Back to Journals » Journal of Pain Research » Volume 19
Toothache and Alzheimer’s Disease: A Mendelian Randomization Study of the NAAG-CD33 Neuroimmune Axis
Authors Wang Q, Xie Y, Wu X, Jin S, Zhen W, Yu S, Sun Y, Sun W, Xu J, Zhang H ![]()
Received 16 February 2026
Accepted for publication 9 May 2026
Published 26 May 2026 Volume 2026:19 603587
DOI https://doi.org/10.2147/JPR.S603587
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr David Keith
Qing Wang,* Yuxuan Xie,* Xing Wu,* Siyu Jin,* Wenyu Zhen, Sensen Yu, Yuqiang Sun, Wansu Sun, Jianguang Xu, Hengguo Zhang
College & Hospital of Stomatology, Anhui Medical University, Anhui Provincial Key Laboratory of Oral Diseases Research, Hefei 230032, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Hengguo Zhang; Wansu Sun, College & Hospital of Stomatology, Anhui Medical University, Anhui Provincial Key Laboratory of Oral Diseases Research, Hefei, 230032, People’s Republic of China, Email [email protected]; [email protected]
Background: Toothache, as an oral pain condition, may disrupt neuroimmune signaling and potentially influence neurodegenerative processes. This study investigated the relationship between toothache and Alzheimer’s disease (AD) through a neuroimmune axis involving N-acetylaspartylglutamate (NAAG) and CD33+ myeloid monocytes.
Methods: We conducted a two-sample Mendelian randomization (MR) analysis using genome-wide association study (GWAS) summary statistics from the UK Biobank for toothache, a large-scale plasma metabolomics study for NAAG, and immune cell trait cohorts for CD33+ myeloid monocyte-related traits, including CD33 on HLA DR+/CD11b+ cells and monocytic myeloid-derived suppressor cells. The AD dataset, derived from a large-scale meta-analysis, primarily focused on late-onset sporadic cases. Pairwise associations among toothache, NAAG, CD33-related traits, and AD were explored. Heterogeneity and horizontal pleiotropy were assessed using Cochran’s Q test and MR Egger regression.
Results: Reduced NAAG in toothache individuals (p = 0.024) exhibited a robust association with increased CD33 expression in myeloid monocytes (p = 0.005), which were associated with increased AD risk (p = 0.003). Sensitivity analyses confirmed the robustness of the results.
Conclusion: This study provides preliminary evidence for a potential causal axis linking toothache, reduced NAAG, increased CD33+ myeloid monocytes, and AD. This study bridges the gap between clinical observations of oral pain-related cognitive decline and the neuroimmune mechanisms underlying this process.
Keywords: toothache, alzheimer’s disease, NAAG, CD33, neuroimmune axis
Background
Toothache presents as persistent and severe pain and is often associated with oral diseases. Due to the unique anatomical structure and dense neural innervation of the tooth, pain originating from the pulp, dentin, or periodontium is particularly intense.1,2 Meanwhile, severe pain, including toothache and other conditions such as pain during natural labor, can disrupt neuroimmune signaling and may contribute to neurodegeneration.3,4 Epidemiological studies have shown that poor oral health (toothache, periodontitis, and tooth loss) is associated with an increased risk of cognitive decline and dementia, including Alzheimer’s disease (AD), which is marked by the neuroimmune injury and formation of β-amyloid plaques in the brain.5–7 These results suggest that oral health may have important implications for population-level neurodegenerative risk. Exploring the neuroimmune axis between toothache and AD offers novel mechanistic insights into the pathophysiology of this widespread neurodegenerative condition and enhances our understanding of the molecular basis of the oral-brain connection.
As a severe pain, toothache and its associated trigeminal neuralgia are transmitted from primary neurons to the trigeminal ganglion, then cross over at the trigeminal nucleus, projecting to the ventral posteromedial (VPM) nucleus of the thalamus, and ultimately reaching the postcentral gyrus (areas 3, 1, and 2) in the cerebral cortex, where pain perception is registered.8 N-acetylaspartylglutamate (NAAG), a neuropeptide abundant in the cerebral cortex, exerts neuroprotective effects by inhibiting excessive excitatory signaling.9 Watanabe et al found that NAAG release in the nucleus accumbens negatively correlates with pain intensity and that NAAG injection reduces pain triggered by neural stimulation, highlighting the critical role of NAAG in modulating pain transmission.10
Intense and persistent trigeminal nociception can compromise blood–brain barrier (BBB) integrity. Sustained activation of trigeminal afferents triggers neurogenic inflammation, during which perivascular nerve endings release vasoactive neuropeptides such as substance P and calcitonin gene-related peptide (CGRP), directly increasing BBB permeability.11 Concurrently, the dysregulated glutamatergic signaling and elevated extracellular glutamate can further disrupt BBB function via excitotoxic mechanisms.12,13
NAAG has been shown to modulate extracellular glutamate levels, partly through activation of the metabotropic glutamate receptor 3 (mGluR3).14 Glutamate is the central nervous system’s primary excitatory neurotransmitter, extensively present across various brain regions.15 Given its inability to cross the BBB, glutamate concentration is inherently isolated from peripheral influences. Dysregulation of glutamate homeostasis has been linked to various neurological disorders, with AD being notably affected.16,17
Microglia, the central nervous system’s principal immune cells, share a myeloid lineage and functional similarities with monocytes, which are derived from hematopoietic stem cells and influenced by neurogenic niches (eg, via ANKRD1 signaling) to counter cognitive aging.18,19 These cells are remarkably versatile and are critical in maintaining brain homeostasis.20 Research indicates pervasive communication between microglia and glutamatergic neurons, suggesting a regulatory relationship.21 A reduction in NAAG may be associated with altered glutamatergic signaling, which has been implicated in microglial activation and dysfunction in neurodegenerative contexts. Moreover, microglia can indirectly influence glutamate homeostasis through inflammatory signaling.22
CD33 is an immunosuppressive receptor highly expressed in myeloid cells (including microglia). Recent studies have identified CD33 as a susceptibility factor for AD.23,24 CD33 expression has been shown to suppress microglial phagocytic activity, thereby impairing the clearance of amyloid-β (Aβ) and other cellular debris and exacerbating neuroinflammatory responses.25 Glutamatergic signaling and CD33-mediated microglial regulation may interact within neuroimmune pathways relevant to neurodegeneration.21 CD33 affects the activity and function of microglia, which play a crucial role in regulating glutamate levels and neuroinflammation.26 This relationship has significant implications for understanding the mechanisms of neurodegenerative diseases.
However, the potential link between toothache and AD through specific neuroimmune-related pathways, as well as the molecular intermediates that may underlie this association, remains largely unexplored. We hypothesize that toothache may be associated with AD through a neuroimmune axis involving NAAG and CD33. To test this hypothesis, we employed two-sample Mendelian Randomization (MR) as the primary analytical framework. Due to the limited availability of live human brain tissue for large-scale mechanistic studies, plasma NAAG levels and CD33 expression on myeloid monocytes were utilized as biological proxies to investigate the systemic components of this neuroimmune axis. MR leverages randomly assigned genetic variants as instrumental variables, minimizing the impact of postnatal environmental confounders and reverse causality. By taking advantage of this “natural randomized trial” design, our study seeks to elucidate the potential causal relationships within the proposed toothache-NAAG-CD33-AD pathway, offering novel insights into how oral sensory signals may be transduced into systemic neuroimmune responses.
