerate the progression of kidney disease. As many of the key pathological characteristics of human DKD are missing in the rat models induced by HFD alone, the regimen of STZ + HFD is more commonly used. Both rats and mice have more severe hyperglycemia in this regimen, but signs of obesity and insulin resistance (ie, hyperinsulinemia) are almost less pronounced than HFD induction alone, which may be related to STZ-mediated glucose metabolism reduction, protein/lipid catabolism excess and insulin release reduction.79,88–90
UNx + STZ + HFD Induced Animal Models of T2D-DKD
Model Mechanism
The mechanism of UNx, STZ and HFD inducing DKD was described previously. There is no uniformity in the induction sequence and specific contents of these three, where high-fat diet can be used either throughout the experimental cycle or after UNx and/or STZ to induce metabolic disorders and insulin resistance.
Species and Strains
SD rats, Wistar rats, B6 mice.
Phenotypes
Sugano et al found diffuse glomerulosclerosis at week 25 of the experiment in male SD rats treated with a single low-dose STZ (40 mg/kg, i.p.), followed by UNx and HFD intervention after 9 and 23 days, respectively.91 Similar results were described by Bayrasheva et al in male Wistar rats treated with another procedure, ie, HFD started 3 weeks after UNx, then NAD (230 mg/kg, i.p.) and high-dose STZ (65 mg/kg, i.p.) administrated at week 8.32 The renal tissue of these rats showed a significant enlargement of the mesangium and sclerosis in both glomerulus and tubulointerstitium at week 30 of the experiment. Further, it has been suggested that NAD + high-dose STZ results in more stable metabolic syndrome in rats compared to low-dose STZ.32,91 Dusabimana et al performed HFD-feeding after UNx on B6 mice, and then administered a single medium-dose STZ (100mg/kg, i.p.) 3 weeks after surgery; at the end of the experiment, these mice developed varying degrees of hyalinosis, glomerular atrophy, capsule shrinkage and chronic glomerulonephritis, with a significant decrease in the number of podocytes, also, they developed serious interstitial injury, including tubular dilatation, loss and necrosis of brush border, ECM accumulation and interstitial fibrosis.92
Advantages and Limitations
The above studies suggest that the protocol of UNx + STZ + HFD accelerates the course of nephropathy in rodents. Nevertheless, the phenotypic findings of these models are not consistent while advanced renal pathological changes are not fully accessible in most other similar researches.
Spontaneous Animal Models of T2D-DKD
Models in this category are established by breeding animals that develop DKD due to genetic abnormalities. These models are reliable because they have similar pathogenesis and renal abnormalities to humans. However, their use in current DKD research is not as widespread as other types because of relative difficulty in feeding and breeding, long modeling period and high cost. Otsuka Long-Evans Tokushima Fatty (OLETF) rats and Goto Kakizaki (GK) rats are the more popular strains among them.
OLETF Rats
Phenotypes
OLETF rats are a spontaneous T2D-DKD model characterized by (i) diabetic phenotypes inherited only in males; (ii) having a transition from non-insulin-dependent DM (NIDDM) to insulin-dependent DM (IDDM), ie, islet cell proliferation and peripheral blood insulin resistance lead to hyperglycemia and hyperinsulinemia at early stage, while pancreatic β-cells begin to deteriorate and eventually develop hypoinsulinemia and IDDM after 40-week-old. (iii) early mild obesity with hyperphagia and weight loss began after 28-week-old; (iv) hyperlipidemia; (v) DM occurred around 18–25-week-old, and blood glucose maintained above 200 mg/dL after ~32-week-old; (vi) progressive microalbuminuria appeared at ~16-week-old, and renal pathological changes started at ~23-week-old and worsened at ~29-week-old with segmental glomerulopathy, further, mesangial expansion, GBM thickening and glomerulosclerosis emerged above 40-week-old, which fully develop into diabetic glomerulopathy with tuberous sclerosis after 65 week-old, in addition to renal hypertrophy at ~55-weeks-old.93–96
Advantages and Limitations
The progression of DKD and renal pathological changes in OLETF rats highly resemble human T2D-DKD, both exhibiting exudative glomerulopathy characterized by fibrin cap, decreased capsule membrane and aneurysmal dilatation of intra-glomerular vessels.95 However, OLETF rats have relatively mild hyperglycemia and late onset of DKD. More importantly, later islet β-cells destruction may require insulin-dependent survival. Thus, OLETF rats are mainly used for early T2D-DKD studies at present.
GK Rats
Phenotypes
GK rats are a non-obese model of early T2D-DKD, produced from Wistar rats by repeatedly selective breeding over many generations using glucose intolerance as an indicator of selection.97 DM in GK rats results from a combination of several pathogenic mechanisms, including impaired individual development of islet cells, abnormal insulin release after glucose loading, insulin resistance, basal hyperinsulinemia, and abnormal glucose metabolism.98–102 These rats typically develop moderately persistent hyperglycemia at 3–4 weeks old, with a glycemic range of 12.9–17.0 mmol/L.103,104 Their renal lesions are mild, with basement membrane thickening and glomerular hypertrophy seen in early and middle life (statistically significant at 35 weeks of age); Interstitial monocyte/macrophage infiltration increases with progression of nephropathy.104,105 It is not until late in life (24 months of age) that a mild increase in urinary protein excretion as well as significant renal pathological changes can be observed: segmental glomerulosclerosis, mesangial proliferation, also, tubulointerstitial fibrosis, atrophy and dilatation and inflammatory cell infiltration.105
Advantages and Limitations
Unlike most other animal models of T2D-DKD, GK rats do not develop obesity, hypertension and hyperlipidemia, thus allowing a more direct study of the relationship between long-term hyperglycemia and DKD without the interference of other risk factors. Since the development of proteinuria in these rats shows an age-related rather than a glycemic correlation in early to middle stage, and the increasing trend of proteinuria is not significant across all ages, this model may be more appropriate for studying early non-proteinuric T2D-DKD in humans.103–105 In addition, administration of deoxycorticosterone acetate (DOCA) or angiotensinǁ (Angǁ) + high-salt diet to superimpose a hypertensive phenotype can lead to progressive proteinuria and glomerulosclerosis in these rats.103,106 Besides, HFD-feeding can worsen their glucose metabolism.107 Consequently, GK rats seem to be an ideal model to study the effects of other risk factors, especially hypertension, on diabetic renal function, kidney morphology and pathological progression.
Genetic Animal Models of T2D-DKD
There are various genetic animal models of T2D-DKD, among them, the models correlated with leptin signaling pathway abnormalities, such as ob/ob mice, db/db mice, Zucker diabetic fat (ZDF) rats and obese ZSF1 rats, as well as some special transgenic models like KKAy/Ta mice, are the most frequently used.
Ob/Ob Mice
Model Mechanism
Ob/ob mice are characterized by a single autosomal recessive mutation (Lepob/ob) on the obesity gene encoding leptin, resulting in a complete lack of leptin production and subsequent hyperphagia, obesity, hyperlipidemia, hyperglycemia and insulin resistance.108
Mouse Strains
For current DKD research, the mutation is mostly induced under the black and tan, brachyuric (BTBR) mouse strain which shows congenital insulin resistance, because other backgrounds, such as B6-ob/ob mice and C57BLKS/J (BKS)-ob/ob mice, are not ideal in the development of nephropathy, although they can develop hyperglycemia.109
Phenotypes
Insulin resistance is detectable in these mice at 4 weeks of age, with islet hypertrophy, hyperinsulinemia and persistent hyperglycemia (up to 350–400 mg/dl), followed by podocyte loss and progressive proteinuria at 8 weeks of age, and mesangial expansion at 10 weeks of age; the nephropathy in BTBR-ob/ob mice is progressive and comes into advanced stage at ~18–20 weeks of age, manifested by >10-fold increase in proteinuria, diffuse/nodular mesangial sclerosis, focal small arterial hyalinization, mesangial vascular lysis, increased mesangial matrix, GBM thickening and focal mild interstitial fibrosis.110 In addition, high protein diet (HPD, 30% kcal protein) feeding can exacerbate glomerular pathology and interstitial fibrosis, and thus shorten the modeling time.111
Advantages and Limitations
BTBR-ob/ob mice are precious because they come close to meeting all the proposed criteria of AMDCC and largely reproduce the basic structural changes and functional features of human diabetic glomerular injury, where high mesangial dilatation and interstitial lysis are uncommon in other animal models. Meanwhile, their renal tissues show large aggregates of monocytes/macrophages, similar to humans, which is valid for studying the relationship between inflammation and DKD. Furthermore, continuous infusion of leptin into BTBR-ob/ob mice via an osmotic minipump can completely reverse the late-stage lesions, making it possible to consider leptin replacement as a strategy to study the underlying reversible mechanisms of DKD, as well as providing ideas for regression.112 Nevertheless, the greatest limitations of this model are strain dependence and leptin deficiency. BTBR-ob/ob mice are sterile, so breeding to sufficient numbers for experimental use requires a lot of time and money. More importantly, pronounced leptin deficiency is not a feature of human diabetes, which limits the overall similarity of these mice to the clinical reality.
Db/Db Mice
Model Mechanism
A single autosomal recessive mutation (LepRdb/db) in the leptin receptor gene (LepR) from Gly to Thr causes defects in the hypothalamic leptin signaling pathway in these mice, followed by obesity, insulin resistance and T2D. Unlike ob/ob mice, db/db mice exhibit circulating leptin excess and leptin resistance.
Mouse Strains
The mutation was first identified in BKS strain which possesses 84% of the B6 allele and 16% of the DBA/2J allele and is more sensitive to the development of DKD.113,114 By far, BKS strain is the most widely used for db mutations, followed by B6, DBA/2J and FVB. Recently, db/db mice were also successfully established in fibrosis-prone CD1 background.115
Phenotypes
In contrast to B6-db/db mice with mild hyperglycemia and marked hyperinsulinemia, BKS-db/db mice show severe progressive hyperglycemia and moderate hyperinsulinemia at 8 weeks of age due to atrophy and necrosis of partial islet β-cells and reduced pancreatic hormone secretion, with peak glucose levels up to 44 mM at ~16 weeks of age; their urinary albumin excretion is significantly increased at ~8 weeks of age; renal pathological changes mainly occur after 12 weeks of age, manifested by renal hypertrophy, glomerulomegaly, expansion of the mesangial matrix, loss of podocytes and thickening of GBM; arterial hyaline degeneration is occasionally seen in older mice (~21 weeks of age), in addition, tubular lesions are dominated by vacuolization.116–119 Males possess more severe hyperglycemia and nephropathy than females.116 However, due to the slow progression of nephropathy and mild pathological changes in BKS-db/db mice, attempts have been made to address this problem by combining other induction modalities or using susceptible strains. It has been shown that HFD (60% kcal fat) leads to renal failure, as well as early death in BKS-db/db mice, while HPD (60% kcal protein) significantly exacerbates glomerular and tubular pathological alterations without increasing mortality.120 HPD-fed BKS-db/db mice develop glomerulosclerosis and tubulointerstitial fibrosis, although the glycemic rise is reduced.121 Similar pathological results can be found in db/db mice post-UNx or in female FVB-db/db mice.122–124 In contrast, despite higher albuminuria levels, DBA/2J-db/db mice do not differ significantly from BKS-db/db mice on glucose levels and renal pathology.117 CD1-db/db mice are a novel mouse model of T2D-DKD with progressive renal fibrosis and cardiac fibrosis. Males appear to have elevated blood glucose levels and decreased fasting insulin levels compared with those of BKS-db/db mice; plasma cystatin C levels tend to be elevated in CD1-db/db mice from 16 to 24-weeks‐of‐age; moreover, males show significantly progressive kidney and heart fibrosis from 16 to 24-weeks‐of‐age, while weak fibrosis in females; however, these mice do not develop nodular lesions or vascular lysis.115 The CD1 mouse is an outbred strain and therefore could show genetic diversity that could produce an unstable phenotype in the kidney as compared with inbred strains, such as the BKS strain.115
Advantages and Limitations
Db/db mice are one of the most widely used models of T2D-DKD. The relatively consistent high albuminuria levels and significant mesangial matrix expansion make them a good model for studying early T2D-DKD. However, leptin resistance is a rare disorder in human T2D-DKD, also, the presence of immune complex deposits in the glomeruli and the non-persistent increase in albuminuria are not consistent with human phenotypes.115,116
ZDF Rats
Model Mechanism
ZDF rats, also known as fa/fa rats, are produced by selective breeding of Zucker rats with leptin receptor mutations (LepR fa/fa).
