Back to Archived Journals » Reports in Electrochemistry » Volume 6
Toward rechargeable lithium–oxygen batteries: recent advances
Authors Imanishi N
Received 23 July 2015
Accepted for publication 20 March 2016
Published 21 June 2016 Volume 2016:6 Pages 15—29
DOI https://doi.org/10.2147/RIE.S65134
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor A.M Kannan
Nobuyuki Imanishi
Department of Chemistry, Mie University, Tsu, Mie, Japan
Abstract: This review summarizes research and development trends in lithium–oxygen (Li–O2) batteries. Development of a post–lithium-ion battery is highly expected to be a means to solve the global energy problem. The metal–oxygen cell, especially the Li–O2 cell, attracts attention, since it has much larger theoretical energy density than conventional battery systems. First, this review discusses the energy density that a battery should have from the viewpoint of power sources for electric vehicles. Then the fundamental features of various kinds of metal–oxygen batteries are explained and challenges faced by them are described. The remaining part of the review focuses on research subjects of the Li–O2 batteries. Early works which led to the present lithium–oxygen cells are briefly presented. Then, systems using various electrolytes and their major issues are introduced. First, irreversible decomposition of the electrolyte and Li2O2 deposition at oxygen cathode of an organic system are described. The effort that is directed toward the solution of dendritic growth of lithium metal anode is also introduced. Thereafter, new materials, mechanisms, and strategy that were reported recently are described with regard to each of the three electrolyte systems, respectively. These are expected to be breakthroughs to solve the serious problems, such as irreversible decomposition of electrolyte. In conclusion, the issues that should be solved with the direction of future development are mentioned.
Keywords: energy density, lithium anode, dendrite, peroxide, superoxide
Introduction to electrochemistry of batteries
A battery is a kind of device that transforms chemical energy into electrical one at high efficiency. The battery in which reverse energy transformation occurs is called a secondary or storage battery. Batteries as electric power sources are characterized mainly by two aspects – energy density and output power. With respect to energy density, let us consider an example of a classical battery system called Daniell cell. The cell reaction is described by the following equation:
Standard electrode potentials of zinc and copper are −0.76 V and +0.34 V, respectively, with respect to the standard hydrogen electrode. When the activity of the ions in each electrolyte is unity, the potentials of these electrodes become equal to the standard electrode potentials. As a consequence, the cell electromotive force (emf) of Daniell cell can be given by the following equation:
F is Faraday’s constant: F=96485 C mol−1. The maximum electrical work when a given amount of electrons, n, travel through the circuit equals the Gibbs energy reaction and depends on the emf (E) as shown in the following equation:
Given that the emf is 1.10 V, Gibbs energy change for the reaction is calculated to be −214 kJ per mole of zinc metal or copper ion. The energy is almost in the order of kinetic energy of a car driving on a motor way.
As shown here, to increase the energy produced in a cell, selection of electrode materials with a large difference in their electrochemical potentials is necessary. The lowest electrochemical potential among common elements is shown for lithium and the highest for fluorine. In an electrochemical cell with lithium metal and fluorine gas electrodes, the emf will be 5.94 V under standard conditions, which is the largest among the combinations of common elements.
Note that the voltage is applied to the electrolyte and it should not exceed the decomposition voltage of the electrolyte that is called electrochemical window. Aqueous batteries using aqueous electrolytes such as dry batteries and Ni–MH batteries usually operate below 1.5 V, and hence the decomposition voltage of water is 1.23 V. Lead–acid batteries deviate this rule, as they operate at a voltage of approximately 2 V. The decomposition reactions of the aqueous electrolyte on lead metal and lead oxide electrodes are slow enough to keep the system kinetically stable.
Some organic electrolytes show electrochemically stable and reversible behavior even on lithium metal. These electrolytes usually from carbonate-based solvent actually decompose on lithium metal but form a solid conductive film comprising kinds of decomposed substances. The film is called solid electrolyte interphase and isolates the electrolyte from lithium metal to hinder further decomposition. The electrolytes also show good electrochemical stability at high potential above 4 V with respect to lithium metal. These findings resulted in the commercialization of three V-class lithium primary batteries in 1970s and lithium-ion battery (LIB) with four V-class transition metal layered oxides launched in 1991. Lithium metal batteries and LIBs are indispensable to the today’s energy-related areas and mobile applications, because they exhibit remarkably higher energy densities than conventional aqueous batteries owing to their high voltages.
Here, we have dealt with the energy density quantitatively in LIBs and have checked the availability for electric vehicle (EV) applications. Theoretical gravimetric energy density is calculated by the maximum electric work divided by the sum of weights of electrodes. W is total mass of n equivalent moles of positive and negative electrode materials.
At present, LIB typically consists of graphite negative electrode, aprotic organic electrolyte, and LiCoO2 positive electrode. Each electrode reaction and whole cell reaction are described as follows:
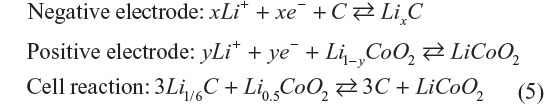
The maximum capacity of graphite anode is 372 mA·h·g−1 of graphite at x=1/6. On the other hand, 137 mA·h·g−1 of LiCoO2 at y=0.5 is the highest for the positive electrode. Given the cell emf is 3.8 V, the calculated gravimetric energy density will be 380 W·h·kg−1 of electrodes assuming that the materials do not react excess or less. A value of 380 W·h·kg−1 exhibits a potential electrical energy that the battery has, and is not actually achievable due to presence of other battery components. Practical energy density taking weight of other components into consideration generally results in less than one-third of the theoretical value. LIBs for large-size applications tend to fall into this category. For the EV commercialized by Nissan Motors Ltd. (Yokohama, Japan), Nissan LEAF EV, it is reported that with a fully charged battery the driving range is 228 km without air-conditioning and 130 km with it, where the total car weight is 1,520 kg and the battery weight is around 240 kg. The specific energy density of LIBs used for Nissan LEAF is around 100 W·h·kg−1, which is approximately 30% of the calculated specific energy density of the LIB.
The calculated energy density of the gasoline is considered to be approximately 13 kW·h·kg−1. And that for automotive applications is estimated to be 1,700 W·h·kg−1 using the reaction heat of gasoline and tank-to-wheel efficiency of 12.6%, while the battery-to-wheel efficiency is approximately 90%.1 This revealed that the calculated energy density of LIBs is only one-fifth of that of gasoline, and thus the driving range of the EVs with LIBs is much shorter than that of gasoline-powered cars. To develop EVs with a comparable driving range to gasoline-powered cars, the battery system with theoretical energy density higher than 1,700 W·h·kg−1 is necessary.
LIBs have been extensively used for small mobile applications, because they show high discharge/charge reversibility close to 100%. History of storage batteries starting from lead–acid batteries to LIBs is a transition of electrode reaction from dissolution/deposition to topochemical type. Topochemical reaction in LIBs performs small structural change and hence almost perfect reversibility. However, 300 W·h·kg−1 is the possible limitation of gravimetric energy density in practical LIBs because of the weight of host matrix containing electrochemically inert atoms.
A primary lithium battery using a lithium metal anode commercialized in 1976 showed a theoretical energy density as high as 2,119 W·h·kg−1. This appeared to progress backward from topochemical to dissolution/deposition type; however, using lithium metal anode can overcome the current limitation of energy densities. The highest energy density is configured from lithium metal and fluorine gas, although such a battery has not been yet proposed. As a next choice to toxic fluorine, the group 16 elements have become a cathode candidate for a high-energy-density battery system.
Metal–oxygen batteries with non-lithium anodes
Energy densities of various combinations of element atoms as electrodes are listed in Table 1. As stated in the previous section, 1,700 W·h·kg−1 is the target value that a storage battery should achieve in order to drive 500 km per a full charge. Most of the metal–oxygen batteries meet this criterion regardless of anode metals and some are already commercialized as a primary battery. For example, zinc–oxygen battery has a lengthy commercial record as one for a hearing aid and magnesium–oxygen battery has been used as a seawater battery. These metal–oxygen batteries are not rechargeable in most cases, since either coulombic or energy efficiency is too low. The coulombic efficiency of zinc–oxygen batteries is only 30% and self-discharge ratio is approximately 25% in half a month. The energy efficiency of lead–acid batteries is as high as 60%–70%, which results in lower energy efficiency of the battery-powered vehicles than that of the gasoline-powered ones. Because LIBs and nickel–metal hydride batteries have energy efficiencies higher than 85%, they are used to drive EVs. It becomes an important goal for the metal–air batteries to improve these efficiencies.
