")
Back to Journals » OncoTargets and Therapy » Volume 9
Toward a noncytotoxic glioblastoma therapy: blocking MCP-1 with the MTZ Regimen
Authors Salacz M, Kast R , Saki N , Brüning A, Karpel-Massler G, Halatsch M
Received 12 November 2015
Accepted for publication 4 February 2016
Published 27 April 2016 Volume 2016:9 Pages 2535—2545
DOI https://doi.org/10.2147/OTT.S100407
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Faris Farassati
Michael E Salacz,1,2 Richard E Kast,3 Najmaldin Saki,4 Ansgar Brüning,5 Georg Karpel-Massler,6 Marc-Eric Halatsch6
1Department of Internal Medicine, 2Department of Neurosurgery, University of Kansas, Kansas City, KS, USA; 3IIAIGC Study Center, Burlington, VT, USA; 4Health Research Institute, Research Center of Thalassemia and Hemoglobinopathy, Ahvaz Jundishapur University of Medical Sciences, Ahvaz, Iran; 5Molecular Biology Laboratory, University Hospital Munich, Munich, Germany; 6Department of Neurosurgery, University of Ulm, Ulm, Germany
Abstract: To improve the prognosis of glioblastoma, we developed an adjuvant treatment directed to a neglected aspect of glioblastoma growth, the contribution of nonmalignant monocyte lineage cells (MLCs) (monocyte, macrophage, microglia, dendritic cells) that infiltrated a main tumor mass. These nonmalignant cells contribute to glioblastoma growth and tumor homeostasis. MLCs comprise of approximately 10%–30% of glioblastoma by volume. After integration into the tumor mass, these become polarized toward an M2 immunosuppressive, pro-angiogenic phenotype that promotes continued tumor growth. Glioblastoma cells initiate and promote this process by synthesizing 13 kDa MCP-1 that attracts circulating monocytes to the tumor. Infiltrating monocytes, after polarizing toward an M2 phenotype, synthesize more MCP-1, forming an amplification loop. Three noncytotoxic drugs, an antibiotic – minocycline, an antihypertensive drug – telmisartan, and a bisphosphonate – zoledronic acid, have ancillary attributes of MCP-1 synthesis inhibition and could be re-purposed, singly or in combination, to inhibit or reverse MLC-mediated immunosuppression, angiogenesis, and other growth-enhancing aspects. Minocycline, telmisartan, and zoledronic acid – the MTZ Regimen – have low-toxicity profiles and could be added to standard radiotherapy and temozolomide. Re-purposing older drugs has advantages of established safety and low drug cost. Four core observations support this approach: 1) malignant glioblastoma cells require a reciprocal trophic relationship with nonmalignant macrophages or microglia to thrive; 2) glioblastoma cells secrete MCP-1 to start the cycle, attracting MLCs, which subsequently also secrete MCP-1 perpetuating the recruitment cycle; 3) increasing cytokine levels in the tumor environment generate further immunosuppression and tumor growth; and 4) MTZ regimen may impede MCP-1-driven processes, thereby interfering with glioblastoma growth.
Keywords: cognition-sparing, high-grade glioma, immunosuppression, macrophage, microglia, monocyte
Introduction
Re-purposing of older, already-marketed drugs is not a new concept. With technological advances giving us a greater understanding of molecular pathways in cancer cells, the idea of re-purposing (redirecting, repositioning, etc) a previously approved drug, with a known safety/toxicity profile for lower cost, risk, and time, compared with developing novel pharmaceuticals has appeal.1–4
Our paper represents a contribution to the ongoing efforts in developing treatments for glioblastoma, one of the most treatment-refractory of all human cancers. Challenges to treatment include difficulty in drug delivery across the blood–brain barrier, the wide heterogeneity of glioblastoma cells within individual tumors,5–7 and the multiplicity of essential signaling and metabolic pathways within individual tumor cells, the so-called “Nile Distributary Problem”.4,8 One area of growing interest is modifying tumor–immune system signaling pathways, specifically the cross talk between the tumor cells and the immune cells resident in the tumor microenvironment including monocytes, dendritic cells, macrophages, and microglia, collectively termed monocyte lineage cells (MLCs).
Early in development, the embryonic brain becomes populated by bone marrow-derived MLCs, to finally comprise ~10%–20% of the adult brain, appearing as microglia. Circulating marrow-derived monocytes can also populate an adult brain, particularly during pathological states, including glioblastoma, but also in the setting of infection, trauma, or inflammatory disease such as multiple sclerosis, appearing then in brain as either macrophages or microglia (described in the magisterial review of microglia by Harry9). Some of the first micrographs of microglia by Klatzo from 1952 have not been surpassed.10 Although normal steady-state microglia have been called “quiescent”, they are in fact physiologically active in multiple homeostatic roles essential to brain function by scavenging dead cell remnants, phagocytosis, antigen presentation, synaptic maintenance, neurite pruning, repair and angiogenesis, and extracellular signaling.11,12 Microglia are in continuous reciprocal humoral communication with nearby neurons and astrocytes. Importantly for understanding glioblastoma, microglia are normally sessile but become motile on activation.
MLCs, including microglia, have activation states termed M1 or M2, but these might be better viewed as spectral extremes. The M1-phenotype is associated with high levels of cytokine interleukin (IL)-1beta, IL-12, IL-23, TNF-alpha, nitric oxide synthase, CD40, MHC I, and MHC II expression; these M1 cells are traditionally called pro-inflammatory state/activated microglia. In contrast, M2 cells have low levels of these inflammatory mediators, are profibrotic, synthesize transforming growth factor-beta and IL-10, tend to suppress inflammation, and promote neovascularization.11,12 In relation to cancer, the M1 phenotype functions more in an immune-boosting/antitumor role, whereas the M2 phenotype is more anti-inflammatory and tumor-supportive.
The critical importance of this tumor–microglia relationship to tumor growth has been observed in a number of recent papers.12–17 Furthermore, this ongoing “reciprocal supportive interplay”17 facilitates more than just enhanced vigor of tumor cell division and enhanced blood supply but potentially also contributes to glioblastoma migratory invasion into the surrounding brain.18 Hewedi et al recognize “glioblastoma therapies should address this key CD68-positive [ie, monocyte/macrophage] population”.19 We propose targeting MCP-1, a 13 kDa signaling peptide, synonymous with CC-chemokine ligand 2, which binds to outer cell membrane receptors CCR2 and/or CCR4 on MLCs and facilitates glioblastoma–microglia interaction.
This paper reviews and extends this concept, fills in details, and draws the following simple and straightforward conclusions:
- The “reactive gliosis”20 seen involved with and surrounding the main glioblastoma tumor mass is not an epiphenomenon but rather is a crucial core element of glioblastoma pathology.
- It is primarily the malignant glioblastoma cell that generates pathological signals, including MCP-1, to systemic monocytes/microglia.
- Otherwise normal but pathologically activated MLCs are important for robust glioblastoma cell growth; these cells are attracted by increased MCP-1 signaling within the tumor mass. MCP-1 is also an important signal for preferred MLC differentiation to the M2 anti-inflammatory/tumor-promoting phenotype.
- These tumor-infiltrating and pathologically functioning MLCs contribute to a feedback loop involving MCP-1 secretion which further perpetuates glioblastoma production of MCP-1.
- MCP-1 represents a critical mechanism of tumor–microglia communication which contributes significantly to tumorigenesis and potential invasion and is a potential therapeutic target.
- Three noncytotoxic drugs, minocycline, telmisartan, and zoledronic acid, the MTZ Regimen, are all approved for use in humans for noncancer conditions, are all lipophilic and highly brain-penetrant, are all capable of targeting the pathologic MCP-1 secretion, and together will provide MCP-1 and consequently MLC-focused treatment.