Rat Strain
Zucker (male).
Phenotypes
Only males develop T2D and DKD. Their physiological and metabolic characteristics, such as obesity, dyslipidemia, abnormal glucose tolerance, insulin resistance, hypertension and progressive renal injury, are similar to those of humans.125 Meanwhile, their impaired and reduced numbers of β-cells may be caused by glucose/lipid toxicity.125,126 The combination of reduced insulin secretion and insulin resistance results in marked DM in male ZDF rats at ~8 weeks of age, with progressive elevation of blood glucose (200–500 mg/dL).125,127,128 Significant albuminuria occurs after 12 weeks of age; then, after 20–22 weeks of age, mesangial matrix expansion, focal segmental glomerulosclerosis, tubular atrophy and dilatation, and tubulointerstitial fibrosis can be observed.127–130 In addition, HFD (60% kcal fat) can exacerbate nephropathy in these rats and shorten the experimental duration.130,131
Advantages and Limitations
ZDF rats are a widely used model in studies of metabolic syndrome and diabetes and can also develop some degree of nephropathy. However, ZDF rats have obvious shortcomings: (i) as the disease progresses, both sexes develop unexplained severe hydronephrosis unrelated to human DKD and lead to severe complications including renal abscess, pyelonephritis and necrotizing granuloma, which makes the cause of nephropathy complex and difficult to explain;125 (ii) whether animal models show a high degree of similarity to human disease at the molecular levels is one of the criteria for judging their merit, and Siwy et al found that ZDF rats are more consistent with human peptide regulation in cardiovascular disease (CVD) than in chronic kidney disease (CKD) at the molecular levels, suggesting that they may be a more appropriate model for human CVD rather than CKD.132
Obese ZSF1 Rats
Model Mechanism
The obese ZSF1 rat (ZSF1 fa/facp) is the F1 offspring produced by crossing lean female ZDF (ZDF +/fa) rats with lean male spontaneously hypertensive heart failure (SHHF/Mcc-facp, +/fa) rats.133
Rat Strains
Female ZDF × Male SHHF.
Phenotypes
In contrast to ZDF rats, obese ZSF1 rats do not develop hydronephrosis and have more severe hypertension with cardiac hypertrophy and atherosclerosis. They exhibit DM, metabolic syndrome and renal damage at ~8 weeks of age; their renal disease is progressively worse, proceeding to late-stage of DKD after ~44 weeks of age and eventually developing into ESRD; most of them die at 12 months of age.133–135
Advantages and Limitations
Obese ZSF1 rats almost fulfill the standards suggested by AMDCC and have similarities to human DKD in terms of glomerular gene expression changes.134,136 Therefore, they are considered as an excellent preclinical translational model of T2D-DKD and a reasonable model to study the effects of comprehensive risk factors (ie, hyperglycemia, obesity, hyperlipidemia and hypertension) on renal function.
KKAy/Ta Mice
Model Mechanism
KKAy/Ta mice are established by introducing the yellow obese Ay gene into the KK/Ta strain which is congenitally glucose intolerance and insulin resistant. KK/Ta mice spontaneously develop a dominant diabetic state with age or consumption of high calorie diet, manifesting as mild obesity, polyphagia, compensatory hyperinsulinemia, hyperglycemia, urine glucose and microalbuminuria, which are more severe in males.137 Therefore, male KK/Ta mice are commonly used in the study of metabolic syndrome and obesity-related DM. Traditional KK/Ta mice with a long period of DKD development and insignificant phenotypic characteristics do not have an advantage among the many models available today, so they are more often used as blank controls or to provide a susceptible genetic background, such as KK/Ta-Akita (described previously) and KKAy/Ta.
Mouse Strain
KK/Ta.
Phenotypes
KKAy/Ta mice spontaneously appear obesity, polyphagia, glucose intolerance, insulin resistance, hyperinsulinemia, impaired insulin secretion and hyperlipidemia.138–141 They develop hyperglycemia with predominantly random blood glucose (RBG) elevation (22.6–25.9 mmol/L) at ~12 weeks of age; their fasting blood glucose (FBG) is mildly raised and not significantly different from non-diabetic controls after 20 weeks of age; progressive proteinuria appears after 8 weeks of age and increases significantly after 12 weeks of age; renal tissue shows significant pathological changes at ~20 weeks of age with diffuse mesangial expansion, GBM thickening and segmental sclerosis.140,141 Moreover, chronic tubulointerstitial injury, for example, varying degrees of interstitial fibrosis and tubular atrophy, nodular glomerular hyaline degeneration and focal segmental/global glomerulosclerosis, can be seen at ~40 weeks of age.139 HFD exacerbates FBG and metabolic disturbances, as well as accelerates the progression of nephropathy in KKAy/Ta mice.138
Advantages and Limitations
KKAy/Ta mice are a powerful polygenic spontaneous model of T2D-DKD; however, their hyperglycemia and lipid metabolic disturbances show some degree of spontaneous recovery after 16 weeks of age; also, proteinuria increases <10-fold and no statistically significant differences in microalbuminuria are observed in these mice.138,140 In the absence of additional intervention, these mice do not show the characteristics of advanced DKD.
T2DN/Mcwi (T2DN) Rats
Model Mechanism
The T2DN rat is a transgenic subline of GK rat, developed by introducing the mitochondrial and some passenger loci genes from the Fawn Hooded Hypertensive (FHH) rat into the GK strain.142
Rat Strain
T2DN/Mcwi.
Phenotypes
Despite their high genetic similarity (97% at locus 681) and similar glucose and blood pressure, nephropathy manifests differently between T2DN and GK rats. T2DN rats have hypercholesterolemia and hypertriglyceridemia, and develop albuminuria at 6 months of age with renal histological abnormalities such as focal glomerulosclerosis, mesangial matrix expansion, and GBM thickening. These features develop over time, so that almost all T2DN rats show a ˃ 10-fold increase in albuminuria, ˃ 50% decrease in GFR, severe diffuse alloglomerulosclerosis, KW-like nodules, arterial hyalinization, tubular necrosis, interstitial inflammation, and fibrosis at 18 months old.142–144
Advantages and Limitations
The evolution of nephropathy in T2DN rats from 3 to 18 months old resembles the natural progression of human DKD, encompassing almost all histological features, and is, therefore well suited to test the effectiveness of new therapeutic approaches to delay disease progression. Furthermore, T2DN rats are significantly sexually dimorphic, with females showing no insulin resistance, hyperlipidemia, milder glycemic and nephrotic phenotypes,145 suggesting that this model may be a valuable tool in the study of sex differences in T2D-DKD.
Complication Models of DKD
Animal Models of DKD with High Blood Pressure
eNOS−/−Db/Db Mice
Model Mechanism
Endothelial nitric oxide synthase (eNOS, NOS III) is closely associated with the development and progression of human DKD. Inhibitory activity or genetic deficiency of eNOS reduces nitric oxide (NO) synthesis, which plays an important role in controlling the integrity of interendothelial junctions (IEJs) and regulating vascular permeability.146,147 Increased production of NO synthesized by eNOS in the early-stage of human DKD may lead to renal vasodilation, hyperfiltration, and microalbuminuria.148,149 However, with disease progression, there is a decrease in renal eNOS, leading to activation of the renin-angiotensin-aldosterone system (RAAS), elevated tissue factor (TF, factor III) levels and excessive activation of the exogenous coagulation that promotes renal thrombosis.150 Severe proteinuria, decreased renal function and hypertension are also considered to correlate with progressive NO deficiency.148,151 Animal models of DKD have been generated through genetic engineering to induce eNOS deficiency. eNOS KO can make mice more prone to develop typical and severe DKD with a moderate hypertensive phenotype, although not able to have a significant effect on blood glucose levels.152–162
Mouse Strain
BKS.
Phenotypes
The eNOS−/−/LepRdb/db double KO is used to induce late-stage T2D-DKD. This model inherits obesity, high fasting blood glucose (FBG), insulin resistance and hypercholesterolemia of the db/db parents, as well as the moderate hypertension and diminished vascular repair of the eNOS−/− parents.153 Despite having lower FBG than db/db mice, eNOS−/−/db/db mice exhibit higher and gradually increasing levels of urinary albumin at 8 weeks of age and have a progressive glomerular pathology similar to that of human T2D-DKD. The pathology initially involves GBM thickening and mesangial dilatation, followed by mesangial lysis leading to microaneurysms, repeated vascular injury leading to nodular lesions similar to KW lesions, and eventually diffuse/nodular glomerulosclerosis; rare fibrin thrombi in glomerular arteries and occasionally arterial hyaline degeneration are also found in these mice.153 Their early elevated GFR decreases significantly at 20 weeks of age162 and by more than 50% at 26 weeks of age.152 Their tubular damage is severe with tubular dilatation, cast formation, epithelial flattening, and increased apoptosis, but their tubular fibrosis is relatively mild.152,153
Advantages and Limitations
eNOS−/−db/db mice are a robust animal model of advanced DKD with stable hypertension. On the other hand, Stec’s team has confirmed that these mice in the BKS background exhibit urinary metabolome changes similar to those seen in human DKD making them a useful tool for pathological metabolomic studies.163 However, eNOS−/−db/db mice have a short lifespan and typically die between 24 and 28 weeks of age without insulin treatment.152,162
Animal Models of DKD with Atherogenesis
The development of DM and DKD is closely related to atherosclerosis. Chronic hyperglycemia-induced injuries, such as increased intravascular ROS production, enhanced inflammatory state, lipid oxidation, and compositional changes, including elevated total cholesterol, total triglycerides, chylomicron/VLDL and intermediate density lipoprotein (IDL)/LDL ratios, can not only accelerate endothelial damage, but also promote plaque formation and smooth muscle cell proliferation, subsequently leading to atherosclerotic lesions in diabetic patients.164 Clinically, the majority of T2D-DKD patients have macroangiopathy as the disease progresses. Atherosclerosis, especially intrarenal atherosclerosis, is one of the main causes of impairing renal function.165 In addition, the superimposed effect of nephropathy and atherosclerosis greatly increases the cardiovascular risk and mortality of patients. However, the mechanism of interaction between these two conditions is not clear; therefore, animal models with accurate translation of these phenotypes are urgently needed.