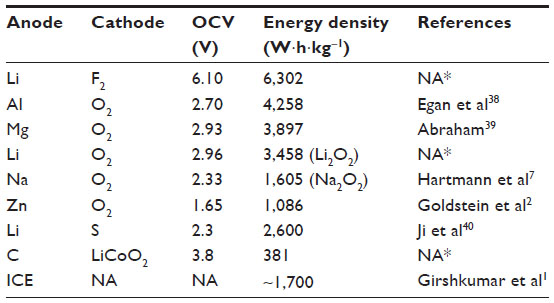 | Table 1 Theoretical gravimetric energy densities of various metal–oxygen batteries compared with those of lithium–fluorine, lithium–sulfur, conventional lithium-ion battery, and internal combustion engine |
As for the zinc–air battery, the efforts for making charge possible have been undertaken in almost all metal–air batteries. Mechanical charging or installation of the third electrode specifically for charging have been examined to improve its low energy and coulombic efficiencies. A full-sized transit bus employing rechargeable zinc–oxygen and nickel–cadmium hybrid battery system was demonstrated in the 1990s.2 This battery-powered bus was designed to drive a daily range of 150–250 km with passengers on board and air-conditioned. The zinc–oxygen battery in this system showed gravimetric energy densities of 200 W·h·kg−1 and volumetric energy density of 221 W·h·L−1. These features are comparable or still lower than those of current LIBs.
Magnesium is a divalent metal with 2,205 mA·h·g−1 and 3,833 mA·h·cm−3 of specific capacities. The electrochemical potential in aqueous media is −2.37 V with respect to the standard hydrogen electrode. Aqueous magnesium–air batteries have a disadvantage of high anode corrosion and large polarization. Organic magnesium–oxygen batteries should exhibit the theoretical voltage around 2.3 V, which leads to 3,059 mW·h·g−1 of the gravimetric energy density including oxygen. Generation of insulating and stable MgO in discharge causes a remarkable decrease in capacity and high polarization by its insolubility. Shiga et al3 reported that additive 2,2,6,6-tetramethylpiperidine-oxyl (TEMPO)–anion complex enhanced the decomposition of MgO. This complex was capable of decomposing MgO at 60°C by chemical oxidation of the oxide anions. Reduced form of TEMPO resulted after the decomposition was electrochemically oxidized by oxygen electrode and was regenerated to the complex, so that TEMPO–anion complex worked as a mediator for the cathode reaction. The discharge/charge performance of the organic magnesium–oxygen battery working at 60°C showed a discharge capacity of 737 mA·h·g−1 in the first cycle, while the charge capacity was 460 mA·h·g−1, resulting in an improved coulombic efficiency of 62.4%.
Aluminum–oxygen batteries have theoretical energy densities of 7,778 W·h·kg−1 and 21,000 W·h·L−1 excluding oxygen. The gravimetric energy density is the second largest next to lithium–oxygen (Li–O2) batteries and the volumetric one is the largest. These features make this battery quite attractive, but the battery is principally not rechargeable due to the high chemical stability of Al2O3. Mechanical charging is one way to charge the battery, but energy corresponding to 15 kW·h·kg−1 is needed to regenerate aluminum metal from Al2O3,4 so it does not meet the criteria of energy conservation. The most important subject relevant to this issue is finding an aluminum-conducting electrolyte. 1-ethyl-3-methylimidazolium chloride is proved a good ionic liquid to dissolve aluminum ions.5 LiCl is also added to form Li2O2 as a discharge product and to avoid precipitation of Al2O3. The concentration of LiCl determines the energy density of the battery. When 30% of LiCl can be dissolved, it is calculated to be 390 W·h·kg−1. Hybrid-type aluminum-oxygen batteries are also proposed to solve the issue of Al2O3 precipitation.6 The battery is constructed from KOH/CH3OH as catholyte and KOH/H2O as anolyte. These electrolyte solutions are separated by anion exchange membrane and hydroxide anions migrate as a charge carrier between the electrodes. The electrode reactions are described as follows:
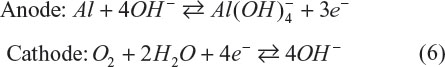
This hybrid aluminum–oxygen battery shows a high energy density of 2,081 W·h·kg−1 (or 5,819 W·h·L−1).
As seen here, it is the simplest technique to use an alkaline metal anode in order to achieve good reversibility. Here, sodium–oxygen battery is briefly described which is an analogous system of the Li–O2 battery, being the main topic of the review. Hartmann et al reported sodium–oxygen battery discharging/charging reversibly with sodium superoxide as the discharge product.7 According to their report, in ether-based electrolyte, NaO2 is exclusively formed during discharge process, while Na2O2 is the main discharge product in carbonate-based electrolyte. This remarkable difference is explained by the stability of ethers against nucleophilic attack by superoxide anion O2−. The discharge and charge voltage plateaus are observed at approximately 2.2 and 2.4 V, respectively, which indicate quite low overvoltages. These electrochemical features are considered to be related to the one-electron oxygen–reduction process that may be kinetically favored. The coulombic efficiencies are still not high enough for practical applications, but sodium–oxygen battery provides interesting chemistry that may contribute to the realization of rechargeable metal–air batteries.
Li–O2 batteries: history and electrolyte
As seen in the previous section, the simplest technique to secure reversibility under the present circumstances is to use an alkaline metal anode. Therefore, the Li–O2 batteries which were extensively studied are described. The concept of primary Li–O2 batteries was proposed by Littauer and Tsai8,9 in 1970s, where alkaline aqueous solution was used. The battery was called lithium–water cell because water was the active material of the cathode that produced hydrogen during discharge. If dissolved oxygen or other oxidizing agent is present in aqueous electrolyte, it also serves as a cathode active material to complete the cell reaction. Another system, lithium–water–air cell, working on a similar mechanism showed higher emf and decreased the electrolysis of water. Lithium metal in both systems reacts with the alkaline aqueous solution and in situ formed surface film on lithium metal retards the rapid corrosion. The lithium–water cells are capable of achieving high specific power density; a current density of approximately 120 mA·cm−2 was achieved at a cell voltage of 2.0 V. The current efficiency increases with an increase in current density and the corrosion reaction can be virtually ignored when the lithium metal is anodically polarized by approximately 300 mV and beyond. However, at the open-circuit potential of 2.9–3.0 V, corrosion rate is relatively large, resulting in low energy conversion efficiency and little rechargeability.
The rechargeable lithium–air cell was reported by Semkow and Sammells in 1987.10 The cell configuration was quite close to a solid oxide fuel cell with the fuel electrode substituted into lithium–alloy electrode.
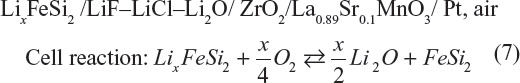
The cell was operated between 600°C and 850°C which was largely dictated by O2− conductivity of the stabilized zirconia. The cell emf is 2.06 and 2.25 V at 650°C and 800°C, respectively. A current density of 200 mA·cm−2 could be applied under the cell polarization by~1 V. A capacity of 200 mA·h·cm−2 was successfully cycled at 20 mA·cm−2 and 650°C. This system delivers a theoretical energy density of 4,266 W·h·kg−1, which was the highest among secondary batteries at that time.
A similar concept was reported by Batalov and Arkhipov in 1997;11 the cell adopted the idea of protected lithium electrode. Li/LiBe2O3/LiCl-Li2CO3 (1:1)/Li0.1NiO, O2 (or La0.7Sr0.3MnO3, O2).
