Back to Journals » Cancer Management and Research » Volume 12
Transcriptome-Wide 5-Methylcytosine Functional Profiling of Long Non-Coding RNA in Hepatocellular Carcinoma
Authors He Y, Shi Q, Zhang Y, Yuan X, Yu Z
Received 14 May 2020
Accepted for publication 19 July 2020
Published 4 August 2020 Volume 2020:12 Pages 6877—6885
DOI https://doi.org/10.2147/CMAR.S262450
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ahmet Emre Eşkazan
Yuting He,1,2,* Qingmiao Shi,1,2,* Yize Zhang,1,2,* Xin Yuan,1,2 Zujiang Yu1,2
1Gene Hospital of Henan Province, Precision Medicine Center, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan 450052, People’s Republic of China; 2Department of Infectious Diseases, The First Affiliated Hospital of Zhengzhou University, Zhengzhou, Henan 450052, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Zujiang Yu;Yuting He
The First Affiliated Hospital of Zhengzhou University, No. 1 Jianshe East Road, Erqi District, Zhengzhou City, Henan Province 450052, People’s Republic of China
Tel/ Fax +86-371-66271133
; +86-371-67967126
Email [email protected]; [email protected]
Background: Growing evidence indicates that methylation status is associated with the pathogenesis of numerous types of cancers. Among these, hepatocellular carcinoma (HCC) is a deadly disease threatening global human health. Although 5-methylcytosine (m5C) has been identified as an important regulatory modification, its distribution in solid tumors, including HCC, remains unclear. The present study aimed to explore the distribution of m5C in HCC.
Materials and Methods: Six pairs of human HCC tissues and adjacent non-tumor tissues were collected to analyze the transcriptome-wide m5C methylation of long non-coding RNA (lncRNA). RNA MeRIP-seq was performed to identify m5C peaks on lncRNA and differences in m5C distribution between HCC and adjacent tissues. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment pathway analyses were explored to predict the possible roles of m5C.
Results: Using m5C peak sequencing, we observed that a sequence motif was necessary for m5C methylation in HCC lncRNA. Unsupervised hierarchical cluster analysis confirmed that lncRNA m5C methylation occurred more frequently in HCC than adjacent non-tumor tissues. RNA sequencing data demonstrated that more genes were up-regulated by methylation in HCC, while methylation down-regulated more genes in adjacent non-tumor tissues. GO and KEGG pathway analyses revealed that genes having a significant correlation with m5C sites in lncRNA were involved in HCC signaling pathways.
Conclusion: Our results revealed the substantially different amounts and distributions of m5C in HCC compared to adjacent non-tumor tissue. We further predicted the cellular functions in HCC that m5C may participate in to provide evidence implicating m5C lncRNA epigenetic regulation in the tumorigenesis and progression in HCC.
Keywords: m5C, lncRNA, hepatocellular carcinoma, tumorigenesis
Introduction
Posttranscriptional RNA modifications have emerged as an additional layer of epigenetic regulation.1 Over one hundred different chemical modifications in RNA have been described, including pseudouridine, N6-methyladenosine (m6A),2 N1-methyladenosine (m1A),3 and 5-methylcytosine (m5C).4 Accumulating evidence suggests that RNA modifications play critical roles across a variety of biological functions that can contribute to disease pathogenesis. For instance, m6A modification of Wilms tumor 1-associated protein (WTAP) positively correlates with liver cancer oncogenesis through the HuR-ETS1-p21/p27 axis,5 and m1A-related gene dysregulation is associated with tumorigenesis in gastrointestinal cancers,6 with ErbB2 and mTOR signaling pathways likely modulated by m1A regulators.