A well-developed research database supports each of the three MTZ Regimen drugs’ ability to block or reduce MCP-1 signaling and thus MLC trophic functions and pathologic activation. Additionally, each of these drugs is relatively nontoxic in humans, with significant post-marketing data available, and does not appear to have significant drug–drug interactions alone or in combination. We propose this approach either alone or in combination with other anticancer therapies as a novel glioblastoma treatment approach.
Glioblastoma are heavily infiltrated by blood monocytes
Glioblastomas are heavily infiltrated by cells of the immune system, and these cells are pathophysiologically active. In his seminal 1952 paper describing glioblastoma histology, Klatzo observed, “In the majority of tumors studied there was extensive microglial reaction […] resting forms were infrequently seen”.10 We have more recently come to understand that these immune cell populations are composed of bone marrow-origin monocytes,21,22 monocyte-derived macrophages, and microglia,19,23–29 and neutrophils.21,30–35 Estimates vary that these bone marrow-derived cells or nonmalignant microglia comprise 10%–30% of the total glioblastoma tumor mass,12,23,26,36,37 but there is general agreement that MLCs make up a considerable percent of total mass38 and MLCs have an established role in glioblastoma growth.39 The monocyte-related component arrives both by migration of circulating monocytes/macrophages27 and by mitoses or centripetal migration of brain-resident microglia40 and monocytes.38
Reactive gliosis in brain tissue adjacent to a growing glioblastoma20 is not an incidental finding or epiphenomenon. Reactive gliosis is rather an integral feature of glioblastoma and a crucial link in glioblastoma’s growth.20 Brain-resident microglia infiltrate and become integrated with malignant glioblastoma cells41 as do circulating MLCs.
M1 versus M2 phenotypes
Macrophages can be divided into M1 and M2 subtypes, which have contrasting activity. M1 macrophages are traditionally thought of as immunostimulatory with potential antitumor activity, whereas M2 exhibit an immunosuppressive phenotype with tumor-promoting and angiotrophic activity.17 Human glioblastomas are richly infiltrated by MLCs expressing both M1 and M2 markers.14,24 Although some studies indicate that these MLCs are more of M2 immunosuppressive, tumor trophic class,26 other studies show that these tissue macrophages are of indeterminate class, with some having attributes of both M1 and M2 phenotypes, whereas others have attributes of neither phenotype.24,42
Furthermore, these M1/M2 phenotypes are not terminally differentiated cells; in glioblastoma cell-conditioned culture medium, M1 macrophages can be induced to transform to an M2 phenotype,43 a finding that was later independently confirmed.28 A differentiation to M2-phenotypic macrophages has also recently been described to be promoted by glioma cell-derived extracellular vesicles.44
In another set of experiments, it has been shown that doxycycline (chemically similar, but more hydrophilic and less brain-penetrant compared with minocycline) inhibits M2-type polarization of human and bone marrow-derived mouse macrophages.45 Clearly in viral infections of the brain, for example, West Nile virus, circulating monocytes are recruited to brain tissue by virus-mediated increases in MCP-1, and these monocytes appear in brain tissue as microglia.46
Disproportionate immunosuppression of glioblastoma
“Gliomas attract brain-resident (microglia) and peripheral macrophages and reprogram these cells into immunosuppressive, proinvasive cells.”40 Myeloid cell infiltration and the function changes induced in them after contact with or proximity to malignant glioblastoma cells form a central element of the otherwise unexplained immunosuppression common in glioblastoma patients.22,32,34,47,48 Of particular note, normal human monocytes exposed in vitro to glioblastoma cells take on an immunosuppressive M2 phenotype, synthesizing IL-10 and transforming growth factor-beta.49,50
The MCP-1 amplification loop
MCP-1 agonism at CCR2 stimulates migration of monocytes/macrophages. Glioblastoma cells themselves synthesize and release MCP-1.51,52 While MCP-1 remains important in MLC recruitment and M2 phenotype promotion, there are other molecules which appear to contribute to these processes as well. One molecule that appears to be primarily secreted from the stem cell subpopulation of glioblastoma cells is periostin, which contributes to M2 differentiation.27 Several other studies have demonstrated that malignant glioblastoma cells will also synthesize and secrete granulocyte–macrophage colony-stimulating factor that can also drive transformation from M1 to M2 tumor-infiltrating MLCs.40 That said, while it is difficult to compare relative contributions of different molecules having similar effects, it appears that MCP-1 encompasses MLC recruiting, M2 transformation, and generation of a positive feedback loop to the glioblastoma tumor cells, while the others promote only portions of the larger process. Brain-resident microglia have been shown to migrate into a growing glioblastoma tumor; this migration is driven in part by granulocyte–macrophage colony-stimulating factor synthesized by glioblastoma malignant cells themselves,40,53–58 and in part by MCP-1 synthesized by other already-resident microglia.
An oddity of human glioblastoma tissue is that it attracts immunosuppressive, regulatory T-cells (Tregs).59 Tregs express CCR2 receptors and are tropic along an increasing MCP-1 gradient.59 Glioma cell lines D-54, U-87, U-251, and LN-229 synthesize large amounts of MCP-1 that was shown to be chemotactic to Tregs.60 Glioblastoma patients with higher circulating plasma MCP-1 titers have a poorer prognosis and faster disease course compared to patients with low plasma titers.61 Human glioblastoma biopsy tissue overexpresses MCP-1.62 Glioblastoma cell lines overexpressing MCP-1 demonstrated enhanced motility in response to MCP-1 exposure.62
Having established the likely importance of MCP-1 in macrophages’ promotion of glioblastoma growth, the next section discusses how several currently marketed noncytotoxic drugs can defeat this growth-facilitating aspect.
Resident peritumoral microglia secrete ample MCP-1 which in turn is chemotactic for circulating monocytes, thereby recruiting yet more MLCs. Thus, MCP-1 synthesis by glioblastoma-resident macrophages/microglia forms a positive amplification loop.62 Since glioblastoma cells themselves express MCP-1 receptors, MCP-1 mRNA, and MCP-1 protein,62–65 they thereby initiate this pathological amplification loop. Wurdinger et al rhetorically wonder “how to break [this] vicious cycle […] the interactive process between glioma cells and microglia and macrophages […]?”66 The MTZ Regimen now attempts to simply answer that question (Figure 1).