LiBe2O3 is a solid lithium-conducting electrolyte. LiCl–Li2CO3 is a binary molten electrolyte. The cell operated at 580°C–600°C and showed a cell voltage of 2 V at 100 mA·cm−2. The molten salt Li–O2 cell could operate at high current density, but the long-term cycling performances of this type of cell have not been reported.
In 1996, Abraham and Jang12 reported the first paper for a rechargeable Li–O2 cell operating at room temperature. They used a polyacrylonitrile-based gel-polymer electrolyte of LiPF6 plasticized with ethylene carbonate and propylene carbonate (PC). The electrical conductivity of approximately 10−3 S·cm−1 at room temperature is comparable to that of nonaqueous lithium-ion–conducting solutions with a separator. The initial open-circuit voltage of the Li–O2 cell was 2.85 V, suggesting that a reaction process involves reduction of oxygen molecule. The analysis of the discharged carbon electrode using a Raman spectroscopy showed that the reaction product is mainly Li2O2. This result indicates that further two-electron reduction reaction beyond Li2O2 hardly occurred on carbon without catalyst. The cell capacity strongly depended on the thickness of the carbon cathode. When the thickness varied between 75 and 100 μm, 1,400–1,600 mA·h·g−1 carbon was obtained at 0.1 mA·cm−2 and room temperature. When cobalt-catalyzed carbon cathode of 250 μm thickness was used, the cell was first discharged to yield a capacity of 580 mA·h·g−1, and in the following charge, a capacity of 630 mA·h·g−1 was obtained as shown in Figure 1. The same amount of capacity could be discharged in the second cycle and good reversibility was obtained between 1.5 and 4.0 V.
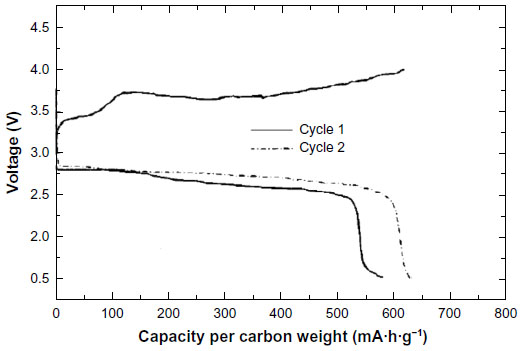 | Figure 1 The first report of a rechargeable lithium–oxygen battery. |
After the first report by Abraham and Jang, Read13,14 studied the oxygen transport properties in various organic electrolytes under the concept that Li–O2 cell performance strongly depends on it. They revealed that the solubility of oxygen and viscosity of organic solvent have a critical effect on the discharge capacity of an oxygen reduction reaction (ORR) on carbon electrode. It was also pointed out that electrolyte formulation has the largest influence on the nature of the deposit formed during discharge. Ether-based electrolytes perform good rate capability in the Li–O2 cell. The high capacities for ORR of 1,633 mA·h·g−1 at 0.5 mA·cm−2 and 2,494 mA·h·g−1 at 0.05 mA·cm−2 were observed for a Super P carbon black in a LiBr in 1,3-dioxolane-1,2 dimethoxyethane electrolyte, which is several times higher than that of the cathode in LIBs.
The report in 2006 by Ogasawara et al15 demonstrated for the first time that repeated discharge/charge of 100 times is possible at high capacity for the nonaqueous Li–O2 cell that uses an electrolytic manganese dioxide (EMD) catalyst for the oxygen electrode. After this paper was published, research activities on the rechargeable Li–O2 batteries have significantly increased. Figure 2 shows the first discharge and charge performance and discharge capacities with cycles for the cell: Li/LiPF6 in PC/Super P carbon-electrolytic manganese oxide/O2.
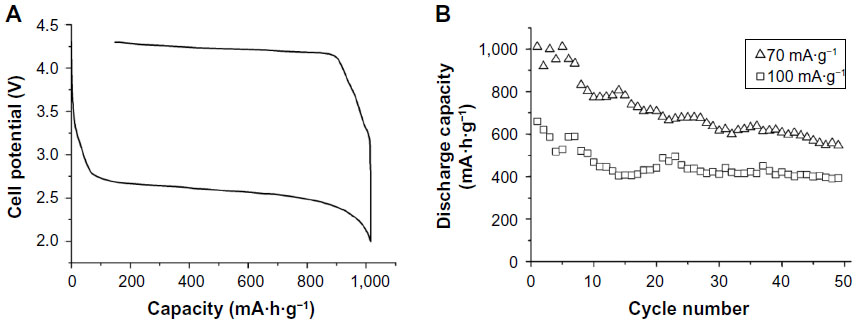 | Figure 2 The first report in 2006 showing practical performance of the nonaqueous lithium–oxygen cell. |
They concluded that the electrochemical reaction of 2Li+ + 2e− + O2 = Li2O2 in a nonaqueous lithium battery with an O2 cathode is reversible. The formation of Li2O2 was confirmed by an in situ differential mass spectrometry. The appropriate choice of catalyst enlarges reversible capacity and also reduces the charging voltage. Their results indicate that MnO2 catalysts are particularly effective at promoting high capacities, especially the alpha-MnO2 polymorph in the nanomaterial form shows capacities as high as 3,000 mA·h·g−1 of carbon.
In Li–O2 batteries with organic liquid or gel electrolyte, performance of oxygen cathode has been a center of discussions. However, in order to develop reliable Li–O2 batteries, irreversible reactions of lithium metal electrode such as with solvent, oxygen, and water, must be avoided by protecting lithium surface from these reactive substances. A composite lithium anode with a double-layer protection was preliminary addressed by Visco et al in 200416 who proposed rechargeable aqueous lithium–air battery. This electrode concept adopts a water-stable NASICON-type lithium-conducting solid glass ceramic as a protective layer that covers and isolates the lithium metal from direct contact with the electrolytes. Aluminum-doped lithium titanium phosphates, Li1+xAlxTi2−x(PO4)3 (x≈0.3) (LATP), used for the glass ceramic are unstable to reduction by lithium metal. So a lithium-stable conductive interlayer is necessary to electrically connect lithium metal and the glass ceramic. As one example, they use thin layer of Cu3N which reacts with lithium metal to produce admixture of Li3N and Cu3N or Cu. The protected electrode is so stable that lithium metal can be cycled in aqueous circumstance, which enables development of aqueous Li–O2 batteries and other lithium metal batteries. Conductivity and stability are key properties of the glass ceramics and the conductive interlayer to realize reversible lithium stripping/deposition for a long life span at high current density.
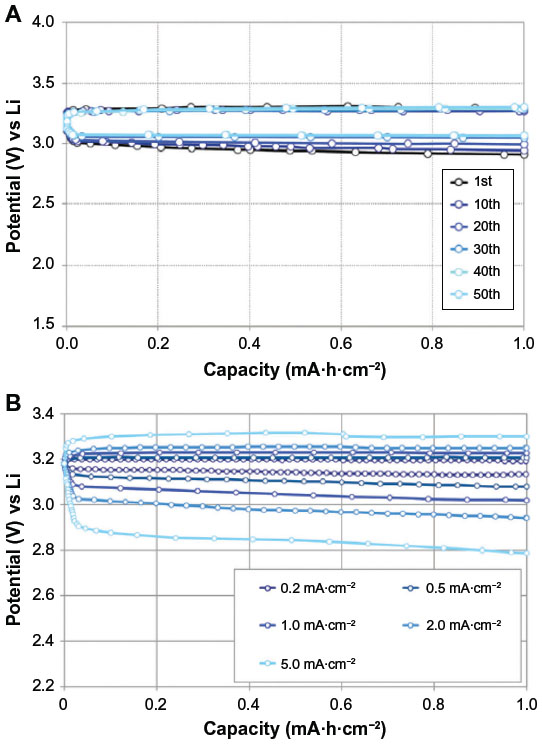
In addition to nonaqueous and aqueous types, solid-state rechargeable Li–O2 batteries are also studied. Kumar and Kumar17 and Kumar et al18 used lithium aluminum germanium phosphate glass ceramics, Li1+xAlxGe2−x(PO4)3 (LAGP), which has the same NSAICON-type structure as lithium titanium phosphates. The electrical conductivity of LAGP was reported as high as 4.48×10−4 S · cm−1 at 23°C.19 This type of solid lithium-ion conductor is unstable in contact with a lithium metal, so that a polyethylene oxide–based polymer electrolyte was used as an interlayer between lithium metal and LAGP to avoid direct contact to lithium metal. Since the polymer electrolyte has low lithium-ion conductivity at room temperature, the cell was operated at a higher temperature. The area-specific capacity of the cell was 5.7 mA·h·cm−2 at 0.1 mA·cm−2 and 95°C. The discharge product Li2O2 was presumed from the equilibrium open circuit voltage of 3.1 V, although no further supporting evidence was given. The charge process showed gradually increasing voltage profile to the limit of 4.05 V. The charge and discharge columbic efficiency was quite high as nearly 100%, and the cell was subjected to 40 cycles at current densities ranging from 0.05 to 0.25 mA·cm−2.