Methods
Data
Genome-wide association study (GWAS) summary statistics for NAAG were obtained from a large-scale plasma metabolomics study conducted within the Canadian Longitudinal Study of Aging (CLSA). This study included 8,299 unrelated individuals of European ancestry aged 45–85 years who underwent genome-wide genotyping and circulating metabolite profiling.27 NAAG was analyzed as a continuous plasma metabolite trait.
GWAS data for CD33 expression were derived from an immune cell trait study conducted in 1,629 individuals from the Sardinian population, in which 539 flow cytometry–defined peripheral blood immune cell traits were assessed, including absolute cell counts, mean fluorescence intensities (MFIs) of surface markers, and morphological parameters.28 Two CD33-related traits were selected according to the original trait annotations provided by the data source: CD33 expression on CD33dim HLA-DR+ CD11b+ cells and monocytic myeloid-derived suppressor cells. These traits are hereafter referred to as CD33 (1) and CD33 (2), respectively. Given their shared myeloid lineage and transcriptional similarity, peripheral blood myeloid mononuclear cells were used as proxies for microglial CD33-related immune activity, consistent with established MR frameworks.29–31 A detailed rationale for this proxy-based approach is provided in the Discussion section.
Summary statistics for AD were obtained from a large-scale meta-analysis, available via the IEU OpenGWAS database. This dataset integrated clinical AD cases from the International Genomics of Alzheimer’s Project (IGAP) and proxy cases from the UK Biobank. The final analysis comprised 455,258 individuals of European ancestry (71,880 cases and 383,378 controls). AD cases were identified through a combination of clinically diagnosed individuals, primarily those with late-onset sporadic AD, and participants reporting a parental history of AD. This family history-based proxy phenotype has been validated as a powerful tool for investigating the genetic architecture of sporadic AD. Notably, while the discovery GWAS adjusted for age and sex as covariates to minimize potential confounding, no explicit age-stratified analysis was performed within the cohorts.
All GWAS datasets were restricted to individuals of European ancestry to minimize population stratification. Non-overlapping samples were ensured across datasets to satisfy the assumptions of two-sample MR. A detailed summary of the GWAS statistics is provided in Table 1.
 |
Table 1 Detailed Information on GWAS Summary Data |
Screening of Instrumental Variables
Following the framework established by Emdin et al,32 instrumental variables were required to meet three assumptions: relevance, independence, and exclusion restriction. To meet MR standards, we followed a detailed process for selecting instrumental variables. The selection criteria were as follows: (1) We initially chose Single Nucleotide Polymorphisms (SNPs) with a strong association with the exposure (p < 5.0×10–6 for Toothache & NAAG; p < 5.0×10–8 for NAAG & CD33 (1); p < 5.0×10–7 for NAAG & CD33 (2); p < 5.0×10–8 for CD33 & AD). Thresholds were established by evaluating multiple cutoffs (10−6, 10−7, 10−8) and selecting the one that provided the best trade-off between SNP availability and the robustness of MR estimates, consistent with recommended practices in MR research. (2) We calculated the linkage disequilibrium (LD) for these SNPs, retaining only those below the r-threshold and above the kb-threshold (r2=0.001, kb=5000, toothache & NAAG; r2=0.1, kb=5000, NAAG & CD33 (1); r2=0.1, kb=1000, NAAG & CD33 (2); r2=0.001, kb=5000, CD33 & AD). (3) To minimize bias, we removed all palindromic SNPs. (4) The “MR-PRESSO” R package was used to identify and exclude pleiotropic SNPs. (5) Additionally, we used Ldlink (https://ldlink.nih.gov/) to eliminate any confounding factors and risk variables (Table 2). (6) Instrument strength was assessed using the F-statistic, calculated as F = (β/SE)2, where β represents the estimated effect of each SNP on the exposure and SE represents the corresponding standard error. All retained SNPs had F-statistics greater than 10, indicating sufficient instrument strength and a low likelihood of weak instrument bias. The remaining SNPs were used for MR analysis (Supplementary Table 1–3). A schematic representation of the research workflow is provided for clarity (Figure 1).
 |
Table 2 SNPs Associated with Known Confounders |
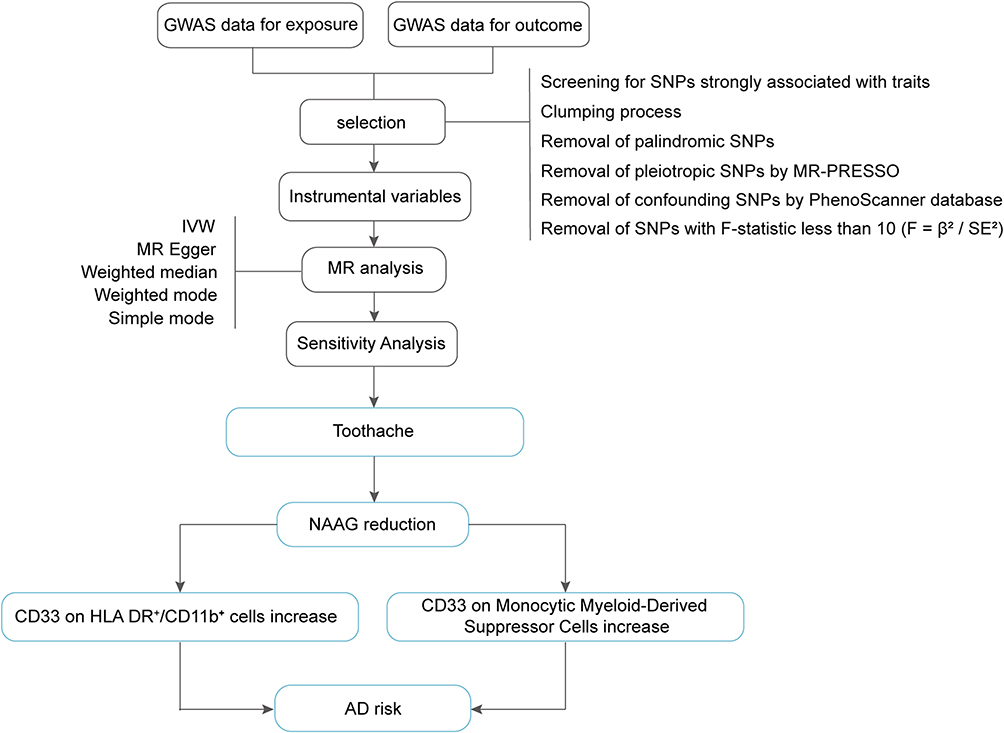 |
Figure 1 Flowchart of the Mendelian randomization process in this study. Abbreviations: GWAS, Genome-wide association study; MR, Mendelian randomization; NAAG, N-acetylglucosylglutamate; AD, Alzheimer’s Disease. |
Because GWAS sample size and signal strength differed across exposures, a uniform SNP selection threshold was not applied. Instead, commonly used significance cutoffs (p = 10−6 to 10−8) were evaluated, and exposure–outcome–specific thresholds were selected to balance instrument availability and estimate stability, consistent with prior MR studies.
Mendelian Randomization
Causal effects between exposures and outcomes were estimated using five complementary MR methods: inverse variance weighted (IVW), MR Egger, weighted median, weighted mode, and simple mode. IVW was designated as the primary method due to its higher statistical efficiency under the assumption of no horizontal pleiotropy, whereas the other methods were applied to assess robustness. Consistency in effect direction across all five methods, combined with an IVW p-value below 0.05, was used to indicate a potential association between exposure and outcome. These associations are interpreted as genetically supported evidence rather than definitive causality. All analyses were conducted along a predefined causal pathway; nominal p-values were reported and interpreted in conjunction with consistency across MR methods and biological plausibility.