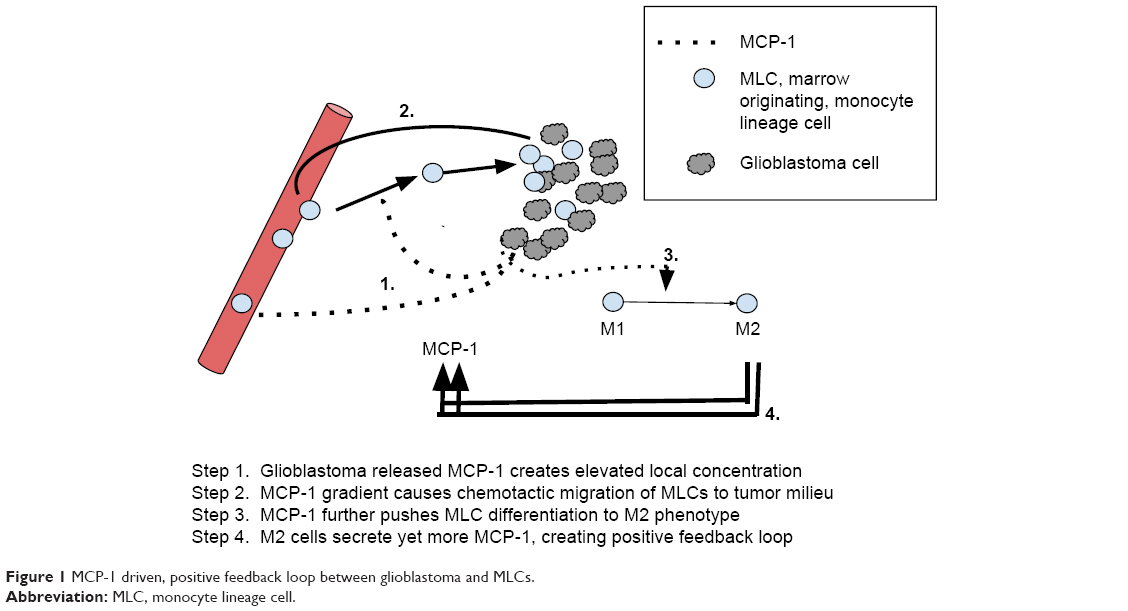 | Figure 1 MCP-1 driven, positive feedback loop between glioblastoma and MLCs. |
Preclinical experiments pointed to tumor-associated macrophages as a primary mediator of escape from antiangiogenic therapies and prompted a study of the consequences of anti-VEGF monoclonal antibody bevacizumab treatment of recurrent glioblastoma.67 They found on autopsy that patients treated with bevacizumab had greatly increased numbers of glioblastoma tissue macrophages compared with those not exposed to bevacizumab.67 Of particular note to our intended use of macrophage-directed treatment, Lu-Emerson et al also found that extent of tumor infiltration with macrophages correlated with shorter overall survival.67
The drugs
Minocycline
Minocycline is a 457 Da, intermediate-half-life (10–20 hours), lipophilic, tetracycline class antibiotic68–70 with excellent blood–brain penetration and brain tissue levels.71 Minocycline is active against a variety of Gram-positive and Gram-negative bacteria, Rickettsia, Chlamydia, as well as protozoans Plasmodia, Toxoplasma, and others.71 As an aside, significant neuroprotective attributes of minocycline have been the subject of dozens of reports in the last few years, this observation may provide future directions for research.72
In culture, normal microglia and glioma cell lines secreted ample MCP-1 which could be inhibited by 25 μM minocycline.73 Lipopolysaccharide-stimulated MCP-1 protein production in monocytes was suppressed by minocycline in vitro by inhibition of an IkB phosphorylation step, keeping NFkB within cytoplasm.74 The related tetracycline class antibiotic doxycycline inhibited M2 polarization in a laser injury model of choroidal neovascularization.45
MCP-1 levels are elevated in human Japanese encephalitis-infected tissues from humans and experimentally infected animals.75 Correspondingly, high tissue infiltration of monocytes is seen, a process that can be suppressed by minocycline.75 Simian immunodeficiency virus-infected macaques had grossly upregulated cerebral MCP-1 and monocyte infiltration that were both partially but significantly mitigated by minocycline.76 Ex vivo West Nile Virus exposure stimulates microglia to synthesize several cytokines, including MCP-1. This finding was shown to be inhibited by minocycline.77 Retinal ischemia–reperfusion injury and cell death was not prevented by minocycline, but the injury-associated centripetal migration of monocytes/macrophages and increase in MCP-1 both were abrogated.78
Empirical preclinical studies of minocycline in glioma models have provided evidence of benefit. Growth and core tumor hypoxia of the gliosarcoma line 9L was inhibited by minocycline by unclear means.79 Weingart et al found that systemically administered minocycline did not extend survival of 9L gliosarcoma-implanted rats but local delivery after primary resection did,80,81 findings confirmed by others in 2014.82 Growth of C6 gliomas was inhibited by minocycline.83 An experimental alkylating drug was more effective in killing C6 orthotropic transplants when minocycline was given concomitantly.84 Minocycline alone inhibited growth in an orthotropic murine glioma model.85 U-87 glioma orthotropic transplants in nude mice grew slower when mice were given intraperitoneal minocycline. GL261 murine glioma cells grew slower in syngeneic mice when intraperitoneal minocycline was given.29
Two clinical studies are in progress, NCT01580969, a Phase II study using minocycline and bevacizumab with radiation for recurrent glioblastoma, and NCT02272270, a Phase I study adding minocycline to standard Stupp Protocol (temozolomide and radiation) for initial post-resection treatment of primary glioblastoma.
Side effects
Skin and other structures can become hyperpigmented after cumulative doses exceed 100 g or duration >1 year.86 Autoimmune disturbances such as serum sickness-like disease, drug-induced lupus, and autoimmune hepatitis occur but are rare.87 Autoimmune reactions tend not to occur until after >2 years of use.88
Telmisartan
Telmisartan is a brain-penetrant, highly lipophilic, 515 Da, intermediate-half-life (~24 hours) angiotensin II receptor blocker marketed to treat high blood pressure.89
Telmisartan profoundly downregulated fibronectin mRNA and suppressed MCP-1 over-synthesis in kidneys after experimental unilateral ureter obstruction.90 In similar fashion, telmisartan decreased elevated MCP-1 and fibronectin associated with diabetic kidneys.91
Cerebral MCP-1 increases with age in spontaneously hypertensive stroke-resistant rats well beyond the minimal increases seen in normotensive rats as they age. Low-dose telmisartan (in doses low enough to not alter hypertension) diminished this progressive increase in cerebral MCP-1 immunohistochemical staining in these spontaneously hypertensive rats with normal aging.92,93 In other experiments, telmisartan given in higher doses to normalize these rats’ blood pressure lowered cerebral MCP-1 even further.92,93 Exposure to homocysteine increased levels of MCP-1 mRNA in, and consequent monocyte binding to, human umbilical vein endothelial cells, an effect blocked by telmisartan and other peroxisome proliferator-activated receptor (PPAR)-gamma agonists.94
Hepatic MCP-1 and macrophage infiltration into diet-induced steatohepatitis in susceptible mice was inhibited by telmisartan.95 Telmisartan diminished macrophage population, MCP-1 protein, and CCR2 expression in the aorta of spontaneously hypertensive rats.96 It also lowered serum MCP-1 level.96
Hypertensive humans with increased carotid intima-media thickness had increased serum MCP-1 (thought to contribute to the carotid inflammatory atherosclerotic process) that diminished under telmisartan treatment.97 However, although confirming that hypertensive patients had elevated circulating MCP-1, others found no decrease in MCP-1 after successful telmisartan lowering of their blood pressure.98 Tumor-infiltrating monocytes/macrophages, as defined by leukocytes that were CD14 and CD68 positive, are known to heavily infiltrate inflammatory breast cancer tissue.99 These monocytes/macrophages secreted significant amounts of MCP-199 forming thereby a macrophage amplification circuit that as we indicate here is also operative in glioblastoma. Telmisartan attenuated the elevated myocardial MCP-1 in streptozotocin diabetic rats.100 Telmisartan decreased MCP-1 mRNA in tumor necrosis factor-alpha-stimulated human umbilical vein endothelial cells.101
Ex vivo lipopolysaccharide-stimulated circulating human monocytes secreted MCP-1 and other inflammation mediators that were decreased by co-incubation with telmisartan.102,103 Otherwise normal people with arterial hypertension treated with telmisartan exhibited lowered gene expression of both MCP-1 and PPAR-gamma in circulating monocytes compared to pretreatment levels.104 In other in vitro experiments, patients with essential hypertension treated for 3 months with telmisartan exhibited reduced circulating monocyte MCP-1 mRNA compared to their pretreatment starting values.104
Angiotensin II signals via angiotensin 1 receptors. Angiotensin II binding to angiotensin 1 receptors induces rat glomerular endothelia to increase synthesis of MCP-1.105 This was blocked by telmisartan.105
Zoledronic acid
Zoledronic acid is a long-half-life, 272 Da bisphosphonate, that although not a low-side effect drug,106 has seen increasing use as treatment adjuvant in a variety of cancers.107 Bisphosphonate drugs are commonly known and clinically used for inhibition of osteoclast activity with consequent reduction in bone resorption. Zoledronic acid use in advanced cancer patients has been shown to reduce bone events and hypercalcemia.107,108 Zoledronic acid depressed normal osteoblast secretion of MCP-1.109
In human osteosarcoma, tumor growth directly correlates with and occurs commensurately with osteoclast activation with corresponding osteolysis. Both cell line and human osteosarcomas secrete MCP-1 that forms one of the osteoclast-stimulating paths. Zoledronic acid inhibits both synthesis and secretion of osteosarcoma MCP-1 with consequent diminution of peritumoral osteoclast activity.110 Of particular note to our thinking about glioblastoma, zoledronic acid inhibition of macrophage/osteoclast function reduced growth of the malignant osteosarcoma clone itself.110 Clinical trials of zoledronic acid in breast cancer have prolonged overall survival.111
The following four data sets are paradigmatic of our suggested use of zoledronic acid in glioblastoma:
- Rietkotter et al showed that zoledronic acid reduced viability of circulating monocytes and central nervous system microglia112 and microglia exposure to zoledronic acid in ex vivo brain slices also reduced invasion by breast cancer cells without affecting the breast cancer cells themselves.112
- Indicative of our view of cancer generally as a systemic disease, Hiroshima et al showed that peritoneal macrophages from mice bearing flank-injected pancreatic tumor cells enhanced this cancer’s growth when in vitro and in vivo when injected into other tumor-bearing mice.113
- Ohba et al showed that MCP-1-secreting osteosarcoma cells were blocked from secreting MCP-1 by zoledronic acid.110
- In discussing the correlation between increased tumor-infiltrated macrophages and poorer prognosis, Rogers and Holen presaged our views, stating in 2011, “Tumor cells communicate with the cells of their microenvironment via a series of molecular and cellular interactions to aid their progression to a malignant state and ultimately their metastatic spread”. They reviewed data collected prior to 2011 supporting their contention that zoledronic acid exerted an anti-breast cancer effect by defeating MLC’s promotion of the malignant clone’s growth.114 In this paper we make a similar argument in the setting of glioblastoma.