Hassoun et al20 have proposed a different type of solid-state Li–O2 battery. The composite polymer electrolyte of poly(ethylene oxide)–LiCF3SO3 and nanosized ZrO2 powder were used as a solid electrolyte. Open circuit voltage of 3.2 V was shown for the cell – Li/polymer electrolyte/Super P carbon black and O2. Pure oxygen gas was provided from rubber balloon connected to the cathode. Electrochemical analyses showed successive reduction from oxygen molecule through superoxide and peroxide to oxide anion, while it is verified that the reaction advances to opposite direction when charging. The discharged product was confirmed to be Li2O2 and LiOH by X-ray diffraction (XRD) analysis. The latter was considered to result from reaction of Li2O with residual water impurities. Discharge and charge proceeded at a constant voltage of 2.8 and 3.2 V, respectively, and a voltage gap of 400 mV was the smallest among the data found in the literature. This fact suggests that decomposition of lithium peroxide can be fast as its formation and the polarization at charge is held down.
Subjects and measures of Li–O2 batteries
The theoretical energy density of nonaqueous-type Li–O2 batteries is approximately 3,500 W·h·kg−1, when considering lithium and oxygen. This is approximately ten times that of LIBs. The battery has a simple structure consisting of lithium metal anode, nonaqueous electrolyte, and oxygen electrode, that is, carbon materials in most cases. The battery can utilize considerable part from the technologies of LIBs. The actual capacities or energy densities reported are usually much lower than the theoretical values. This is mainly due to the performance of oxygen electrode and hence charging overvoltage is so large and capacity retention with cycling is quite low. Oxygen reduction and evolution reactions in nonaqueous solution must be understood to solve these issues. Laoire et al studied the stability of one-electron reduction form, superoxide anion, in various nonaqueous solvents with different donor numbers (DNs).21 Since lithium cation is a strong Lewis acid, it forms more complexes with solvent molecules compared to O2−, which is a soft Lewis base. In high DN solvent as dimethylsulfoxide (DMSO), solvent-coordinated Li+ shows lower Lewis acidity than that in other solvents. Superoxide anion is stabilized in the solution, because it is a soft base and has an affinity toward the DMSO-solvated Li+. The ability to dissolve discharge product, O2− in this case, is an important feature of the electrolyte, while the solvent stability against these species with high nucleophilicity is also required at the same time.
In 2010, Mizuno et al22 studied the decomposition of organic electrolyte of Li(CF3SO2)2N (LiTFSI) in PC using transmission electron microscope with energy dispersive X-ray spectroscopy and Fourier transform infrared spectroscopy (FTIR) spectroscopy.22 Cycling performance of a Li–O2 cell with PC-based electrolyte is shown in Figure 3. Energy dispersive X-ray spectroscopy analysis for the discharged product showed both signals of carbon and oxygen, implying the formation of carbonate. FTIR spectra of the discharged products indicated the formation of Li2CO3 and lithium alkyl carbonates RO–(C=O)–OLi (R=alkyl group). The responses of the ideal discharge product, Li2O2, were not clearly observed. Cyclic voltammogram studies of PC-based electrolyte with oxygen electrode proved that one-electron reductant, O2 radical (superoxide anion), occurred during discharge and was mostly consumed by attacking PC. In the following charge process, CO2 was found to be the main component in the evolved gas from gas chromatography - mass spectrometry measurement. These results suggested that a carbonate solvent was first decomposed by the superoxide anion, and then carbonate species such as RO–(C=O)–OLi and Li2CO3 were obtained in discharge and CO2 was obtained in charge. It is concluded that the voltage gap between discharge and charge is closely related with the side reaction from the decomposed carbonate species. Mizuno et al continued to find a stable solvent against O2 radical by using molecular orbital calculation. The calculation showed that the electronic distribution in a solvent molecule and all the atoms in PP13TFSA except for H atom were charged to either zero or negative, resulting in the stabilization of O2 radical in the solvents.23
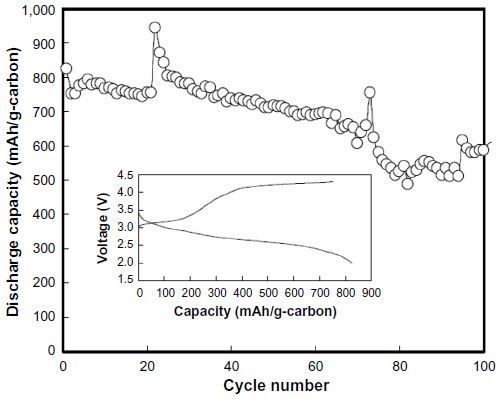 | Figure 3 Cycling performance of a Li–O2 cell. |
Thotiyl et al studied the stability of electrolytes on carbon cathodes.24 Combined with differential electrochemical mass spectrometry (DEMS) and FTIR, decomposition of carbons and electrolytes is analyzed as a function of oxygen electrode potential or capacity. DMSO and tetraglyme electrolytes were examined and compared. During discharge, carbon electrode is relatively stable and shows little decomposition. The decomposition extent depends on the hydrophobicity of the surface. The hydrophilic carbon is more unstable to form Li2CO3 and enhances more decomposition of the electrolyte. The electrolyte decomposition is the major side reaction during discharge in which Li2CO3 and lithium carboxylates are formed. During charge, both carbon electrode and electrolytes were decomposed to Li2CO3. While Li2CO3 is oxidatively decomposed during charge, more Li2CO3 are formed by the decomposition. Consequently, Li2CO3 was accumulated in the oxygen electrode. DMSO showed better stability than tetraglyme electrolyte with respect to the formation of Li2CO3 and lithium carboxylates. They found that the carbon decomposition only occurs above 3.5 V and with the presence of Li2O2. Li2O2 is not considered to cause the carbon decomposition, since pure chemical reaction between carbon and Li2O2 was not observed. They concluded that its oxidized species, O2−, arising during charge may enhance the carbon decomposition.
One of the methods to alleviate the damage on the carbon materials is to reduce the charging overvoltage with a catalyst. Grimaud et al investigated double perovskites (Ln0.5Ba0.5)CoO3−δ (Ln=Pr, Sm, Gd, and Ho) as catalysts for the oxygen evolution reaction (OER) upon water oxidation in alkaline solution.25 The OER activities of these double perovskites are exceptionally high, since the electron filling of 3d state in an eg, symmetry of transition metal cations is close to unity as suggested in their previous work.26 (Pr0.5Ba0.5)CoO3−δ exhibited the highest OER activity in the series and its origin was discussed with respect to the electronic structure. The cobalt oxidation state was found to increase with increasing lanthanide ionic radius. The density functional theory computed O p-band center shifts closer to the Fermi level with increasing cobalt oxidation state. They pointed out that the moving of O p-band toward the Fermi level results in an increasing intrinsic OER activity. Moving too close to the Fermi level decreases the structural stability during OER. (Ln0.5Ba0.5)CoO3−δ were found to be stable under OER potentials and cyclic voltammetry data of (Pr0.5Ba0.5)CoO3−δ showed no significant changes in the 25 cycles. On the other hand, Ba0.5Sr0.5Co0.8Fe0.2O3−δ with higher O p-band center readily becomes amorphous under OER conditions. They concluded that controlling a transition metal oxide having the O p-band close to the Fermi level is a promising strategy to create highly active oxide catalysts for OER of rechargeable metal–oxygen batteries.
