Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 18
Transcriptome Sequencing Identifies PCK1 and IRS4 as Key Regulators of Gestational Diabetes Mellitus in Placenta: A Pilot Study
Authors Wang W ![]() , Zhang Q, Lu J, Zhou L
, Zhang Q, Lu J, Zhou L
Received 22 May 2025
Accepted for publication 14 October 2025
Published 27 October 2025 Volume 2025:18 Pages 3985—3997
DOI https://doi.org/10.2147/DMSO.S542175
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Rebecca Baqiyyah Conway
Wenwen Wang,1,* Qianrui Zhang,2,* Jing Lu,2 Li Zhou1
1Department of Obstetrics and Gynecology, Capital Medical University Affiliated Beijing Tongren Hospital, Beijing, People’s Republic of China; 2Department of Endocrinology, Beijing Diabetes Institute, Beijing Key Laboratory of Diabetes Research and Care, Beijing Tongren Hospital, Capital Medical University, Beijing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Jing Lu, Email [email protected] Li Zhou, Email [email protected]
Aim: To identify key molecular regulators of GDM in placental tissues using transcriptome sequencing.
Methods: This study employed transcriptome sequencing to investigate placental tissue from GDM patients (n=7) and healthy controls (n=6), aiming to identify key molecular regulators and pathways underlying GDM pathogenesis.
Results: We identified 505 differentially expressed genes (DEGs), including 216 upregulated and 289 downregulated genes. Notably, PCK1 (phosphoenolpyruvate carboxykinase 1) and IRS4 (insulin receptor substrate 4) emerged as critical regulators. Quantitative real time PCR validation confirmed significant downregulation of PCK1 and upregulation of IRS4 in GDM placentas. Functional enrichment analysis revealed that PCK1 is associated with the CLINK/HLA-G/INAVA/KLRC2/PCK1/RBP4/SLAMF6 pathway, which was suppressed in GDM, while IRS4 correlated with the dysregulated AGRP/IRS4/RXRG axis.
Conclusion: These findings highlight the roles of placental PCK1 and IRS4 in GDM. Further mechanistic studies, including protein-level validation and animal models, are warranted to explore these pathways as potential diagnostic or therapeutic targets for GDM.
Keywords: gestational diabetes mellitus, placenta, transcriptome sequencing, PCK1, IRS4
Introduction
Gestational Diabetes Mellitus (GDM) is one of the most prevalent metabolic disorders during pregnancy, affecting approximately 24% of pregnancies in Beijing, with incidence rates rising due to increasing obesity and advanced maternal age.1 Characterized by the onset of glucose intolerance during pregnancy, GDM is strongly associated with short-term complications such as preeclampsia, elevated cesarean delivery rates, and fetal macrosomia.2 Moreover, it significantly elevates long-term metabolic risks for both mother and offspring: mothers face a seven-fold increased risk of developing type 2 diabetes (T2DM), while offspring exhibit higher probabilities of adult-onset obesity and glucose metabolism disorders.3 Although insulin resistance (IR) and insufficient compensatory β-cell function are recognized as central pathological mechanisms of GDM, emerging evidence highlights the placenta—a critical metabolic and endocrine interface between mother and fetus—as a key player in GDM pathogenesis.4–7 Placental dysfunction may exacerbate maternal metabolic imbalance through the secretion of inflammatory factors and adipokines, which directly impair maternal insulin sensitivity, alongside localized reprogramming of glucose and lipid metabolism. However, significant gaps remain in understanding how placenta-specific molecular networks regulate GDM progression.
The interplay of genetic and epigenetic factors is considered a crucial driver of GDM. Studies indicate that downregulation of glucose transporter family members (eg, GLUT1/GLUT3) correlates with impaired placental glucose uptake,8 while dysregulation of gluconeogenic enzymes (eg, PCK1) may worsen maternal hyperglycemia by disrupting placental energy metabolism.9,10 Notably, current GDM research predominantly focuses on maternal peripheral or umbilical cord blood biomarkers, with limited systematic analysis of placenta-specific gene expression profiles. Prior studies often narrow their scope to single pathways or candidate genes, lacking integrative exploration of multidimensional regulatory networks, which hinders a holistic understanding of placental metabolic reprogramming in GDM.
To address these gaps, this study systematically investigated placental tissues from GDM patients and healthy controls using transcriptome sequencing and bioinformatics analysis. Our objectives were as follows: (1) to identify differentially expressed genes (DEGs) in placental tissues and uncover key molecular signatures linked to GDM; and (2) to elucidate the molecular mechanisms by which candidate genes such as PCK1 and IRS4 regulate GDM. This work not only provides novel insights into the pathogenesis of GDM but also establishes a theoretical foundation for developing placenta-specific diagnostic biomarkers and therapeutic targets. These advancements hold significant clinical implications for improving long-term health outcomes in both mothers and offspring.