The MTZ trial
We are planning a pilot clinical trial of the MTZ Regimen in primary glioblastoma, using oral minocycline 100 mg three times daily, oral telmisartan 80 mg twice daily, and intravenous zoledronic acid 4 mg once every 28 days to be given starting postprimary resection, to run concurrent with standard radiation and temozolomide. The primary end points will be tolerability and time to progression, and secondary end point, overall survival.
Discussion
This paper recounts past research data establishing that normal host monocytes are pathologically recruited to and by the glioblastoma tumor. Glioblastomas synthesize and secrete MCP-1 that recruits these monocytes among other effects. Glioblastoma, like other tumors, is not simply a growing autonomous parasitic entity, although it does have elements of this. Glioblastoma is more accurately understood as a malignant clone that secretes cytokines and other hormone-like molecules that signal to normal host cells and organs to support the malignant clone’s survival and growth. Glioblastoma is thus integrated into total body physiology.
Given that MCP-1 plays an important role in the destructive inflammatory process of nonalcoholic steatohepatitis,115 we could consider MTZ Regimen or elements of this as part of its treatment.
Psychiatry also is intently interested in microglia–neuron interactions potentially modifiable by minocycline.116 A single case report of minocycline 50 mg po twice daily stopping difficult-to-control seizures in a grade III glioma was interpreted as evidence of its anti-inflammatory effects.117 Going on data and theory that glial activation contributes to pathophysiology of psychotic disorders, three studies, one from 2010 and two from 2014, independently confirmed that minocycline reduced the negative sign/symptom picture that is commonly seen as residue in otherwise well-treated, “schizophrenia”.118–120 “Schizophrenia” is mentioned in quotes because it is an outmoded term comprising a dozen different pathophysiological entities with prominent psychosis that, as yet, remain unnamed.121
The malignant MCP-1-centered synergy between the glioblastoma cell and infiltrating microglia or circulating monocytes goes some of the way toward explaining the rarity of metastases outside the central nervous system, even though circulating malignant glioblastoma cells have been easily demonstrated in human disease.122
It is standard military doctrine to destroy a pathway crucial to the enemy along several points along that pathway. Hitting such a pathway at one point only allows too many chances for the enemy to develop repairs, workarounds, or alternatives that can substitute for or reconstitute the blocked pathway. Destroying multiple points along a crucial pathway significantly impedes an enemy from doing this.
In reviewing the trophic and permissive function of brain microglia for breast cancer metastases Pukrop et al observed “inhibition of proinvasive microglia as a promising anti-metastatic strategy”.123 We provided it here.
MCP-1-triggered hypoxia-related gene transcription and translation via a signal transducer and activator of transcription 3 phosphorylation represents an intermediate step in pluripotent epiblast cells (outer embryonic layer destined to later differentiate into ectoderm and mesoderm layers).124 The potential importance of this in glioblastoma and other cancers is that MCP-1 has hypoxia-adapting signaling attributes to cells other than MLCs. In accord with these findings are those of Jung et al who found that minocycline inhibited hypoxia induction factor-1α transcription and translation.125
Baer et al recount details of a reciprocal feedback system between MLCs and tumor-associated new vessel formation, where “physical contact of macrophages with growing blood vessels coordinates vascular fusion of emerging sprouts”.126 This feedback (feed-forward) system between MLCs and developing blood vessels is reminiscent of the feed-forward system of Schruefer et al who describe a vessel formation enhancing reciprocal relationship between centripetal-migrating neutrophils and the developing tumor vessel neo-intima that was based on IL-8.31,127 We can expect interventions directed against both MLCs and neutrophils each to contribute to suppression of tumor neoangiogenesis.
Radiation of brain tissue during initial glioblastoma treatment often results in memory or intellectual impairments of varying degree.128 As a potential added benefit, minocycline reduced memory impairment in radiated rats129 and might do so in brain-radiated glioblastoma patients.
As an added benefit of telmisartan, Januel et al showed empirically that those glioblastoma patients coincidentally taking angiotensin-blocking drugs or angiotensin-converting enzyme inhibitors have longer overall survival than those not on these antihypertensive drugs.130
Conclusion
Malignant cells co-opt normal cells and physiological systems to serve and enhance growth of the malignant clone/clones. Glioblastoma, like other cancers, can be thought of as an organ system, actively and reciprocally interacting and coordinating with other organ systems of the body, including the immune system. In this case, this collaboration has destructive consequences for the host, yet the elements of this pathologic relationship incorporate normal physiological systems and communication molecules such as MCP-1.
Current thinking in oncology is not oriented to considering a) the collective effects of drugs or b) effect on and treating contributions of nonmalignant helper cells of the body – in this case MLCs. We tend rather to think of individual effects of the various drugs in a mix. We now propose a paradigm shift to reflect reality more accurately. The reality is that glioblastoma like other cancers has a multiplicity of cross-covering growth paths, most of which must be addressed for successful treatment. Malignant clone-directed treatments must be augmented by simultaneous different treatments directed at halting or limiting the participation of normal host cells and organ systems that subserve and enhance growth of the malignant clone.
Our paper reviewed data showing that growth of glioblastoma cells is connected with and facilitated by surrounding normal microglia and other MLCs, abnormally recruited to and activated by the malignant clone. Our views are in accord with data and views of others. We offer the MTZ Regimen to defeat the role of MLCs that have been abnormally stimulated by MCP-1.
Acknowledgments
This article came about during the authors’ discussions over the last 2 years under the aegis of the International Initiative for Accelerated Improvement of Glioblastoma Care, The IIAIGC Study Center. This research was not funded.