In 2012, Peng et al27 reported an excellent cycle performance for a Li–O2 cell composed of a DMSO-based electrolyte and a nanoporous gold (NPG) electrode, where NPG was used for the oxygen electrode to avoid the decomposition of carbon due to the reasons described previously.24 The cycle performances for the Li/0.1 M LiClO4 in DMSO/NPG/O2 cell are shown in Figure 4. Nearly 95% of the initial discharge capacity was retained after 100 cycles. On discharge, Li2O2 was the main product together with a little amount of Li2CO3 and HCO2Li observed in the FTIR spectra. The DEMS analysis of evolved gases showed that no CO2, SO2, or SO3 by electrolyte decomposition was found. It is confirmed by FTIR and Raman analyses that the formed Li2O2 was removed on charging. DEMS data indicate that only O2 was the product on charge and electrolyte was not decomposed in the presence of Li2O2. They also investigated the effect of NPG by replacing it with carbon black (super P) electrode. On discharge, FTIR showed that the carbon electrode produced large amount of Li2CO3 and HCO2Li and their proportion is estimated to be approximately 15%. Hydrophilic carbon itself is unstable on discharge and the carbon possibly promotes electrolyte decomposition on discharge. NPG is not only stable but also provides higher rate performance for Li2O2 decomposition on charging.
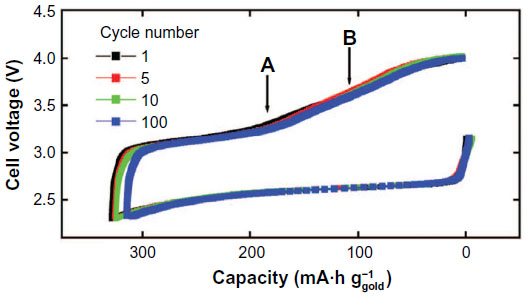 | Figure 4 Charge–discharge curves for a Li–O2 cell with a 0.1 M LiClO4–DMSO electrolyte and a nanoporous gold cathode at a current density of 500 mA·g-1 (based on the mass of Au). |
Tetraglyme glycol dimethyl ether [CH3O(CH2CH2O)4CH3] (TEGDME) is another candidate used as electrolyte in Li–O2 batteries. Ether-based electrolytes compared to organic carbonates are more stable toward reduced O2 species. A Li–O2 cell containing a lithium metal anode, an electrolyte of 1 M LiPF6 in TEGDME and a porous Super P carbon black cathode was discharged to 2 V and charged to 4.6 V at 70 mA·g−1 under 1 atm of O2.28 The XRD data demonstrated the presence of Li2O2 at the end of the first discharge. However, the FTIR spectra provided clear evidence of electrolyte decomposition. The 1H NMR study for the discharge products suggested the presence of HCOOD and CH3COOD. The mass spectroscopy analysis showed CO2 and H2O evolution during discharge. Laoire et al29 also examined the charge and discharge performance of the Li/LiPF6 in TEGDME/carbon black, O2 cell. The XRD pattern of the carbon electrode discharged at 2.0 V showed the presence of Li2O2, but the rechargeability was poor. Cyclic ethers, such as 1,3-dioxolen and 2-methyl-tetrahydrofuran, were in turn used to replace TEGDME in Li–O2 cells.28 The XRD data for the discharge products of the oxygen electrode with these electrolytes indicated the formation of Li2O2. However, the FTIR and NMR data demonstrated the presence of HCOOLi and CH3COOLi. In conclusion, electrolytes based on linear and cyclic ethers all exhibited electrolyte decomposition in Li–O2 cells.
Issues of lithium metal electrode are discussed next. Lithium metal is an ideal anode material because the potential is one of the lowest and the specific capacity is ten times that of graphite electrode. However, the practical use of lithium metal for reversible electrode has been a challenge due to the significantly low reversibility and high risk of flaming in organic solvents. Historically, a dissolution–deposition-type electrode was used in the first rechargeable battery, which was a lead–acid battery. Dissolution–deposition-type electrode is intrinsically low in reversibility due to large volume change, but the theoretical capacity is potentially high when compared to topochemical-type electrode. Study of lithium metal electrode is a necessary step for going beyond the limit of LIBs.
Lithium electrode is known to have an issue of depositing in dendritic morphology. In addition to its tendency in crystal growth, there are many parameters that should be considered to explain the dendritic growth. The growth rate of the nuclei depends on lithium transport ability from the electrolyte, whereas the nature of solid electrolyte interphase formed on lithium surface determines the current distribution homogeneity. The choice of electrolyte, therefore, is critical to suppress dendrite formation. Qian et al30 reported 4 M lithium bis(fluorosulfonyl)imide (LiFSI) in 1,2-dimethoxyethane (DME) as the electrolyte. The concentration of LiFSI to DME is approximately 1:2.41 in molar ratio. A remarkable feature of this electrolyte is the stability against reduction at the potential of lithium metal. Small linear ethers as DME are more stable than conventional carbonate solvent. The highly concentrated electrolyte has less number of free DME molecules that are more reactive than the ones coordinating ions. The highly compact feature of the SEI layer prevents further corrosion of the lithium metal electrode. The large amount of lithium cations can deliver high current densities in more homogeneous manner. Owing to these benefits, the highly concentrated LiFSI in DME electrolytes show excellent performances at high current densities as indicated in Figure 5. Lithium and copper cell could be cycled at 4 mA·cm−2 for more than 1,000 cycles with an average coulombic efficiency of 98.4%.
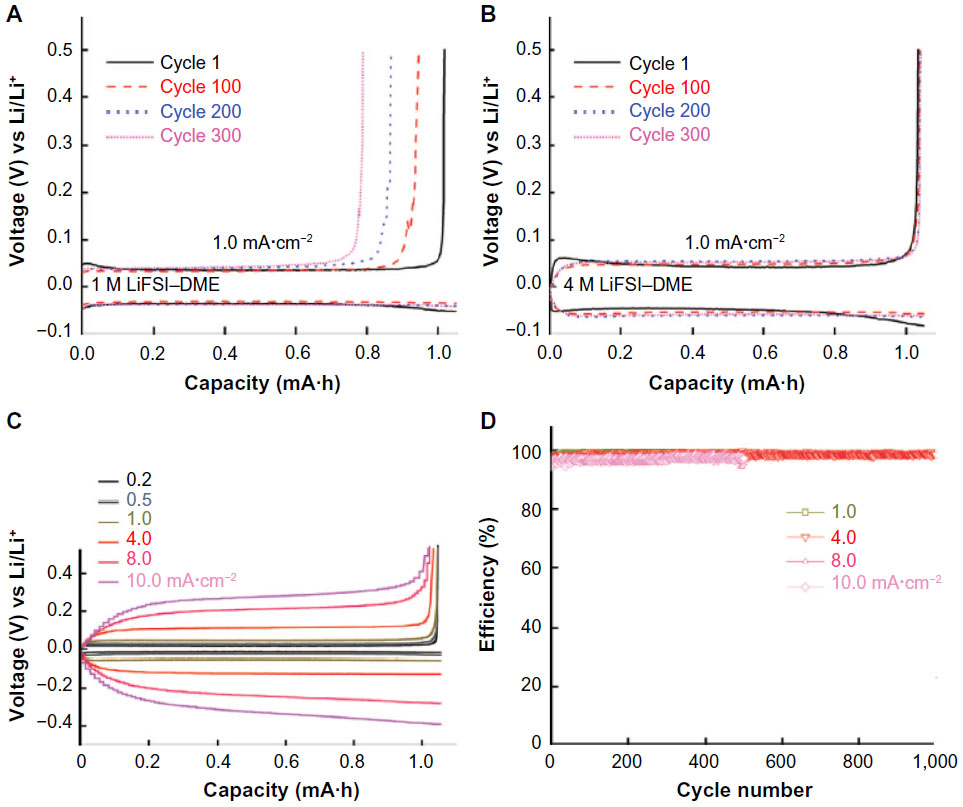 | Figure 5 Discharge–charge cycle performances of lithium metal electrode in DME-based electrolytes. |
Several researches have focused on electrolytic salts to produce flat electrodeposition of lithium without dendritic growth. It is reported that a single-ion lithium-conducting electrolyte can increase the lifetime of lithium metal anode compared to normal liquid electrolytes with mobile anions. Other researchers suggest a mechanically strong separator with uniform pores can stop the dendrites. Lu et al31 reported lithium-substituted Nafion membrane can show synergies between interconnected nanopores and single-ion-conductivity. The lithium transference number estimated is almost unity for the Nafion membrane soaked with ethylene carbonate–diethyl carbonate. This membrane electrolyte with two lithium electrodes facilitates stable lithium plating/deposition for more than 300 hours at 0.32 mA·cm−2. It could be galvanostatically charged at 0.05 mA·cm−2 without short circuit for more than 2,000 hours, while liquid electrolyte with LiPF6 shows quick short circuit at around 50 hours which is no more than 1/40 time of the single-ion electrolyte. The authors concluded that single-ion conducting nanoporous polymer membrane shows good ability to suppress dendrites and promote smooth lithium deposition.