IDOL-AAV-DJ/8-Ob/Ob Mice
Model Mechanism
IDOL is a key substance in reducing LDL receptor and thereby increasing LDL cholesterol (LDL-C) and total cholesterol levels. It targets the LDL receptor ubiquitin-mediated lysosomal degradation and increases LDL-C levels when overexpressed, which promotes atherogenesis.166
Mouse Strain
BTBR.
Phenotypes
After injecting male BTBR-ob/ob mice with the AAV-DJ/8 viral genome containing IDOL mutants to overexpress IDOL under the control of the liver-specific human thyroxine-binding globulin promoter, and feeding these animals a semi-purified, high-fat, cholesterol-containing diet (40% kcal fat and 1.25% kcal cholesterol) post-injection to promote atherosclerosis, Bornfeldt’s team successfully generated a new T2D-DKD model with atherosclerosis.166 Compared to BTBR-ob/ob controls, this model exhibits higher blood cholesterol (non-HDL-C), plasma triglyceride and urinary albumin levels, more severe glomerular macrophage accumulation and local inflammation; also, it shows early atherosclerosis formation – aortic smooth muscle fibrous cap lesions containing Mac-2-positive macrophages.166 The rest of phenotypes of this model are similar to those of BTBR-ob/ob mice.
Advantages and Limitations
IDOL-AAV-DJ/8-ob/ob mice provide a new tool to study the mechanisms of interaction between T2D-DKD and atherosclerosis, as well as to develop new therapeutic strategies. It should be noted that the mortality rate of BTBR-ob/ob mice increases after 24 weeks old, and studies should be performed before this age. On the other hand, atherosclerosis in the BTBR strain is relatively mild, and lesions are only observed in the aorta. It remains to be explored whether other strains that are more susceptible to atherosclerosis, such as the B6 strain, can generate relevant models.
Animal Models of Non-Proteinuric DKD
The concept of nonproteinuric DKD was first proposed by Tsalamandris et al.167 This type of DKD is common in clinical practice and manifests as normal-range proteinuria, ie, ACR < 30 mg/g or UAER < 30 mg/day, but with renal insufficiency. It is now commonly referred to as normoalbuminuric DKD (NADKD), nonalbuminuric diabetic nephropathy, or DKD without albuminuria.165 The latest diagnostic criteria for DKD proposed by the American Diabetes Association (ADA) in 2015 includes UAER > 30 mg/24h or eGFR <60 mL/(min⋅1.73 m2).168 Therefore, DM patients with normal-range proteinuria and renal insufficiency meet the criteria for DKD. In recent years, numerous studies have shown that atherosclerosis and tubulointerstitial damage (but not glomerular damage) are strongly associated with NADKD.165,169
HFD-Fed ApoE−/− Mice
Model Mechanism
Unlike classical DKD models that revolve around urinary albumin and glomerular lesions, the typical pathologies of the NADKD model should be focus on atherosclerosis, renal insufficiency, and tubulointerstitial damage. ApoE is a polymorphic protein that is an important ligand for apolipoproteins, which have an elimination receptor-mediated role. In the absence of ApoE, cholesterol accumulates in the vessel wall, causing thickening and sclerosis, as well as reduced elasticity and lumen narrowing, further developing into atherosclerosis.170 HFD not only causes metabolic disturbances in ApoE−/− mice, dominated by significantly elevated levels of triglycerides, total cholesterol, and LDL, but also promotes the development of atherosclerosis.
Mouse Strain
B6.
Phenotypes
Tomita’s team crossed 8-week-old female and male ApoE+/- mice to produce ApoE−/− mice. Subsequently, 10-week-old male ApoE−/− mice were selected and fed with HFD (60% kcal fat) for 24 weeks to generate the NADKD model.169 Compared to normal diet (ND)-fed ApoE+/+ mice, HFD-fed ApoE−/− mice exhibit obesity-related hyperglycemia, aortic atherosclerosis, decreased CD31-positive peritubular capillary density and renal blood flow, and renal dysfunction defined by high levels of plasma cystatin-C at ~34-week-old; their Renal pathology reveals glomerular hypertrophy and mesangial expansion with fibrosis; however, podocyte ultrastructure is essentially normal with mild proteinuria.169 In contrast, these mice have severe proximal tubular epithelial cell damage with follicular alterations, apoptosis, as well as tubulointerstitial fibrosis and inflammation.169
Advantages and Limitations
HFD-fed ApoE−/− mice are considered to successfully reflect the renal phenotypes of human NADKD. However, renal dysfunction in these mice is mild and it is not clear whether the lesions are progressive. Therefore, it is preferable to use these mice for studies in the early-stage of the disease.