Sensitivity Analyses
To assess the robustness of the MR estimates and potential violations of MR assumptions, multiple sensitivity analyses were performed. Heterogeneity was assessed using the Cochran's Q test for MR Egger and IVW methodologies. A Q_pval greater than 0.1 indicated no heterogeneity among the studies. Additionally, MR Egger was used to evaluate pleiotropy; an Egger_intercept close to 0 or a p-value (between the intercept and 0) greater than 0.05 indicated no pleiotropy in the results. We also performed a leave-one-out analysis to assess the influence of individual SNPs on the overall findings (Figure 2). Collectively, these analyses indicated that the observed associations were not influenced by heterogeneity or by individual SNPs.
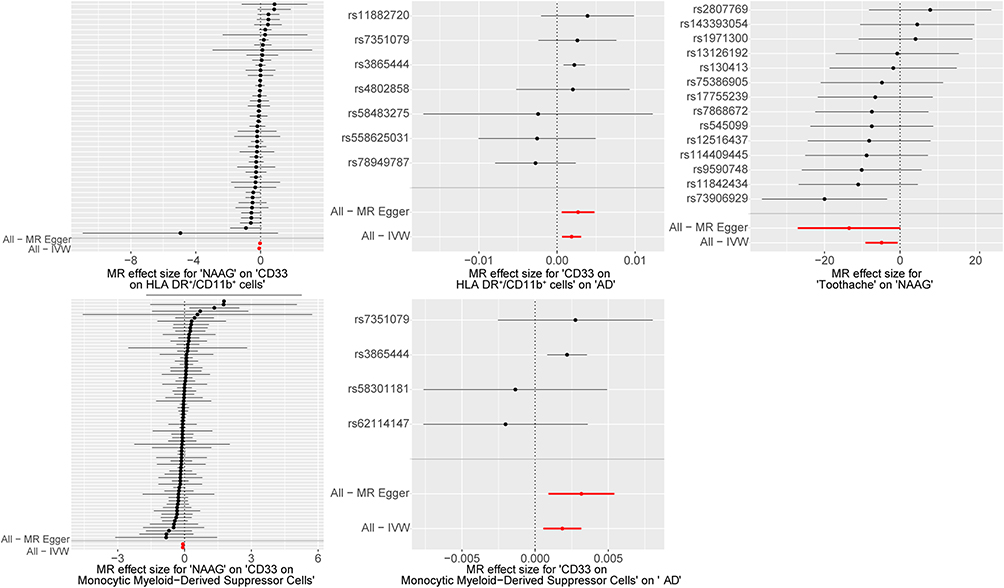 |
Figure 2 Forest plots for leave-one-out analysis in sensitivity analyses. MR analysis after removing SNPs one by one. The red lines represent the effect sizes of the MR Egger and IVW analyses for all SNPs. Positive values indicate increased outcome risk/levels, whereas negative values indicate decreased risk/levels. Due to visual crowding from the large number of instrumental variables, individual SNP labels have been omitted from the y-axes of the left panels. To facilitate accurate cross-referencing, the top-to-bottom sequence of data points in these panels corresponds exactly to the vertical order of SNPs listed in Supplementary Table 1−3. Abbreviations: NAAG, N-acetylglucosylglutamate; AD, Alzheimer’s disease. |
All analyses were performed in R (version 4.3.3) using the TwoSampleMR package (version 0.5.10) and MR-PRESSO (version 1.0) for horizontal pleiotropy assessment and outlier correction. The analysis code will be available in the Supplementary Code.
Results
Toothache and Decreased NAAG Level
We conducted MR analysis using 14 SNPs, carefully screened to exclude outliers, palindromic, or confounding SNPs. The weighted median method (p = 0.029, beta = −6.798, 95% CI = −12.886 to −0.710) and IVW (p = 0.024, beta = −4.894, 95% CI = −9.139 to −0.649) revealed significant causal evidence supporting a potential causal link between toothache and reduced NAAG levels (Figures 3 and 4). These results are statistically significant, consistently indicating that toothache is associated with reduced NAAG levels. Detailed results are provided in Table 3. Additionally, Cochran’s Q test for heterogeneity (Q_pval = 0.7185 for MR Egger; Q_pval = 0.6509 for IVW) and the MR Egger test (p [between intercept and 0] = 0.2163) confirmed the absence of heterogeneity and horizontal pleiotropy.