Fundamental study on lithium metal deposition is indispensable to control the morphology and suppress the dendritic growth. Sano et al32 investigated lithium deposition in room-temperature ionic liquids that are highly stable at the potential of lithium metal and minimized the effect of surface electro-decomposed film. They discussed lithium nucleation and growth as a function of current density and lithium-ion diffusion rate by changing viscosity of the applied ionic liquids. According to their report, lithium nucleus decreased in size and more highly distributed with an increase in current density. This phenomenon is explained using the theory of thermodynamics. Formation free energy change of a lithium cluster can be expressed as a function of electrode overvoltage in lithium deposition reaction; and critical radius of the cluster is determined from a profile of the free energy change. When large current density is passed, higher overvoltage is applied and the critical radius becomes smaller. Therefore, in order to realize uniform deposits, relatively large overvoltage is needed at early stage of electrodeposition. In contrast, mass transfer plays a more important role in the process of nucleus growth. When lithium diffusion in electrolyte is slow and electrodeposition becomes diffusion-controlled, the nucleus grows in a dendritic morphology regardless of the number of nuclei. Their conclusion suggests that the current density should be maintained at not too large and too small to have moderate overvoltage and avoid dendrites. Consequently, lithium anode performance is strongly affected by the properties of electrolytes and external conditions as temperature.
Recent advances and future prospects
Aqueous Li–O2 batteries are advantageous in that the oxygen electrode reaction inherently includes electrolyte decomposition to form hydroxide anions. The system suffers none of the decomposed species that needs large polarization to be oxidized as seen in organic Li–O2 batteries. However, inclusion of water into the battery reaction as shown in the following equation significantly decreases the gravimetric energy density:
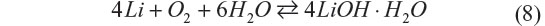
Management of water utilization is a critical issue to achieve stable electrochemical performance of the battery, and it is extremely difficult when the liquid amount must be minimized. Aqueous electrolytes in Li–O2 have distinct merits and drawbacks simultaneously. Recently, Matsui et al33 reported a novel electrochemistry for the aqueous Li–O2 battery, which has the potential to solve these issues.33 It is based on the oxygen–peroxide couple using a catalyst-free carbon-based electrode. Figure 6 shows the cycling performance and rate capability of a Li–O2 cell with the catalyst-free carbon black–polytetrafluoroethylene electrode and 10 M LiCl and saturated LiOH aqueous electrolyte. The XRD data for the discharge products of the oxygen electrode matched the simulated pattern of Li2O2. It was commonly believed that the reaction at the oxygen electrode is four-electron reduction of dioxygen to hydroxide anion, but the result implies that the reaction stops when two electrons are transferred. The four-electron transfer process consists of two two-electron steps, and the second step, irrespective of whether it is electrochemical or chemical, did not occur at a reasonable rate in this aqueous Li–O2 cell. Catalyst-free carbon-based electrodes are known to be good reversible electrodes for the oxygen–peroxide couple in alkaline solution.11 According to these data, the typical electrochemical reaction of the oxygen–peroxide couple in aqueous systems is as shown in the following equations:
 | Figure 6 The catalyst-free carbon black–polytetrafluoroethylene electrode was tested in 10 M LiCl and saturated LiOH aqueous electrolyte. |
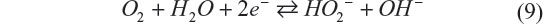
Then hydroperoxide anions react with lithium cations to form lithium peroxide.
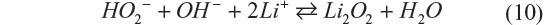
This reaction should occur depending on the concentrations of the species and acidity/basicity of their solvation states by solvent molecules. The next question is why Li2O2 was not hydrolyzed. Li2O2 should disproportionate in aqueous electrolyte into LiOH and O2. The electrolyte saturated with LiCl and LiOH gives very small number of free water molecules. Such circumstance with extremely low activity of water molecules is thought to result in specifically stabilized Li2O2. These findings are quite interesting, since the issues originated from water participating in the reaction; for example, low gravimetric energy densities can be eliminated without sacrificing the advantages of the aqueous solution. Ensuring long-term stability of Li2O2 in the aqueous solution is of crucial significance to this new concept Li–O2 battery.
One of the critical issues in organic Li–O2 batteries is the solvent degradation by superoxide anion formed in the course of oxygen reduction. Another issue is the Li2O2 precipitation on porous carbon oxygen electrode. Li2O2 not only enhances carbon decomposition during charge but also physically blocks the mass and electron transportation by covering the electrode surface. It is, therefore, a good target to find a solvent that can dissolve Li2O2 or LiO2 that shows stability to these oxide species. Khetan et al34 developed a thermodynamic model that can describe these two effects. Salt solubility is determined by a balance of binding energy between cations and anions and their solvation energies. If the solvent has high donor and acceptor numbers, LiO2 or Li2O2 tends to dissolve as a solvated lithium cations and O2− or O22− anions. They analyzed the commonly used solvents such as DMSO, dimethylacetamide (DMA), dimethylformamide (DMF), N-methylpyrrolidone (NMP), DME and others in terms of their solvation ability and rate of nucleophilic attack based on their thermodynamic model. As shown in Figure 7, there exists an anticorrelation between these two functionalities. Solvents with high DNs have better solvation capabilities, as they are prone to oxidation during charging. It is concluded that good solvation ability and high stability against nucleophile are fundamental trade-off characters and finding an all-around solvent is not straightforward. Identifying a right blend of solvents is one way to achieve these two requirements. Computations and model development are considered to greatly contribute to this work.