Materials and Methods
Patient Selection and Specimen Collection
Pregnant women diagnosed with gestational diabetes mellitus (GDM) and healthy controls were recruited from Beijing Tongren Hospital, Capital Medical University, between January 2025 and March 2025. GDM diagnosis adhered to the Chinese Guidelines for the Prevention and Treatment of Type 2 Diabetes (2020), with a 75-g oral glucose tolerance test (OGTT) performed at 24–28 weeks of gestation. GDM was confirmed if any of the following criteria were met: fasting plasma glucose (FPG) ≥5.1 mmol/L and <7.0 mmol/L, 1-hour OGTT glucose ≥10.0 mmol/L, or 2-hour OGTT glucose ≥8.5 mmol/L and <11.1 mmol/L. Healthy controls comprised normoglycemic women matched for gestational age, normal body mass index (BMI), singleton pregnancy, and absence of comorbidities such as hypertensive disorders, cardiovascular diseases, infectious diseases, severe hepatic/renal dysfunction, endocrine disorders, or other pregnancy complications. A total of 6 healthy controls and 7 GDM cases were included in the study.
Placental tissues were collected within 10 minutes after delivery. Approximately 1 cm³ of tissue was excised from the fetal surface of the placenta within a 2-cm radius from the umbilical cord insertion site. Samples were rinsed with sterile saline and immediately stored at −80°C for subsequent analysis. This research was approved by the Ethics Committee of Beijing Tongren Hospital, Capital Medical University[TRECKY2021-169]. This study complies with the Declaration of Helsinki.
RNA Sequencing
Sample Quality Assessment and Library Preparation
RNA purity and concentration were measured using a NanoDrop 2000 microspectrophotometer, while RNA integrity was evaluated via the LabChip GX Touch microfluidic capillary system.
Library Construction
- mRNA Enrichment: Total RNA was isolated, and eukaryotic mRNA was enriched using Oligo(dT) magnetic beads.
- Fragmentation: mRNA was fragmented by incubation with Fragmentation Buffer.
- cDNA Synthesis: First-strand cDNA was synthesized from fragmented mRNA using random hexamer primers, followed by second-strand synthesis with DNA polymerase I, RNase H, and dNTPs. Double-stranded cDNA was purified to remove residual enzymes and primers.
- Library Preparation: Purified cDNA underwent end repair, A-tailing, and adapter ligation. Fragments of ~350 bp were size-selected using magnetic beads.
- Amplification: PCR was performed to amplify the adapter-ligated DNA, generating the final cDNA library.
Library Quality Control
Library concentration was initially quantified using a Qubit 3.0 fluorometer, and fragment size distribution was assessed with an Agilent 2100 Bioanalyzer. Libraries meeting quality thresholds were further validated for precise quantification via Bio-Rad CFX 96 real-time PCR.
Sequencing
Qualified libraries were pooled based on effective concentrations and sequenced on an Illumina platform using paired-end 150-bp reads. Sequencing by Synthesis (SBS) technology was employed, wherein fluorescently labeled dNTPs, DNA polymerase, and sequencing primers were incorporated into flow cells. During cluster amplification, fluorescence signals emitted by incorporated nucleotides were captured, converted into base calls, and assembled into sequence reads for downstream analysis.
Quantitative Real-Time PCR
Quantitative Real-Time
PCR was performed using isolated placental specimen. Briefly, a total of 500 ng RNA of each sample was converted into cDNA via FastQuant RT Super Mix according to the protocol (TIANGEN, China). Real-time PCR was then performed using the LightCycler® 480 Real-Time PCR System (Roche, USA) by SYBR Green I Master Mix (Roche, USA). β-actin was used as a reference gene. All primer sequences used in the experiment were listed in Table 1. The amplification efficiency for each primer pair was calculated from a standard curve generated by serial dilutions of cDNA and was confirmed to be between 90% and 110%, meeting the assumption of the 2−ΔΔCT method. The relative mRNA levels were then analyzed using this method. Each experiment was performed in triplicate.
 |
Table 1 Human Primers for Quantitative Real-Time PCR |
Data Analysis
Data Quality Control
Raw sequencing data (FASTQ files) generated by high-throughput sequencers were processed through CASAVA base calling to extract sequence reads and their corresponding quality scores. To ensure analytical reliability, raw reads were filtered to remove adapter-contaminated sequences, reads with ambiguous bases (N > 0), and low-quality reads (Qphred ≤20 in >50% of bases). Cleaned data were further evaluated for Q30 scores and GC content. All downstream analyses were performed using high-quality filtered reads.
Sequence Alignment
The reference genome and gene annotation files were retrieved from the Ensembl database (Human genome assembly GRCh38.p13). A genome index was constructed using HISAT2 v2.0.5, and paired-end clean reads were aligned to the reference genome with the same tool. HISAT2 was selected for its ability to generate splice-aware alignments using gene annotation-guided databases, ensuring superior accuracy compared to non-spliced alignment methods.
Gene Expression Quantification
Gene expression levels were quantified as FPKM (Fragments Per Kilobase of transcript per Million mapped reads), which normalizes read counts by gene length and sequencing depth. This metric accounts for transcript length and library size variations, providing a robust estimate of gene expression.
Differential Expression Analysis
For datasets with biological replicates, differential expression analysis between groups was conducted using DESeq2. This tool employs a negative binomial model to identify statistically significant expression changes, with Benjamini-Hochberg correction applied to control the false discovery rate (FDR). Genes with adjusted P ≤ 0.05 were classified as differentially expressed. Samples lacking replicates were analyzed using DESeq.
Functional Enrichment Analysis
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were performed on differentially expressed genes using the clusterProfiler package. Terms or pathways with P < 0.05 were deemed significantly enriched.