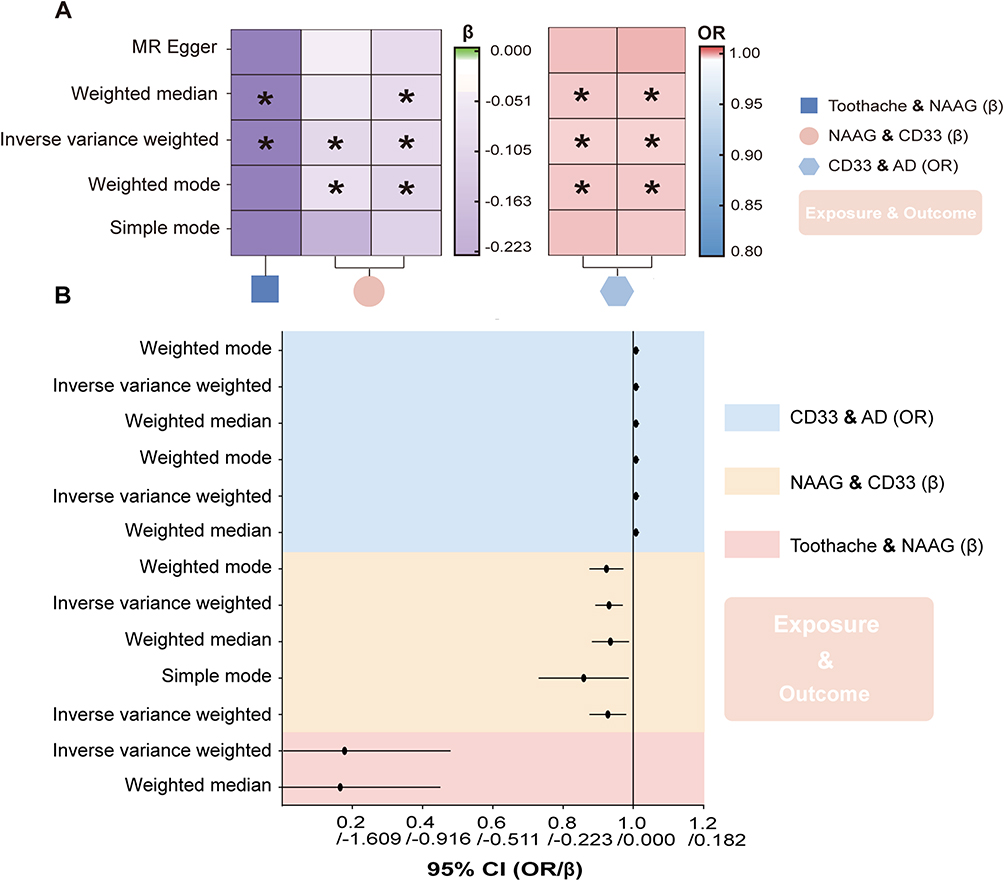 |
Figure 3 Causal effects of exposure on outcome. (A) Heatmap illustrating causal relationships between exposure and outcomes. The left panel displays Beta values for continuous traits using a green-to-purple color scale (centered at Beta = 0, with green indicating positive associations and purple indicating negative associations). The right panel shows Odds Ratios (ORs) for binary outcomes using a red-to-blue color scale (anchored at OR = 1, with red indicating increased risk and blue indicating protective effects). Asterisks (*) indicate nominal significance with a p-value < 0.05. (B) Forest plot showing 95% confidence intervals for significant Mendelian randomization analysis results. The corresponding OR and β values are provided, and the 95% CI reflects the variability of the estimated effect size. Abbreviations: NAAG, N-acetylglucosylglutamate; AD, Alzheimer’s Disease. |
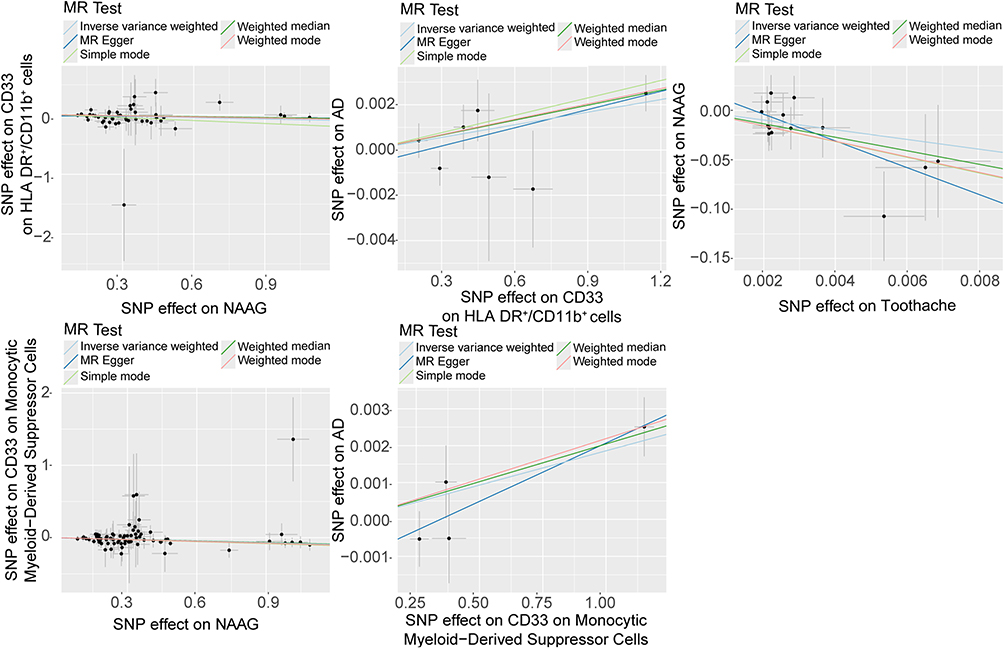 |
Figure 4 Scatter plots of causality. Perform linear regression on the results of the five different methods in MR analysis. The slope is equal to the beta. Abbreviations: NAAG, N-acetylglucosylglutamate; AD, Alzheimer’s disease. |
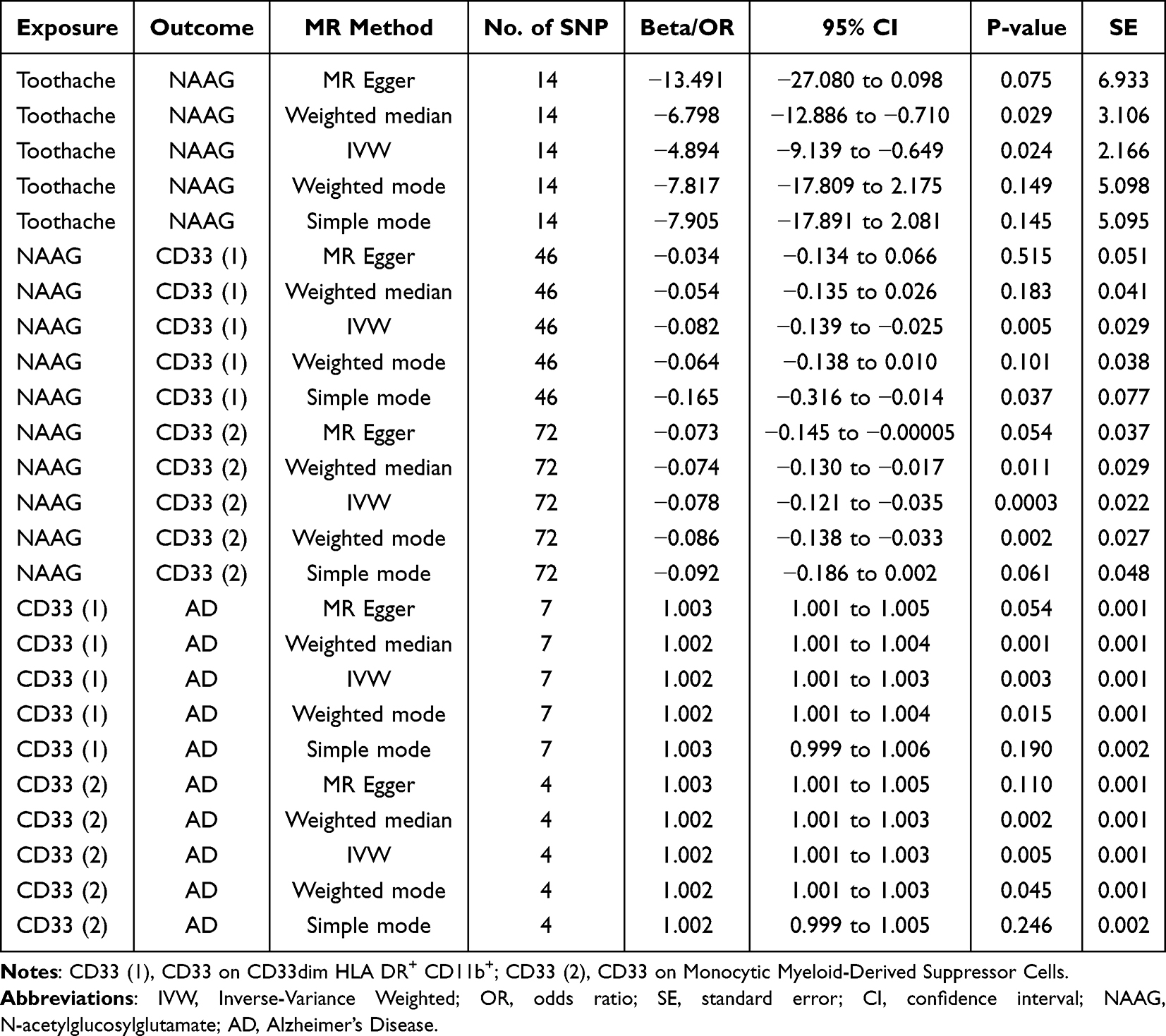 |
Table 3 MR Estimates for the Association Between Exposures and Outcome |
NAAG Reduction and CD33+ Myeloid Monocytes
For the CD33 (1) group, 46 SNPs were included in the analysis, with all outlier, palindromic, and confounding SNPs excluded. Results from the IVW method (p = 0.005, beta = −0.082, 95% CI = −0.139 to −0.025) and the Simple Mode method (p = 0.037, beta = −0.165, 95% CI = −0.316 to −0.014) indicate that reduced NAAG levels are associated with increased CD33 expression (Figures 3 and 4). Cochran’s Q test (Q_pval = 0.5045 for MR Egger; Q_pval = 0.4911 for IVW) and the MR Egger intercept test (p = 0.2576) further confirmed the absence of heterogeneity and horizontal pleiotropy.