Author contributions
All authors made substantial contributions to conception and design, acquisition of data, or analysis and interpretation of data; took part in either drafting the article or revising it critically for important intellectual content; gave final approval of the version to be published; and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Pantziarka P, Bouche G, Meheus L, Sukhatme V, Sukhatme VP, Vikas P. The Repurposing Drugs in Oncology (ReDO) Project. Ecancermedicalscience. 2014;8:442. doi:10.3332/ecancer.2014.442. | ||
Pantziarka P, Bouche G, Meheus L, Sukhatme V, Sukhatme VP. Repurposing drugs in your medicine cabinet: untapped opportunities for cancer therapy? Future Oncol. 2015;11(2):181–184. doi:10.2217/fon.14.244. | ||
Pantziarka P, Sukhatme V, Bouche G, Meheus L, Sukhatme VP. Repurposing Drugs in Oncology (ReDO)-itraconazole as an anti-cancer agent. Ecancermedicalscience. 2015;9:521. doi:10.3332/ecancer.2015.521. | ||
Kast RE, Karpel-Massler G, Halatsch ME. CUSP9* treatment protocol for recurrent glioblastoma: aprepitant, artesunate, auranofin, captopril, celecoxib, disulfiram, itraconazole, ritonavir, sertraline augmenting continuous low dose temozolomide. Oncotarget. 2014;5(18):8052–8082. | ||
Safa AR, Saadatzadeh MR, Cohen-Gadol AA, Pollok KE, Bijangi-Vishehsaraei K. Glioblastoma stem cells (GSCs) epigenetic plasticity and interconversion between differentiated non-GSCs and GSCs. Genes Dis. 2015;2(2):152–163. | ||
Tang C, Guo J, Chen H, et al. Gene mutation profiling of primary glioblastoma through multiple tumor biopsy guided by (1)H-magnetic resonance spectroscopy. Int J Clin Exp Pathol. 2015;8(5):5327–5335. | ||
Lemée JM, Clavreul A, Menei P. Intratumoral heterogeneity in glioblastoma: don’t forget the peritumoral brain zone. Neuro Oncol. 2015;17(10):1322–1332. | ||
Kast RE. Agomelatine or ramelteon as treatment adjuncts in glioblastoma and other M1- or M2-expressing cancers. Contemp Oncol (Pozn). 2015;19(2):157–162. doi:10.5114/wo.2015.51421. | ||
Harry GJ. Microglia during development and aging. Pharmacol Ther. 2013;139(3):313–326. doi:10.1016/j.pharmthera.2013.04.013. | ||
Klatzo I. A study of glia by the Golgi method. Lab Invest. 1952;1(3):345–350. | ||
Ginhoux F, Prinz M. Origin of microglia: current concepts and past controversies. Cold Spring Harb Perspect Biol. 2015;7(8):a020537. doi:10.1101/cshperspect.a020537. | ||
Lisi L, Stigliano E, Lauriola L, Navarra P, Dello Russo C. Proinflammatory-activated glioma cells induce a switch in microglial polarization and activation status, from a predominant M2b phenotype to a mixture of M1 and M2a/B polarized cells. ASN Neuro. 2014;6(3):171–183. doi:10.1042/AN20130045. | ||
Markovic DS, Glass R, Synowitz M, Rooijen Nv, Kettenmann H. Microglia stimulate the invasiveness of glioma cells by increasing the activity of metalloprotease-2. J Neuropathol Exp Neurol. 2005;64(9):754–762. | ||
Hattermann K, Sebens S, Helm O, et al. Chemokine expression profile of freshly isolated human glioblastoma-associated macrophages/microglia. Oncol Rep. 2014;32(1):270–276. doi:10.3892/or.2014.3214. | ||
Glass R, Synowitz M. CNS macrophages and peripheral myeloid cells in brain tumours. Acta Neuropathol. 2014;128(3):347–362. doi:10.1007/s00401-014-1274-2. | ||
Vinnakota K, Hu F, Ku MC, et al. Toll-like receptor 2 mediates microglia/brain macrophage MT1-MMP expression and glioma expansion. Neuro Oncol. 2013;15(11):1457–1468. doi:10.1093/neuonc/not115. | ||
Zhou W, Bao S. Reciprocal supportive interplay between glioblastoma and tumor-associated macrophages. Cancers (Basel). 2014;6(2):723–740. doi:10.3390/cancers6020723. | ||
Coniglio SJ, Eugenin E, Dobrenis K, et al. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol Med. 2012;18:519–527. doi:10.2119/molmed.2011.00217. | ||
Hewedi IH, Radwan NA, Shash LS, Elserry TH. Perspectives on the immunologic microenvironment of astrocytomas. Cancer Manag Res. 2013;5:293–299. doi:10.2147/CMAR.S48942. | ||
Couldwell WT, Fraser G, De Vellis G, Antel JP, Villemure JG, Yong VW. Malignant glioma-derived soluble factors regulate proliferation of normal adult human astrocytes. J Neuropathol Exp Neurol. 1992;51(5):506–513. | ||
Atai NA, Bansal M, Lo C, et al. Osteopontin is up-regulated and associated with neutrophil and macrophage infiltration in glioblastoma. Immunology. 2011;132(1):39–48. doi:10.1111/j.1365-2567.2010.03335.x. | ||
da Fonseca AC, Badie B. Microglia and macrophages in malignant gliomas: recent discoveries and implications for promising therapies. Clin Dev Immunol. 2013;2013:264124. doi:10.1155/2013/264124. | ||
Feng X, Szulzewsky F, Yerevanian A, et al. Loss of CX3CR1 increases accumulation of inflammatory monocytes and promotes gliomagenesis. Oncotarget. 2015;6(17):15077–15094. | ||
Szulzewsky F, Pelz A, Feng X, et al. Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS One. 2015;10(2):e0116644. doi:10.1371/journal.pone.0116644. | ||
Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia. 2011;59(8):1169–1180. doi:10.1002/glia.21136. | ||
Alves TR, Lima FR, Kahn SA, et al. Glioblastoma cells: a heterogeneous and fatal tumor interacting with the parenchyma. Life Sci. 2011;89(15–16):532–539. doi:10.1016/j.lfs.2011.04.022. | ||
Zhou W, Ke SQ, Huang Z, et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol. 2015;17(2):170–182. doi:10.1038/ncb3090. | ||
Komohara Y, Ohnishi K, Kuratsu J, Takeya M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J Pathol. 2008;216(1):15–24. doi:10.1002/path.2370. | ||
Hu F, Ku MC, Markovic D, et al. Glioma-associated microglial MMP9 expression is upregulated by TLR2 signaling and sensitive to minocycline. Int J Cancer. 2014;135(11):2569–2578. doi:10.1002/ijc.28908. | ||
Kast RE, Scheuerle A, Wirtz CR, Karpel-Massler G, Halatsch ME. The rationale of targeting neutrophils with dapsone during glioblastoma treatment. Anticancer Agents Med Chem. 2011;11(8):756–761. | ||
Kast RE, Lefranc F, Karpel-Massler G, Halatsch ME. Why dapsone stops seizures and may stop neutrophils’ delivery of VEGF to glioblastoma. Br J Neurosurg. 2012;26(6):813–817. doi:10.3109/02688697.2012.674577. | ||
Liang J, Piao Y, Holmes L, et al. Neutrophils promote the malignant glioma phenotype through S100A4. Clin Cancer Res. 2014;20(1):187–198. doi:10.1158/1078-0432.CCR-13-1279. | ||
Fossati G, Ricevuti G, Edwards SW, Walker C, Dalton A, Rossi ML. Neutrophil infiltration into human gliomas. Acta Neuropathol. 1999;98(4):349–354. | ||
Sippel TR, White J, Nag K, et al. Neutrophil degranulation and immunosuppression in patients with GBM: restoration of cellular immune function by targeting arginase I. Clin Cancer Res. 2011;17(22):6992–7002. doi:10.1158/1078-0432.CCR-11-1107. | ||