In particular, m5C is one of the most well-known RNA modifications that was first identified in stable and highly abundant ribosomal RNAs (rRNAs) and transfer RNAs (tRNAs).7−9 m5C sites in tRNAs usually occur around the variable region and anticodon loop. As a heavily m5C modified RNA class, tRNA modifications stabilize tRNA secondary structure, confer metabolic stability, and affect aminoacylation and codon recognition.10,11 m5C sites identified in rRNA play an important role in rRNA processing, structure, and translation.12 With the development of effective techniques like high-throughput sequencing, it is now possible to identify and quantify m5C modifications in low-abundance RNA species, such as mRNA and long non-coding RNAs (lncRNAs).13 A recent study reported that m5C sites were mainly enriched in CG motifs of mRNA, showing conserved, tissue-specific, and dynamic properties in the mammalian transcriptome to regulate mRNA export.14 To date, the distribution and function of m5C across diverse classes of RNA have been explored.15,16 However, knowledge surrounding the prevalence and transcriptome-wide distribution of m5C in lncRNA is still very limited.
lncRNAs are a class of RNA molecules that are more than 200 nucleotides long and that derive from non-coding regions of the genome.17 The functions and mechanisms of most lncRNAs are unclear. In recent years, lncRNAs have obtained widespread attention as key regulators of gene expression in various physiological and pathological processes.18 Epigenetic regulation is one of the main mechanisms controlling lncRNAs expression and tissue specificity. As an important epigenetic modification, RNA methylation has been identified as an essential marker of oncogenesis.
Hepatocellular carcinoma (HCC), the fourth leading cause of cancer-related deaths globally, is primarily caused by viral hepatitis infections and exposure to aflatoxin and aristolochic acid.19–21 Although recent decades have yielded breakthroughs in HCC prevention, early detection, diagnosis, and treatment, the global disease burden remains significant due to malignancy and heterogeneity. However, several recent studies have revealed that methylation modifications participate in HCC development and progression, suggesting that methylation could be a vital therapeutic target for HCC.22–24
In this study, we sought to obtain a deeper understanding of m5C methylation in HCC lncRNA. To this end, we globally mapped m5C using RNA MeRIP-seq in human HCC cells and normal adjacent non-tumor tissues, comparing its prevalence and distribution in both cell/tissue types and cellular compartments. We found the degree of m5C modification in HCC to be much greater than that of adjacent tissue. This difference can be characterized by intra-tissue consistency and inter-tissue differences involving all chromosomes. Our findings may provide new insights into the m5C epigenetic regulation of lncRNA in HCC toward the development of new therapies.
Materials and Methods
Sample Collection and RNA Sequencing Samples Preparation
Specimens of resected HCC and adjacent non-tumor tissues controls were collected from HCC patients. In total, six pairs of HCC and adjacent non-tumor tissue samples were obtained. RNA from each sample was isolated from cells using TRIzol reagent (Invitrogen Corporation, Carlsbad, CA) following manufacturer’s instructions. A NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) was used to quantify RNA concentration. RNA with OD260/OD280 between 1.8 and 2.2 was marked as qualified. LncRNA was fragmented with fragmentation buffer into short fragments (about 100 bp) by GenSeq m5C RNA IP Kit (GenSeq Inc, China).
cDNA Library Construction
Based on the MeRIP-seq technique, we obtained m5C peaks from each sample. RNA immunoprecipitation (RIP) was performed by the GenSeq m5C RNA IP Kit (GenSeq Inc, China). The NEBNext Ultra II RNA Library Prep Kit for Illumina (New England Biolabs, Inc.) was applied to construct the cDNA library. The quality and purity of the cDNA library for each sample were validated using an Agilent Bioanalyzer 2100 system. The cDNA libraries were sequenced on a HiSeq 4000 platform (Illumina, San Diego, CA, USA) with 150 bp paired-end reads.
Sequencing Data Analysis
Libraries were sequenced and visualized on a HiSeq 4000 (Illumina). ‘Q30’ identifies the mapping quality, and reads higher than 30 were considered as reliable mapping and retained. Library mapping was drawn through STAR software (http://gingeraslab.cshl.edu/STAR).25 Sequencing datasets were aligned to the reference genome (//http.ncbi.nlm.nih.gov/genomes/Helicoverpa-armigera/) using HISAT software (2.0.4). Methylation peaks and their differences between HCC and adjacent non-tumor tissue were identified by MACS software and DiffReps, respectively.26,27
GO and KEGG Pathway Databases Analysis
GO (http://www.geneontology.org) and KEGG (https://david.ncifcrf.gov) pathway enrichment analyses were performed using DAVID to reveal the functions of these differentially methylated genes. GO analysis was performed based on differentially methylated genes to analyze the biological processes, cellular components, and molecular functions of these genes. P < 0.05 was considered as the cutoff for significant GO terms. KEGG pathway enrichment for the differentially methylated and expressed genes was also performed to investigate their biological pathway and functions, as previously described.28 P-values <0.05 were considered significant.