In the CD33 (2) group, after excluding 3 SNPs associated with confounders, we included 72 SNPs for MR analysis. The weighted median method (p = 0.011, beta = −0.074, 95% CI = −0.130 to −0.017), IVW (p = 0.0003, beta = −0.078, 95% CI = −0.121 to −0.035), and weighted mode (p = 0.002, beta = −0.086, 95% CI = −0.138 to −0.033) all indicate a negative association between NAAG and CD33 expression, suggesting that reduced NAAG is associated with increased CD33 expression (Figures 3 and 4). Cochran’s Q test (Q_pval = 1.000 for MR Egger; Q_pval = 1.000 for IVW) and the MR Egger test (p [between intercept and 0] = 0.872) indicated no heterogeneity or horizontal pleiotropy.
CD33+ Myeloid Monocytes Serve as a Risk Factor for AD
In the CD33 (1) group, seven SNPs were retained after rigorous screening, excluding a confounding SNP. Analysis using the weighted median method (p = 0.001, OR = 1.002, 95% CI = 1.001–1.004), IVW (p = 0.003, OR = 1.002, 95% CI = 1.001–1.003), and weighted mode (p = 0.015, OR = 1.002, 95% CI = 1.001–1.004) provided genetic support for the associations between genetically predicted CD33 expression and AD (Figures 3 and 4). Cochran’s Q and MR-Egger intercept tests showed no evidence of heterogeneity or horizontal pleiotropy.
In the CD33 (2) group, four SNPs passed screening and were included in the MR analysis. Findings were consistent with the CD33 (1) group, indicating an association between increased CD33 expression and higher AD risk. While the effect sizes appear modest, the consistent directionality observed across both CD33 cohorts underscores the biological relevance of this neuroimmune pathway in AD pathogenesis (Figures 3 and 4). No heterogeneity or pleiotropy was detected.
Discussion
Toothache is common among the elderly, likely due to aging tooth structure, periodontal disease, and age-related immune decline. A recent prospective cohort study reported a statistically significant association between oral pain and an increased risk of AD, as well as adverse structural changes in the brain. Conversely, effective alleviation of oral pain was linked to a substantial reduction in AD risk.33 Research has shown that pain activates microglia and astrocytes, triggering the release of inflammatory cytokines (eg, IL-1, IL-6, TNF-alpha) and other harmful molecules, which progressively cause neuronal damage and contribute to the development of neurodegenerative diseases.34,35 This supports the hypothesis of a potential neuroimmune pathway linking toothache and AD risk. Currently, molecular-level research on the link between toothache and AD is still limited. This study employs MR analyses to bridge this gap by exploring a novel neuroimmune pathway: the toothache-NAAG-CD33-AD axis.
Toothache is a prevalent pain experience that involves complex neurotransmitter dynamics during its transmission from peripheral receptors to the cerebral cortex.36 NAAG has been implicated in nociceptive signaling and shown to exert analgesic effects through activation of mGluR3. Studies indicate that both NAAG and its enzyme inhibitors (eg, JZ-43 and 2-PMPA) exhibit notable analgesic properties within the spinal cord and other pain-regulatory systems, especially in cases of inflammatory and neuropathic pain.37 The strong association between NAAG and cognitive function led us to examine the relationship between toothache and NAAG levels using MR analysis.14,38 Our results indicated a significant reduction in NAAG levels among individuals with toothache (p = 0.024, IVW), consistent with animal study findings by Watanabe et al.10 Preclinical studies in AD-relevant animal models have reported NAAG-related alterations, suggesting a potential role for NAAG in neurodegeneration.39 A possible explanation is that reduced NAAG is associated with altered glutamatergic signaling, potentially increasing vulnerability to excitotoxicity.40,41
Elevated glutamate, as the central nervous system’s primary excitatory neurotransmitter, is associated with neurotoxicity and may activate neuroimmune signaling pathways relevant to AD pathology, potentially contributing to neuronal damage.17,42 Microglia, the brain’s resident immune cells, play a crucial role in managing neuroimmune responses.43,44 Evidence suggests that elevated glutamate levels may activate microglia, triggering an inflammatory cascade that could exacerbate neurodegenerative progression.26,45 In this activated state, microglia may release increased levels of inflammatory cytokines, and immune-modulating receptors such as CD33, which has been implicated in AD risk, may influence microglial immune responses.46
To further investigate the neuroimmune pathways linking NAAG to microglial function, we used CD33 expression GWAS data from two myeloid monocyte sources as a proxy for microglia-relevant CD33-related immune activity. The biological validity of this surrogate is supported by the conserved myeloid lineage and the observed phenotypic convergence, where infiltrating monocytes undergo microglial-like transcriptomic reprogramming in AD pathological environments.47–49 Methodologically, recent proteomic MR studies have suggested that peripheral genetic signals provide directionally consistent and, in certain contexts, robust predictive insights into central nervous system (CNS) pathology comparable to or even complementary to cerebrospinal fluid.29,50 By focusing on two specific myeloid monocyte subpopulations associated with immune regulation and phagocytosis, our approach seeks to mitigate transcriptomic distortions typically associated with post-mortem microglia isolation while enhancing biological relevance to the neuroinflammatory niche.
Following this rationale, we conducted MR analysis with NAAG as the exposure and found a significant association between reduced NAAG levels and increased CD33 expression in CD33-related traits (p = 0.005; p = 0.0003, IVW, respectively)—a relationship that may be influenced by elevated glutamate levels.51 Increased glutamate in neuroinflammatory or neurodegenerative conditions has been shown to strongly activate microglia, potentially promoting a shift toward a disease-associated microglia (DAM) phenotype.21,52 This DAM subtype, prevalent in AD and other neurodegenerative disorders, may be characterized by specific molecular markers, including CD33-related changes.53,54 Under such pathological conditions, heightened CD33 expression may impair microglial phagocytic function and modulate inflammatory signaling, thereby promoting a shift toward a pro-inflammatory profile and potentially exacerbating neuronal injury and AD pathology.24,55 This dysregulated microglial state could contribute to a self-reinforcing cycle of neuroinflammation and neurotoxicity.
CD33 is an immune-modulatory receptor implicated in the neuroinflammatory pathology of AD.56,57 Current evidence underscores CD33 as a pivotal neuroimmune checkpoint in the pathogenesis of AD. Genetic meta-analyses have consistently identified variants at the CD33 locus as robust risk factors, positioning this receptor alongside other key modulators of microglial function.58,59 Mechanistically, CD33 acts as a “brake” on the central immune system; its expression inhibits microglial phagocytosis and the subsequent clearance of Aβ plaques.60 The functional significance of this neuroimmune signaling pathway is further supported by in vivo studies in AD mouse models (eg, 5XFAD), where modulation of CD33 significantly alters microglial responses and plaque accumulation.24 Moreover, the development of therapeutic agents, such as the humanized antibody HuM195 designed to neutralize CD33-mediated inhibitory signaling, underscores the therapeutic potential of targeting this pathway for disease-modifying interventions.61 Consistent with this biological framework, our MR results align directionally with both experimental paradigms and recent genetic studies.