Conclusion
There are various animal models of DKD available, but no single model can represent all the features of human DKD. Therefore, it is essential to choose the appropriate model based on the research needs, as different models develop different phenotypes and indispensably have their limitations. The desired positive results may not be obtained if the animal model does not fit the experiment, which can be avoided in principle. To address this issue, this paper provides a comprehensive overview of the biochemical and histological phenotypes of DKD animal models, based on the key features of human DKD and the criteria of AMDCC (Tables 1–3, respectively). Generally, STZ-induced models and Akita mice are the most commonly used for T1D-DKD studies, while db/db mice for T2D-DKD studies. A few animal models can develop advanced DKD phenotypes, among them, OVE26 mice, T2DN rats, eNOS−/− db/db mice appear to be the most potent. Additionally, female OVE26 mice seem to be advantageous in the study of female DKD, while T2DN rats seem to be suitable for exploring sex differences in T2D-DKD. However, it is also important to pay attention to the adaptability of these models, as some classical genetic mutant models of DKD may no longer exhibit severe renal pathological changes due to adaptive changes.
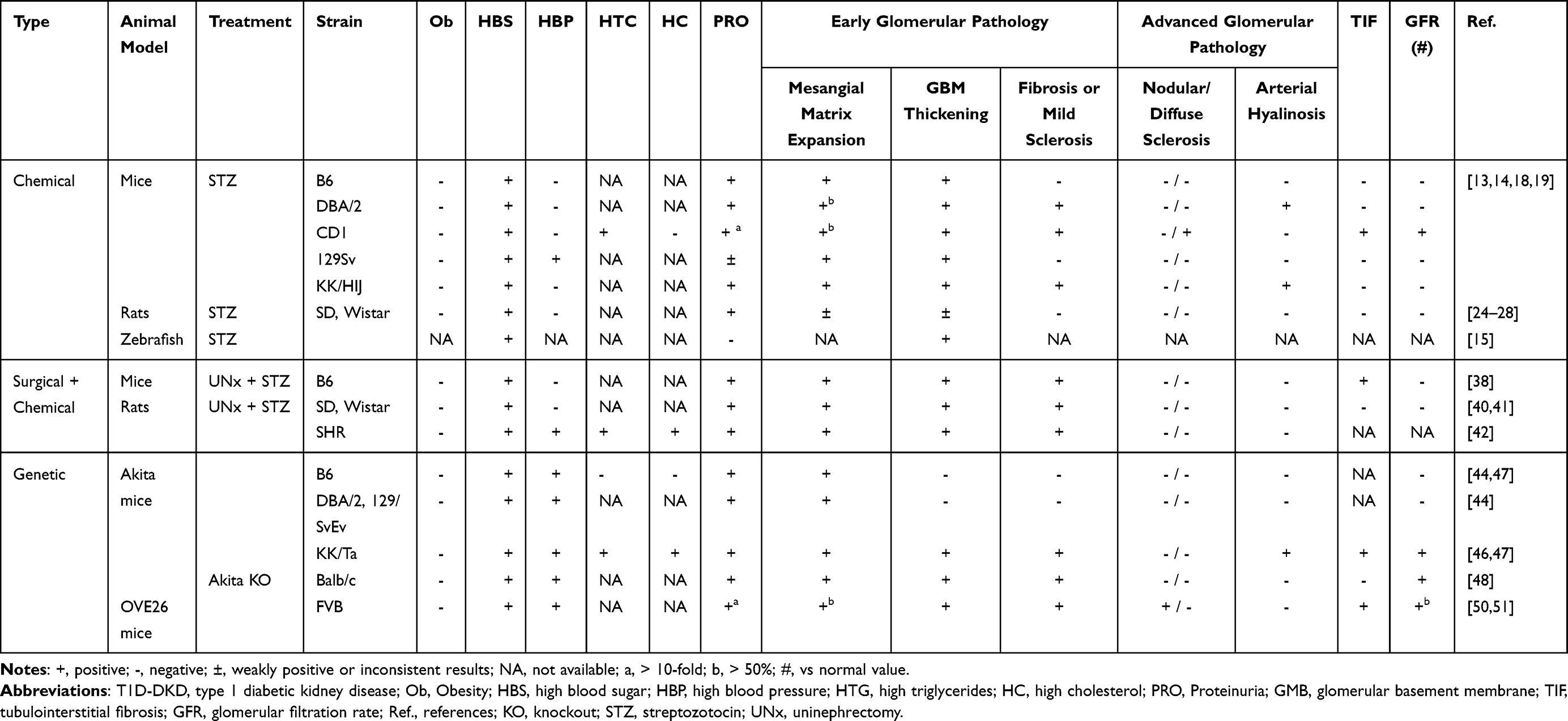 |
Table 1 Biochemical and Histological Phenotypes in Translation Animal Models of T1D-DKD |
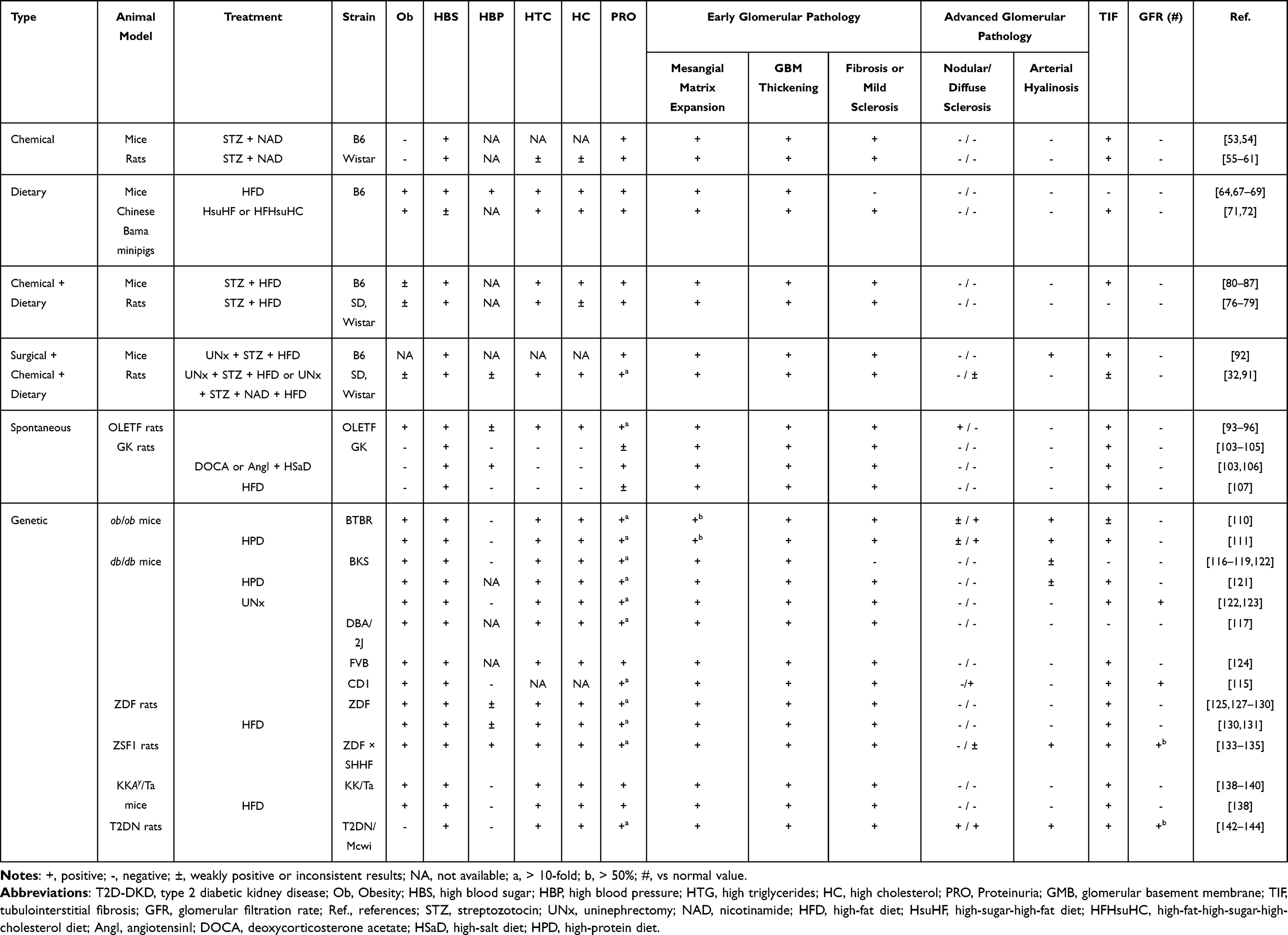 |
Table 2 Biochemical and Histological Phenotypes in Translation Animal Models of T2D-DKD |
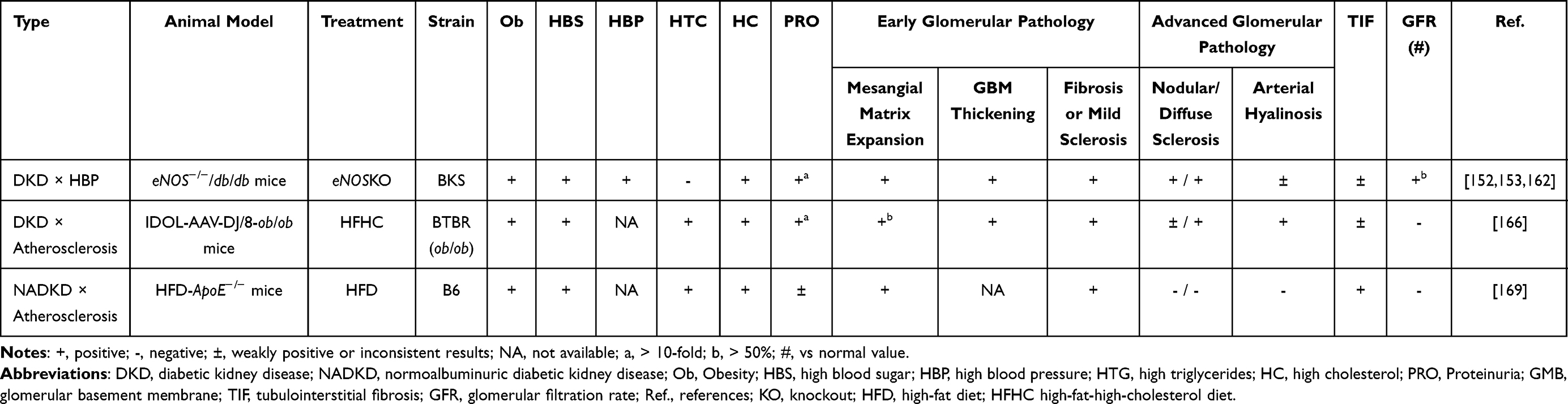 |
Table 3 Biochemical and Histological Phenotypes in Complication Models of DKD and NADKD |
Induction methods are an essential part of DKD modeling. This paper summarizes routine laboratory procedures for DKD (Figures 1 and 2). Standardizing modeling is crucial to improve the success rate and model stability, save resources and make experimental data more reliable. Furthermore, exploration traditional induction means and searching for new induction tools, such as adenovirus (AV), adeno-associated virus (AAV), double-stranded adeno-associated virus (dsAAV), EntransterTM-in vivo, etc., could be the focus of future studies.
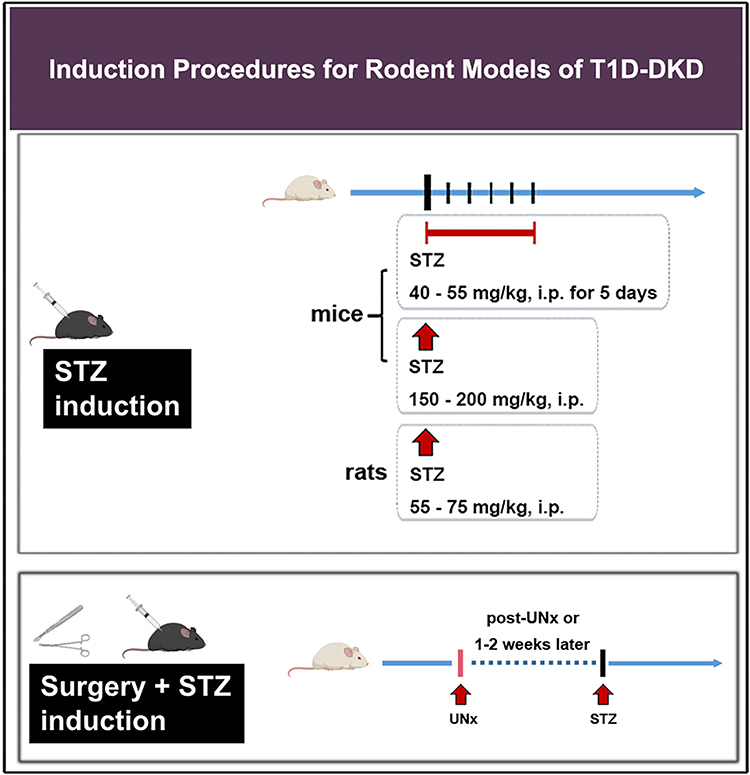 |
Figure 1 Induction procedures for rodent models of T1D-DKD. Abbreviations: T1D-DKD, type 1 diabetic kidney disease; STZ, streptozotocin; UNx, uninephrectomy; i.p. intraperitoneal injection. |
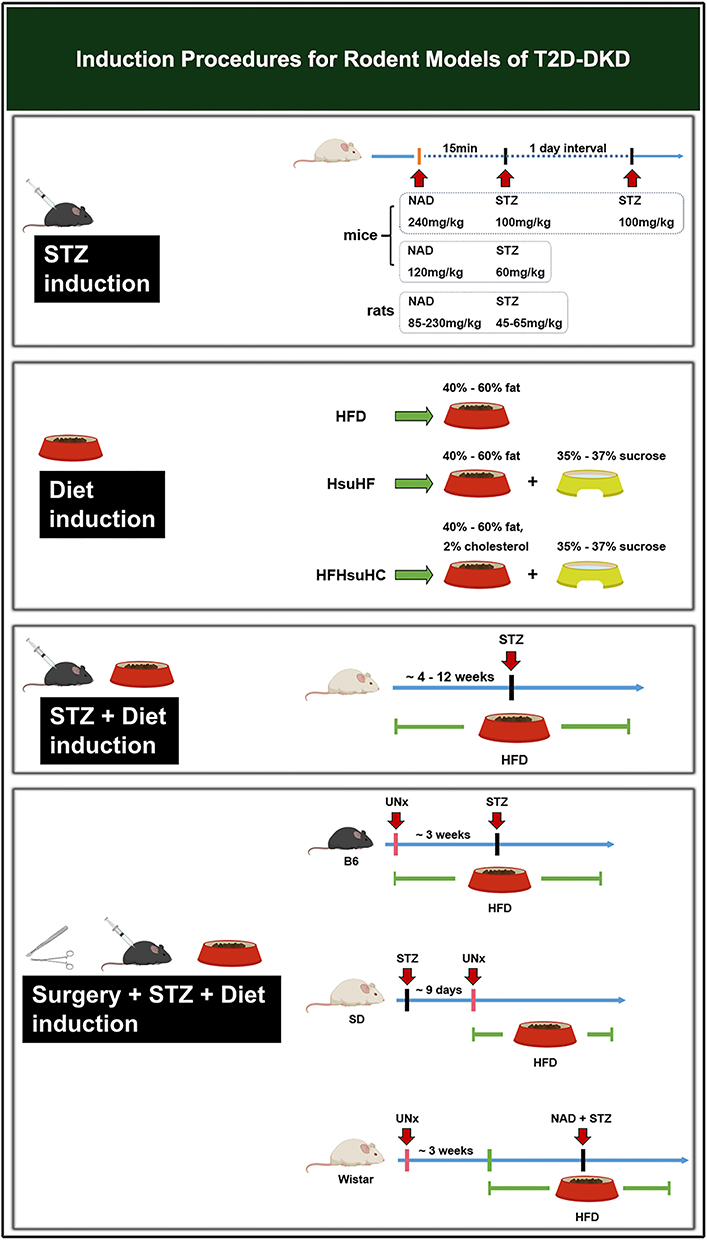 |
Figure 2 Induction procedures for rodent models of T2D-DKD. Abbreviations: T2D-DKD, type 2 diabetic kidney disease; NAD, nicotinamide; STZ, streptozotocin; UNx, uninephrectomy; i.p. intraperitoneal injection; HFD, high-fat diet; HsuHF, high-sugar-high-fat diet; HFHsuHC, high-fat-high-sugar-high-cholesterol diet. |
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was funded by National Natural Science Foundation of China, grant number 82004351.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Akhtar M, Taha NM, Nauman A, Mujeeb IB, Al-Nabet A. Diabetic kidney disease: past and present. Adv Anat Pathol. 2020;27(2):87–97. doi:10.1097/PAP.0000000000000257
2. Groop PH, Thomas MC, Moran JL, et al. The presence and severity of chronic kidney disease predicts all-cause mortality in type 1 diabetes. Diabetes. 2009;58(7):1651–1658. doi:10.2337/db08-1543
3. Afkarian M, Sachs MC, Kestenbaum B, et al. Kidney disease and increased mortality risk in type 2 diabetes. J Am Soc Nephrol. 2013;24(2):302–308. doi:10.1681/ASN.2012070718
4. Orchard TJ, Secrest AM, Miller RG, Costacou T. In the absence of renal disease, 20 year mortality risk in type 1 diabetes is comparable to that of the general population: a report from the Pittsburgh Epidemiology of Diabetes Complications Study. Diabetologia. 2010;53(11):2312–2319. doi:10.1007/s00125-010-1860-3
5. Thomas MC, Cooper ME, Zimmet P. Changing epidemiology of type 2 diabetes mellitus and associated chronic kidney disease. Nat Rev Nephrol. 2016;12(2):73–81. doi:10.1038/nrneph.2015.173
6. Perkovic V, Jardine MJ, Neal B, et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med. 2019;380(24):2295–2306. doi:10.1056/NEJMoa1811744
7. Khan SS, Quaggin SE. Therapies on the horizon for diabetic kidney disease. Curr Diab Rep. 2015;15(12):111. doi:10.1007/s11892-015-0685-3