Ethical Approval
The study has been approved by the Ethics Committee of the First Affiliated Hospital of Zhengzhou University, and the patient’s written informed consent was obtained before the study began. And the study was conducted in accordance with the Declaration of Helsinki.
Results
Transcriptome-Wide m5C Methylation of lncRNA in Human HCC
To gain insight into transcriptome-wide m5C methylation, we performed RNA MeRIP-seq of lncRNA and identified 21,960 m5C peaks in HCC, and 15,325 m5C peaks in adjacent non-tumor tissues (Figure 1A and B). Up to 19,935 annotated genes (HCC tissues) and 14,127 annotated genes (adjacent tissues) were mapped, respectively. Among them, 12,928 m5C peaks were detected within both HCC and its adjacent non-tumor tissues. m5C sites with fold changes (FCs) >2 are considered to be specific to a certain tissues. Comparing HCC with adjacent non-tumor tissue revealed that 39% of identified sites were specific to HCC, whereas 14% were specific to adjacent non-tumor tissues. This showed that the overall occurrence of m5C methylation is higher in HCC than adjacent non-tumor tissue.
 |
Figure 1 Transcriptome-wide m5C methylation and characters of lncRNA in human HCC. (A) Venn diagram of m5C methylation sites identified in lncRNA from human HCC and adjacent non-tumor tissues. (B) Venn diagram of m5C genes in HCC and adjacent tissues. (C) The sequence motif of m5C sites in human HCC and adjacent non-tumor tissues. (D) Percentage of lncRNA harboring different numbers of m5C peaks in the two tissue types, with the majority harboring only one m5C peak. |
UARUCCCA is a Conserved Sequence Motif for m5C-Containing Regions
To determine the presence of an m5C motif, we scanned the sequences of m5C-methylated peaks. UARUCCCA (R = purine) was determined to be the most reliable motif in HCC (Figure 1C). Interestingly, the most reliable motif for m5C peaks in the adjacent non-tumor tissues was also UARUCCCA, indicating that the same sequence motif may be necessary for m5C methylation in HCC lncRNA. We speculate that m5C in HCC and adjacent non-tumor tissues may be caused by the same methylase.
Abundance of m5C Peaks in lncRNA
We next determined the abundance of peaks in lncRNA according to tissue type. Notably, 91.4% of the lncRNA contained one m5C peak, which was slightly lower than the 92.9% of single peak lncRNA in adjacent non-tumor tissues (Figure 1D). Percentages for two peaks (7.7% HCC vs 6.4% adjacent non-tumor tissues), three peaks (0.79% HCC vs 0.58% adjacent non-tumor), and more than three peaks (0.17% HCC vs 0.16% adjacent non-tumor tissues) were also determined.
Heatmap of m5C Levels in Human HCC and Adjacent Non-Tumor Tissues
Quantitatively, the methylation of m5C sites varied between HCC and adjacent non-tumor tissue based on unsupervised hierarchical cluster analysis. A heatmap of methylation levels showed the apparent expression difference between the two tissue types (Figure 2A). Specifically, cluster analysis revealed that the degree of m5C methylation had intra-tissue consistency and inter-tissue differences between HCC and adjacent non-tumor tissue.
 |
Figure 2 m5C levels and distribution of chromosome in human HCC and adjacent non-tumor tissue. (A) A cluster tree at the top indicates the relatedness of the two tissues (x-axis) or methylation levels (y-axis). The heatmap uses a color scale to indicate the relative methylation level at each locus. Each row of colored lines (N = 117) represents the methylation level for each CpG locus: red for hypermethylated and yellow for hypomethylated. Each column (n = 12) corresponds to each tissue. (B) Circos plot showing the distribution of m5C methylation sites on each chromosome. Red represents HCC, blue represents adjacent non-tumor tissue. |
Chromosome Visualization of m5C in lncRNA
In HCC and adjacent non-tumor tissues, we investigated the distribution of m5C methylation sites throughout the whole genome (Figure 2B). We found that m5C sites were dispersed across all chromosomes, which may indicate the extent to which m5C functions in cells. m5C methylation levels and distribution were different on each chromosome between the two tissue types.