For instance, Gu et al reported that higher peripheral blood CD33 mRNA levels were associated with an increased risk of AD (p = 3.25 × 10−5; OR = 1.156, IVW), with a slightly attenuated association for CD33 expression in myeloid monocytes (p = 0.005; OR = 1.055, IVW).62 Similarly, Zhang et al reported a positive association between CD33 expression on peripheral myeloid monocytes and increased AD risk (OR = 1.02).63 The modest effect size observed in our primary analysis (OR = 1.002, p = 0.003 and p = 0.005, IVW, respectively) is likely a result of stringent methodological rigor and the inherent limitations of cell-specific MR. Notably, our study utilized a more contemporary and substantially larger AD outcome dataset, offering greater statistical power and precision compared to previous cohorts. Moreover, by applying a conservative genome-wide significance threshold (P < 5 × 10−8) for instrument selection—compared to the more lenient P < 1 × 10−5 used in earlier studies—we ensured robust genetic instruments, albeit resulting in more conservative effect estimates. Crucially, when applying our CD33 genetic instruments to previous analytical frameworks (same outcome database and P < 1 × 10−5), we obtained consistent results, confirming the stability and reliability of both our exposure data and MR pipeline. In this context, elevated CD33 expression may impair microglial phagocytosis, promoting the accumulation of neuronal debris and Aβ,64,65 thereby amplifying neuroinflammation and exacerbating AD pathology.19 While these results provide preliminary insights into a hypothesized neuroimmune axis, the modest effect size suggests that this pathway should be interpreted as a subtle regulatory signal rather than as a direct clinical target for disease prevention.
Our findings suggest that toothache is associated with a neuroimmune regulatory axis implicated in AD. Glutamate signaling emerges as a potential intermediary in the NAAG-CD33 signaling pathway, suggesting a plausible crosstalk between neuroimmune responses to toothache and AD pathology. MR analyses indicate that toothache liability is associated with lower NAAG levels, which are linked to elevated glutamate, potentially leading to neurotoxicity. Moreover, this reduction in NAAG may promote a shift toward the DAM phenotype, likely involving CD33-related changes, which impair microglial phagocytic activity. High CD33 expression impairs microglial clearance of neurotoxic molecules, such as Aβ, contributing to a self-sustaining cycle of neurotoxicity and inflammation. Notably, this neuroimmune pathway and the resulting neuroinflammation seem to be more closely associated with sporadic AD, which is primarily influenced by a combination of environmental and genetic risk factors. In contrast, familial AD is predominantly driven by specific high-penetrance genetic mutations. These findings are consistent with three MR analyses and are supported by previous studies linking neuroimmune dysregulation to AD risk, positioning the NAAG-CD33 pathway as a candidate mediating axis for the effect of toothache on AD pathogenesis by disrupting CNS homeostasis.
While these findings provide new insights, several limitations must be considered. First, the GWAS data used in this study were exclusively from European populations, emphasizing the need for validation in more diverse ethnic groups to confirm broader applicability. Second, the toothache phenotype in this study was based on self-reported data, which is inherently subjective and may introduce measurement error or recall bias. Although we implemented a stringent methodological pipeline, we cannot completely exclude the influence of unmeasured socioeconomic or behavioral confounders that could independently affect both oral health and cognitive decline. Third, since NAAG levels and CD33 expression were measured in peripheral blood rather than the CNS, these proxies may attenuate the biological signal, as reflected in the modest effect sizes observed. Finally, our study proposes a hypothesized molecular framework, and future research should focus on larger, multi-ancestry cohorts and CNS-specific mechanistic models to further validate the role of the NAAG-CD33 axis in the complex oral-brain connection.
In conclusion, this study provides preliminary genetic evidence suggesting that toothache liability is associated with Alzheimer’s disease risk through the potential NAAG-glutamate-CD33 neuroimmune axis. These findings highlight the NAAG-glutamate-CD33 pathway as a candidate therapeutic target for further investigation, though further mechanistic studies and longitudinal clinical cohorts are needed to validate these results and fully elucidate its role in AD pathology.
Abbreviations
AD, Alzheimer's disease; VPM, ventral posteromedial; NAAG, N-acetylaspartylglutamate; BBB, blood–brain barrier; CGRP, calcitonin gene-related peptide; mGluR3, metabotropic glutamate receptor 3; MR, Mendelian Randomization; GWAS, Genome-wide association study; CLSA, Canadian Longitudinal Study of Aging; MFIs, mean fluorescence intensities; IGAP, International Genomics of Alzheimer's Project; LD, linkage disequilibrium; IVW, inverse variance weighted; CNS, central nervous system; Aβ, amyloid-β; DAM, disease-associated microglia; SNPs, Single Nucleotide Polymorphisms.
Data Sharing Statement
1. Data for toothache were obtained from the Open GWAS repository (identifier: ukb-b-19191, URL: https://gwas.mrcieu.ac.uk/datasets/ukb-b-19191/).
2. Data for NAAG were obtained from the GWAS Catalog (identifier: GCST90200684, URL: http://ftp.ebi.ac.uk/pub/databases/gwas/summary_statistics/GCST90200001-GCST90201000/GCST90200684/).
3. Data for CD33 on CD33dim HLA DR+ CD11b+ cells were obtained from the Open GWAS repository (identifier: ebi-a-GCST90001948, URL: https://gwas.mrcieu.ac.uk/datasets/ebi-a-GCST90001948/).
4. Data for CD33 on monocytic myeloid-derived suppressor cells were obtained from the Open GWAS repository (identifier: ebi-a-GCST90001952, URL: https://gwas.mrcieu.ac.uk/datasets/ebi-a-GCST90001952/).
5. Data for Alzheimer’s disease were obtained from the Open GWAS repository (identifier: ebi-a-GCST90012878, URL: https://gwas.mrcieu.ac.uk/datasets/ebi-a-GCST90012878/).
Ethics Approval and Consent to Participate
This study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of the Affiliated Hospital of Stomatology, Anhui Medical University (Approval No. 20260021-A). As the study exclusively utilized publicly available, de-identified summary statistics from genome-wide association studies, additional informed consent from participants was not required by the committee.
Acknowledgments
We extend our heartfelt gratitude to the participants and contributors of the IEU OpenGWAS project. Additionally, we express our appreciation to the authors of the two GWAS studies on metabolites and immune cells, whose data were instrumental in our research. We also thank the researchers and support staff involved in the creation and maintenance of these datasets, who have worked tirelessly to ensure data integrity and accessibility for the broader scientific community. We would also like to thank Wenhao Zhang and Yulong Zhang for their valuable assistance with data analysis and helpful suggestions during the revision of the manuscript.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This work was supported by the Outstanding Young Teacher Development Program for Higher Education Institutions of Anhui Province (No. YQYB2024013), the Natural Science Foundation of Anhui Province (No. 2408085MH221), the School Foundation of Anhui Medical University (No. 2023xkj041), and the Anhui Medical University Student Innovation and Entrepreneurship Training Program (No. S202410366087 and S202410366086).
Disclosure
The authors declare no conflicts of interest.