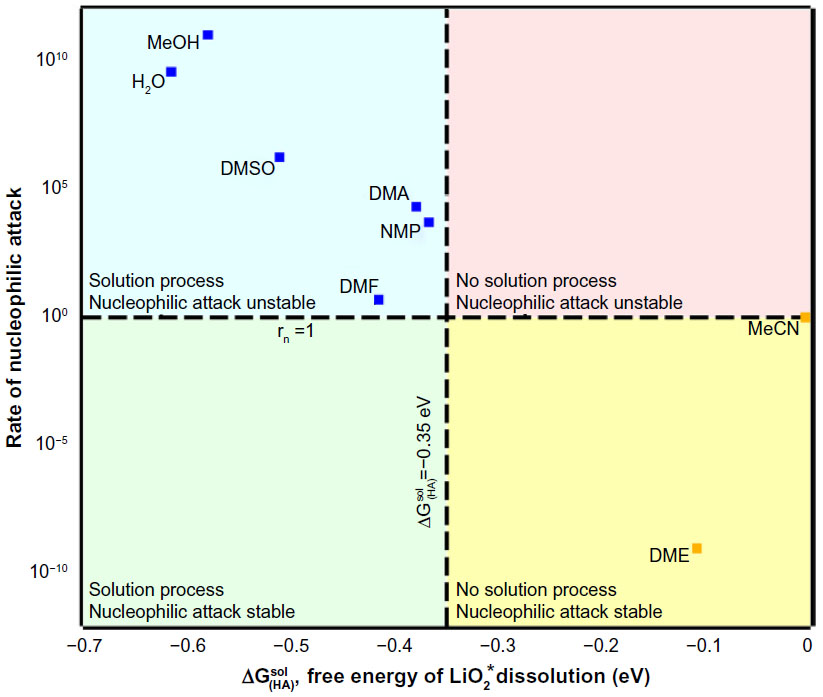 | Figure 7 The rate of nucleophilic attack by superoxide anion is plotted against solvent’s ability to dissolve lithium superoxide. |
Associated with the earlier discussion, understanding the mechanism of formation of Li2O2 is also an important subject. Aetukuri et al35 found that Li2O2 is generally deposited as thin conformal films on the cathode surface. However, after the addition of H2O, larger toroids and platelets are formed. The presence of H2O in the electrolyte changes the morphology of Li2O2 and increase the discharge capacity as H2O concentration increases. They studied linear sweep voltammetry behavior of DME electrolyte containing different amount of H2O. Anhydrous DME sample shows only one peak, while the electrolyte with a certain amount of H2O shows an additional second peak. They attributed the first peak to surface electrochemical growth of Li2O2 and the second peak to the solution-mediated growth of Li2O2. The surface-adsorbed LiO2, formed initially in oxygen reduction process, can be dissolved in the presence solvent with a high donor number, DME, and solvent with a high acceptor number, H2O. Solvated O2− ions again form LiO2 with solvated lithium-ions. These LiO2 disproportionate to form Li2O2 on a larger growing particle. This formation of Li2O2 in solution drastically increases the discharge capacity, but it stops when the H2O is finally consumed and converted to H2O2 and when it reacts with Li anode. The present method is not directly applicable to a practical battery, but the concept suggests that appropriate combination of solvents and additives can improve small discharge capacity due to insolubility of Li2O2.
Effect of water in organic electrolyte is also discussed by a group in the University of Cambridge. Liu et al36 reported a unique approach that the organic Li–O2 battery operated with large quantities of water and soluble redox mediator such as LiI.36 The battery was prepared from lithium metal anode, LiTFSI/DME electrolyte with LiI soaked in a glass fiber separator and reduced graphene oxide cathode. The first discharge and charge were observed at 2.75 and 2.95 V, respectively, as shown in Figure 8. The battery could be cycled 2,000 times without capacity fading with a capacity limit of 1,000 mA·h·g−1 carbon and at 0.02 mA·cm−2. Such performance with excellent energy efficiency has never been observed in a normal organic Li–O2 battery without the LiI additive. After discharge, the graphene oxide cathode in the battery with LiI was completely covered with LiOH particles that is confirmed a dominant phase by X-ray diffraction method. O2 is first reduced on the cathode surface to produce LiO2. LiO2 is considered to chemically transform to LiOH in the co-presence of H2O and LiI. The precipitated LiOH was fully removed after charge process. The role of LiI was deduced as follows. LiI brings the trace amount of water in the cell and converts LiO2 to LiOH. It also provides a chemical pathway to decompose LiOH into Li+, H2O, and O2. Since I− becomes an active material during charge process, the charge overvoltage remains very low. As a consequence, reversible formation and removal of LiOH lead to a highly efficient and rechargeable organic Li–O2 battery.
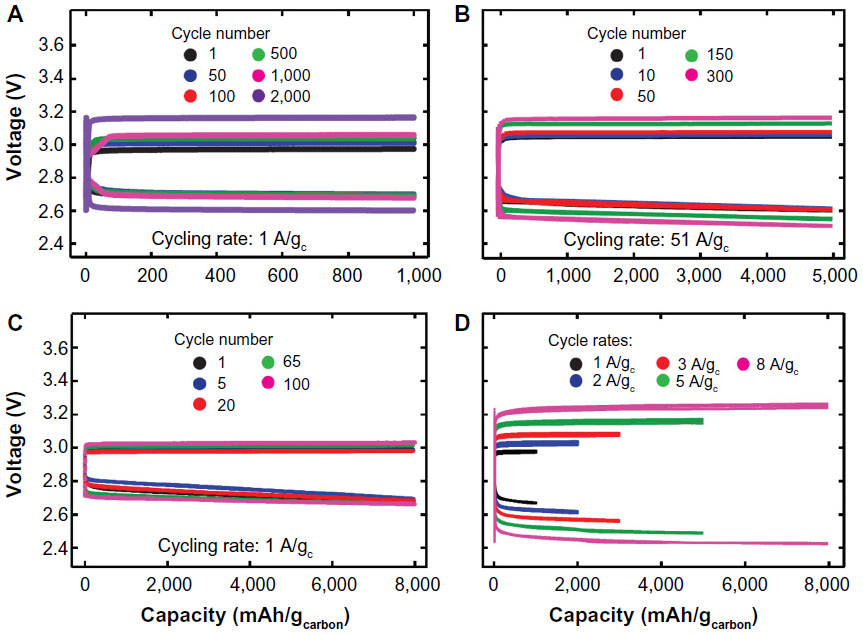 | Figure 8 Discharge/charge curves for Li–O2 batteries using LiTFSI/DME electrolyte with LiI additive at different capacity limits and current densities. |
Lithium metal anode with solid electrolyte is expected to serve as a safer electrode system, since lithium dendrite can be blocked physically. Garnet-type Li7La3Zr2O12 electrolytes are stable in contact with lithium metal. In direct lamination with lithium metal sheet, the ceramic is required to have high sintering density, because lithium metal deposition may occur in the voids or pores that result in a short circuit of the cell. Recently, Suzuki et al37 prepared transparent Al2O3-doped Li7La3Zr2O12 by hot isostatic pressing (HIP) and examined the lithium dendrite formation. The relative densities of 1 wt % Al2O3-doped Li7La3Zr2O12 with and without HIP treatment were 91.5% and 99.1%, respectively. The change in the cell voltage with the polarization period at 0.5 mA·cm−2 and 25°C was observed for the Li/Li symmetrical cell. The cell with HIP-treated transparent solid electrolyte showed a gradual increase in the voltage, but no sudden voltage drop during the polarization. Previous studies on Nb-doped and Ta-doped Li7La3Zr2O12 with 92.8% and 96.7% of relative densities showed quick cell voltage drops in several hundreds of seconds. This result, therefore, suggests that the grain boundary-free solid electrolyte prevents lithium dendrite penetration through the electrolyte.
Conclusion
In this article, the works of the non aqueous and aqueous Li–O2 batteries are reviewed. In the former, since the report by Ogasawara et al,15 electrochemistry of oxygen electrode has been a main research target. Due to the efforts of researchers in this field, the oxygen reduction mechanism and critical role of electrolyte solvents and catalysts have become more obvious as described earlier. Finding a stable organic solvent is still underway, but these accumulated knowledge will surely lead to optimized electrolyte system. On the other hand, there are not much clear visions to solve severe side reaction of carbon materials during discharge and charge. These challenges are closely related to the efficient way of dealing with insoluble and insulating discharge product. Redox mediator together with H2O as additive to organic electrolyte is one way to avoid these issues and improve rechargeability of cathode materials. As the protection of lithium becomes necessary, technical development including protected lithium electrode which maintains the stability of lithium anode will become an important theme.
Regarding lithium, the safety and long-term operation issues in addition to the lithium dendrite problems should be solved. Solvated ionic liquid, which is a highly concentrated glyme electrolyte, can reduce surface film formation on lithium metal – that is, improve coulombic efficiency and yield spherical electrodeposition. In order to secure safety by blocking dendrites physically, researches of solid electrolytes and separators also become important. Lithium metal performance in larger scale must be examined to understand clearly the kind of problem encountered in practical battery systems.
In aqueous system, the choice of battery material is wider. This is mostly because the reaction product of LiOH or Li2O2 is soluble in aqueous electrolyte. However, the aqueous system could not reach a comparable specific energy density to the nonaqueous counterpart, because large amount of water must be contained as active material. The ceramic protective layer limits the output power and increases the weight of a cell.
These two Li–O2 batteries stand on a different basis. However, the goal that they aim at seems to be getting closer as symbolically shown in the use of water in organic electrolyte. Further research works and cooperation between both areas are important to achieve a practical Li–O2 battery.