The Differences in lncRNA Expression According to m5C Methylation
To explore differences in lncRNA expression according to m5C methylation, we researched lncRNA expression in HCC and adjacent non-tumor tissues using the RNA sequencing data. We found that more lncRNA were up-regulated by methylation and tended to have corresponding higher expression levels in HCC tissues (Figure 3A). On the contrary, more lncRNA were down-regulated by methylation in adjacent non-tumor tissues than HCC. Interestingly, we found that some lncRNA with minimal expression in adjacent non-tumor tissues were highly expressed in HCC and up-regulated by methylation (Table 1). These genes may be related to the pathogenesis of HCC and are worth further investigation. Among lncRNAs with m5C modifications, more were down-regulated than up-regulated in the HCC tissues (Figure 3B). Together, the data indicated that m5C tends to correlate with gene expression in a large fraction of transcripts in HCC tissues.
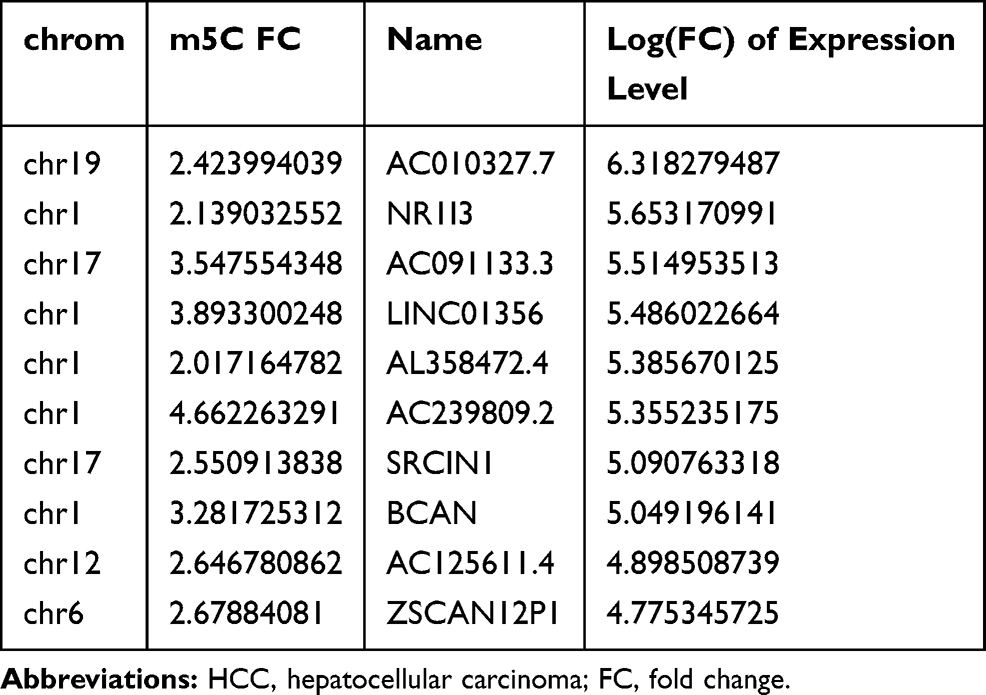 |
Table 1 Top 10 Highly Expressed lncRNAs in HCC |
 |
Figure 3 The differences in lncRNA expression according to m5C methylation. (A) Differentially expressed lncRNAs in HCC and adjacent non-tumor tissues. Genes up-regulated by methylation are red, and genes down-regulated by methylation are blue. (B) Cumulative distribution of lncRNA expression changes between HCC and adjacent non-tumor tissue for m5C up-regulated genes (red) and m5C down-regulated genes (green), whereas blue represents others. |
Gene Ontology (GO) Enrichment Analysis
To examine the function of m5C methylation in HCC and adjacent non-tumor tissues, GO enrichment analysis was performed based on biological processes (BP), cellular components (CC), and molecular functions (MF). For BP, genes with up-methylated m5C sites were significantly enriched in cell development, system development, multicellular organismal development, and neurogenesis (Figure 4A), whereas genes with down-methylated m5C sites were highly enriched in cellular processes such as nervous system development, neurogenesis, and generation of neurons and synaptic transmission (Figure 4D). For MF, genes with up-methylated m5C sites were notably enriched in enzyme binding, sequence-specific DNA binding transcription factor activity, nucleic acid binding transcription factor activity, and sequence-specific DNA binding (Figure 4B). Furthermore, genes with up-methylated m5C sites were enriched in gated channel activity, substrate-specific channel activity, ion channel activity, ligand−gated channel activity, and others (Figure 4E). For CC, genes with up-methylated m5C sites were mainly enriched in cell projection, neuron projection, neuron part, and others, as was the function of genes with down-regulated methylation (Figure 4C and F).