Statistical Analysis
Data are mean±SEM. DEGs used DESeq2 (FDR-adjusted P < 0.05); qPCR used Student’s t-test.
Results
Baseline Characteristics of Study Participants
The maternal age, pre-pregnancy BMI, gestational age at delivery, neonatal birth weight, and OGTT results for both GDM and non-GDM groups are summarized in Table 2.
 |
Table 2 Basic Information of the Pregnant Women |
RNA Sequencing
Sample Quality Assessment
Samples meeting the following criteria were selected for library preparation: RNA concentration ≥30 ng/µL, total RNA ≥0.5 µg, and RNA integrity number (RIN) ≥4. Detailed quality metrics are provided in Table 3.
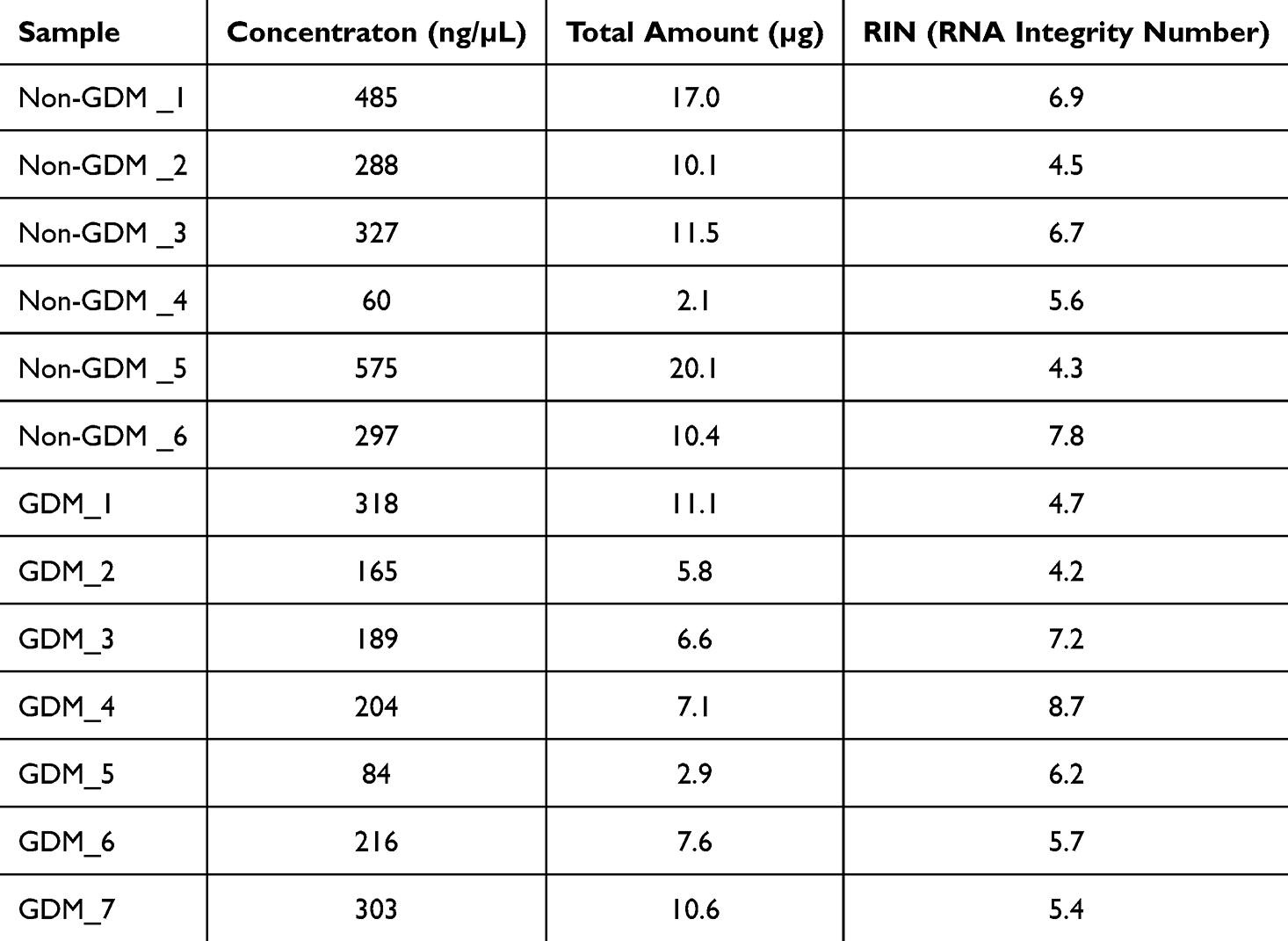 |
Table 3 RNA Quality of the Samples |
Sequencing Quality
High proportions of Q30 bases (base call accuracy ≥99.9%) confirmed robust sequencing quality. Sequencing statistics, including read counts and quality scores, are listed in Table 4.