8. Hu YY, Ye SD. Experimental models of type 2 diabetic nephropathy. Chin Med J. 2013;126(3):574–577.
9. Lee TN, Alborn WE, Knierman MD, Konrad RJ. The diabetogenic antibiotic streptozotocin modifies the tryptic digest pattern for peptides of the enzyme O-GlcNAc-selective N-acetyl-beta-d-glucosaminidase that contain amino acid residues essential for enzymatic activity. Biochem Pharmacol. 2006;72(6):710–718. doi:10.1016/j.bcp.2006.06.005
10. Szkudelski T. The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol Res. 2001;50(6):537–546.
11. Furman BL. Streptozotocin-Induced Diabetic Models in Mice and Rats. Curr Protoc Pharmacol. 2015;70(1):547 1–547 20. doi:10.1002/0471141755.ph0547s70
12. Dufrane D, van Steenberghe M, Guiot Y, Goebbels RM, Saliez A, Gianello P. Streptozotocin-induced diabetes in large animals (pigs/primates): role of GLUT2 transporter and beta-cell plasticity. Transplantation. 2006;81(1):36–45. doi:10.1097/01.tp.0000189712.74495.82
13. Sugimoto H, Grahovac G, Zeisberg M, Kalluri R. Renal fibrosis and glomerulosclerosis in a new mouse model of diabetic nephropathy and its regression by bone morphogenic protein-7 and advanced glycation end product inhibitors. Diabetes. 2007;56(7):1825–1833. doi:10.2337/db06-1226
14. Tesch GH, Allen TJ. Rodent models of streptozotocin-induced diabetic nephropathy. Nephrology. 2007;12(3):261–266. doi:10.1111/j.1440-1797.2007.00796.x
15. Betz B, Conway BR. An update on the use of animal models in diabetic nephropathy research. Curr Diab Rep. 2016;16(2):18. doi:10.1007/s11892-015-0706-2
16. Zamboni F, Cengiz IF, Barbosa AM, et al. Towards the development of a female animal model of T1DM using hyaluronic acid nanocoated cell transplantation: refinements and considerations for future protocols. Pharmaceutics. 2021;13(11). doi:10.3390/pharmaceutics13111925
17. Gurley SB, Clare SE, Snow KP, Hu A, Meyer TW, Coffman TM. Impact of genetic background on nephropathy in diabetic mice. Am J Physiol Renal Physiol. 2006;290(1):F214–F222. doi:10.1152/ajprenal.00204.2005
18. Zheng C, Huang L, Luo W, et al. Inhibition of STAT3 in tubular epithelial cells prevents kidney fibrosis and nephropathy in STZ-induced diabetic mice. Cell Death Dis. 2019;10(11):848. doi:10.1038/s41419-019-2085-0
19. Liu XQ, Jiang L, Li YY, et al. Wogonin protects glomerular podocytes by targeting Bcl-2-mediated autophagy and apoptosis in diabetic kidney disease. Acta Pharmacol Sin. 2022;43(1):96–110. doi:10.1038/s41401-021-00721-5
20. Zheng X, Soroush F, Long J, et al. Murine glomerular transcriptome links endothelial cell-specific molecule-1 deficiency with susceptibility to diabetic nephropathy. PLoS One. 2017;12(9):e0185250. doi:10.1371/journal.pone.0185250
21. Liu H, Takagaki Y, Kumagai A, Kanasaki K, Koya D. The PKM2 activator TEPP-46 suppresses kidney fibrosis via inhibition of the EMT program and aberrant glycolysis associated with suppression of HIF-1α accumulation. J Diabetes Investig. 2021;12(5):697–709. doi:10.1111/jdi.13478
22. Li J, Liu H, Takagi S, et al. Renal protective effects of empagliflozin via inhibition of EMT and aberrant glycolysis in proximal tubules. JCI Insight. 2020;5(6). doi:10.1172/jci.insight.129034
23. Kanasaki K, Shi S, Kanasaki M, et al. Linagliptin-mediated DPP-4 inhibition ameliorates kidney fibrosis in streptozotocin-induced diabetic mice by inhibiting endothelial-to-mesenchymal transition in a therapeutic regimen. Diabetes. 2014;63(6):2120–2131. doi:10.2337/db13-1029
24. Ilatovskaya DV, Blass G, Palygin O, et al. A NOX4/TRPC6 pathway in podocyte calcium regulation and renal damage in diabetic kidney disease. J Am Soc Nephrol. 2018;29(7):1917–1927. doi:10.1681/ASN.2018030280
25. Jiang Y, Xie F, Lv X, et al. Mefunidone ameliorates diabetic kidney disease in STZ and db/db mice. FASEB J. 2021;35(1):e21198. doi:10.1096/fj.202001138RR
26. Malik S, Suchal K, Khan SI, et al. Apigenin ameliorates streptozotocin-induced diabetic nephropathy in rats via MAPK-NF-kappaB-TNF-alpha and TGF-beta1-MAPK-fibronectin pathways. Am J Physiol Renal Physiol. 2017;313(2):F414–F422. doi:10.1152/ajprenal.00393.2016
27. Xiang E, Han B, Zhang Q, et al. Human umbilical cord-derived mesenchymal stem cells prevent the progression of early diabetic nephropathy through inhibiting inflammation and fibrosis. Stem Cell Res Ther. 2020;11(1):336. doi:10.1186/s13287-020-01852-y
28. Zhu L, Han J, Yuan R, Xue L, Pang W. Berberine ameliorates diabetic nephropathy by inhibiting TLR4/NF-kappaB pathway. Biol Res. 2018;51(1):9. doi:10.1186/s40659-018-0157-8
29. Giralt-Lopez A, Molina-Van den Bosch M, Vergara A, et al. Revisiting experimental models of diabetic nephropathy. Int J Mol Sci. 2020;21(10). doi:10.3390/ijms21103587
30. Tay YC, Wang Y, Kairaitis L, Rangan GK, Zhang C, Harris DC. Can murine diabetic nephropathy be separated from superimposed acute renal failure? Kidney Int. 2005;68(1):391–398. doi:10.1111/j.1523-1755.2005.00405.x
31. Kraynak AR, Storer RD, Jensen RD, et al. Extent and persistence of streptozotocin-induced DNA damage and cell proliferation in rat kidney as determined by in vivo alkaline elution and BrdUrd labeling assays. Toxicol Appl Pharmacol. 1995;135(2):279–286. doi:10.1006/taap.1995.1234
32. Bayrasheva VK, Babenko AY, Dobronravov VA, et al. Uninephrectomized high-fat-fed nicotinamide-streptozotocin-induced diabetic rats: a model for the investigation of diabetic nephropathy in type 2 diabetes. J Diabetes Res. 2016;2016:8317850. doi:10.1155/2016/8317850
33. Schnaper HW. Remnant nephron physiology and the progression of chronic kidney disease. Pediatr Nephrol. 2014;29(2):193–202. doi:10.1007/s00467-013-2494-8
34. Taal MW, Brenner BM. Renoprotective benefits of RAS inhibition: from ACEI to angiotensin II antagonists. Kidney Int. 2000;57(5):1803–1817. doi:10.1046/j.1523-1755.2000.00031.x
35. Wu LL, Cox A, Roe CJ, Dziadek M, Cooper ME, Gilbert RE. Transforming growth factor beta 1 and renal injury following subtotal nephrectomy in the rat: role of the renin-angiotensin system. Kidney Int. 1997;51(5):1553–1567. doi:10.1038/ki.1997.214
36. Abrass CK. Diabetic nephropathy. Mechanisms of mesangial matrix expansion. West J Med. 1995;162(4):318–321.
37. Mezzano SA, Ruiz-Ortega M, Egido J. Angiotensin II and renal fibrosis. Hypertension. 2001;38(3 Pt 2):635–638. doi:10.1161/hy09t1.094234
38. Uil M, Scantlebery AML, Butter LM, et al. Author correction: combining streptozotocin and unilateral nephrectomy is an effective method for inducing experimental diabetic nephropathy in the ‘resistant’ C57Bl/6J mouse strain. Sci Rep. 2019;9(1):3425. doi:10.1038/s41598-018-38075-4
39. Wada J, Zhang H, Tsuchiyama Y, et al. Gene expression profile in streptozotocin-induced diabetic mice kidneys undergoing glomerulosclerosis. Kidney Int. 2001;59(4):1363–1373. doi:10.1046/j.1523-1755.2001.0590041363.x
40. Mao ZM, Shen SM, Wan YG, et al. Huangkui capsule attenuates renal fibrosis in diabetic nephropathy rats through regulating oxidative stress and p38MAPK/Akt pathways, compared to α-lipoic acid. J Ethnopharmacol. 2015;173:256–265. doi:10.1016/j.jep.2015.07.036
41. Liu BC, Huang HQ, Luo DD, Ma KL, Liu DG, Liu H. Connective tissue growth factor is associated with the early renal hypertrophy in uninephrectomized diabetic rats. Chin Med J. 2006;119(12):1010–1016.