References
1. Aminoshariae A, Kulild JC. Current concepts of dentinal hypersensitivity. J Endod. 2021;47(11):1696–13. doi:10.1016/j.joen.2021.07.011
2. Bender IB. Pulpal pain diagnosis--a review. J Endod. 2000;26(3):175–179. doi:10.1097/00004770-200003000-00012
3. Zuarez-Easton S, Erez O, Zafran N, Carmeli J, Garmi G, Salim R. Pharmacologic and nonpharmacologic options for pain relief during labor: an expert review. Am J Obstet Gynecol. 2023;228(5):S1246–s1259. doi:10.1016/j.ajog.2023.03.003
4. Fu Y, Gong C, Zhu C, Zhong W, Guo J, Chen B. Research trends and hotspots of neuropathic pain in neurodegenerative diseases: a bibliometric analysis. Front Immunol. 2023;14:1182411. doi:10.3389/fimmu.2023.1182411
5. Thu Ya M, Hasegawa Y, Sta Maria MT, et al. Predicting cognitive function changes from oral health status: a longitudinal cohort study. Sci Rep. 2024;14(1):24153. doi:10.1038/s41598-024-75169-8
6. Wang Q, Zhen W, Hu R, et al. Occlusion dysfunction and Alzheimer’s disease: mendelian randomization study. Front Aging Neurosci. 2024;16: 1423322.
7. Lane CA, Hardy J, Schott JM. Alzheimer’s disease. Eur J Neurol. 2018;25(1):59–70. doi:10.1111/ene.13439
8. Bista P, Imlach WL. Pathological mechanisms and therapeutic targets for trigeminal neuropathic pain. Medicines. 2019;6(3). doi:10.3390/medicines6030091
9. Neale JH, Bzdega T, Wroblewska B. N-Acetylaspartylglutamate: the most abundant peptide neurotransmitter in the mammalian central nervous system. J Neurochem. 2000;75(2):443–452. doi:10.1046/j.1471-4159.2000.0750443.x
10. Watanabe M, Sugiura Y, Sugiyama E, et al. Extracellular N-acetylaspartylglutamate released in the nucleus accumbens modulates the pain sensation: analysis using a microdialysis/mass spectrometry integrated system. Mol Pain. 2018;14:1744806918754934. doi:10.1177/1744806918754934
11. Skaper SD, Facci L, Zusso M, Giusti P. Neuroinflammation, mast cells, and glia: dangerous liaisons. Neuroscientist. 2017;23(5):478–498. doi:10.1177/1073858416687249
12. András IE, Toborek M. Extracellular vesicles of the blood-brain barrier. Tissue Barriers. 2016;4(1):e1131804. doi:10.1080/21688370.2015.1131804
13. Vazana U, Veksler R, Pell GS, et al. Glutamate-mediated blood-brain barrier opening: implications for neuroprotection and drug delivery. J Neurosci. 2016;36(29):7727–7739. doi:10.1523/JNEUROSCI.0587-16.2016
14. Neale JH, Olszewski R. A role for N-acetylaspartylglutamate (NAAG) and mGluR3 in cognition. Neurobiol Learn Mem. 2019;158:9–13. doi:10.1016/j.nlm.2019.01.006
15. Petroff OA. GABA and glutamate in the human brain. Neuroscientist. 2002;8(6):562–573. doi:10.1177/1073858402238515
16. Cox MF, Hascup ER, Bartke A, Hascup KN. Friend or Foe? defining the role of glutamate in aging and Alzheimer’s disease. Front Aging. 2022;3:929474. doi:10.3389/fragi.2022.929474
17. Vongthip W, Nilkhet S, Boonruang K, Sukprasansap M, Tencomnao T, Baek SJ. Neuroprotective mechanisms of luteolin in glutamate-induced oxidative stress and autophagy-mediated neuronal cell death. Sci Rep. 2024;14(1):7707. doi:10.1038/s41598-024-57824-2
18. Prinz M, Masuda T, Wheeler MA, Quintana FJ. Microglia and central nervous system-associated macrophages-from origin to disease modulation. Annu Rev Immunol. 2021;39(1):251–277. doi:10.1146/annurev-immunol-093019-110159
19. Wang Z, Liu X, Zhen W, et al. ANKRD1 sustains a neurogenic BMSC niche and counters cognitive aging. Int J Oral Sci. 2026;18(1). doi:10.1038/s41368-026-00428-5.
20. Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol. 2018;18(4):225–242. doi:10.1038/nri.2017.125
21. Czapski GA, Strosznajder JB. Glutamate and GABA in microglia-neuron cross-talk in Alzheimer’s disease. Int J Mol Sci. 2021;22(21):11677. doi:10.3390/ijms222111677
22. Neves D, Salazar IL, Almeida RD, Silva RM. Molecular mechanisms of ischemia and glutamate excitotoxicity. Life Sci. 2023;328:121814. doi:10.1016/j.lfs.2023.121814
23. Wißfeld J, Mathews M, Mossad O, et al. Reporter cell assay for human CD33 validated by specific antibodies and human iPSC-derived microglia. Sci Rep. 2021;11(1):13462. doi:10.1038/s41598-021-92434-2
24. Eskandari-Sedighi G, Crichton M, Zia S, et al. Alzheimer’s disease associated isoforms of human CD33 distinctively modulate microglial cell responses in 5XFAD mice. Mol Neurodegener. 2024;19(1):42. doi:10.1186/s13024-024-00734-8
25. Butler CA, Thornton P, Brown GC. CD33M inhibits microglial phagocytosis, migration and proliferation, but the Alzheimer’s disease-protective variant CD33m stimulates phagocytosis and proliferation, and inhibits adhesion. J Neurochem. 2021;158(2):297–310. doi:10.1111/jnc.15349
26. Zhang X, Wang D, Zhang B, Zhu J, Zhou Z, Cui L. Regulation of microglia by glutamate and its signal pathway in neurodegenerative diseases. Drug Discov Today. 2020;25(6):1074–1085. doi:10.1016/j.drudis.2020.04.001
27. Chen Y, Lu T, Pettersson-Kymmer U, et al. Genomic atlas of the plasma metabolome prioritizes metabolites implicated in human diseases. Nat Genet. 2023;55(1):44–53. doi:10.1038/s41588-022-01270-1
28. Orrù V, Steri M, Sidore C, et al. Complex genetic signatures in immune cells underlie autoimmunity and inform therapy. Nat Genet. 2020;52(10):1036–1045. doi:10.1038/s41588-020-0684-4
29. Yuan Y, Wang S, Lian P, et al. From plasma proteomics Mendelian randomization to neuropathological validation: the potential role of CTSH-GRN-TMEM106B and TREM2-IL-34 networks of microglia in Alzheimer’s disease. Eur J Pharmacol. 2025;1007:178297. doi:10.1016/j.ejphar.2025.178297
30. Raj T, Ryan KJ, Replogle JM, et al. CD33: increased inclusion of exon 2 implicates the Ig V-set domain in Alzheimer’s disease susceptibility. Hum Mol Genet. 2014;23(10):2729–2736. doi:10.1093/hmg/ddt666
31. Bradshaw EM, Chibnik LB, Keenan BT, et al. CD33 Alzheimer’s disease locus: altered monocyte function and amyloid biology. Nat Neurosci. 2013;16(7):848–850. doi:10.1038/nn.3435
32. Emdin CA, Khera AV, Kathiresan S. Mendelian Randomization. JAMA. 2017;318(19):1925–1926. doi:10.1001/jama.2017.17219
33. Li W, Hu X, Zhang Y, et al. Oral pain and Alzheimer’s disease: prospective cohort and cross-sectional analyses. J Dent Res. 2026;220345251412305. 10.1177/00220345251412305
34. Loggia ML, Chonde DB, Akeju O, et al. Evidence for brain glial activation in chronic pain patients. Brain. 2015;138(Pt 3):604–615.