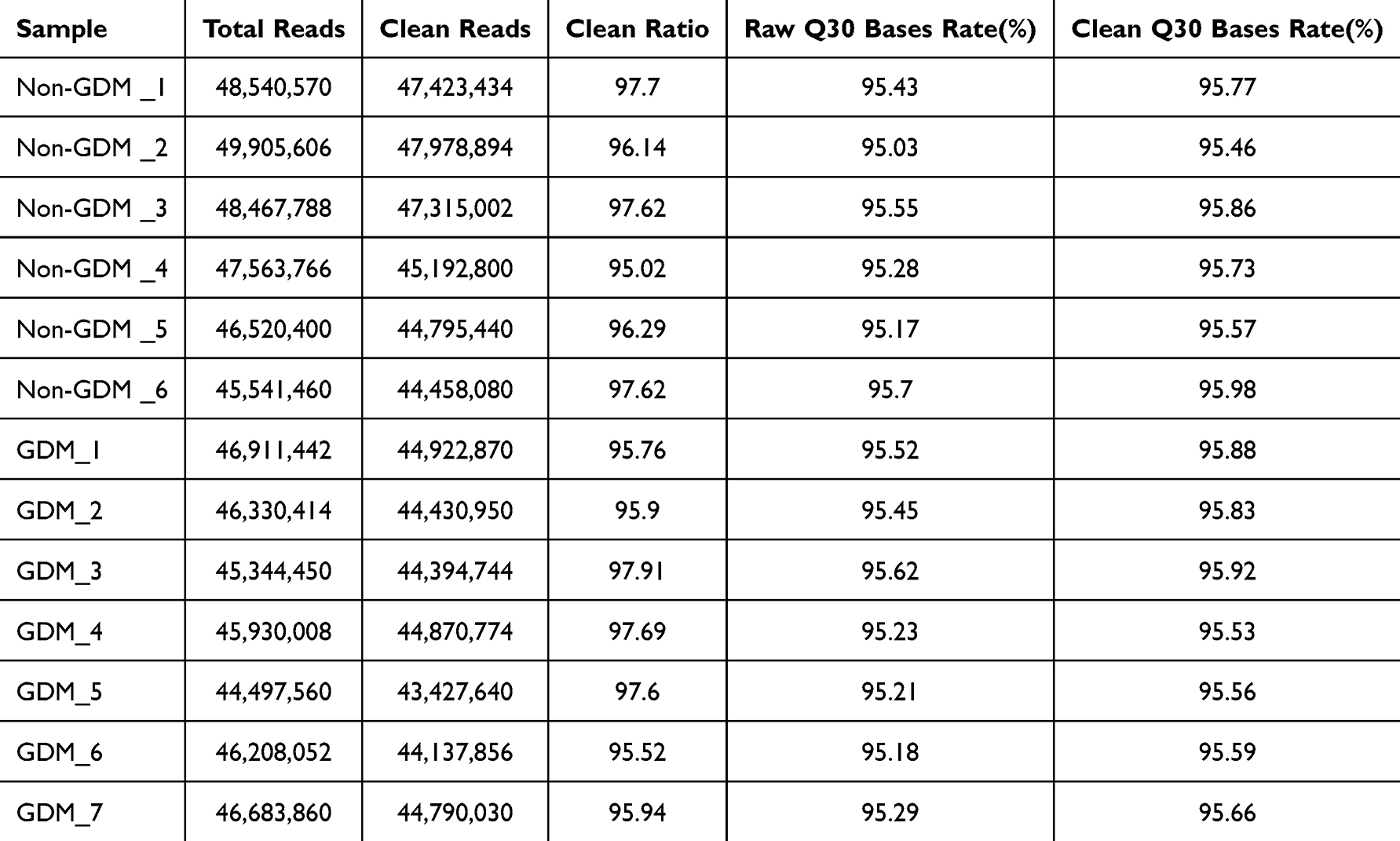 |
Table 4 Sequencing Quality and Reads |
Differentially Expressed Genes (DEGs) in GDM Vs Non-GDM Placenta
A total of 505 DEGs were identified, with 216 upregulated and 289 downregulated in GDM placentas (Figure 1 and Supplementary Table S1).