Iwatsuki K, Kumara E, Yoshimine T, Nakagawa H, Sato M, Hayakawa T. Elastase expression by infiltrating neutrophils in gliomas. Neurol Res. 2000;22(5):465–468. | ||
Lapa C, Linsenmann T, Lückerath K, et al. Tumor-associated macrophages in glioblastoma multiforme-a suitable target for somatostatin receptor-based imaging and therapy? PLoS One. 2015;10(3):e0122269. doi:10.1371/journal.pone.0122269. | ||
Leung SY, Wong MP, Chung LP, Chan AS, Yuen ST. Monocyte chemoattractant protein-1 expression and macrophage infiltration in gliomas. Acta Neuropathol. 1997;93(5):518–527. | ||
Klein R, Roggendorf W. Increased microglia proliferation separates pilocytic astrocytomas from diffuse astrocytomas: a double labeling study. Acta Neuropathol. 2001;101(3):245–248. | ||
Squadrito ML, De Palma M. A niche role for periostin and macrophages in glioblastoma. Nat Cell Biol. 2015;17(2):107–109. doi:10.1038/ncb3095. | ||
Sielska M, Przanowski P, Wylot B, et al. Distinct roles of CSF family cytokines in macrophage infiltration and activation in glioma progression and injury response. J Pathol. 2013;230(3):310–321. doi:10.1002/path.4192. | ||
Rodero M, Marie Y, Coudert M, et al. Polymorphism in the microglial cell-mobilizing CX3CR1 gene is associated with survival in patients with glioblastoma. J Clin Oncol. 2008;26(36):5957–5964. doi:10.1200/JCO.2008.17.2833. | ||
Prosniak M, Harshyne LA, Andrews DW, et al. Glioma grade is associated with the accumulation and activity of cells bearing M2 monocyte markers. Clin Cancer Res. 2013;19(14):3776–3786. doi:10.1158/1078-0432.CCR-12-1940. | ||
Wu A, Wei J, Kong LY, et al. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro Oncol. 2010;12(11):1113–1125. doi:10.1093/neuonc/noq082. | ||
de Vrij J, Maas SL, Kwappenberg KM, et al. Glioblastoma-derived extracellular vesicles modify the phenotype of monocytic cells. Int J Cancer. 2015;137(7):1630–1642. doi:10.1002/ijc.29521. | ||
He L, Marneros AG. Doxycycline inhibits polarization of macrophages to the proangiogenic M2-type and subsequent neovascularization. J Biol Chem. 2014;289(12):8019–8028. doi:10.1074/jbc.M113.535765. | ||
Getts DR, Terry RL, Getts MT, et al. Ly6c+ “inflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. J Exp Med. 2008;205(10):2319–2337. doi:10.1084/jem.20080421. | ||
Chae M, Peterson TE, Balgeman A, et al. Increasing glioma-associated monocytes leads to increased intratumoral and systemic myeloid-derived suppressor cells in a murine model. Neuro Oncol. 2015;17(7):978–991. doi:10.1093/neuonc/nou343. | ||
Gielen PR, Schulte BM, Kers-Rebel ED, et al. Increase in both CD14-positive and CD15-positive myeloid-derived suppressor cell subpopulations in the blood of patients with glioma but predominance of CD15-positive myeloid-derived suppressor cells in glioma tissue. J Neuropathol Exp Neurol. 2015;74(5):390–400. | ||
Rodrigues JC, Gonzalez GC, Zhang L, et al. Normal human monocytes exposed to glioma cells acquire myeloid-derived suppressor cell-like properties. Neuro Oncol. 2010;12(4):351–365. doi:10.1093/neuonc/nop023. | ||
Bloch O, Crane CA, Kaur R, Safaee M, Rutkowski MJ, Parsa AT. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin Cancer Res. 2013;19(12):3165–3175. doi:10.1158/1078-0432.CCR-12-3314. | ||
Thorne AH, Meisen WH, Russell L, et al. Role of cysteine-rich 61 protein (CCN1) in macrophage-mediated oncolytic herpes simplex virus clearance. Mol Ther. 2014;22(9):1678–1687. doi:10.1038/mt.2014.101. | ||
Boukhari A, Alhosin M, Bronner C, et al. CD47 activation-induced UHRF1 over-expression is associated with silencing of tumor suppressor gene p16INK4A in glioblastoma cells. Anticancer Res. 2015;35(1):149–157. | ||
Pyonteck SM, Akkari L, Schuhmacher AJ, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19(10):1264–1272. doi:10.1038/nm.3337. | ||
Kohanbash G, McKaveney K, Sakaki M, et al. GM-CSF promotes the immunosuppressive activity of glioma-infiltrating myeloid cells through interleukin-4 receptor-alpha. Cancer Res. 2013;73(21):6413–6423. doi:10.1158/0008-5472.CAN-12-4124. | ||
Albulescu R, Codrici E, Popescu ID, et al. Cytokine patterns in brain tumour progression. Mediators Inflamm. 2013;2013:979748. doi:10.1155/2013/979748. | ||
Revoltella RP, Menicagli M, Campani D. Granulocyte-macrophage colony-stimulating factor as an autocrine survival-growth factor in human gliomas. Cytokine. 2012;57(3):347–359. doi:10.1016/j.cyto.2011.11.016. | ||
Curran CS, Evans MD, Bertics PJ. GM-CSF production by glioblastoma cells has a functional role in eosinophil survival, activation, and growth factor production for enhanced tumor cell proliferation. J Immunol. 2011;187(3):1254–1263. doi:10.4049/jimmunol.1001965. | ||
Mueller MM, Herold-Mende CC, Riede D, Lange M, Steiner HH, Fusenig NE. Autocrine growth regulation by granulocyte colony-stimulating factor and granulocyte macrophage colony-stimulating factor in human gliomas with tumor progression. Am J Pathol. 1999;155(5):1557–1567. | ||
Vasco C, Canazza A, Rizzo A, et al. Circulating T regulatory cells migration and phenotype in glioblastoma patients: an in vitro study. J Neurooncol. 2013;115(3):353–363. doi:10.1007/s11060-013-1236-x. | ||
Jordan JT, Sun W, Hussain SF, DeAngulo G, Prabhu SS, Heimberger AB. Preferential migration of regulatory T cells mediated by glioma-secreted chemokines can be blocked with chemotherapy. Cancer Immunol Immunother. 2008;57(1):123–131. | ||
Lin Y, Zhang G, Zhang J, et al. A panel of four cytokines predicts the prognosis of patients with malignant gliomas. J Neurooncol. 2013;114(2):199–208. doi:10.1007/s11060-013-1171-x. | ||
Liang Y, Bollen AW, Gupta N. CC chemokine receptor-2A is frequently overexpressed in glioblastoma. J Neurooncol. 2008;86(2):153–163. | ||
Lindemann C, Marschall V, Weigert A, Klingebiel T, Fulda S. Smac mimetic-induced upregulation of CCL2/MCP-1 triggers migration and invasion of glioblastoma cells and influences the tumor microenvironment in a paracrine manner. Neoplasia. 2015;17(6):481–489. doi:10.1016/j.neo.2015.05.002. | ||
Kuratsu J, Yoshizato K, Yoshimura T, Leonard EJ, Takeshima H, Ushio Y. Quantitative study of monocyte chemoattractant protein-1 (MCP-1) in cerebrospinal fluid and cyst fluid from patients with malignant glioma. J Natl Cancer Inst. 1993;85(22):1836–1839. | ||
Desbaillets I, Tada M, de Tribolet N, Diserens AC, Hamou MF, Van Meir EG. Human astrocytomas and glioblastomas express monocyte chemoattractant protein-1 (MCP-1) in vivo and in vitro. Int J Cancer. 1994;58(2):240–247. | ||
Wurdinger T, Deumelandt K, van der Vliet HJ, Wesseling P, de Gruijl TD. Mechanisms of intimate and long-distance cross-talk between glioma and myeloid cells: how to break a vicious cycle. Biochim Biophys Acta. 2014;1846(2):560–575. doi:10.1016/j.bbcan.2014.10.003. | ||