 |
Figure 4 Gene Ontology (GO) term enrichment analysis of m5C genes in HCC lncRNAs. (A-C) The top 10 GO terms for (A) biological processes, (B) molecular functions, and (C) cellular components were significantly enriched for up-methylated m5C genes in HCC. (D–E) The top 10 gene GO terms of (D) biological processes, (E) molecular functions, and (F) cellular components were significantly enriched for the down-methylated m5C genes in HCC. |
Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Analysis
Notably, KEGG pathway analysis indicated that hypermethylated genes were significantly related to endocytosis, thyroid hormone signaling pathway, pathways in cancer, and dilated cardiomyopathy (Figure 5A), whereas down-methylated genes were associated with glutamatergic synapse, glycosaminoglycan biosynthesis-heparan sulfate, transcriptional misregulation in cancer, and retrograde endocannabinoid signaling (Figure 5B).
 |
Figure 5 KEGG pathway analysis of m5C genes in HCC lncRNAs. (A) Bar plot showing the top 10 enrichment scores of significantly enriched pathways for up-methylated m5C genes in HCC. (B) Bar plot showing the top 10 enrichment scores of significantly enriched pathways for down-methylated m5C genes in HCC. |
Discussion
HCC has become the second leading cause of cancer-related deaths, which accounts for approximately 90% of all cases of primary liver cancer.29 HCC pathogenesis is a complex process, with epigenetic alterations, heritable gene expression changes, and chromatin organization shown to drive tumor initiation and progression.30 Recently, epigenetic alterations, in particular methylation, have come into focus for cancer pathogenesis and prognosis. Epigenetic alterations to DNA, RNA methylation, and histone deacetylation are the most frequent epigenetic events that play numerous roles in tumorigenesis.31
Characteristically, DNA methylation could regulate tumor metastasis through up-regulation of DNA methyltransferases and a series of molecular mechanisms in HCC.32 Besides, CPG island hypermethylation in promoter regions was found in HCC tissues, which could silence the expression of tumor suppressor genes and promote tumor development.33 In addition to DNA methylation, RNA modifications were revealed to moderation of RNA structure, function, and stability. Previous study reported that RNA m6A methyltransferase-like 3 was up-regulated in HCC samples by m6A-YTHDF2-dependent posttranscriptional silencing of suppressor of cytokine signaling 2.34 Another research found that ALYREF, a m5C-related regulatory gene, was associated with cell cycle regulation and mitosis in HCC progression.4
m5C is ubiquitous in nature and one of the most important RNA modifications identified in tRNA, rRNA, and mRNA.35 m5C has been implicated in protein translational regulation, RNA processing, and stress responses, and been closely correlated with cell proliferation and differentiation.36,37 The levels of m5C regulation are significantly different according to tissue type.38 For instance, m5C levels in metastatic neoplasms were much lower than that of benign neoplasms or normal tissue. It is worth mentioning that in humans, aberrant regulation of m5C causing CpG events is underrepresented.39 Over one-third of germline point mutations are closely correlated with human genetic diseases, and many somatic mutations leading to cancer involve the loss of CpG.