Disclosure
The author reports no conflicts of interest in this work.
References
Girshkumar G, McCloskey B, Luntz AC, Swanson S, Wilche W. Lithium-air battery:promise and challenges. J Phys Chem Lett. 2010;1:2193–2203. | |
Goldstein J, Brown I, Koretz B. New developments in the Electric Fuel Ltd. zinc/air system. J Power Sources. 1999;80:171–179. | |
Shiga T, Hase Y, Yagi Y, Takahashi N, Takechi K. Catalytic cycle employing a TEMPO-anion complex to obtain a secondary Mg-O2 battery. J Phys Chem Lett. 2014;5:1648–1652. | |
Yang S, Knickle H. Design and analysis of aluminum/air battery system for electric vehicles. J Power Sources. 2002;112:162–173. | |
Carlin RT, Crawford W, Bersch M. Nucleation and morphology studies of aluminum deposited from an ambient-temperature chloroaluminate molten salt. J Electrochem Soc. 1992;139:2720–2727. | |
Wang L, Liu F, Wang W, Yang G, Zheng D, Wub Z, Leung MKH. A high-capacity dual-electrolyte aluminum/air electrochemical cell. RSC Adv. 2014;4:30857–30863. | |
Hartmann P, Bender CL, Vracar M, Dürr AK, Garsuch A, Janek J, Adelhelm P. A rechargeable room-temperature sodium superoxide (NaO2) battery. Nat Mater. 2013;12:228–232. | |
Littauer EL, Tsai KC. Corrosion of lithium in aqueous electrolyte. J Electrochem Soc. 1977;124:850–855. | |
Littauer EL, Tsai KC. Anodic behavior of lithium in aqueous electrolyte I transient passivation. J Electrochem Soc. 1976;123:771–776. | |
Semkow KW, Sammells FA. A lithium oxygen secondary battery. J Electrochem Soc. 1987;134:2084–2085. | |
Batalov NN, Arkhipov EL. Investigation aimed at development of a high-temperature lithium/air storage battery (The 20th International Power Sources Symposium, P30). J Power Sources. 1997;65:287–288. | |
Abraham KM, Jang Z. Polymer electrolyte based rechargeable lithium oxygen battery. J Electrochem Soc. 1996;143:1–5. | |
Read J. Characterization of lithium/oxygen organic electrolyte battery. J Electrochem Soc. 2002;149:A1190–A1195. | |
Read J. Ether-based electrolyte for the lithium/oxygen organic electrolyte battery. J Electrochem Soc. 2006;153:A96–A100. | |
Ogasawara T, Debart A, Holtzapfel M, Novak P, Bruce PG. Rechargeable Li2O2 electrode for lithium batteries. J Am Chem Soc. 2006;128:1390–1393. | |
Visco SJ, Nimon E, Katz B, De Jonghe LC, Chu MY. Lithium metal aqueous batteries. Abstract of the International Meeting on Lithium Batteries 12; June 27-July 2, 2004; Nara, Japan. Abstract 53. | |
Kumar B, Kumar J. Cathode for solid-state lithium-oxygen cells:roles of nasicon glass ceramics. J Electrochem Soc. 2010;157:A611–A616. | |
Kumar B, Kumar J, Leese R, Fellner JP, Rodrigues SJ, Abraham KM. A solid-state rechargeable, long cycle life lithium-air battery. J Electrochem Soc. 2010;157:A50–A54. | |
Kumar B, Thomas D, Kumar J. Space-charge-mediated superionic transport in lithium ion conducting glass-ceramics. J Electrochem Soc. 2009;156:A506–A513. | |
Hassoun J, Croce F, Armand M, Scrosati B. Investigation of the O2 electrochemistry in a polymer electrolyte solid-state cell. Angew Chem Int Ed. 2011;50:2999–3002. | |
Laoire CO, Mukerjee S, Abraham KM, Plichta EJ, Hendrickson MA. Influence of nonaqueous solvents on the electrochemistry of oxygen in the rechargeable lithium-air battery. J Phys Chem C. 2010;114:9178–9186. | |
Mizuno F, Nakanishi S, Kotani Y, Yokoishi S, Iba H. Rechargeable Li-air batteries with carbonate-based liquid electrolytes. Electrochemistry. 2010;78:403–405. | |
Mizuno F, Nakanishi S, Shirasawa A, Takechi K, Shiga T, Nishikoori H, Iba H. Design of non-aqueous liquid electrolytes for rechargeable Li-O2 batteries. Electrochemistry. 2011;79:876–881. | |
Thotiyl MMO, Freunberger SA, Peng Z, Bruce PG. The carbon electrode in nonaqueous Li-O2 cells. J Am Chem Soc. 2013;135:494–500. | |
Grimaud A, May KJ, Carlton CE, et al. Double perovskites as a family of highly active catalysts for oxygen evolution in alkaline solution. Nat Commun. 2013;4:Article number 2439. | |
Suntivich J, May KJ, Gasteiger HA, Goodenough JB, Shao-Horn Y. A perovskite oxide optimized for oxygen evolution catalysis from molecular orbital principles. Science. 2011;334:1383–1385. | |
Peng Z, Freunberger SA, Chen Y, Bruce PG. A reversible and high rate Li-O2 battery. Science. 2012;337:563–566. | |
Freunberger SA, Chen Y, Drewett NE, Hardwick LJ, Bardt F, Bruce PG. The lithium-oxygen battery with ether-based electrolytes. Angew Chem Int Ed. 2011;50:8609–8613. | |
Laoire CO, Mukerjee S, Plichta EJ, Hendrickson MA, Abraham KM. Rechargeable lithium/TEGDME-LiPF6/O2 battery:batteries and energy storage. J Electrochem Soc. 2011;158:A302–A308. | |
Qian J, Henderson WA, Xu W, Bhattacharya P, Engelhard M, Borodin O, Zhang JG. High rate and stable cycling of lithium metal anode. Nat Commun. 2015;6:Article number 6362. | |
Lu Y, Tikekar M, Mohanty R, Hendrickson K, Ma L, Archer LA. Stable cycling of lithium metal batteries using high transference number electrolytes. Adv Energy Mater. 2015;5:Article number 1402073. | |
Sano H, Sakaebe H, Senoh H, Matsumoto H. Effect of current density on morphology of lithium electrodeposited in ionic liquid-based electrolytes. J Electrochem Soc. 2014;161:A1236–A1240. | |
Matsui M, Wada A, Matsuda Y, Yamamoto O, Takeda Y, Imanishi N. A novel aqueous lithium-oxygen cell based on the oxygen-peroxide redox couple. Chem Commun. 2015;51:3189–3192. | |
Khetan A, Luntz AC, Viswanathan V. Trade-offs in capacity and rechargeability in nonaqueous Li-O2 batteries:solution-driven growth versus nucleophilic stability. J Phys Chem Lett. 2015;6:1254–1259. | |
Aetukuri NB, McCloskey BD, García JM, Krupp LE, Viswanathan V, Luntz AC. Solvating additives drive solution-mediated electrochemistry and enhance toroid growth in non-aqueous Li-O2 batteries. Nat Chem. 2015;7:50–56. | |
Liu T, Leskes M, Yu W, et al. Cycling Li-O2 batteries via LiOH formation and decomposition. Science. 2015;350:530–533. | |
Suzuki Y, Kami K, Watanabe K, et al. Transparent cubic garnet-type solid electrolyte of Al2O3-doped Li7La3Zr2O12. Solid State Ionics. 2015;278:172–176. | |
Egan DR, Ponce de León C, Wood RJK, Jones RL, Stokes KR, Walsh FC. Developments in electrode materials and electrolytes for aluminium-air batteries. J Power Sources. 2013;236:293–310. | |
Abraham KM. A brief history of non-aqueous metal-air batteries. ECS Trans. 2008;3:67–71. | |
Ji L, Rao M, Zheng H, et al. Graphene oxide as a sulfur immobilizer in high performance lithium/sulfur cells. J Am Chem Soc. 2011;133:18522–18525. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.