42. Inada Y, Murakami M, Tazawa S, Akahane M. KRH-594, a new angiotensin AT1 receptor antagonist, ameliorates nephropathy and hyperlipidaemia in diabetic spontaneously hypertensive rats. Clin Exp Pharmacol Physiol. 2000;27(4):270–276. doi:10.1046/j.1440-1681.2000.03235.x
43. Li D, Lu Z, Jia J, Zheng Z, Lin S. MiR-124 is related to podocytic adhesive capacity damage in STZ-induced uninephrectomized diabetic rats. Kidney Blood Press Res. 2013;37(4–5):422–431. doi:10.1159/000355721
44. Gurley SB, Mach CL, Stegbauer J, et al. Influence of genetic background on albuminuria and kidney injury in Ins2(+/C96Y) (Akita) mice. Am J Physiol Renal Physiol. 2010;298(3):F788–F795. doi:10.1152/ajprenal.90515.2008
45. Susztak K, Raff AC, Schiffer M, Böttinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55(1):225–233.
46. Jerums G, Premaratne E, Panagiotopoulos S, MacIsaac RJ. The clinical significance of hyperfiltration in diabetes. Diabetologia. 2010;53(10):2093–2104. doi:10.1007/s00125-010-1794-9
47. Fujita H, Fujishima H, Chida S, et al. Reduction of renal superoxide dismutase in progressive diabetic nephropathy. J Am Soc Nephrol. 2009;20(6):1303–1313. doi:10.1681/asn.2008080844
48. Yu L, Su Y, Paueksakon P, et al. Integrin α1/Akita double-knockout mice on a Balb/c background develop advanced features of human diabetic nephropathy. Kidney Int. 2012;81(11):1086–1097. doi:10.1038/ki.2011.474
49. Epstein PN, Overbeek PA, Means AR. Calmodulin-induced early-onset diabetes in transgenic mice. Cell. 1989;58(6):1067–1073. doi:10.1016/0092-8674(89)90505-9
50. Zheng S, Noonan WT, Metreveli NS, et al. Development of late-stage diabetic nephropathy in OVE26 diabetic mice. Diabetes. 2004;53(12):3248–3257. doi:10.2337/diabetes.53.12.3248
51. Zheng S, Huang Y, Yang L, Chen T, Xu J, Epstein PN. Uninephrectomy of diabetic OVE26 mice greatly accelerates albuminuria, fibrosis, inflammatory cell infiltration and changes in gene expression. Nephron Exp Nephrol. 2011;119(1):e21–e32. doi:10.1159/000327586
52. Wang W, Jiang S, Tang X, et al. Sex differences in progression of diabetic nephropathy in OVE26 type 1 diabetic mice. Biochim Biophys Acta Mol Basis Dis. 2020;1866(1):165589. doi:10.1016/j.bbadis.2019.165589
53. Hsu JD, Wu CC, Hung CN, Wang CJ, Huang HP. Myrciaria cauliflora extract improves diabetic nephropathy via suppression of oxidative stress and inflammation in streptozotocin-nicotinamide mice. J Food Drug Anal. 2016;24(4):730–737. doi:10.1016/j.jfda.2016.03.009
54. Perez Gutierrez RM, Garcia Campoy AH, Paredes Carrera SP, Muniz Ramirez A, Mota Flores JM, Flores Valle SO. 3’-O-beta-d-glucopyranosyl-alpha,4,2’,4’,6’-pentahydroxy-dihydrochalcone, from bark of eysenhardtia polystachya prevents diabetic nephropathy via inhibiting protein glycation in STZ-nicotinamide induced diabetic mice. Molecules. 2019;24(7). doi:10.3390/molecules24071214
55. Abd El Motteleb DM, Abd El Aleem DI. Renoprotective effect of Hypericum perforatum against diabetic nephropathy in rats: insights in the underlying mechanisms. Clin Exp Pharmacol Physiol. 2017;44(4):509–521. doi:10.1111/1440-1681.12729
56. Chandran R, George BP, Abrahamse H, Parimelazhagan T. Therapeutic effects of Syzygium mundagam bark methanol extract on type-2 diabetic complications in rats. Biomed Pharmacother. 2017;95:167–174. doi:10.1016/j.biopha.2017.08.061
57. Corremans R, D’Haese PC, Vervaet BA, Verhulst A. L-NAME administration enhances diabetic kidney disease development in an STZ/NAD rat model. Int J Mol Sci. 2021;22(23). doi:10.3390/ijms222312767
58. Kishore L, Kaur N, Singh R. Renoprotective effect of Bacopa monnieri via inhibition of advanced glycation end products and oxidative stress in STZ-nicotinamide-induced diabetic nephropathy. Ren Fail. 2016;38(9):1528–1544. doi:10.1080/0886022X.2016.1227920
59. Maheshwari RA, Balaraman R, Sen AK, Seth AK. Effect of coenzyme Q10 alone and its combination with metformin on streptozotocin-nicotinamide-induced diabetic nephropathy in rats. Indian J Pharmacol. 2014;46(6):627–632. doi:10.4103/0253-7613.144924
60. Rai U, Kosuru R, Prakash S, Tiwari V, Singh S. Tetramethylpyrazine alleviates diabetic nephropathy through the activation of Akt signalling pathway in rats. Eur J Pharmacol. 2019;865:172763. doi:10.1016/j.ejphar.2019.172763
61. Sathibabu Uddandrao VV, Brahmanaidu P, Ravindarnaik R, Suresh P, Vadivukkarasi S, Saravanan G. Restorative potentiality of S-allylcysteine against diabetic nephropathy through attenuation of oxidative stress and inflammation in streptozotocin-nicotinamide-induced diabetic rats. Eur J Nutr. 2019;58(6):2425–2437. doi:10.1007/s00394-018-1795-x
62. Yan LJ. The nicotinamide/streptozotocin rodent model of type 2 diabetes: renal pathophysiology and redox imbalance features. Biomolecules. 2022;12(9). doi:10.3390/biom12091225
63. Zheng Y, Ley SH, Hu FB. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol. 2018;14(2):88–98. doi:10.1038/nrendo.2017.151
64. Lee ES, Kwon MH, Kim HM, et al. Dibenzoylmethane ameliorates lipid-induced inflammation and oxidative injury in diabetic nephropathy. J Endocrinol. 2019;240(2):169–179. doi:10.1530/JOE-18-0206
65. Buettner R, Scholmerich J, Bollheimer LC. High-fat diets: modeling the metabolic disorders of human obesity in rodents. Obesity. 2007;15(4):798–808. doi:10.1038/oby.2007.608
66. Samane S, Christon R, Dombrowski L, et al. Fish oil and argan oil intake differently modulate insulin resistance and glucose intolerance in a rat model of dietary-induced obesity. Metabolism. 2009;58(7):909–919. doi:10.1016/j.metabol.2009.02.013
67. Sangartit W, Ha KB, Lee ES, et al. Tetrahydrocurcumin ameliorates kidney injury and high systolic blood pressure in high-fat diet-induced type 2 diabetic mice. Endocrinol Metab. 2021;36(4):810–822. doi:10.3803/EnM.2021.988
68. Lee ES, Kang JS, Kim HM, et al. Dehydrozingerone inhibits renal lipotoxicity in high-fat diet-induced obese mice. J Cell Mol Med. 2021;25(18):8725–8733. doi:10.1111/jcmm.16828
69. Locatelli M, Macconi D, Corna D, et al. Sirtuin 3 deficiency aggravates kidney disease in response to high-fat diet through lipotoxicity-induced mitochondrial damage. Int J Mol Sci. 2022;23(15). doi:10.3390/ijms23158345
70. Nunes S, Alves A, Preguica I, et al. Crescent-like lesions as an early signature of nephropathy in a rat model of prediabetes induced by a hypercaloric diet. Nutrients. 2020;12(4). doi:10.3390/nu12040881
71. Liu Y, Wang Z, Yin W, et al. Severe insulin resistance and moderate glomerulosclerosis in a minipig model induced by high-fat/ high-sucrose/ high-cholesterol diet. Exp Anim. 2007;56(1):11–20. doi:10.1538/expanim.56.11
72. Liu Y, Li H, Wang S, Yin W, Wang Z. Ibrolipim attenuates early-stage nephropathy in diet-induced diabetic minipigs: focus on oxidative stress and fibrogenesis. Biomed Pharmacother. 2020;129:110321. doi:10.1016/j.biopha.2020.110321
73. Bakker PJ, Butter LM, Kors L, et al. Nlrp3 is a key modulator of diet-induced nephropathy and renal cholesterol accumulation. Kidney Int. 2014;85(5):1112–1122. doi:10.1038/ki.2013.503
74. Panasevich MR, Meers GM, Linden MA, et al. High-fat, high-fructose, high-cholesterol feeding causes severe NASH and cecal microbiota dysbiosis in juvenile Ossabaw swine. Am J Physiol Endocrinol Metab. 2018;314(1):E78–E92. doi:10.1152/ajpendo.00015.2017
75. Feng Y, Yang S, Ma Y, Bai XY, Chen X. Role of Toll-like receptors in diabetic renal lesions in a miniature pig model. Sci Adv. 2015;1(5):e1400183. doi:10.1126/sciadv.1400183
76. Zhu Q, Li XH, Chen HY, Jin QY. The effects of compound centella formula on OxInflammation and silent information regulator 1 in a high-fat diet/streptozotocin-induced diabetic kidney disease rat model. Exp Ther Med. 2021;22(3):962. doi:10.3892/etm.2021.10394
77. Wang Z, Fu W, Huo M, et al. Spatial-resolved metabolomics reveals tissue-specific metabolic reprogramming in diabetic nephropathy by using mass spectrometry imaging. Acta Pharm Sin B. 2021;11(11):3665–3677. doi:10.1016/j.apsb.2021.05.013
78. Wu T, Yang X, Cong Y, et al. Effects of Qidantang Granule on early stage of diabetic kidney disease in rats. Aging. 2022;14(11):4888–4896. doi:10.18632/aging.204121
79. Chen HW, Yang MY, Hung TW, Chang YC, Wang CJ. Nelumbo nucifera leaves extract attenuate the pathological progression of diabetic nephropathy in high-fat diet-fed and streptozotocin-induced diabetic rats. J Food Drug Anal. 2019;27(3):736–748. doi:10.1016/j.jfda.2018.12.009
80. Huang W, Man Y, Gao C, et al. Short-chain fatty acids ameliorate diabetic nephropathy via GPR43-mediated inhibition of oxidative stress and NF-kappaB signaling. Oxid Med Cell Longev. 2020;2020:4074832. doi:10.1155/2020/4074832
81. Kim DH, Choi BH, Ku SK, Park JH, Oh E, Kwak MK. Beneficial effects of sarpogrelate and rosuvastatin in high fat diet/streptozotocin-induced nephropathy in mice. PLoS One. 2016;11(4):e0153965. doi:10.1371/journal.pone.0153965
82. Song W, Wei L, Du Y, Wang Y, Jiang S. Protective effect of ginsenoside metabolite compound K against diabetic nephropathy by inhibiting NLRP3 inflammasome activation and NF-kappaB/p38 signaling pathway in high-fat diet/streptozotocin-induced diabetic mice. Int ImmunopharmacoL. 2018;63:227–238. doi:10.1016/j.intimp.2018.07.027
83. Han J, Pang X, Shi X, Zhang Y, Peng Z, Xing Y. Ginkgo biloba extract EGB761 ameliorates the extracellular matrix accumulation and mesenchymal transformation of renal tubules in diabetic kidney disease by inhibiting endoplasmic reticulum stress. Biomed Res Int. 2021;2021:6657206. doi:10.1155/2021/6657206
84. Han YC, Tang SQ, Liu YT, et al. AMPK agonist alleviate renal tubulointerstitial fibrosis via activating mitophagy in high fat and streptozotocin induced diabetic mice. Cell Death Dis. 2021;12(10):925. doi:10.1038/s41419-021-04184-8
85. An X, Zhang Y, Cao Y, Chen J, Qin H, Yang L. Punicalagin protects diabetic nephropathy by inhibiting pyroptosis based on TXNIP/NLRP3 pathway. Nutrients. 2020;12(5). doi:10.3390/nu12051516
86. Li Y, Xue M, Hu F, et al. Klotho prevents epithelial-mesenchymal transition through Egr-1 downregulation in diabetic kidney disease. BMJ Open Diabetes Res Care. 2021;9(1). doi:10.1136/bmjdrc-2020-002038
87. Glastras SJ, Chen H, Teh R, et al. Mouse models of diabetes, obesity and related kidney disease. PLoS One. 2016;11(8):e0162131. doi:10.1371/journal.pone.0162131
88. Hakim ZS, Patel BK, Goyal RK. Effects of chronic ramipril treatment in streptozotocin-induced diabetic rats. Indian J Physiol Pharmacol. 1997;41(4):353–360.