It has been reported that genes have a high occurrence of methylation in gastric cancer, breast cancer, and Hodgkin’s and non-Hodgkin lymphomas, ranging from 60% to 90%.40 Xue et al demonstrated that TRDMT1, an important member of the m5C family, plays a crucial role in inhibiting the proliferation and migration of HEK293 cells.41 Li et al showed that m5C enhanced the extent of overall DNA modifications, which lays a foundation for somatic mutation.42 However, m5C expression levels and its function in HCC remains poorly defined. We estimated the degree of cellular methylation by measuring the ratio of m5C in HCC tissues to adjacent non-tumor tissues.
For this reason, we focused our research on m5C methylation in HCC. Our results show that the number and distribution of m5C on HCC and adjacent non-tumor tissues are markedly different. In short, the m5C sites of lncRNA in HCC and the genes mapped by m5C sites far exceed those in adjacent tissues. These differences in methylation are spread across all chromosomes. Cluster analysis results show that the degree of methylation can clearly distinguish HCC from adjacent tissues, which further confirms the potential relationship between m5C and HCC. Interestingly, we found that some genes minimally expressed in adjacent non-tumor tissues are not only highly expressed in HCC, but also have an increased degree of methylation. The role of methylation of these genes in the pathogenesis of HCC deserves further discussion.
As a potentially critical layer of bioregulation, lncRNAs were reported to be aberrantly expressed through histone modification or DNA methylation, or transcription factors, and posttranscriptional modification in HCC.43 Dys-regulated lncRNAs might change the expression and activities of mRNAs, miRNA and proteins, resulting in epigenetic alteration in HCC. For instance, the up-regulated lncRNA DANCR in HCC could bind to 3 UTR of CTNNB1 mRNA, blocking the inhibitory effects of mir-214 miR320a and miR199a, thus increasing the stemness-like characteristics of HCC.44 Besides, lncRNA PVT1 could promote cell proliferation, cell cycle progression, and increased stem-like characteristics through stabilizing the nucleolar protein NOP2 in HCC.45 Transcriptional expression levels of lncRNAs in HCC have been widely studied. However, the mechanism of m5C methylation in lncRNAs promoting HCC progress is unclear, and deeper exploration would be helpful for understanding the pathogenesis of HCC.
Conclusion
These findings have significant implications for understanding the mechanisms of m5C regulation in HCC. Further exploration of epigenetic alterations in the pathology of HCC is still needed.
Funding
This study was supported by the National Natural Science Foundation of China (81902832 and U1904164); Tianqing Liver Diseases Research Fund (TQGB20200073); and the Science and Technology Research Project of Henan Province (192102310117). The funding body had no role in the design of the study, in the collection, analysis, and interpretation of the data, or in the manuscript writing. In addition, we thank the patients who participated in this study and Cloud-Seq Biotech Ltd. Co. (Shanghai, China) for the MeRIP-Seq service.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2017;18(1):31–42. doi:10.1038/nrm.2016.132
2. Meyer KD, Jaffrey SR. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat Rev Mol Cell Biol. 2014;15(5):313–326. doi:10.1038/nrm3785
3. Li X, Xiong X, Wang K, et al. Transcriptome-wide mapping reveals reversible and dynamic N(1)-methyladenosine methylome. Nat Chem Biol. 2016;12(5):311–316. doi:10.1038/nchembio.2040
4. He Y, Yu X, Li J, Zhang Q, Zheng Q, Guo W. Role of m5C-related regulatory genes in the diagnosis and prognosis of hepatocellular carcinoma. Am J Transl Res. 2020;12(3):912–922.