Lu-Emerson C, Snuderl M, Kirkpatrick ND, et al. Increase in tumor-associated macrophages after antiangiogenic therapy is associated with poor survival among patients with recurrent glioblastoma. Neuro Oncol. 2013;15(8):1079–1087. doi:10.1093/neuonc/not082. | ||
Elewa HF, Hilali H, Hess DC, Machado LS, Fagan SC. Minocycline for short-term neuroprotection. Pharmacotherapy. 2006;26(4):515–521. | ||
Saivin S, Hovin G. Clinical pharmacokinetics of doxycycline and minocycline. Clin Pharmacokinet. 1988;15:355–366. | ||
Kim HS, Suh YH. Minocycline and neurodegenerative diseases. Behav Brain Res. 2009;196(2):168–179. doi:10.1016/j.bbr.2008.09.040. | ||
Ochsendorf F. Minocycline in acne vulgaris: benefits and risks. Am J Clin Dermatol. 2010;11(5):327–341. doi:10.2165/11319280-000000000-00000. | ||
Garrido-Mesa N, Zarzuelo A, Galvez J. Minocycline: far beyond an antibiotic. Br J Pharmacol. 2013;169(2):337–352. doi:10.1111/bph.12139. | ||
Mitchell RM, Lee SY, Randazzo WT, Simmons Z, Connor JR. Influence of HFE variants and cellular iron on monocyte chemoattractant protein-1. J Neuroinflammation. 2009;6:6. doi:10.1186/1742-2094-6-6. | ||
Tai K, Iwasaki H, Ikegaya S, Ueda T. Minocycline modulates cytokine and chemokine production in lipopolysaccharide-stimulated THP-1 monocytic cells by inhibiting IkB kinase alpha/beta phosphorylation. Transl Res. 2013;161(2):99–109. doi:10.1016/j.trsl.2012.10.001. | ||
Dutta K, Kumawat KL, Nazmi A, Mishra MK, Basu A. Minocycline differentially modulates viral infection and persistence in an experimental model of Japanese encephalitis. J Neuroimmune Pharmacol. 2010;5(4):553–565. doi:10.1007/s11481-010-9233-8. | ||
Zink MC, Uhrlaub J, DeWitt J, et al. Neuroprotective and anti-human immunodeficiency virus activity of minocycline. JAMA. 2005;293(16):2003–2011. | ||
Quick ED, Leser JS, Clarke P, Tyler KL. Activation of intrinsic immune responses and microglial phagocytosis in an ex vivo spinal cord slice culture model of West Nile virus infection. J Virol. 2014;88(22):13005–13014. doi:10.1128/JVI.01994-14. | ||
Abcouwer SF, Lin CM, Shanmugam S, Muthusamy A, Barber AJ, Antonetti DA. Minocycline prevents retinal inflammation and vascular permeability following ischemia-reperfusion injury. J Neuroinflammation. 2013;10:149. doi:10.1186/1742-2094-10-149. | ||
Teicher BA, Dupuis NP, Emi Y, Ikebe M, Kakeji Y, Menon K. Increased efficacy of chemo- and radio-therapy by a hemoglobin solution in the 9L gliosarcoma. In Vivo. 1995;9(1):11–18. | ||
Weingart JD, Sipos EP, Brem H. The role of minocycline in the treatment of intracranial 9L glioma. J Neurosurg. 1995;82(4):635–640. | ||
Frazier JL, Wang PP, Case D, et al. Local delivery of minocycline and systemic BCNU have synergistic activity in the treatment of intracranial glioma. J Neurooncol. 2003;64(3):203–209. | ||
Bow H, Hwang LS, Schildhaus N, et al. Local delivery of angiogenesis-inhibitor minocycline combined with radiotherapy and oral temozolomide chemotherapy in 9L glioma. J Neurosurg. 2014;120(3):662–669. doi:10.3171/2013.11.JNS13556. | ||
Liu WT, Lin CH, Hsiao M, Gean PW. Minocycline inhibits the growth of glioma by inducing autophagy. Autophagy. 2011;7(2):166–175. | ||
Hour MJ, Liu WT, Lu IC, Kuo SC, Gean PW. Aggravated DNA damage as a basis for enhanced glioma cell killing by MJ-66 in combination with minocycline. Am J Cancer Res. 2014;4(5):474–483. | ||
Markovic DS, Vinnakota K, van Rooijen N, et al. Minocycline reduces glioma expansion and invasion by attenuating microglial MT1-MMP expression. Brain Behav Immun. 2011;25(4):624–628. doi:10.1016/j.bbi.2011.01.015. | ||
Eisen D, Hakim MD. Minocycline-induced pigmentation. Incidence, prevention and management. Drug Saf. 1998;18(6):431–440. | ||
Eichenfield AH. Minocycline and autoimmunity. Curr Opin Pediatr. 1999;11(5):447–456. | ||
Elkayam O, Yaron M, Caspi D. Minocycline-induced autoimmune syndromes: an overview. Semin Arthritis Rheum. 1999;28(6):392–397. | ||
Bakheit AH, Abd-Elgalil AA, Mustafa B, Haque A, Wani TA. Telmisartan. Profiles Drug Subst Excip Relat Methodol. 2015;40:371–429. doi:10.1016/bs.podrm.2015.01.003. | ||
Zhang J, Wong MG, Wong M, et al. A cationic-independent mannose 6-phosphate receptor inhibitor (PXS64) ameliorates kidney fibrosis by inhibiting activation of transforming growth factor-beta. PLoS One. 2015;10(2):e0116888. doi:10.1371/journal.pone.0116888. | ||
Gangadharan Komala M, Gross S, Mudaliar H, et al. Inhibition of kidney proximal tubular glucose reabsorption does not prevent against diabetic nephropathy in type 1 diabetic eNOS knockout mice. PLoS One. 2014;9(11):e108994. doi:10.1371/journal.pone.0108994. | ||
Kurata T, Lukic V, Kozuki M, et al. Telmisartan reduces progressive accumulation of cellular amyloid beta and phosphorylated tau with inflammatory responses in aged spontaneously hypertensive stroke resistant rat. J Stroke Cerebrovasc Dis. 2014;23(10):2580–2590. doi:10.1016/j.jstrokecerebrovasdis.2014.05.023. | ||
Kono S, Kurata T, Sato K, et al. Neurovascular protection by telmisartan via reducing neuroinflammation in stroke-resistant spontaneously hypertensive rat brain after ischemic stroke. J Stroke Cerebrovasc Dis. 2015;24(3):537–547. doi:10.1016/j.jstrokecerebrovasdis.2014.09.037. | ||
Xu S, Song H, Huang M, Wang K, Xu C, Xie L. Telmisartan inhibits the proinflammatory effects of homocysteine on human endothelial cells through activation of the peroxisome proliferator-activated receptor-gamma pathway. Int J Mol Med. 2014;34(3):828–834. doi:10.3892/ijmm.2014.1834. | ||
Kudo H, Yata Y, Takahara T, et al. Telmisartan attenuates progression of steatohepatitis in mice: role of hepatic macrophage infiltration and effects on adipose tissue. Liver Int. 2009;29(7):988–996. doi:10.1111/j.1478-3231.2009.02006.x. | ||
Dai Q, Xu M, Yao M, Sun B. Angiotensin AT1 receptor antagonists exert anti-inflammatory effects in spontaneously hypertensive rats. Br J Pharmacol. 2007;152(7):1042–1048. | ||
Liu Z, Zhao Y, Wei F, et al. Treatment with telmisartan/rosuvastatin combination has a beneficial synergistic effect on ameliorating Th17/Treg functional imbalance in hypertensive patients with carotid atherosclerosis. Atherosclerosis. 2014;233(1):291–299. doi:10.1016/j.atherosclerosis.2013.12.004. | ||
Klinghammer L, Urschel K, Cicha I, et al. Impact of telmisartan on the inflammatory state in patients with coronary atherosclerosis-influence on IP-10, TNF-alpha and MCP-1. Cytokine. 2013;62(2):290–296. doi:10.1016/j.cyto.2013.02.001. | ||
Mohamed MM, El-Ghonaimy EA, Nouh MA, Schneider RJ, Sloane BF, El-Shinawi M. Cytokines secreted by macrophages isolated from tumor microenvironment of inflammatory breast cancer patients possess chemotactic properties. Int J Biochem Cell Biol. 2014;46:138–147. doi:10.1016/j.biocel.2013.11.015. | ||