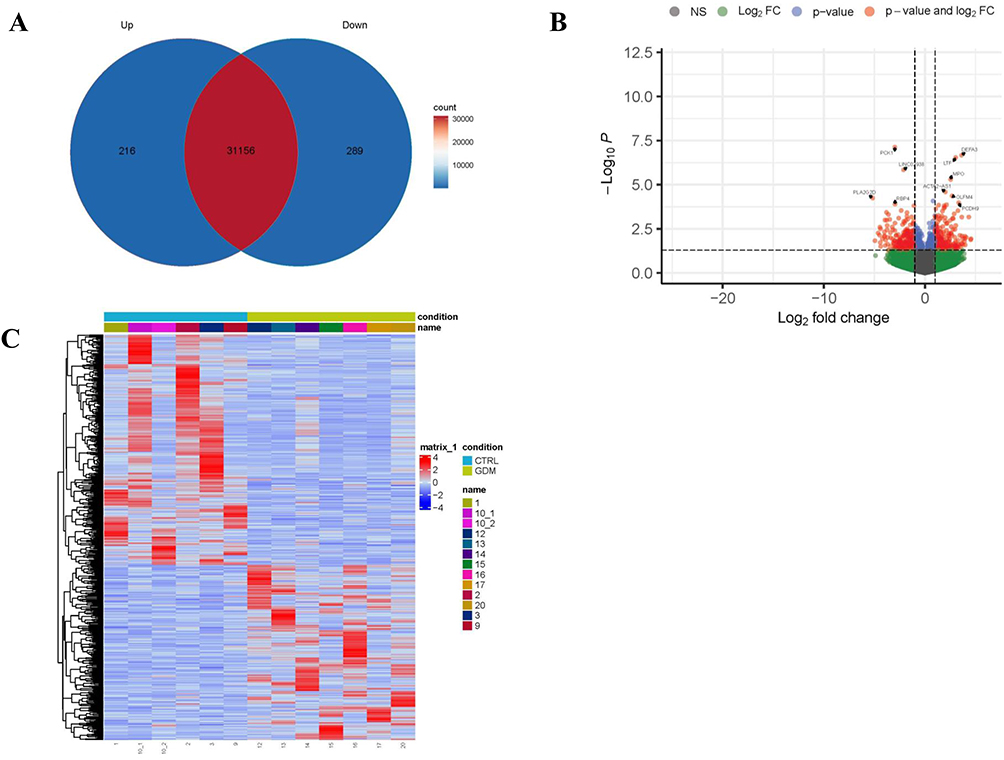 |
Figure 1 Differentially Expressed Genes (DEGs) in GDM vs Non-GDM Placenta. (A) Differentially Expressed Genes. (B) Differential gene volcano map. (C) Differential gene heat map. |
GO Enrichment Analysis of DEGs
Gene Ontology (GO) analysis categorized enriched terms into biological processes (BP), cellular components (CC), and molecular functions (MF) (Figure 2). The y-axis represents functional categories, while the x-axis denotes the number of enriched genes. Key BP terms included regulation of peptidase activity, response to molecule of bacterial origin, response to lipopolysaccharide, humoral immune response, defense response to bacterium, and muscle organ development. Enriched CC terms were vesicle lumen, specific granule, secretory granule lumen, and cytoplasmic vesicle lumen. MF terms predominantly involved heparin binding, glycosaminoglycan binding, and sulfur compound binding.
 |
Figure 2 GO Enrichment Map of DEGs in GDM vs Non-GDM Placenta. (A) The enriched GO(BP) terms. (B) The enriched GO(CC) terms. (C) The enriched GO(MF) terms. |
KEGG Pathway Enrichment Analysis
KEGG pathway enrichment results are displayed in Figure 3. The y-axis lists enriched pathways, the x-axis indicates the gene count per pathway, and color gradients reflect statistical significance. Key pathways included Malaria, Cytoskeleton in muscle cells, Cardiac muscle contraction, Hypertrophic cardiomyopathy, Dilated cardiomyopathy, NOD-like receptor signaling pathway, Motor proteins, and Adipocytokine signaling pathway.
 |
Figure 3 KEGG Enrichment Map of DEGs in GDM vs Non-GDM Placenta. |
Validation of DEGs by qRT-PCR
qRT-PCR analysis validated the differential expression of selected genes in placental tissues. PCK1, SLC2A2, RIMKLA, and RD3 were significantly downregulated in GDM placentas, while LTF, IRS4, and DEFA3 were upregulated, consistent with bioinformatics findings (Figure 4).
 |
Figure 4 Validation of DEGs by qRT-PCR. (A) PCK1 gene expression. (B) SLC2A2 gene expression. (C) RIMKLA gene expression. (D) RD3 gene expression. (E) LTF gene expression. (F) IRS4 gene expression. (G) DEFA3 gene expression. *P < 0.05. |
Integrated analysis of GO and KEGG results highlighted the AGRP/IRS4/RXRG* pathway (associated with upregulated IRS4) and the CLINK/HLA-G/INAVA/KLRC2/PCK1/RBP4/SLAMF6 pathway (linked to downregulated PCK1). AGRP and RXRG expression levels were markedly reduced in the GDM group and inversely correlated with IRS4, whereas SLAMF6, HLA-G, and CLNK showed positive correlations with PCK1 (Figure 5).
 |
Figure 5 Validation of KEGG pathway by qRT-PCR. (A) SLAMF6 gene expression. (B) HLA-G gene expression. (C) CLNK gene expression. (D) AGRP gene expression. (E) RXRG gene expression. *P < 0.05, and ***P < 0.001. |
Discussion
This study integrates transcriptome sequencing and qRT-PCR validation to reveal, for the first time, the aberrant expression of PCK1 and IRS4 in the fetal side of placental tissues and their associated signaling pathways in the pathogenesis of gestational diabetes mellitus (GDM). The results demonstrate significant downregulation of PCK1 and its regulatory pathway (CLINK/HLA-G/INAVA/KLRC2/PCK1/RBP4/SLAMF6) in GDM placentas, alongside dysregulation of the IRS4-associated AGRP/IRS4/RXRG axis. These findings suggest that PCK1 and IRS4 may contribute to GDM progression through dysregulated glucose-lipid metabolism and insulin signaling. This work provides novel insights into placenta-specific molecular networks in GDM and establishes a theoretical foundation for developing diagnostic biomarkers and therapeutic targets.
Phosphoenolpyruvate carboxykinase 1 (PCK1), a rate-limiting enzyme in gluconeogenesis, plays a central role in metabolic regulation.10 Our systematic analysis identifies PCK1 as a key gene linked to GDM, potentially involving multi-organ metabolic dysfunction and signaling pathway abnormalities. Previous studies highlight that PCK1 deficiency or dysregulation disrupts hepatic lipid metabolism, renal mitochondrial function, and systemic glucose homeostasis—pathological processes closely aligned with GDM clinical manifestations.