5. Chen Y, Peng C, Chen J, et al. WTAP facilitates progression of hepatocellular carcinoma via m6A-HuR-dependent epigenetic silencing of ETS1. Mol Cancer. 2019;18(1):127. doi:10.1186/s12943-019-1053-8
6. Zhao Y, Zhao Q, Kaboli PJ, et al. m1A regulated genes modulate PI3K/AKT/mTOR and ErbB pathways in gastrointestinal cancer. Transl Oncol. 2019;12(10):1323–1333. doi:10.1016/j.tranon.2019.06.007
7. Helm M. Post-transcriptional nucleotide modification and alternative folding of RNA. Nucleic Acids Res. 2006;34(2):721–733. doi:10.1093/nar/gkj471
8. Schaefer M, Pollex T, Hanna K, Lyko F. RNA cytosine methylation analysis by bisulfite sequencing. Nucleic Acids Res. 2009;37(2):e12. doi:10.1093/nar/gkn954
9. Agris PF. Bringing order to translation: the contributions of transfer RNA anticodon-domain modifications. EMBO Rep. 2008;9(7):629–635. doi:10.1038/embor.2008.104
10. Motorin Y, Helm M. tRNA stabilization by modified nucleotides. Biochemistry. 2010;49(24):4934–4944. doi:10.1021/bi100408z
11. Squires JE, Preiss T. Function and detection of 5-methylcytosine in eukaryotic RNA. Epigenomics. 2010;2(5):709–715. doi:10.2217/epi.10.47
12. Chow CS, Lamichhane TN, Mahto SK. Expanding the nucleotide repertoire of the ribosome with post-transcriptional modifications. ACS Chem Biol. 2007;2(9):610–619. doi:10.1021/cb7001494
13. David R, Burgess A, Parker B, et al. Transcriptome-wide mapping of RNA 5-methylcytosine in arabidopsis mRNAs and noncoding RNAs. Plant Cell. 2017;29(3):445–460. doi:10.1105/tpc.16.00751
14. Yang X, Yang Y, Sun BF, et al. 5-methylcytosine promotes mRNA export - NSUN2 as the methyltransferase and ALYREF as an mC reader. Cell Res. 2017;27(5):606–625. doi:10.1038/cr.2017.55
15. Squires JE, Patel HR, Nousch M, et al. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 2012;40(11):5023–5033. doi:10.1093/nar/gks144
16. Huang T, Chen W, Liu J, Gu N, Zhang R. Genome-wide identification of mRNA 5-methylcytosine in mammals. Nat Struct Mol Biol. 2019;26(5):380–388. doi:10.1038/s41594-019-0218-x
17. Kung JT, Colognori D, Lee JT. Long noncoding RNAs: past, present, and future. Genetics. 2013;193(3):651–669. doi:10.1534/genetics.112.146704
18. Hu X, Li Q, Zhang J. The long noncoding RNA LINC00908 facilitates hepatocellular carcinoma progression via interaction with Sox-4. Cancer Manag Res. 2019;11:8789–8797. doi:10.2147/CMAR.S216774
19. Fitzmaurice C, Allen C, Barber RM, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the global burden of disease study. JAMA oncol. 2017;3(4):524–548. doi:10.1001/jamaoncol.2016.5688
20. Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, Roberts LR. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol. 2019;16(10):589–604. doi:10.1038/s41575-019-0186-y
21. El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132(7):2557–2576. doi:10.1053/j.gastro.2007.04.061
22. Yeh CC, Goyal A, Shen J, et al. Global level of plasma DNA methylation is associated with overall survival in patients with hepatocellular carcinoma. Ann Surg Oncol. 2017;24(12):3788–3795. doi:10.1245/s10434-017-5913-4
23. Zhang X, He H, Zhang X, Guo W, Wang Y. RUNX3 promoter methylation is associated with hepatocellular carcinoma risk: a meta-analysis. Cancer Invest. 2015;33(4):121–125. doi:10.3109/07357907.2014.1003934
24. Wu X, Yao X, Cao Q, et al. CDH1Clinicopathological and prognostic significance of hypermethylation in hepatocellular carcinoma: a meta-analysis. Cancer Manag Res. 2019;11:857–864. doi:10.2147/CMAR.S179710
25. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics (Oxford, England). 2013;29(1):15–21. doi:10.1093/bioinformatics/bts635
26. Zhang Y, Liu T, Meyer CA, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9(9):R137. doi:10.1186/gb-2008-9-9-r137