89. Rajkumar L, Srinivasan N, Balasubramanian K, Govindarajulu P. Increased degradation of dermal collagen in diabetic rats. Indian J Exp Biol. 1991;29(11):1081–1083.
90. Rossmeisl M, Rim JS, Koza RA, Kozak LP. Variation in type 2 diabetes--related traits in mouse strains susceptible to diet-induced obesity. Diabetes. 2003;52(8):1958–1966. doi:10.2337/diabetes.52.8.1958
91. Sugano M, Yamato H, Hayashi T, et al. High-fat diet in low-dose-streptozotocin-treated heminephrectomized rats induces all features of human type 2 diabetic nephropathy: a new rat model of diabetic nephropathy. Nutr Metab Cardiovasc Dis. 2006;16(7):477–484. doi:10.1016/j.numecd.2005.08.007
92. Dusabimana T, Park EJ, Je J, et al. Geniposide improves diabetic nephropathy by enhancing ULK1-mediated autophagy and reducing oxidative stress through AMPK activation. Int J Mol Sci. 2021;22(4). doi:10.3390/ijms22041651
93. Kawano K, Hirashima T, Mori S, Saitoh Y, Kurosumi M, Natori T. Spontaneous long-term hyperglycemic rat with diabetic complications. Otsuka Long-Evans Tokushima Fatty (OLETF) strain. Diabetes. 1992;41(11):1422–1428. doi:10.2337/diab.41.11.1422
94. Shin JH, So BI, Song YS, et al. Histopathological analyses of diabetic nephropathy in sucrose-fed Otsuka Long-Evans Tokushima fatty rats. Endocr Res. 2015;40(1):29–36. doi:10.3109/07435800.2014.915848
95. Kawano K, Mori S, Hirashima T, Man ZW, Natori T. Examination of the pathogenesis of diabetic nephropathy in OLETF rats. J Vet Med Sci. 1999;61(11):1219–1228. doi:10.1292/jvms.61.1219
96. Nozako M, Koyama T, Nagano C, et al. An atherogenic paigen-diet aggravates nephropathy in type 2 diabetic OLETF rats. PLoS One. 2015;10(11):e0143979. doi:10.1371/journal.pone.0143979
97. Goto Y, Kakizaki M, Masaki N. Production of spontaneous diabetic rats by repetition of selective breeding. Tohoku J Exp Med. 1976;119(1):85–90. doi:10.1620/tjem.119.85
98. Movassat J, Saulnier C, Serradas P, Portha B. Impaired development of pancreatic beta-cell mass is a primary event during the progression to diabetes in the GK rat. Diabetologia. 1997;40(8):916–925. doi:10.1007/s001250050768
99. Ismail I, Lewis M, Peters JR, Scanlon MF. Hypothalamic mediation of reduced GH secretion in diabetic rats: evidence for reduced cholinergic inhibition of somatostatin release. J Neuroendocrinol. 1995;7(4):311–318. doi:10.1111/j.1365-2826.1995.tb00763.x
100. Ostenson CG, Khan A, Abdel-Halim SM, et al. Abnormal insulin secretion and glucose metabolism in pancreatic islets from the spontaneously diabetic GK rat. Diabetologia. 1993;36(1):3–8. doi:10.1007/bf00399086
101. Bisbis S, Bailbe D, Tormo MA, et al. Insulin resistance in the GK rat: decreased receptor number but normal kinase activity in liver. Am J Physiol. 1993;265(5 Pt 1):E807–E813. doi:10.1152/ajpendo.1993.265.5.E807
102. Picarel-Blanchot F, Berthelier C, Bailbé D, Portha B. Impaired insulin secretion and excessive hepatic glucose production are both early events in the diabetic GK rat. Am J Physiol. 1996;271(4 Pt 1):E755–E762. doi:10.1152/ajpendo.1996.271.4.E755
103. Janssen U, Riley SG, Vassiliadou A, Floege J, Phillips AO. Hypertension superimposed on type II diabetes in Goto Kakizaki rats induces progressive nephropathy. Kidney Int. 2003;63(6):2162–2170. doi:10.1046/j.1523-1755.2003.00007.x
104. Phillips AO, Baboolal K, Riley S, et al. Association of prolonged hyperglycemia with glomerular hypertrophy and renal basement membrane thickening in the goto kakizaki model of non–insulin-dependent diabetes mellitus. Am J Kidney Dis. 2001;37(2):400–410. doi:10.1053/ajkd.2001.21322
105. Sato N, Komatsu K, Kurumatani H. Late onset of diabetic nephropathy in spontaneously diabetic GK rats. Am J Nephrol. 2003;23(5):334–342. doi:10.1159/000072915
106. Olearczyk JJ, Quigley JE, Mitchell BC, et al. Administration of a substituted adamantyl urea inhibitor of soluble epoxide hydrolase protects the kidney from damage in hypertensive Goto–Kakizaki rats. Clin Sci. 2009;116(1):61–70. doi:10.1042/cs20080039
107. Kuwabara WMT, Panveloski-Costa AC, Yokota CNF, et al. Comparison of Goto-Kakizaki rats and high fat diet-induced obese rats: are they reliable models to study Type 2 diabetes mellitus? PLoS One. 2017;12(12):e0189622. doi:10.1371/journal.pone.0189622
108. Suriano F, Vieira-Silva S, Falony G, et al. Novel insights into the genetically obese (ob/ob) and diabetic (db/db) mice: two sides of the same coin. Microbiome. 2021;9(1):147. doi:10.1186/s40168-021-01097-8
109. Qi Z, Fujita H, Jin J, et al. Characterization of susceptibility of inbred mouse strains to diabetic nephropathy. Diabetes. 2005;54(9):2628–2637. doi:10.2337/diabetes.54.9.2628
110. Hudkins KL, Pichaiwong W, Wietecha T, et al. BTBR Ob/Ob mutant mice model progressive diabetic nephropathy. J Am Soc Nephrol. 2010;21(9):1533–1542. doi:10.1681/asn.2009121290
111. Björnson Granqvist A, Ericsson A, Sanchez J, et al. High-protein diet accelerates diabetes and kidney disease in the BTBR ob/ob mouse. Am J Physiol Renal Physiol. 2020;318(3):F763–F771. doi:10.1152/ajprenal.00484.2019
112. Alpers CE, Hudkins KL. Mouse models of diabetic nephropathy. Curr Opin Nephrol Hypertens. 2011;20(3):278–284. doi:10.1097/MNH.0b013e3283451901
113. Hummel KP, Dickie MM, Coleman DL. Diabetes, a new mutation in the mouse. Science. 1966;153(3740):1127–1128. doi:10.1126/science.153.3740.1127
114. Mao HZ, Roussos ET, Péterfy M. Genetic analysis of the diabetes-prone C57BLKS/J mouse strain reveals genetic contribution from multiple strains. Biochim Biophys Acta. 2006;1762(4):440–446. doi:10.1016/j.bbadis.2006.01.002
115. Mizunuma Y, Kanasaki K, Nitta K, et al. CD-1(db/db) mice: a novel type 2 diabetic mouse model with progressive kidney fibrosis. J Diabetes Investig. 2020;11(6):1470–1481. doi:10.1111/jdi.13311
116. Sharma K, McCue P, Dunn SR. Diabetic kidney disease in the db/db mouse. Am J Physiol Renal Physiol. 2003;284(6):F1138–F1144. doi:10.1152/ajprenal.00315.2002
117. Østergaard MV, Pinto V, Stevenson K, Worm J, Fink LN, Coward RJ. DBA2J db/db mice are susceptible to early albuminuria and glomerulosclerosis that correlate with systemic insulin resistance. Am J Physiol Renal Physiol. 2017;312(2):F312–F321. doi:10.1152/ajprenal.00451.2016
118. Boquist L, Hellman B, Lernmark A, Täljedal IB. Influence of the mutation “diabetes” on insulin release and islet morphology in mice of different genetic backgrounds. J Cell Biol. 1974;62(1):77–89. doi:10.1083/jcb.62.1.77
119. Meade CJ, Brandon DR, Smith W, Simmonds RG, Harris S, Sowter C. The relationship between hyperglycaemia and renal immune complex deposition in mice with inherited diabetes. Clin Exp Immunol. 1981;43(1):109–120.