By catalyzing oxaloacetate conversion to phosphoenolpyruvate, PCK1 not only regulates hepatic gluconeogenesis to maintain glucose balance but also modulates lipid metabolism via non-canonical pathways.10–12 Animal models reveal that hepatic PCK1 deletion activates the PI3K/AKT/PDGF signaling axis, promoting lipid synthesis and hepatic stellate cell activation, thereby inducing metabolic dysfunction-associated fatty liver disease (MAFLD).11 Notably, MAFLD and GDM share a common pathophysiological basis—insulin resistance. Our data suggest that reduced placental PCK1 expression in GDM may similarly exacerbate local lipid accumulation and inflammation. Furthermore, PCK1 deficiency-driven activation of the hexosamine biosynthesis pathway aligns with the accumulation of advanced glycation end products in GDM, implicating PCK1 as a critical nexus linking glucose-lipid metabolic disturbances.12
Pregnancy-specific metabolic reprogramming demands precise maternal glucose regulation. Emerging roles of PCK1 in renal physiology offer new perspectives on multi-organ damage in GDM. Renal tubule-specific PCK1 deletion impairs ammonia synthesis and elevates lactate levels, mirroring the metabolic acidosis tendency observed in GDM.13 Importantly, PCK1 safeguards mitochondrial ribosome function in renal tubular epithelial cells,14 a mechanism potentially disrupted by gestational hyperglycemia-induced mitochondrial dysfunction. We hypothesize that placental trophoblasts may employ a PCK1-dependent metabolic regulatory mechanism, with its dysfunction directly impairing vascular remodeling.
Recent tumor research has provided new insights into the pathological mechanism of PCK1.15–18 PCK1 sustains tumorigenesis by metabolic reprogramming (such as activating oncogenic pathways like PI3K/AKT), which shows remarkable similarity to the aberrant PI3K/AKT activation triggered by PCK1 deficiency in Gestational Diabetes Mellitus (GDM). This property—where the same molecule mediates pathological processes through reprogramming in different diseases—highlights the core value of PCK1 as a hub in the metabolic-signaling network. Based on this, targeting downstream pathways of PCK1 (such as PI3K/AKT or the placenta-specific CLINK network) may represent a potential strategy for improving metabolic complications (eg, fatty liver) in GDM and provide common therapeutic approaches across diseases. Future research should delve into the epigenetic regulatory mechanisms of PCK1 in the placenta and explore the similarities and differences between its functions in the placental and tumor microenvironments, to advance its clinical translation as a diagnostic biomarker or therapeutic target.
The molecular mechanism of downregulation of PCK1 expression in the placental tissue of GDM patients is unclear. As a key rate-limiting enzyme in gluconeogenesis, dysfunction of PCK1 directly disrupts glucose homeostasis and contributes to the diabetic pathology.19–21 Notably, while PCK1 is typically upregulated in classical type 2 diabetes mellitus (T2DM) due to insulin resistance, the downregulation observed in GDM may stem from unique metabolic reprogramming triggered by the placental microenvironment. This distinction suggests that GDM warrants its distinction as a pathological entity independent of T2DM for clinical intervention.
While IRS family members are central to insulin signaling, the role of IRS4 remains poorly characterized. Our study identifies aberrant IRS4 expression in GDM placentas, inversely correlating with the AGRP/RXRG pathway, suggesting its involvement in non-canonical metabolic regulation. Unlike the ubiquitous expression of IRS1/2, the tissue-specific distribution of IRS4 (eg, brain, kidney, skeletal muscle) may underpin its unique placental function.22 Although IRS4 reportedly regulates GLUT4 trafficking via PI3K/Grb2 interactions, its direct impact on placental glucose uptake requires further validation.
It is noteworthy that IRS4 has been reported in tumor contexts to promote malignant progression by activating the PI3K/Akt and Ras-MAPK pathways.23–26 However, in placental tissue, it may regulate metabolic homeostasis through a distinct AGRP pathway. This tissue-specific mechanism of action merits further investigation.
AgRP neurons—critical regulators of energy metabolism—exhibit tight coupling with insulin sensitivity. Experimental ablation of AgRP neurons in adult mice reduces feeding behavior, while optogenetic or chemogenetic activation induces compulsive foraging.27 Beyond appetite regulation, AgRP neurons coordinate autonomic responses to energy status. For instance, insulin receptor inactivation in AgRP neurons impairs insulin-mediated suppression of hepatic glucose production, whereas acute AgRP neuron activation suppresses brown adipose tissue (BAT) glucose metabolism, reducing peripheral insulin sensitivity.28 We posit that placental IRS4 may dysregulate AGRP/RXRG-mediated energy sensing, akin to hypothalamic circuits, potentially via IRS4-dependent modulation of lipid metabolites that activate RXRG. Additionally, RXRG polymorphisms are associated with type 2 diabetes susceptibility in Han Chinese populations,29 further supporting its metabolic relevance. Collectively, IRS4 may regulate placental energy sensing and metabolic reprogramming via the AGRP/RXRG axis, though precise molecular interactions require validation through receptor-binding assays and chromatin immunoprecipitation.