35. Adamu A, Li S, Gao F, Xue G. The role of neuroinflammation in neurodegenerative diseases: current understanding and future therapeutic targets. Front Aging Neurosci. 2024;16:1347987. doi:10.3389/fnagi.2024.1347987
36. Yu F, Li M, Wang Q, et al. Spatiotemporal dynamics of brain function during the natural course in a dental pulp injury model. Eur J Nucl Med Mol Imaging. 2022;49(8):2716–2722.
37. Yamamoto T, Hirasawa S, Wroblewska B, et al. Antinociceptive effects of N-acetylaspartylglutamate (NAAG) peptidase inhibitors ZJ-11, ZJ-17 and ZJ-43 in the rat formalin test and in the rat neuropathic pain model. Eur J Neurosci. 2004;20(2):483–494. doi:10.1111/j.1460-9568.2004.03504.x
38. Zink CF, Barker PB, Sawa A, et al. Association of missense mutation in FOLH1 with decreased NAAG Levels and impaired working memory circuitry and cognition. Am J Psychiatry. 2020;177(12):1129–1139. doi:10.1176/appi.ajp.2020.19111152
39. Morland C, Nordengen K. N-Acetyl-Aspartyl-Glutamate in brain health and disease. Int J Mol Sci. 2022;23(3): 1268.
40. Jessen F, Fingerhut N, Sprinkart AM, et al. N-acetylaspartylglutamate (NAAG) and N-acetylaspartate (NAA) in patients with schizophrenia. Schizophr Bull. 2013;39(1):197–205. doi:10.1093/schbul/sbr127
41. Thomas AG, Olkowski JL, Slusher BS. Neuroprotection afforded by NAAG and NAALADase inhibition requires glial cells and metabotropic glutamate receptor activation. Eur J Pharmacol. 2001;426(1–2):35–38. doi:10.1016/S0014-2999(01)01198-0
42. Targa Dias Anastacio H, Matosin N, Ooi L. Neuronal hyperexcitability in Alzheimer’s disease: what are the drivers behind this aberrant phenotype? Transl Psychiatry. 2022;12(1):257.
43. Kettenmann H, Hanisch UK, Noda M, Verkhratsky A. Physiology of microglia. Physiol Rev. 2011;91(2):461–553.
44. Guo ML, Roodsari SK, Cheng Y, Dempsey RE, Hu W. Microglia NLRP3 inflammasome and neuroimmune signaling in substance use disorders. Biomolecules. 2023;13(6):922. doi:10.3390/biom13060922
45. Davalos D, Grutzendler J, Yang G, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8(6):752–758. doi:10.1038/nn1472
46. Malik M, Simpson JF, Parikh I, et al. CD33 Alzheimer’s risk-altering polymorphism, CD33 expression, and exon 2 splicing. J Neurosci. 2013;33(33):13320–13325. doi:10.1523/JNEUROSCI.1224-13.2013
47. Mrdjen D, Pavlovic A, Hartmann FJ, et al. High-dimensional single-cell mapping of central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease. Immunity. 2018;48(2):380–395.e386. doi:10.1016/j.immuni.2018.01.011
48. Drost N, Houtman J, Cseresnyés Z, et al. The amyloid-beta rich CNS environment alters myeloid cell functionality independent of their origin. Sci Rep. 2020;10(1):7152. doi:10.1038/s41598-020-63989-3
49. Shukla AK, McIntyre LL, Marsh SE, et al. CD11a expression distinguishes infiltrating myeloid cells from plaque-associated microglia in Alzheimer’s disease. Glia. 2019;67(5):844–856. doi:10.1002/glia.23575
50. Zhang X, Dong Y, Zou Z, et al. Association of human plasma and cerebrospinal fluid metabolomes with vascular dementia and its subtypes: a Mendelian randomization study. Brain Res. 2026;1871:150060. doi:10.1016/j.brainres.2025.150060
51. Olszewski RT, Janczura KJ, Bzdega T, et al. NAAG peptidase inhibitors act via mGluR3: animal models of memory, Alzheimer’s, and ethanol intoxication. Neurochem Res. 2017;42(9):2646–2657. doi:10.1007/s11064-017-2181-4
52. Balbi M, Bonanno G, Bonifacino T, Milanese M. The physio-pathological role of Group I metabotropic glutamate receptors expressed by microglia in health and disease with a focus on amyotrophic lateral sclerosis. Int J Mol Sci. 2023;24(6). doi:10.3390/ijms24065240
53. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–1318. doi:10.1126/science.1110647
54. Zhang L, Wang Y, Liu T, Mao Y, Peng B. Novel microglia-based therapeutic approaches to neurodegenerative disorders. Neurosci Bull. 2023;39(3):491–502. doi:10.1007/s12264-022-01013-6
55. Griciuc A, Serrano-Pozo A, Parrado AR, et al. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron. 2013;78(4):631–643. doi:10.1016/j.neuron.2013.04.014
56. Biber K, Bhattacharya A, Campbell BM, et al. Microglial drug targets in AD: opportunities and challenges in drug discovery and development. Front Pharmacol. 2019;10:840. doi:10.3389/fphar.2019.00840
57. Hampel H, Caraci F, Cuello AC, et al. A path toward precision medicine for neuroinflammatory mechanisms in Alzheimer’s disease. Front Immunol. 2020;11:456. doi:10.3389/fimmu.2020.00456
58. Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43(5):429–435. doi:10.1038/ng.803
59. Carrasquillo MM, Belbin O, Hunter TA, et al. Replication of EPHA1 and CD33 associations with late-onset Alzheimer’s disease: a multi-centre case-control study. Mol Neurodegener. 2011;6(1):54. doi:10.1186/1750-1326-6-54
60. Eskandari-Sedighi G, Jung J, Macauley MS. CD33 isoforms in microglia and Alzheimer’s disease: friend and foe. Mol Aspects Med. 2023;90:101111. doi:10.1016/j.mam.2022.101111
61. Wong E, Malviya M, Jain T, et al. HuM195 and its single-chain variable fragment increase Aβ phagocytosis in microglia via elimination of CD33 inhibitory signaling. Mol Psychiatry. 2024;29(7):2084–2094. doi:10.1038/s41380-024-02474-z
62. Gu X, Dou M, Cao B, Jiang Z, Chen Y. Peripheral level of CD33 and Alzheimer’s disease: a bidirectional two-sample Mendelian randomization study. Transl Psychiatry. 2022;12(1):427. doi:10.1038/s41398-022-02205-4
63. Zhang H, Cao F, Zhou Y, Wu B, Li C. Peripheral immune cells contribute to the pathogenesis of Alzheimer’s disease. Mol Neurobiol. 2025;62(1):264–270. doi:10.1007/s12035-024-04266-6
64. Griciuc A, Federico AN, Natasan J, et al. Gene therapy for Alzheimer’s disease targeting CD33 reduces amyloid beta accumulation and neuroinflammation. Hum Mol Genet. 2020;29(17):2920–2935. doi:10.1093/hmg/ddaa179
65. Puigdellívol M, Allendorf DH, Brown GC. Sialylation and Galectin-3 in microglia-mediated neuroinflammation and neurodegeneration. Front Cell Neurosci. 2020;14:162. doi:10.3389/fncel.2020.00162
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.