27. Shen L, Shao NY, Liu X, Maze I, Feng J, Nestler EJ. diffReps: detecting differential chromatin modification sites from ChIP-seq data with biological replicates. PLoS One. 2013;8(6):e65598. doi:10.1371/journal.pone.0065598
28. He Y, Dang Q, Li J, et al. Prediction of hepatocellular carcinoma prognosis based on expression of an immune-related gene set. Aging. 2020;12(1):965–977. doi:10.18632/aging.102669
29. Zhao ZB, Chen F, Bai XF. Long noncoding RNA MALAT1 regulates hepatocellular carcinoma growth under hypoxia via sponging MicroRNA-200a. Yonsei Med J. 2019;60(8):727–734. doi:10.3349/ymj.2019.60.8.727
30. Zhou C, Zhang W, Chen W, et al. Integrated analysis of copy number variations and gene expression profiling in hepatocellular carcinoma. Sci Rep. 2017;7(1):10570. doi:10.1038/s41598-017-11029-y
31. Ravacci GR, Ishida R, Torrinhas RS, et al. Potential premalignant status of gastric portion excluded after Roux en-Y gastric bypass in obese women: a pilot study. Sci Rep. 2019;9(1):5582. doi:10.1038/s41598-019-42082-4
32. Han TS, Ban HS, Hur K, Cho HS. The epigenetic regulation of HCC metastasis. Int J Mol Sci. 2018;19:12. doi:10.3390/ijms19123978
33. Tischoff I, Tannapfe A. DNA methylation in hepatocellular carcinoma. World J Gastroenterol. 2008;14(11):1741–1748. doi:10.3748/wjg.14.1741
34. Chen M, Wei L, Law CT, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology (Baltimore, Md). 2018;67(6):2254–2270. doi:10.1002/hep.29683
35. Zou F, Tu R, Duan B, et al. Drosophila YBX1 homolog YPS promotes ovarian germ line stem cell development by preferentially recognizing 5-methylcytosine RNAs. Proc Natl Acad Sci U S A. 2020;117(7):3603–3609. doi:10.1073/pnas.1910862117
36. Van Haute L, Lee SY, McCann BJ, et al. NSUN2 introduces 5-methylcytosines in mammalian mitochondrial tRNAs. Nucleic Acids Res. 2019;47(16):8720–8733. doi:10.1093/nar/gkz559
37. Liu RJ, Long T, Li J, Li H, Wang ED. Structural basis for substrate binding and catalytic mechanism of a human RNA: m5Cmethyltransferase NSun6. Nucleic Acids Res. 2017;45(11):6684–6697. doi:10.1093/nar/gkx473
38. Gama-Sosa MA, Slagel VA, Trewyn RW, et al. The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res. 1983;11(19):6883–6894. doi:10.1093/nar/11.19.6883
39. Hendrich B, Hardeland U, Ng HH, Jiricny J, Bird A. The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature. 1999;401(6750):301–304. doi:10.1038/45843
40. Bethge N, Lothe RA, Honne H, et al. Colorectal cancer DNA methylation marker panel validated with high performance in Non-Hodgkin lymphoma. Epigenetics. 2014;9(3):428–436. doi:10.4161/epi.27554
41. Xue S, Xu H, Sun Z, et al. Depletion of TRDMT1 affects 5-methylcytosine modification of mRNA and inhibits HEK293 cell proliferation and migration. Biochem Biophys Res Commun. 2019;520(1):60–66. doi:10.1016/j.bbrc.2019.09.098
42. Li VS, Tang MS, Kohn H. The effect of C(5) cytosine methylation at CpG sequences on mitomycin-DNA bonding profiles. Bioorg Med Chem. 2001;9(4):863–873. doi:10.1016/S0968-0896(00)00301-1
43. Klingenberg M, Matsuda A, Diederichs S, Patel T. Non-coding RNA in hepatocellular carcinoma: mechanisms, biomarkers and therapeutic targets. J Hepatol. 2017;67(3):603–618. doi:10.1016/j.jhep.2017.04.009
44. Yuan SX, Wang J, Yang F, et al. Long noncoding RNA DANCR increases stemness features of hepatocellular carcinoma by derepression of CTNNB1. Hepatology (Baltimore, Md). 2016;63(2):499–511. doi:10.1002/hep.27893
45. Wang F, Yuan JH, Wang SB, et al. Oncofetal long noncoding RNA PVT1 promotes proliferation and stem cell-like property of hepatocellular carcinoma cells by stabilizing NOP2. Hepatology (Baltimore, Md). 2014;60(4):1278–1290. doi:10.1002/hep.27239
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.