The upstream mechanisms leading to increased IRS4 levels in GDM placental tissue remain unclear. Recent studies in gastric cancer have demonstrated that the immune receptor PILRB binds to IRS4 and recruits the deubiquitinating enzyme OTUB1.26 This complex specifically removes K48-linked ubiquitination from IRS4, thereby blocking its proteasomal degradation pathway. Consequently, this leads to aberrant activation of the PI3K/AKT signaling pathway. Given the observed dysregulation of IRS4 expression in GDM placental tissue in this study, a similar mechanism regulating protein stability may be involved. This finding provides new insights for elucidating the role of IRS4 in GDM.
In conclusion, placenta-specific biomarkers such as PCK1 and IRS4 offer transformative clinical potential by directly capturing molecular dysfunction at the primary disease site. Unlike systemic blood markers, they enable: (1) Early risk prediction through non-invasive detection in maternal circulation (eg, placental exosomes) before 24 weeks, allowing pre-OGTT interventions; (2) Precision prognostics post-delivery—persistently dysregulated biomarkers (eg, PCK1↓) can stratify mothers’ T2DM progression risk and predict offspring metabolic vulnerability; and (3) Targeted therapy development by identifying druggable placental pathways (eg, PCK1/CLINK or IRS4/AGRP axes) for localized treatments with reduced systemic side effects. These advantages position placental biomarkers as tools for proactive, personalized GDM management.
Limitations
This study has several limitations: (1) The small sample size necessitates larger cohorts to enhance reliability; (2) Functional validation of DEGs is restricted to mRNA levels, warranting proteomic and functional studies (eg, knockout/overexpression models); (3) Mechanistic details of PCK1- and IRS4-associated pathways remain unresolved, requiring further exploration. Future work will integrate human placental organoids to bridge translational gaps. (4) Sampling focused on the fetal side, maternal-facial heterogeneity warrants future study.
Data Sharing Statement
The transcriptome sequencing datasets and PCR datasets generated and analyzed during this study are available from the corresponding author (Li Zhou) upon reasonable request.
Ethics Approval and Consent to Participate
This study was approved by the Ethics Committee of Beijing Tongren Hospital, Capital Medical University. All participants provided written informed consent prior to sample collection.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work. Wenwen Wang and Qianrui Zhang as first authors contributed equally. The corresponding authors contributed equally.
Disclosure
The authors declare no competing interests.
References
1. Wang C, Jin L, Tong M, et al. Prevalence of gestational diabetes mellitus and its determinants among pregnant women in Beijing. J Matern Fetal Neonatal Med. 2022;35(7):1337–1343. doi:10.1080/14767058.2020.1754395
2. Moon JH, Jang HC. Gestational diabetes mellitus: diagnostic approaches and maternal-offspring complications. Diabetes Metab J. 2022;46(1):3–14. doi:10.4093/dmj.2021.0335
3. Vounzoulaki E, Khunti K, Abner SC, Tan BK, Davies MJ, Gillies CL. Progression to type 2 diabetes in women with a known history of gestational diabetes: systematic review and meta-analysis. BMJ. 2020;369:m1361. doi:10.1136/bmj.m1361
4. Yang Y, Guo F, Peng Y, et al. Transcriptomic profiling of human placenta in gestational diabetes mellitus at the single-cell level. Front Endocrinol. 2021;12:679582. doi:10.3389/fendo.2021.679582
5. Li YX, Long DL, Liu J, et al. Gestational diabetes mellitus in women increased the risk of neonatal infection via inflammation and autophagy in the placenta. Medicine. 2020;99(40):e22152. doi:10.1097/MD.0000000000022152
6. Sa R, Ma J, Yang J, et al. High TXNIP expression accelerates the migration and invasion of the GDM placenta trophoblast. BMC Pregnancy Childbirth. 2023;23(1):235. doi:10.1186/s12884-023-05524-6
7. Hamilton J, Weksberg R, Hanley A. Exposure to gestational diabetes mellitus (GDM) alters DNA methylation in placenta and fetal cord blood. Diabet Res Clin Pract. 2021;174:108690. doi:10.1016/j.diabres.2021.108690
8. Stanirowski PJ, Szukiewicz D, Majewska A, et al. Placental expression of glucose transporters GLUT-1, GLUT-3, GLUT-8 and GLUT-12 in pregnancies complicated by gestational and type 1 diabetes mellitus. J Diabetes Investig. 2022;13(3):560–570. doi:10.1111/jdi.13680
9. Hu S, Ma S, Li X, et al. Relationships of SLC2A4, RBP4, PCK1, and PI3K Gene Polymorphisms with Gestational Diabetes Mellitus in a Chinese Population. Biomed Res Int. 2019;2019:7398063. doi:10.1155/2019/7398063