120. Zhang HM, Dang H, Kamat A, Yeh CK, Zhang BX. Geldanamycin derivative ameliorates high fat diet-induced renal failure in diabetes. PLoS One. 2012;7(3):e32746. doi:10.1371/journal.pone.0032746
121. Nørgaard SA, Briand F, Sand FW, et al. Nephropathy in diabetic db/db mice is accelerated by high protein diet and improved by the SGLT2 inhibitor dapagliflozin. Eur J Pharmacol. 2019;860:172537. doi:10.1016/j.ejphar.2019.172537
122. Maekawa M, Maekawa T, Sasase T, et al. Pathophysiological analysis of uninephrectomized db/db mice as a model of severe diabetic kidney disease. Physiol Res. 2022;71(2):209–217. doi:10.33549/physiolres.934784
123. Aibara Y, Nakashima A, Kawano KI, et al. Daily low-intensity pulsed ultrasound ameliorates renal fibrosis and inflammation in experimental hypertensive and diabetic nephropathy. Hypertension. 2020;76(6):1906–1914. doi:10.1161/hypertensionaha.120.15237
124. Wang Z, Jiang T, Li J, et al. Regulation of renal lipid metabolism, lipid accumulation, and glomerulosclerosis in FVBdb/db mice with type 2 diabetes. Diabetes. 2005;54(8):2328–2335. doi:10.2337/diabetes.54.8.2328
125. Vora JP, Zimsen SM, Houghton DC, Anderson S. Evolution of metabolic and renal changes in the ZDF/Drt-fa rat model of type II diabetes. J Am Soc Nephrol. 1996;7(1):113–117. doi:10.1681/asn.V71113
126. Tokuyama Y, Sturis J, DePaoli AM, et al. Evolution of beta-cell dysfunction in the male Zucker diabetic fatty rat. Diabetes. 1995;44(12):1447–1457. doi:10.2337/diab.44.12.1447
127. Kubota M, Watanabe R, Yamaguchi M, et al. Rice endosperm protein slows progression of fatty liver and diabetic nephropathy in Zucker diabetic fatty rats. Br J Nutr. 2016;116(8):1326–1335. doi:10.1017/s0007114516003512
128. Wang Z, Liu Q, Dai W, Hua B, Li H, Li W. Pioglitazone downregulates Twist-1 expression in the kidney and protects renal function of Zucker diabetic fatty rats. Biomed Pharmacother. 2019;118:109346. doi:10.1016/j.biopha.2019.109346
129. Chander PN, Gealekman O, Brodsky SV, et al. Nephropathy in Zucker diabetic fat rat is associated with oxidative and nitrosative stress: prevention by chronic therapy with a peroxynitrite scavenger ebselen. J Am Soc Nephrol. 2004;15(9):2391–2403. doi:10.1097/01.Asn.0000135971.88164.2c
130. Kundu A, Gali S, Sharma S, et al. Tenovin-1 ameliorates renal fibrosis in high-fat-diet-induced diabetic nephropathy via antioxidant and anti-inflammatory pathways. Antioxidants. 2022;11(9). doi:10.3390/antiox11091812
131. Pugsley MK, Brooks MB, Fishman CE, et al. Use of the ZDF rat to model dietary fat induced hypercoagulability is limited by progressive and fatal nephropathy. J Pharmacol Toxicol Methods. 2021;107:106933. doi:10.1016/j.vascn.2020.106933
132. Siwy J, Zoja C, Klein J, et al. Evaluation of the Zucker diabetic fatty (ZDF) rat as a model for human disease based on urinary peptidomic profiles. PLoS One. 2012;7(12):e51334. doi:10.1371/journal.pone.0051334
133. Tofovic SP, Kusaka H, Kost CK, Bastacky S. Renal function and structure in diabetic, hypertensive, obese ZDFxSHHF-hybrid rats. Ren Fail. 2000;22(4):387–406. doi:10.1081/jdi-100100882
134. Dower K, Zhao S, Schlerman FJ, et al. High resolution molecular and histological analysis of renal disease progression in ZSF1 fa/faCP rats, a model of type 2 diabetic nephropathy. PLoS One. 2017;12(7):e0181861. doi:10.1371/journal.pone.0181861
135. Bilan VP, Salah EM, Bastacky S, et al. Diabetic nephropathy and long-term treatment effects of rosiglitazone and enalapril in obese ZSF1 rats. J Endocrinol. 2011;210(3):293–308. doi:10.1530/joe-11-0122
136. Zhang C, Dower K, Zhang B, Martinez RV, Lin LL, Zhao S. Computational identification and validation of alternative splicing in ZSF1 rat RNA-seq data, a preclinical model for type 2 diabetic nephropathy. Sci Rep. 2018;8(1):7624. doi:10.1038/s41598-018-26035-x
137. Taketomi S, Ikeda H, Ishikawa E, Iwatsuka H. Determination of overall insulin sensitivity in diabetic mice, KK. Horm Metab Res. 1982;14(1):14–18. doi:10.1055/s-2007-1018909
138. Liu Y, Huang H, Gao R, Dynamic Phenotypes LY. Molecular mechanisms to understand the pathogenesis of diabetic nephropathy in two widely used animal models of type 2 diabetes mellitus. Front Cell Dev Biol. 2020;8:172. doi:10.3389/fcell.2020.00172
139. Mori Y, Ajay AK, Chang JH, et al. KIM-1 mediates fatty acid uptake by renal tubular cells to promote progressive diabetic kidney disease. Cell Metab. 2021;33(5):1042–1061.e7. doi:10.1016/j.cmet.2021.04.004
140. Ito T, Tanimoto M, Yamada K, et al. Glomerular changes in the KK-Ay/Ta mouse: a possible model for human type 2 diabetic nephropathy. Nephrology. 2006;11(1):29–35. doi:10.1111/j.1440-1797.2006.00543.x
141. Oda K, Miyamoto S, Kodera R, Wada J, Shikata K. Suramin prevents the development of diabetic kidney disease by inhibiting NLRP3 inflammasome activation in KK-Ay mice. J Diabetes Investig. 2023;14(2):205–220. doi:10.1111/jdi.13930
142. Nobrega MA, Fleming S, Roman RJ, et al. Initial characterization of a rat model of diabetic nephropathy. Diabetes. 2004;53(3):735–742. doi:10.2337/diabetes.53.3.735
143. Kojima N, Slaughter TN, Paige A, Kato S, Roman RJ, Williams JM. Comparison of the development diabetic induced renal disease in strains of goto-kakizaki rats. J Diabetes Metab. 2013;Suppl 9(5). doi:10.4172/2155-6156.S9-005
144. Palygin O, Spires D, Levchenko V, et al. Progression of diabetic kidney disease in T2DN rats. Am J Physiol Renal Physiol. 2019;317(6):F1450–F1461. doi:10.1152/ajprenal.00246.2019
145. Spires DR, Palygin O, Levchenko V, et al. Sexual dimorphism in the progression of type 2 diabetic kidney disease in T2DN rats. Physiol Genomics. 2021;53(6):223–234. doi:10.1152/physiolgenomics.00009.2021
146. Sessa WC. eNOS at a glance. J Cell Sci. 2004;117(Pt 12):2427–2429. doi:10.1242/jcs.01165
147. Predescu D, Predescu S, Shimizu J, Miyawaki-Shimizu K, Malik AB. Constitutive eNOS-derived nitric oxide is a determinant of endothelial junctional integrity. Am J Physiol Lung Cell Mol Physiol. 2005;289(3):L371–L381. doi:10.1152/ajplung.00175.2004
148. Prabhakar SS. Role of nitric oxide in diabetic nephropathy. Semin Nephrol. 2004;24(4):333–344. doi:10.1016/j.semnephrol.2004.04.005
149. Hiragushi K, Sugimoto H, Shikata K, et al. Nitric oxide system is involved in glomerular hyperfiltration in Japanese normo- and micro-albuminuric patients with type 2 diabetes. Diabetes Res Clin Pract. 2001;53(3):149–159. doi:10.1016/s0168-8227(01)00260-1
150. Oe Y, Miyazaki M, Takahashi N. Coagulation, protease-activated receptors, and diabetic kidney disease: lessons from eNOS-deficient mice. Tohoku J Exp Med. 2021;255(1):1–8. doi:10.1620/tjem.255.1
151. Yamada K, Nakano H, Nakayama M, et al. Endothelium-dependent relaxation in peripheral vasculature and kidney of non-insulin-dependent diabetes mellitus. J Diabetes Complications. 1995;9(4):203–207. doi:10.1016/1056-8727(95)80002-v
152. Zhao HJ, Wang S, Cheng H, et al. Endothelial nitric oxide synthase deficiency produces accelerated nephropathy in diabetic mice. J Am Soc Nephrol. 2006;17(10):2664–2669. doi:10.1681/asn.2006070798
153. Mohan S, Reddick RL, Musi N, et al. Diabetic eNOS knockout mice develop distinct macro- and microvascular complications. Lab Invest. 2008;88(5):515–528. doi:10.1038/labinvest.2008.23
154. Nakagawa T, Sato W, Glushakova O, et al. Diabetic endothelial nitric oxide synthase knockout mice develop advanced diabetic nephropathy. J Am Soc Nephrol. 2007;18(2):539–550. doi:10.1681/asn.2006050459
155. Dschietzig TB, Krause-Relle K, Hennequin M, et al. Relaxin-2 does not ameliorate nephropathy in an experimental model of type-1 diabetes. Kidney Blood Press Res. 2015;40(1):77–88. doi:10.1159/000368484
156. Kosugi T, Heinig M, Nakayama T, Matsuo S, Nakagawa T. eNOS knockout mice with advanced diabetic nephropathy have less benefit from renin-angiotensin blockade than from aldosterone receptor antagonists. Am J Pathol. 2010;176(2):619–629. doi:10.2353/ajpath.2010.090578
157. Shi Y, Huang C, Zhao Y, et al. RIPK3 blockade attenuates tubulointerstitial fibrosis in a mouse model of diabetic nephropathy. Sci Rep. 2020;10(1):10458. doi:10.1038/s41598-020-67054-x
158. Harloff M, Prüschenk S, Seifert R, Schlossmann J. Activation of soluble guanylyl cyclase signalling with cinaciguat improves impaired kidney function in diabetic mice. Br J Pharmacol. 2022;179(11):2460–2475. doi:10.1111/bph.15425
159. Yuen DA, Stead BE, Zhang Y, et al. eNOS deficiency predisposes podocytes to injury in diabetes. J Am Soc Nephrol. 2012;23(11):1810–1823. doi:10.1681/asn.2011121170
160. Kanetsuna Y, Takahashi K, Nagata M, et al. Deficiency of endothelial nitric-oxide synthase confers susceptibility to diabetic nephropathy in nephropathy-resistant inbred mice. Am J Pathol. 2007;170(5):1473–1484. doi:10.2353/ajpath.2007.060481
161. Li F, Wang CH, Wang JG, et al. Elevated tissue factor expression contributes to exacerbated diabetic nephropathy in mice lacking eNOS fed a high fat diet. J Thromb Haemost. 2010;8(10):2122–2132. doi:10.1111/j.1538-7836.2010.03976.x
162. Li Y, Chung S, Li Z, et al. Fatty acid receptor modulator PBI-4050 inhibits kidney fibrosis and improves glycemic control. JCI Insight. 2018;3(10). doi:10.1172/jci.insight.120365
163. Stec DF, Wang S, Stothers C, et al. Alterations of urinary metabolite profile in model diabetic nephropathy. Biochem Biophys Res Commun. 2015;456(2):610–614. doi:10.1016/j.bbrc.2014.12.003
164. Jha JC, Ho F, Dan C, Jandeleit-Dahm K. A causal link between oxidative stress and inflammation in cardiovascular and renal complications of diabetes. Clin Sci. 2018;132(16):1811–1836. doi:10.1042/cs20171459
165. Chen C, Wang C, Hu C, et al. Normoalbuminuric diabetic kidney disease. Front Med. 2017;11(3):310–318. doi:10.1007/s11684-017-0542-7
166. Bornfeldt KE, Kramer F, Batorsky A, et al. A novel type 2 diabetes mouse model of combined diabetic kidney disease and atherosclerosis. Am J Pathol. 2018;188(2):343–352. doi:10.1016/j.ajpath.2017.10.012
167. Tsalamandris C, Allen TJ, Gilbert RE, et al. Progressive decline in renal function in diabetic patients with and without albuminuria. Diabetes. 1994;43(5):649–655. doi:10.2337/diab.43.5.649
168. American Diabetes Association. Standards of medical care in diabetes-2015 abridged for primary care providers. Clin Diabetes. 2015;33(2):97–111. doi:10.2337/diaclin.33.2.97
169. Tomita I, Kume S, Sugahara S, et al. SGLT2 inhibition mediates protection from diabetic kidney disease by promoting ketone body-induced mTORC1 inhibition. Cell Metab. 2020;32(3):404–419.e6. doi:10.1016/j.cmet.2020.06.020
170. Li Y, Zhang CG, Wang XH, Liu DH. Progression of atherosclerosis in ApoE-knockout mice fed on a high-fat diet. Eur Rev Med Pharmacol Sci. 2016;20(18):3863–3867.
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
The Mechanosensation-Metabolism-Inflammation Axis: The Central Role of Piezo1 and TRPV4 in Hypertension-Related Atherosclerosis
Mu YY, Liu TT, Xu JL, Li PC, Qian Y, Zhang W
International Journal of General Medicine 2026, 19:610549
Published Date: 13 May 2026