Guo Z, Zhang R, Li J, Xu G. Effect of telmisartan on the expression of adiponectin receptors and nicotinamide adenine dinucleotide phosphate oxidase in the heart and aorta in type 2 diabetic rats. Cardiovasc Diabetol. 2012;11:94. doi:10.1186/1475-2840-11-94. | ||
Sonoki K, Iwase M, Ohdo S, et al. Telmisartan and N-acetylcysteine suppress group V secretory phospholipase A2 expression in TNF-alpha stimulated human endothelial cells and reduce associated atherogenicity. J Cardiovasc Pharmacol. 2012;60(4):367–374. doi:10.1097/FJC.0b013e3182646ccc. | ||
Pang T, Benicky J, Wang J, Orecna M, Sanchez-Lemus E, Saavedra JM. Telmisartan ameliorates lipopolysaccharide-induced innate immune response through peroxisome proliferator-activated receptor-gamma activation in human monocytes. J Hypertens. 2012;30(1):87–96. doi:10.1097/HJH.0b013e32834dde5f. | ||
Matsumura T, Kinoshita H, Ishii N, et al. Telmisartan exerts antiatherosclerotic effects by activating peroxisome proliferator-activated receptor-gamma in macrophages. Arterioscler Thromb Vasc Biol. 2011;31(6):1268–1275. doi:10.1161/ATVBAHA.110.222067. | ||
Marketou ME, Kontaraki JE, Tsakountakis NA, et al. Differential effect of telmisartan and amlodipine on monocyte chemoattractant protein-1 and peroxisome proliferator-activated receptor-gamma gene expression in peripheral monocytes in patients with essential hypertension. Am J Cardiol. 2011;107(1):59–63. doi:10.1016/j.amjcard. 2010.08.048. | ||
Pan Q, Yang XH, Cheng YX. Angiotensin II stimulates MCP-1 production in rat glomerular endothelial cells via NAD(P)H oxidase-dependent nuclear factor-kappa B signaling. Braz J Med Biol Res. 2009;42(6):531–536. | ||
Berenson JR. Recommendations for zoledronic acid treatment of patients with bone metastases. Oncologist. 2005;10(1):52–62. | ||
Perry CM, Figgitt DP. Zoledronic acid: a review of its use in patients with advanced cancer. Drugs. 2004;64(11):1197–1211. | ||
Li EC, Davis LE. Zoledronic acid: a new parenteral bisphosphonate. Clin Ther. 2003;25(11):2669–2708. | ||
Kaiser T, Teufel I, Geiger K, et al. Bisphosphonates modulate vital functions of human osteoblasts and affect their interactions with breast cancer cells. Breast Cancer Res Treat. 2013;140(1):35–48. doi:10.1007/s10549-013-2613-z. | ||
Ohba T, Cole HA, Cates JM, et al. Bisphosphonates inhibit osteosarcoma-mediated osteolysis via attenuation of tumor expression of MCP-1 and RANKL. J Bone Miner Res. 2014;29(6):1431–1445. doi:10.1002/jbmr.2182. | ||
He M, Fan W, Zhang X. Adjuvant zoledronic acid therapy for patients with early stage breast cancer: an updated systematic review and meta-analysis. J Hematol Oncol. 2013;6(1):80. doi:10.1186/1756-8722-6-80. | ||
Rietkotter E, Menck K, Bleckmann A, et al. Zoledronic acid inhibits macrophage/microglia-assisted breast cancer cell invasion. Oncotarget. 2013;4(9):1449–1460. | ||
Hiroshima Y, Maawy A, Hassanein MK, et al. The tumor-educated-macrophage increase of malignancy of human pancreatic cancer is prevented by zoledronic acid. PLoS One. 2014;9(8):e103382. doi:10.1371/journal.pone.0103382. | ||
Rogers TL, Holen I. Tumour macrophages as potential targets of bisphosphonates. J Transl Med. 2011;9:177. doi:10.1186/1479-5876-9-177. | ||
Cynis H, Kehlen A, Haegele M, et al. Inhibition of glutaminyl cyclases alleviates CCL2-mediated inflammation of non-alcoholic fatty liver disease in mice. Int J Exp Pathol. 2013;94(3):217–225. doi:10.1111/iep.12020. | ||
Pae CU. The potential role of monocyte chemoattractant protein-1 for major depressive disorder. Psychiatry Investig. 2014;11(3):217–222. doi:10.4306/pi.2014.11.3.217. | ||
Nowak M, Strzelczyk A, Reif PS, et al. Minocycline as potent anticonvulsant in a patient with astrocytoma and drug resistant epilepsy. Seizure. 2012;21(3):227–228. doi:10.1016/j.seizure.2011.12.009. | ||
Levkovitz Y, Mendlovich S, Riwkes S, et al. A double-blind, randomized study of minocycline for the treatment of negative and cognitive symptoms in early-phase schizophrenia. J Clin Psychiatry. 2010;71(2):138–149. doi:10.4088/JCP.08m04666yel. | ||
Liu F, Guo X, Wu R, et al. Minocycline supplementation for treatment of negative symptoms in early-phase schizophrenia: a double blind, randomized, controlled trial. Schizophr Res. 2014;153(1–3):169–176. doi:10.1016/j.schres.2014.01.011. | ||
Ghanizadeh A, Dehbozorgi S, Omrani Sigaroodi M, Rezaei Z. Minocycline as add-on treatment decreases the negative symptoms of schizophrenia; a randomized placebo-controlled clinical trial. Recent Pat Inflamm Allergy Drug Discov. 2014;8(3):211–215. | ||
Nasrallah H, Tandon R, Keshavan M. Beyond the facts in schizophrenia: closing the gaps in diagnosis, pathophysiology, and treatment. Epidemiol Psychiatr Sci. 2011;20(4):317–327. | ||
Best MG, Sol N, Zijl S, Reijneveld JC, Wesseling P, Wurdinger T. Liquid biopsies in patients with diffuse glioma. Acta Neuropathol. 2015;129(6):849–865. doi:10.1007/s00401-015-1399-y. | ||
Pukrop T, Dehghani F, Chuang HN, et al. Microglia promote colonization of brain tissue by breast cancer cells in a Wnt-dependent way. Glia. 2010;58(12):1477–1489. doi:10.1002/glia.21022. | ||
Hasegawa Y, Tang D, Takahashi N, Hayashizaki Y, Forrest AR. CCL2 enhances pluripotency of human induced pluripotent stem cells by activating hypoxia related genes. Sci Rep. 2014;4:5228. doi:10.1038/srep05228. | ||
Jung HJ, Seo I, Jha BK, Suh SI, Suh MH, Baek WK. Minocycline inhibits angiogenesis in vitro through the translational suppression of HIF-1α. Arch Biochem Biophys. 2014;545:74–82. doi:10.1016/j.abb.2013.12.023. | ||
Baer C, Squadrito ML, Iruela-Arispe ML, De Palma M. Reciprocal interactions between endothelial cells and macrophages in angiogenic vascular niches. Exp Cell Res. 2013;319(11):1626–1634. doi:10.1016/j.yexcr.2013.03.026. | ||
Schruefer R, Lutze N, Schymeinsky J, Walzog B. Human neutrophils promote angiogenesis by a paracrine feedforward mechanism involving endothelial interleukin-8. Heart Circ Physiol. 2005;288:H1186–H1192. | ||
Reddy K, Gaspar LE, Kavanagh BD, et al. Prospective evaluation of health-related quality of life in patients with glioblastoma multiforme treated on a phase II trial of hypofractionated IMRT with temozolomide. J Neurooncol. 2013;114(1):111–116. doi:10.1007/s11060-013-1159-6. | ||
Zhang L, Li K, Sun R, et al. Minocycline ameliorates cognitive impairment induced by whole-brain irradiation: an animal study. Radiat Oncol. 2014;9(1):281. | ||
Januel E, Ursu R, Alkhafaji A, et al. Impact of renin-angiotensin system blockade on clinical outcome in glioblastoma. Eur J Neurol. 2015;22(9):1304–1309. doi:10.1111/ene.12746. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.