10. Bertinat R, Holyoak T, Gatica R, Jara N, González-Chavarría I, Westermeier F. The neglected PCK1/glucagon (inter)action in nutrient homeostasis beyond gluconeogenesis: disease pathogenesis and treatment. Mol Metab. 2025;94:102112. doi:10.1016/j.molmet.2025.102112
11. Ye Q, Liu Y, Zhang G, et al. Deficiency of gluconeogenic enzyme PCK1 promotes metabolic-associated fatty liver disease through PI3K/AKT/PDGF axis activation in male mice. Nat Commun. 2023;14(1):1402. doi:10.1038/s41467-023-37142-3
12. Verissimo T, Dalga D, Arnoux G, et al. PCK1 is a key regulator of metabolic and mitochondrial functions in renal tubular cells. Am J Physiol Renal Physiol. 2023;324(6):F532–F543. doi:10.1152/ajprenal.00038.2023
13. Hasegawa K, Sakamaki Y, Tamaki M, Wakino S. PCK1 protects against mitoribosomal defects in diabetic nephropathy in mouse models. J Am Soc Nephrol. 2023;34(8):1343–1365. doi:10.1681/ASN.0000000000000156
14. Fantin VR, Sparling JD, Slot JW, Keller SR, Lienhard GE, Lavan BE. Characterization of insulin receptor substrate 4 in human embryonic kidney 293 cells. J Biol Chem. 1998;273(17):10726–10732. doi:10.1074/jbc.273.17.10726
15. Qin W, Duan Y, Hu Z, et al. PCK1 inhibits cGAS-STING activation by consumption of GTP to promote tumor immune evasion. J Exp Med. 2025;222(5):e20240902. doi:10.1084/jem.20240902
16. Liu N, Zhu XR, Wu CY, Liu YY, Chen MB, Gu JH. PCK1 as a target for cancer therapy: from metabolic reprogramming to immune microenvironment remodeling. Cell Death Discov. 2024;10(1):478. doi:10.1038/s41420-024-02240-8
17. Abate E, Mehdi M, Addisu S, Degef M, Tebeje S, Kelemu T. Emerging roles of cytosolic phosphoenolpyruvate kinase 1 (PCK1) in cancer. Biochem Biophys Rep. 2023;35:101528. doi:10.1016/j.bbrep.2023.101528
18. Zhang X, Tao G, Jiang J, et al. PCK1 activates oncogenic autophagy via down-regulation Serine phosphorylation of UBAP2L and antagonizes colorectal cancer growth. Cancer Cell Int. 2023;23(1):68. doi:10.1186/s12935-023-02894-x
19. Desjardins EM, Steinberg GR. Emerging role of AMPK in brown and beige adipose tissue (BAT): implications for obesity, insulin resistance, and type 2 diabetes. Curr Diab Rep. 2018;18(10):80. doi:10.1007/s11892-018-1049-6
20. Hu Q, Chen H, Zuo Y, et al. Role of PCK1 gene on oil tea-induced glucose homeostasis and type 2 diabetes: an animal experiment and a case-control study. Nutr Metab. 2019;16:12. doi:10.1186/s12986-019-0337-8
21. Oh KJ, Han HS, Kim MJ, Koo SH. Transcriptional regulators of hepatic gluconeogenesis. Arch Pharm Res. 2013;36(2):189–200. doi:10.1007/s12272-013-0018-5
22. Dörpholz G, Murgai A, Jatzlau J, et al. IRS4, a novel modulator of BMP/Smad and Akt signalling during early muscle differentiation. Sci Rep. 2017;7(1):8778. doi:10.1038/s41598-017-08676-6
23. Hao P, Huang Y, Peng J, et al. IRS4 promotes the progression of non-small cell lung cancer and confers resistance to EGFR-TKI through the activation of PI3K/Akt and Ras-MAPK pathways. Exp Cell Res. 2021;403(2):112615. doi:10.1016/j.yexcr.2021.112615
24. Zhang Y, Xiong X, Zhu Q, et al. FER-mediated phosphorylation and PIK3R2 recruitment on IRS4 promotes AKT activation and tumorigenesis in ovarian cancer cells. Elife. 2022;11:e76183. doi:10.7554/eLife.76183
25. Guo X, Huang S, Zhang Y, et al. Evodiamine inhibits growth of vemurafenib drug-resistant melanoma via suppressing IRS4/PI3K/AKT signaling pathway. J Nat Med. 2024;78(2):342–354. doi:10.1007/s11418-023-01769-9
26. Wang X, Liu Y, Zhao Q, et al. PILRB potentiates the PI3K/AKT signaling pathway and reprograms cholesterol metabolism to drive gastric tumorigenesis and metastasis. Cell Death Dis. 2024;15(9):642. doi:10.1038/s41419-024-07026-5
27. Gropp E, Shanabrough M, Borok E. Agouti-related peptide-expressing neurons are mandatory for feeding. Nat Neurosci. 2005;8:1289–1291. doi:10.1038/nn1548
28. Luquet S, Perez FA, Hnasko TS, Palmiter RD. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science. 2005;310(5748):683–685. doi:10.1126/science.1115524
29. Yu H, Wang S, Hu W, et al. Association between single-nucleotide polymorphisms of rxrg and genetic susceptibility to type 2 diabetes in South China. Curr Mol Med. 2020;20(6):408–414. doi:10.2174/1566524020666191206163951
 © 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2025 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
Gestational Diabetes Mellitus Impedes Fetal Lung Development Through Exosome-Dependent Crosstalk Between Trophoblasts and Lung Epithelial Cells
Chen P, Gu M, Wan S, Jiang X, Zhang F, Li Y, Zhou Q, Lu Y, Li L, Wang X
International Journal of Nanomedicine 2023, 18:641-657
Published Date: 8 February 2023